")
Back to Archived Journals » Research and Reports in Biology » Volume 6
Advances in the understanding of the BBSome complex structure and function
Authors Hernandez-Hernandez V, Jenkins D
Received 19 June 2015
Accepted for publication 27 August 2015
Published 15 October 2015 Volume 2015:6 Pages 191—201
DOI https://doi.org/10.2147/RRB.S65700
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Zvi Kelman
Victor Hernandez-Hernandez, Dagan Jenkins
Genetics and Genomic Medicine Programme, UCL Institute of Child Health, London, UK
Abstract: Bardet–Biedl syndrome (BBS) is an autosomal recessive condition characterized by important clinical features, including obesity, blindness, renal cystic disease, and intellectual disability. BBS is caused by mutations in >20 genes, a subset of which form the so-called BBSome. The BBSome is a complex that coats intracellular vesicles and interacts with key proteins, such as small GTPases, that regulate the trafficking of these vesicles to the base of cilia. Cilia are microtubular protusions present on the surface of most cells that are defective in a key group of disorders known as ciliopathies, of which BBS is one. BBSome components particularly localize to the basal body of cilia, and also centrosomes, where they interact with pericentriolar material proteins that regulate their function. The BBSome also facilitates the transport of key cargo within cilia by acting as an adaptor protein for intraflagellar transport complexes, and as such BBS mutations lead to a variety of functional defects in cilia in a tissue- and cell-type-specific manner. This might include defects in photoreceptor trafficking linked to the connecting cilium, abnormal hedgehog signaling within bone, and aberrant calcium signaling in response to fluid flow along renal tubules, although the precise mechanisms are still not completely understood. Taken together, the BBSome is an important complex that may be targeted for treatment of a variety of common and important disorders, and understanding the precise function of the BBSome will be essential to capitalize on this translationally.
Keywords: retinitis pigmentosa GTPase regulator, polycystin, disrupted in schizophrenia 1, Hedgehog signaling, calcium signaling, photoreceptors
Introduction
The BBSome complex in human genetic disease
Bardet–Biedl syndrome (BBS) is a genetic disorder with an autosomal recessive mode of inheritance. Major features of this condition are truncal obesity, polydactyly, rod-cone dystrophy hypogenitalism in males, learning difficulties, and renal anomalies. Patients are diagnosed as having BBS if they exhibit four of the major criteria or three major and two minor criteria; brachydactyly/syndactyly, abnormal speech, polydypsia/polyuria, ataxia, diabetes mellitus, developmental delay, congenital heart disease, abnormal dentition/palatal defects, and hepatic fibrosis all constitute minor features.1,2 So far, 19 different genes have been found to be mutated in BBS, with different contributions, from >20%, ie, BBS1 or <1% for the most unusual ones (Table 1).
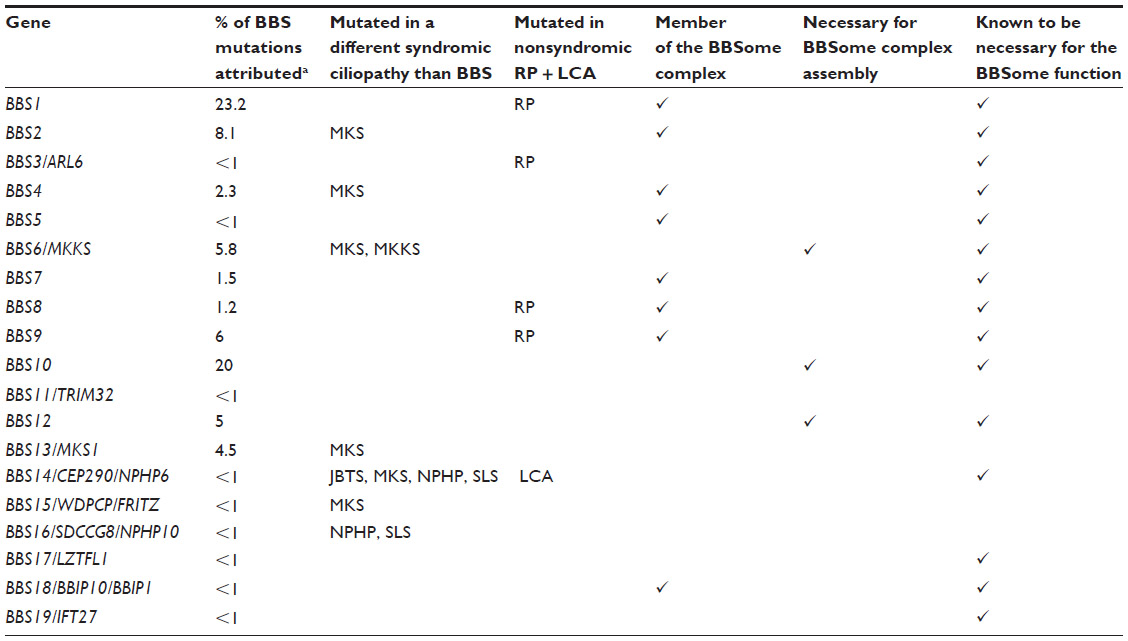 | Table 1 Genes mutated in BBS, contribution to other syndromes and BBSome links |
BBS was one of the first diseases demonstrated to be a ciliopathy. Cilia are microtubular protrusions present on the surface of most cells. Mutations that disrupt the function of cilia have been identified in many human diseases, and abnormal ciliogenesis is associated with a characteristic set of human phenotypes, the “ciliopathies”.1 These phenotypes include a variety of tissue patterning defects that are attributable to roles of nonmotile cilia in regulating a number of signal transduction pathways,2–4 as well as defects caused by the abnormal formation and/or degeneration of specialized cilia such as photoreceptors (eg, retinitis pigmentosa). Approximately 100 causative genes have been identified in ciliopathies. Although the prevalence of individual ciliopathies varies considerably, collectively, the ciliopathies are much more common and certain disorders, such as polycystic kidney disease, affect up to 1 in 1,000 people. In some cases, genes found to be mutated in BBS also contribute to other syndromic cilipathies and nonsyndromic retinal degeneration (Table 1).
BBS proteins localize to centrosomes and basal bodies in mammalian cells and tissues, including sensory neurons and photoreceptors. Many of the encoded proteins are uncharacterized biochemically and are defined by the structural domains that they contain, such as tetratricopeptide repeats and tripartite motifs. A subset of these proteins forms the protein complex known as the BBSome.3 The stage for discovery of the BBSome was set by elucidation of the molecular genetic basis for BBS. In this review, we will see how some of the proteins encoded by the different BBS genes are part of this BBSome complex and some others contribute directly or indirectly to its function (summarized in Table 1).
BBSome structure and assembly
The first work proposing the existence of the BBSome demonstrated the presence of a stable heptameric complex consisting of BBS1, BBS2, BBS4, BBS5, BBS7, BBS8, and BBS9. Using BBS4-LAP technology in mammalian cells, the authors found that the seven BBS proteins pull-down together and bind with pericentriolar material 1 (PCM1) and Rabin8, a guanosine exchange factor for RAB8A.3,4 An additional member of the complex, BBIP10/BBS18/BBIP1, was subsequently identified making the BBSome an octameric complex.5 These early studies showed that the BBSome was implicated in vesicular protein transport to the cilium. Further studies in Chlamydomonas flagella show a similar complex, including BBS1, BBS4, BBS5, BBS7, and BBS8, orthologous to the previously found mammalian BBSome.6 Evolutionary analysis of proteins linked to ciliopathies supported these results showing that the seven BBS proteins are conserved in a modular fashion.7
However, not all the genes found to be mutated in patients with BBS are part of the BBSome complex. The complete picture on how the BBSome and these other BBS proteins interact is still incomplete, but we know that some of them directly regulate cellular localization of the BBSome.
BBS3/ARL6 is a small ARF-like GTPase that localizes to the basal body and ciliary membrane. ARL6 interacts with BBS1 in its active GTP-bound form (Figure 1) through its N-terminal domain, and this interaction is necessary for the transport of the BBSome to the cilium; in human retinal pigmented epithelial cells, knockdown of ARL6 prevents localization of BBS1 to the cilium, which can be rescued following transfection of ARL6GTP, but not ARL6GDP.6 The BBSome complex is pulled-down in its entirety in ARL6-deficient cells and so it has been proposed that its function is to regulate trafficking of the BBSome. In its GTP-bound form, ARL6 binds the BBSome and forms a BBSome/ARL6 coat on the surface of membrane lipids of cellular vesicles. This BBSome-ARL6 coat has been shown to sort membrane proteins into cilia by recognizing ciliary targeting sequences by the BBSome.8–11 The BBSome associates with the cellular membranes, thanks to the pleckstrin domains of BBS5, that bind phosphoinositides.3 How this membrane–BBSome interaction works is not yet clear, but it has been reported that in the retina, BBS5 can be phosphorylated by light stimulation of protein kinase C, releasing Arrestin1 into the outer segment membranes.12 It is possible that a similar mechanism could be in place for primary cilia to regulate ciliary BBSome function.
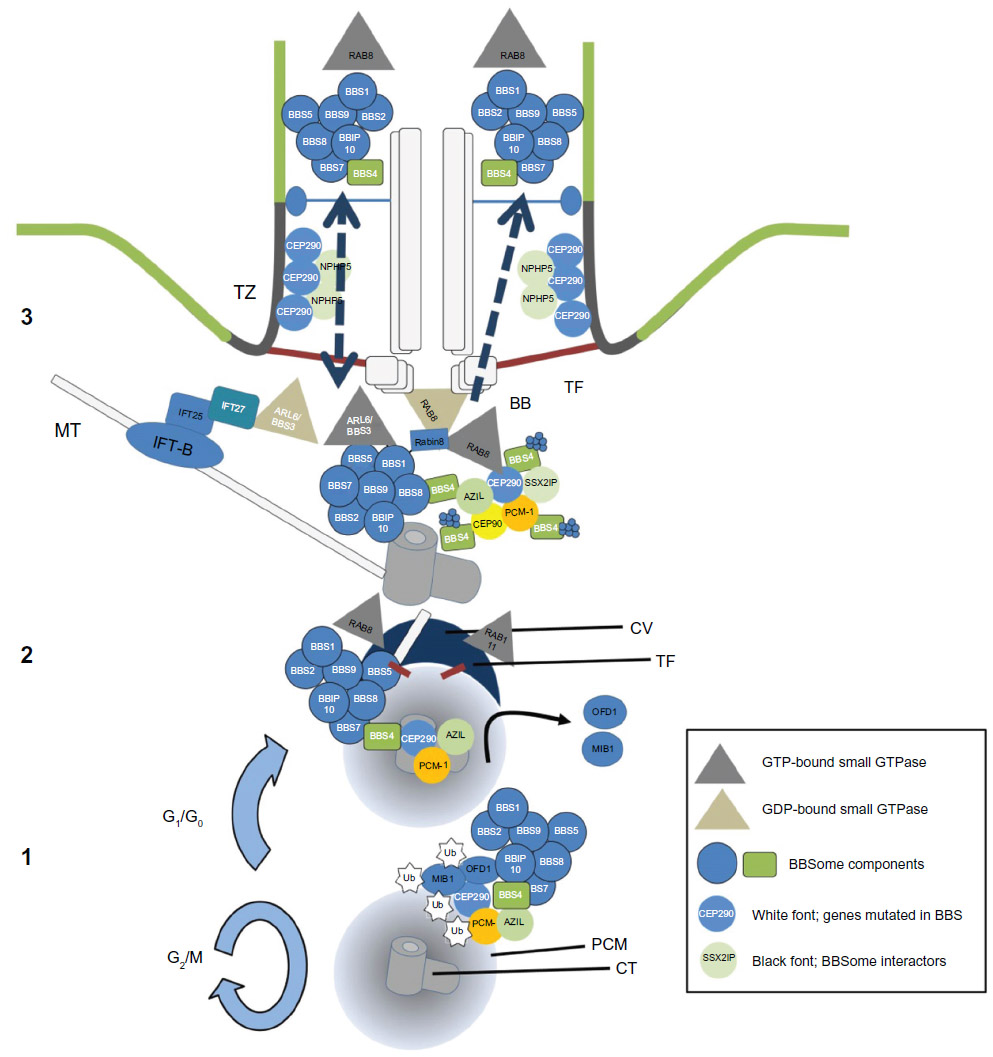 | Figure 1 The BBSome. |
The BBSome is also regulated by the interaction of ARL6 with another protein mutated in patients with BBS, IFT27/Rab4l/BBS19. IFT27 is a Rab-like GTPase that associates with IFT25 to interact with the intraflagellar transport complex-B. IFT27 can bind nucleotide-free ARL6 (ARL6GDP), and cells lacking IFT27 hyper-accumulate ARL6 and the BBSome in cilia. This accumulation is due to the requirement for IFT27 to export the BBSome from the cilium, although its transport into the cilium is not dependent on IFT27 function. This mechanism is not specific for the BBSome as other ciliary proteins, such as GPR161 and leucine-zipper transcription factor-like 1 (LZTFL1), also accumulate in cilia in IFT27-deprived cells.10,13 The LZTFL1, encoded by BBS17, is an occasional cause of disease in a subset of patients with BBS.14,15 LZTFL1 interacts with the BBSome through its C-terminal half that binds BBS9. Its expression is cytosolic, and it is not enriched in the centriolar region or the cilium. Moreover, when LZTFL1 expression is inhibited, cilia length or cilia formation are not disrupted. Only when LZTFL1 is deleted is its ciliary function observed, increasing translocation of BBSome proteins to the cilium. Even in cells depleted for BBS3 and BBS5, LZTFL1 downregulation is able to restore BBSome localization to the cilium.16
The chaperonin-like proteins, BBS6, BBS10, and BBS12, another group of BBS proteins, are only required for the assembly of the BBSome. They mediate the association of the BBSome proteins with a CCT/TRiC chaperonin complex that folds all the components in the right place.17 BBS7 is stabilized by the CCT/TRiC complex in partnership with BBS6, BBS10, and BBS12. BBS7 is then coupled with BBS2 and BBS9 to form the BBSome core complex. BBS1, BBS8, BBS5, and finally BBS4 are added individually to complete the complex.18 It is still unclear how BBIP10 is incorporated into the complex. No other function has been found for these BBS chaperones indicating that disruption of BBSome assembly and its mobilization to the basal body/centriolar satellites (CSs) causes similar phenotypes.
Functions of the BBS proteins
The BBSome and the CSs
The recruitment of the BBSome to the base of the cilium depends first on its localization to the centrioles. This localization depends on the interaction of certain BBSome components, especially BBS4, with proteins expressed in CSs. The CSs are dense granules clustered around the centrosomes and centrioles in the PCM.
CEP290 is a gene found to be mutated in several ciliopathies from Joubert and Senior–Løken syndromes to Meckel–Gruber syndrome, Leber congenital amaurosis, or BBS. It is still unclear why mutations in CEP290 can give such a diversity of phenotypes. Structurally, CEP290 is part of the CSs, where it interacts with CEP72, and it has been proposed that depletion of these proteins disrupts the localization of BBS4 to the cilium before the final step of BBSome formation. As such, mutations in CEP290 interfere with the formation of the BBSome and its recruitment to the cilium.19 When a hypomorphic mutation in murine CEP290 (Cep290rd16) is crossed onto the Bbs6 (Mkks−/−) mutant background, the Bbs6 mutant phenotype is partially rescued when compared with single mutants. On the other hand, Cep290rd16 and Bbs4−/− double mutants show aggravated phenotypes, including additive effects on obesity and photoreceptor degeneration.20,21 It is possible that these apparently discrepant results could be explained by the particular CEP290 protein domains that interact differentially with BBS4 or BBS6.
PCM1 and disrupted in Schizophrenia 1 (DISC1) are centriolar proteins, and like CEP290, they regulate BBSome function through their interactions with BBS1, BBS4, and BBS8. PCM1 and DISC1 are core constituents of the CSs and have been shown to cause genetic susceptibility to schizophrenia and other psychiatric diseases.22 PCM1, DISC1, and BBS4 form a complex that mutually regulates their localization to the CSs. Disruption of either BBS4 or DISC1 expression leads to a decreased enrichment of PCM1 to the CSs.23,24
AZI1 (also known as CEP131) is another CS that interacts with the BBSome through BBS4. AZI1 co-localizes with BBS4 to the CSs, and when it is depleted by siRNA-mediated knockdown, the ciliary localization of BBSome proteins like BBS4, BBS8, and BBS9 is increased, and the proportion of ciliated cells is also increased.25 Accordingly, AZI1 overexpression reduces BBS8 and BBS9 inside the cilium.26
The CS protein SSX2IP is another common interactor between the BBSome and CEP290.27 Loss of SSX2IP leads to shortened cilia and is needed to target BBS2, BBS4, BBS8, CEP290, SSTR3, and RAB8A to the primary cilium. CEP90 is another CS that is required for BBS4 localization to the CSs.28 SSX2IP, CEP90, and PCM1 associate with each other, and their localization to the CSs is interdependent; all three proteins are necessary for cilia formation.27–29
Nephrocystin-5 (NPHP5) protein localizes to the distal ends of the centrioles (Figure 1). When mutated, it causes Leber congenital amaurosis and Senior–Løken syndromes, but NPHP5 mutations have not been reported in BBS. We know that NPHP5 interacts with CEP290, and mutations in NPHP5 impair their interaction.30 When one of the two is depleted in cells, both disrupt the transport of BBSome components to the cilium. Interestingly, NPHP5 depletion only affects the ciliary localization of BBS2 and BBS5, but not BBS1, BBS7, BBS8, BBS9, BBIP10, or even BBS4, a BBSome component that is known to interact with CEP290.
In contrast, when CEP290 (or CEP72) are depleted, the BBS4 and BBS8 fail to relocalize from the CSs to the cilium.19 Therefore, NPHP5 and CEP290 interact together in the transition zone and both are able to bind and alter localization of BBSome proteins, but their interaction with the BBSome seems to have different effects on BBSome stability.31
PCM1, CEP290, and AZI1 CSs seem to be the link between the BBSome, ciliogenesis, and the cell cycle. Depletion of Cep290 results in the accumulation of PCM1 at the centrosomes.19,32 When ciliogenesis is naturally or stress induced, PCM1 preferentially binds AZl1, releasing CEP290 from the CSs, accumulating in the centrosome/basal body. When CEP290 is at its optimal concentration within the basal body, the AZl1–PCM1 complex concentrates RAB8A near the centrosomes and allows CEP290 to recruit RAB8A into the cilium.33 In that context, lack of expression of BBS4 reduces PCM1 localization in the CSs, possibly reducing the concentration of CEP290 to the basal body disrupting ciliogenesis.
The link between Rab GTPases and the BBSome
There are two proposed models for the transport of ciliary proteins inside the ciliary membrane: lateral diffusion and targeted vesicle transport. The second type of delivery and membrane sorting depends on the small GTPases RAB11 and RAB8A. RAB11 controls vesicle exit from the recycling endosomes,34 and RAB8A is involved in vesicular trafficking between the trans-golgi network and the plasma membrane.35,36 In addition to these functions, their cooperation regulate ciliogenesis and transport of proteins to the cilium.37,38
The BBSome interacts with the RAB8A interactor, Rabin8, through a direct interaction with BBS1 alone. BBS1 binds the C-terminal end of Rabin8 (Figure 1). Since Rabin8 is not found in the cilium or the CSs, it seems that this interaction with BBS1 must occur in the basal body.3,4 Rabin8 is a guanosine exchange factor for RAB8A. Rabin8 forms a key bridge between the vesicular membranes and the BBSome, forming a direct interaction only with BBS1. Rabin8 drives ciliogenesis by loading GTP on to RAB8A allowing it to enter the cilium and to elongate its membrane. Tandem affinity purification to identify Rabin8-associated proteins from 293 and IMCD3 cells has shown only weak signals for BBS1 (and BBS7) suggesting that BBSome interactions can be transient.39 As discussed previously, CEP290 has been linked with ciliary RAB8A function, and it is necessary for the localization of RAB8A to the basal body.27,31,33
IFT27, a RAB-like GTPase, and BBS3, which are both mutated in patients with BBS, form a direct protein–protein interaction and regulate BBSome entry into the cilium. However, so far only ARL6 has been shown to bind the BBSome. Both ARL6 and Rabin8 interact with BBS1. However, while ARL6 is distributed in a punctate manner along the ciliary axoneme, Rabin8 and BBS1 appear to co-localize at the basal body. Moreover, polycystin-1 (PC-1), a protein mutated in autosomal dominant polycystic kidney disease is able to interact with all subunits of the BBSome. Depletion of BBS1, but not any of the other BBSome subunits, impairs ciliary trafficking of PC-1 in kidney epithelial cells.40 In the same manner, over-expression of BBS3/ARL6 mutant (T31R) that locks ARL6 in its inactive GDP-bound form generates shorter cilia and loss of localization of PC-1 to primary cilia, although a separate report did not see the same effect on cilia length.8 As BBSome assembly appears to be normal in ARL6 knockdown cells, while translocation of the complex to the cilium is impaired, it is possible that interactions of the BBSome with Rabin8 and ARL6 are functionally separable.8,11 It is noteworthy that, although data implicating the RAB8–RAB11–Rabin8 complex in ciliary regulation are growing in number and quality,38 publications about the role of the BBSome with different small GTPases are limited.
The BBSome and the cell cytoskeleton
Research into ciliopathies has focused almost exclusively on the cilium. The initial indication that cilia are of fundamental importance came from the observation that the majority of proteins mutated in a group of clinically overlapping syndromes – subsequently defined as ciliopathies – do indeed localize to cilia and/or affect cilia formation/function. However, ciliopathy proteins represent a range of protein classes involved in processes as diverse as microtubule-based transport, vesicle transport/small GTPases, molecular chaperone activity, and cytoskeletal regulation. In terms of subcellular distribution, ciliopathy proteins are found in many locations within the cell. One example is the polycystins which regulate cell–cell adherens junctions in cells grown in conditions that do not permit cilia formation. We reported the first evidence linking BBSome components to cytoskeletal abnormalities. For example, BBS4 knockdown leads to microtubule-organizing center disorganization and failed microtubule anchoring to the CS, and silencing of BBS6 in a range of cell types leads to multinucleate and multicentrosomal cells with defects in cytokinesis.24,41 Cytoskeletal phenotypes were not limited to microtubules, and we also found perturbations in actin cytoskeleton polymerization.42,43
We now know of additional BBSome interactors (ALDOB, EXOC7, FLOT1, KRT18, or PAX2) that are expressed not only in the CSs but also in the microtubule-organizing center.44 The cellular consequences of potential nonciliary-related phenotypes seem to be diverse. The planar cell polarity pathway is disrupted in BBS gene-trap mice, affecting Vangl2 expression and causing early neural tube defects and cochlear stereociliary disruption.43,45–47 They also seem to be responsible for the disorganization of the dendritic microtubule network and trapping of olfactory ciliary proteins in dendrites and cell bodies,48 suggesting a role in the central nervous system.
How the BBS genes and the BBSome regulate cytoskeletal architecture still remains unclear. It is possible that the BBSome is required for cytoplasmic microtubule polymerization and acetylation. BBIP10 has been proposed to physically interact with Histone-deacetylase 6 to regulate microtubule stability and to couple acetylation of axonemal microtubules and ciliary membrane growth.5 It has also been reported that some BBSome subunits are expressed in focal adhesions. The focal adhesions provide a structural link between the actin cytoskeleton and the extracellular matrix. The polymerization of actin is highly regulated by the small GTPase RHOA,49 and the active GTP-bound form of RHOA is upregulated in BBS mutant cells. Actin cytoskeletal defects in BBS mutants can be rescued by inhibiting this pathway with the ROCK inhibitor Y-27632.42 Moreover, disruption of the cellular cytoskeleton is not unique to BBS cells and is shared with other ciliopathies,50 suggesting that some of the phenotypes observed in ciliopathies may be directly linked to regulation of the cytoskeleton rather than the ciliary function.
BBS syndrome as a model ciliopathy
Further observations indicate that BBS proteins function at the basal body stems from the identification of 668 proteins that are conserved in the ciliated organism Homo sapiens and Chlamydomonas reinhardtii (C. reinhardtii) but absent from Arabidopsis thaliana, a plant that lacks cilia. This cilia-enriched set of proteins contained all known BBS proteins, and it was found that bbs gene expression was activated in C. reinhardtii when induced to form flagella.51 These observations led to the discovery of mutations in BBS5. Second, all Caenorhabditis elegans bbs genes are expressed exclusively in ciliated neurons where they regulate intraflagellar transport (IFT).52,53 All types of cilia lack the necessary machinery for protein synthesis. Therefore, all proteins required for ciliary function must first be transported to the membrane and/or base of cilia in specific vesicles.1 Many of these proteins are subsequently transported from the base to within the cilium by the process of IFT, whereby kinesin or dynein molecular motors drive anterograde (from base to tip) or retrograde (tip to base) IFT along microtubules that comprise the ciliary axoneme. In addition to determining the ciliary composition, IFT of certain signaling molecules is necessary for their biochemical processing and therefore for proper regulation of signal transduction.5 IFT along the connecting cilium is also required to replenish components of the outer segments of photoreceptor cells, which degrade following exposure to light.6,7 Therefore, defects in IFT represent a mechanism of pathogenesis that is relevant to many, if not all, ciliopathies, including retinal degeneration/blindness and a variety of birth defects that are found in patients with BBS.
The BBSome acts as a scaffold linking IFT components and key cargo, including PCM1, to dynein molecular motors, thereby acting as key regulators of retrograde IFT within cilia.54–56 Remarkably, almost all mutations found in the 100 or so genes in ciliopathies cause a loss of function and are inherited in a recessive mode. This is likely to reflect the fact that many ciliopathy proteins form structural complexes, such as those forming the BBSome. It is therefore perhaps not surprising that mutations in more than one protein within such complexes could lead to additive functional defects. Support for this contention comes from examples of possible triallelic inheritance in BBS, with three mutations in two genes being necessary to produce the disease.57,58 A minor subset of families has been described that carry mutations in more than one BBS gene. In some cases, three mutations correlated with a more severe clinical phenotype than members of the same family who carried mutations in just one gene. In very rare examples, phenotypically normal individuals were found who were homozygous for deleterious mutations.59,60 Other examples of triallelic inheritance have been reported.
These observations suggest that multiallelic inheritance could be important in modifying the penetrance and expressivity of ciliopathies. A crucial protein complex underlying this phenomenon is the BBSome.
Molecular mechanisms underlying ciliary phenotypes
Key phenotypes in BBS that illustrate current knowledge about some of the fundamental mechanisms involved in pathogenesis include polydactyly/limb malformations, renal cystic disease, and retinal degeneration/blindness. How BBSome disruption is linked to the phenotypes observed in patients and animal models is intrinsically linked with the function of cilia in sensing environmental cues. Therefore, the disparate roles of the BBSome and cilia in producing organ-specific phenotypes relates to tissue-specific context for ciliary function. Little is known about the mechanisms that cause BBS specifically, although inference can be made from two areas of investigation. First, insight can be gleaned from investigations of classical ciliopathies. Second, biochemical investigations of the BBSome have revealed direct interactions with key effector proteins implicated in key human diseases. We have reviewed much of this in detail elsewhere,61–63 and so it will only be summarized briefly here.
Classic autosomal dominant polycystic disease is characterized by cysts affecting the renal collecting duct epithelium. It is caused by mutations in PKD1 or PKD2, which encode the transmembrane proteins, PC-1 or polycystin-2 (PC-2). Landmark studies have shown that PC-1 and PC-2 interact to form a calcium channel that is present within the ciliary membrane.64–73 Bending of the cilium was shown to activate this channel leading to calcium influx. This can lead to calcium waves that spread across an epithelial cell monolayer by the mechanism of calcium-induced calcium release from the endoplasmic reticulum. In the context of the renal epithelium, it is therefore speculated that urine flow along the collecting ducts may lead to orchestrated cilia bending, thereby generating co-ordinated waves of calcium signaling across the epithelium.
A similar mechanism has been proposed in relation to the laterality defects that are observed in some patients with BBS.65,68 Two populations of cilia are found in the embryonic node, which is an epithelial invagination present on the early embryo. A population of motile primary cilia is found within the center of the node, and a second population of immotile cilia is found laterally. Studies in which embryos were cultured in a fluid flow chamber showed that reversing the direction of fluid flow, which normally travels from right to left, have suggested a model whereby the central cilia generate unidirectional flow owing to the angle at which they are embedded within the epithelium, leading to bending of cilia specifically to the left of the node. These lateral cilia, which contain polycystin paralogs, including PKD2 and PKD1l1, initiate calcium signaling. This initiates a series of further signaling cascades which establish asymmetric patterns of Nodal signaling within the lateral plate mesoderm, which gives rise to organ asymmetries. Interestingly, multiple components of the BBSome have been shown to interact directly with PC-1 and -2. Although the exact relevance of this interaction is unclear, it is tempting to speculate that the BBSome plays a part in regulating localization of this calcium channel to cilia.
We already commented that PC-1 is able to interact with all subunits of the BBSome, and depletion of BBS1 or locking of BBS3/ARL6 in its inactive GDP-bound impairs ciliary trafficking of PC-1 in kidney epithelial cells and generates shorter cilia.40 We cannot be sure whether the failed polycystin localization is directly affected by its interactions with the BBSome subunits or if it is secondary to defects in primary cilia assembly. Patients with BBS sometimes present with situs inversus owing to early left–right patterning defects. This could be linked to the role of PC-1, the embryonic organizer in mammals, where it seems to control calcium signaling on the left side only. When bbs9 or bbs8 are knocked down in zebrafish, the motile cilia of Kupffer’s vesicle – the zebrafish equivalent of the node – appear fewer and shorter. Heart looping is also dependent on left–right patterning and is randomized in bbs morphants.74,75 On the other hand, mouse models for BBS show no obvious left–right patterning defects, and the penetrance of renal cysts is low.45,76–79 How BBSome disruption exactly modifies calcium signaling, cystogenesis or left-right patterning development still needs to be unveiled.
Another molecular pathway for which cilia are essential is the Hedgehog (Hh) pathway, and this has been reviewed extensively elsewhere.62 Hh signaling is a unique signaling pathway which centers around two transmembrane proteins, known as Smoothened (SMO) and Patched-1, named according to the loss-of-function phenotypes observed in Drosophila mutants for each of the encoding genes. SMO is derived from G-protein coupled receptors (GPCRs). However, other than signaling via small G-proteins, it appears to have retained no functional relationship to GPCRs. The precise mechanism by which SMO induces downstream signaling is still ambiguous, but it has become clear that its transport through the cilium by the process of IFT is essential for proper activation and regulation of the pathway. Patched-1 is a 12-pass transmembrane protein that is related to the Niemann-Pick protein pump. In the absence of Hh ligand, it serves to repress Hh signaling through inhibition of SMO. Like SMO, it too localizes to the ciliary membrane and serves to exclude SMO from cilia. This activity is inhibited following binding of Hh ligand.
The Hh pathway plays well-established roles in many aspects of embryonic development. The Hh ligands are well-established morphogens that pattern embryonic tissues by setting-up concentration-dependent signaling responses. Within the embryo, Hh ligand is expressed specifically within the posterior aspect of the limb bud, in a group of cells known as the zone of polarizing activity. This establishes an antero-posterior gradient of Hh signaling that determines the correct number and identity of each of the digits. Given the essential role of cilia in regulating Hh signaling, and the numerous examples of limb patterning defects in ciliopathy models, it is speculated that the polydactyly that is observed in some patients with BBS is the result of abnormal Hh signaling within the limb bud.
The Hh pathway has also been implicated in the regulation of adiposity. A genome-wide RNAi-based screen identified Hh pathway components as key regulators of triglyceride content in fruit flies and identified Hh signaling as a fat-specific pathway in Drosophila.80 Crucially, adipose-specific activation of the Hh pathway generated a lean phenotype in mice and revealed a role for Hh signaling in driving atrophy of white fat. Subsequent work revealed a cilium-dependent Smo-Ca(2+)-Ampk axis that triggers rapid Warburg-like metabolic reprogramming within adipose tissue.81 Collectively, this raises the possibility that obesity, which is found in BBS, is the result of reduced cilium-dependent Hh signaling in a cell autonomous manner. This being said, other work has shown that Bbs mutant mice exhibit reduced localization of neuropeptide Y receptors, which are GPCRs, to hypothalamic neurons.82 This was accompanied by excessive food intake in the mouse models. This raises the possibility that obesity in BBS is driven non-cell autonomously by GPCR signaling to regulate satiety. Further studies will be needed to bring these two datasets together, in particular, a complete analysis of Hh signaling in Bbs mutant mice and in adipose tissue is required.
In patients with BBS, disruption of the BBSome could perturb cilia function in the limb buds, producing the typical postaxial polydactyly. However, the nature of this perturbation, whether it involves disruption of cilia formation or specific aspects of IFT, is unclear. It is noteworthy that polydactyly has not been reported in any of the available mouse models of BBS.45,76–79
Conclusion and perspectives
BBS is a complex disorder, both genetically and in terms of pathogenic mechanisms. While we can infer several potential mechanisms of pathogenesis based on what we know about ciliary biology and ciliopathies in general, several of which have been discussed in this review, their exact contribution to the BBS phenotype is unclear. For instance, renal cystic disease is much less well characterized in BBS than in other disorders, such as classic polycystic kidney disease, and so the exact role of polycystin signaling in BBS is open for debate. Furthermore, while BBSome components are clearly important for IFT in neurons, for example, the exact contribution of defective IFT along the connecting cilium of photoreceptors in retinal degeneration in BBS is currently unclear and other mechanisms, such as altered vesicle trafficking in response to light stimulation, might be more important. What does seem to be clear is that the mechanisms of pathogenesis underlying different clinical features are likely to be tissue and cell type specific.
This phenotypic complexity is mirrored by the vast array of functional and biochemical interactions that the BBSome makes. Since the initial discovery of the BBSome, the list of interactors has grown and some unexpected functional relationships have been revealed. One example is the partial rescue of the Cep290 mutant mouse phenotype on the Bbs6 mutant background, but enhancement of this phenotype when crossed to Bbs4 mutant mice. It is also intriguing, given the many roles of the BBSome in ciliary function, that loss of BBSome trafficking to cilia does not prevent cilia from forming. Likewise, enrichment of the BBSome within cilia (eg, in LZTFL1 null mutant cells) also has no overt morphological effects on cilia. Therefore, the role of the BBSome in regulating ciliary function seems to be rather subtle. What is clear, however, is the importance of small GTPases, such as ARL6 and RAB8A, in regulating BBSome function. Because GTPases are classical molecular switches which have been targeted therapeutically in other disorders, including various forms of cancer, this raises the intriguing possibility that modulating the activity of specific GTPases could also be used to manipulate BBSome function therapeutically.
Acknowledgment
This work was supported by grants from the Medical Research Council (MR/L009978/1 and MR/M004597/1) to DJ.
Disclosure
The authors report no conflicts of interest in this work.
References
Baker K, Beales PL. Making sense of cilia in disease: the human ciliopathies. Am J Med Genet C Semin Med Genet. 2009;151C(4):281–295. | |
Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet. 1999;36(6):437–446. | |
Nachury MV, Loktev AV, Zhang Q, et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129(6):1201–1213. | |
Nachury MV. Tandem affinity purification of the BBSome, a critical regulator of Rab8 in ciliogenesis. Methods Enzymol. 2008;439:501–513. | |
Loktev AV, Zhang Q, Beck JS, et al. A BBSome subunit links ciliogenesis, microtubule stability, and acetylation. Dev Cell. 2008;15(6):854–865. | |
Lechtreck KF, Johnson EC, Sakai T, et al. The Chlamydomonas reinhardtii BBSome is an IFT cargo required for export of specific signaling proteins from flagella. J Cell Biol. 2009;187(7):1117–1132. | |
Hodges ME, Scheumann N, Wickstead B, Langdale JA, Gull K. Reconstructing the evolutionary history of the centriole from protein components. J Cell Sci. 2010;123(pt 9):1407–1413. | |
Jin H, White SR, Shida T, et al. The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell. 2010;141(7):1208–1219. | |
Mourao A, Nager AR, Nachury MV, Lorentzen E. Structural basis for membrane targeting of the BBSome by ARL6. Nat Struct Mol Biol. 2014;21(12):1035–1041. | |
Liew GM, Ye F, Nager AR, et al. The intraflagellar transport protein IFT27 promotes BBSome exit from cilia through the GTPase ARL6/BBS3. Dev Cell. 2014;31(3):265–278. | |
Wiens CJ, Tong Y, Esmail MA, et al. Bardet-Biedl syndrome-associated small GTPase ARL6 (BBS3) functions at or near the ciliary gate and modulates Wnt signaling. J Biol Chem. 2010;285(21):16218–16230. | |
Smith TS, Spitzbarth B, Li J, et al. Light-dependent phosphorylation of Bardet-Biedl syndrome 5 in photoreceptor cells modulates its interaction with arrestin1. Cell Mol Life Sci. 2013;70(23):4603–4616. | |
Eguether T, San Agustin JT, Keady BT, et al. IFT27 links the BBSome to IFT for maintenance of the ciliary signaling compartment. Dev Cell. 2014;31(3):279–290. | |
Marion V, Stutzmann F, Gérard M, et al. Exome sequencing identifies mutations in LZTFL1, a BBSome and smoothened trafficking regulator, in a family with Bardet–Biedl syndrome with situs inversus and insertional polydactyly. J Med Genet. 2012;49(5):317–321. | |
Schaefer E, Lauer J, Durand M, et al. Mesoaxial polydactyly is a major feature in Bardet-Biedl syndrome patients with LZTFL1 (BBS17) mutations. Clin Genet. 2014;85(5):476–481. | |
Seo S, Zhang Q, Bugge K, et al. A novel protein LZTFL1 regulates ciliary trafficking of the BBSome and smoothened. PLoS Genet. 2011;7(11):e1002358. | |
Seo S, Baye LM, Schulz NP, et al. BBS6, BBS10, and BBS12 form a complex with CCT/TRiC family chaperonins and mediate BBSome assembly. Proc Natl Acad Sci U S A. 2010;107(4):1488–1493. | |
Zhang Q, Yu D, Seo S, Stone EM, Sheffield VC. Intrinsic protein-protein interaction-mediated and chaperonin-assisted sequential assembly of stable bardet-biedl syndrome protein complex, the BBSome. J Biol Chem. 2012;287(24):20625–20635. | |
Stowe TR, Wilkinson CJ, Iqbal A, Stearns T. The centriolar satellite proteins Cep72 and Cep290 interact and are required for recruitment of BBS proteins to the cilium. Mol Biol Cell. 2012;23(17):3322–3335. | |
Zhang Y, Seo S, Bhattarai S, et al. BBS mutations modify phenotypic expression of CEP290-related ciliopathies. Hum Mol Genet. 2014; 23(1):40–51. | |
Rachel RA, May-Simera HL, Veleri S, et al. Combining Cep290 and Mkks ciliopathy alleles in mice rescues sensory defects and restores ciliogenesis. J Clin Invest. 2012;122(4):1233–1245. | |
Soares DC, Carlyle BC, Bradshaw NJ, Porteous DJ. DISC1: structure, function, and therapeutic potential for major mental illness. ACS Chem Neurosci. 2011;2(11):609–632. | |
Kamiya A, Tan PL, Kubo K, et al. Recruitment of PCM1 to the centrosome by the cooperative action of DISC1 and BBS4: a candidate for psychiatric illnesses. Arch Gen Psychiatry. 2008;65(9):996–1006. | |
Kim JC, Badano JL, Sibold S, et al. The Bardet-Biedl protein BBS4 targets cargo to the pericentriolar region and is required for microtubule anchoring and cell cycle progression. Nat Genet. 2004; 36(5):462–470. | |
Hall EA, Keighren M, Ford MJ, et al. Acute versus chronic loss of mammalian Azi1/Cep131 results in distinct ciliary phenotypes. PLoS Genet. 2013;9(12):e1003928. | |
Chamling X, Seo S, Searby CC, Kim G, Slusarski DC, Sheffield VC. The centriolar satellite protein AZI1 interacts with BBS4 and regulates ciliary trafficking of the BBSome. PLoS Genet. 2014;10(2):e1004083. | |
Klinger M, Wang W, Kuhns S, et al. The novel centriolar satellite protein SSX2IP targets Cep290 to the ciliary transition zone. Mol Biol Cell. 2014;25(4):495–507. | |
Kim K, Lee K, Rhee K. CEP90 is required for the assembly and centrosomal accumulation of centriolar satellites, which is essential for primary cilia formation. PLoS One. 2012;7(10):e48196. | |
Hori A, Ikebe C, Tada M, Toda T. Msd1/SSX2IP-dependent microtubule anchorage ensures spindle orientation and primary cilia formation. EMBO Rep. 2014;15(2):175–184. | |
Barbelanne M, Song J, Ahmadzai M, Tsang WY. Pathogenic NPHP5 mutations impair protein interaction with Cep290, a prerequisite for ciliogenesis. Hum Mol Genet. 2013;22(12):2482–2494. | |
Barbelanne M, Hossain D, Chan DP, Peranen J, Tsang WY. Nephrocystin proteins NPHP5 and Cep290 regulate BBSome integrity, ciliary trafficking and cargo delivery. Hum Mol Genet. 2015;24(8):2185–2200. | |
Villumsen BH, Danielsen JR, Povlsen L, et al. A new cellular stress response that triggers centriolar satellite reorganization and ciliogenesis. EMBO J. 2013;32(23):3029–3040. | |
Kim J, Krishnaswami SR, Gleeson JG. CEP290 interacts with the centriolar satellite component PCM-1 and is required for Rab8 localization to the primary cilium. Hum Mol Genet. 2008;17(23):3796–3805. | |
Welz T, Wellbourne-Wood J, Kerkhoff E. Orchestration of cell surface proteins by Rab11. Trends Cell Biol. 2014;24(7):407–415. | |
Huber LA, Pimplikar S, Parton RG, Virta H, Zerial M, Simons K. Rab8, a small GTPase involved in vesicular traffic between the TGN and the basolateral plasma membrane. J Cell Biol. 1993;123(1):35–45. | |
Zerial M, McBride H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol. 2001;2(2):107–117. | |
Wang J, Morita Y, Mazelova J, Deretic D. The Arf GAP ASAP1 provides a platform to regulate Arf4- and Rab11-Rab8-mediated ciliary receptor targeting. EMBO J. 2012;31(20):4057–4071. | |
Wang J, Deretic D. The Arf and Rab11 effector FIP3 acts synergistically with ASAP1 to direct Rabin8 in ciliary receptor targeting. J Cell Sci. 2015;128(7):1375–1385. | |
Westlake CJ, Baye LM, Nachury MV, et al. Primary cilia membrane assembly is initiated by Rab11 and transport protein particle II (TRAPPII) complex-dependent trafficking of Rabin8 to the centrosome. Proc Natl Acad Sci U S A. 2011;108(7):2759–2764. | |
Su X, Driscoll K, Yao G, et al. Bardet-Biedl syndrome proteins 1 and 3 regulate the ciliary trafficking of polycystic kidney disease 1 protein. Hum Mol Genet. 2014;23(20):5441–5451. | |
Kim JC, Ou YY, Badano JL, et al. MKKS/BBS6, a divergent chaperonin-like protein linked to the obesity disorder Bardet-Biedl syndrome, is a novel centrosomal component required for cytokinesis. J Cell Sci. 2005;118(pt 5):1007–1020. | |
Hernandez-Hernandez V, Pravincumar P, Diaz-Font A, et al. Bardet-Biedl syndrome proteins control the cilia length through regulation of actin polymerization. Hum Mol Genet. 2013;22(19):3858–3868. | |
May-Simera HL, Ross A, Rix S, Forge A, Beales PL, Jagger DJ. Patterns of expression of Bardet-Biedl syndrome proteins in the mammalian cochlea suggest noncentrosomal functions. J Comp Neurol. 2009; 514(2):174–188. | |
Oeffner F, Moch C, Neundorf A, Hofmann J, Koch M, Grzeschik KH. Novel interaction partners of Bardet-Biedl syndrome proteins. Cell Motil Cytoskeleton. 2008;65(2):143–155. | |
Ross AJ, May-Simera H, Eichers ER, et al. Disruption of Bardet-Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat Genet. 2005;37(10):1135–1140. | |
Mei X, Westfall TA, Zhang Q, Sheffield VC, Bassuk AG, Slusarski DC. Functional characterization of Prickle2 and BBS7 identify overlapping phenotypes yet distinct mechanisms. Dev Biol. 2014;392(2):245–255. | |
Tadenev AL, Kulaga HM, May-Simera HL, Kelley MW, Katsanis N, Reed RR. Loss of Bardet-Biedl syndrome protein-8 (BBS8) perturbs olfactory function, protein localization, and axon targeting. Proc Natl Acad Sci U S A. 2011;108(25):10320–10325. | |
Kulaga HM, Leitch CC, Eichers ER, et al. Loss of BBS proteins causes anosmia in humans and defects in olfactory cilia structure and function in the mouse. Nat Genet. 2004;36(9):994–998. | |
Akhshi TK, Wernike D, Piekny A. Microtubules and actin crosstalk in cell migration and division. Cytoskeleton (Hoboken). 2014;71(1):1–23. | |
Dawe HR, Adams M, Wheway G, et al. Nesprin-2 interacts with meckelin and mediates ciliogenesis via remodelling of the actin cytoskeleton. J Cell Sci. 2009;122(pt 15):2716–2726. | |
Li JB, Gerdes JM, Haycraft CJ, et al. Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell. 2004;117(4):541–552. | |
Ansley SJ, Badano JL, Blacque OE, et al. Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature. 2003;425(6958):628–633. | |
Blacque OE, Reardon MJ, Li C, et al. Loss of C. elegans BBS-7 and BBS-8 protein function results in cilia defects and compromised intraflagellar transport. Genes Dev. 2004;18(13):1630–1642. | |
Haycraft CJ, Swoboda P, Taulman PD, Thomas JH, Yoder BK. The C. elegans homolog of the murine cystic kidney disease gene Tg737 functions in a ciliogenic pathway and is disrupted in osm-5 mutant worms. Development. 2001;128(9):1493–1505. | |
Pazour GJ, Dickert BL, Vucica Y, et al. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J Cell Biol. 2000;151(3):709–718. | |
Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet. 2010;11(5):331–344. | |
Beales PL, Badano JL, Ross AJ, et al. Genetic interaction of BBS1 mutations with alleles at other BBS loci can result in non-Mendelian Bardet-Biedl syndrome. Am J Hum Genet. 2003;72(5):1187–1199. | |
Katsanis N, Ansley SJ, Badano JL, et al. Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science. 2001; 293(5538):2256–2259. | |
Katsanis N, Eichers ER, Ansley SJ, et al. BBS4 is a minor contributor to Bardet-Biedl syndrome and may also participate in triallelic inheritance. Am J Hum Genet. 2002;71(1):22–29. | |
Badano JL, Kim JC, Hoskins BE, et al. Heterozygous mutations in BBS1, BBS2 and BBS6 have a potential epistatic effect on Bardet-Biedl patients with two mutations at a second BBS locus. Hum Mol Genet. 2003;12(14):1651–1659. | |
Jenkins D. Hedgehog signalling: emerging evidence for non-canonical pathways. Cell Signal. 2009;21(7):1023–1034. | |
Winyard P, Jenkins D. Putative roles of cilia in polycystic kidney disease. Biochim Biophys Acta. 2011;1812(10):1256–1262. | |
Jenkins D, Beales PL. Principles and Practice of Medical Genetics. 6th ed. Amsterdam: Elsevier; 2013. | |
Pennekamp P, Karcher C, Fischer A, et al. The ion channel polycystin-2 is required for left-right axis determination in mice. Curr Biol. 2002; 12(11):938–943. | |
Schottenfeld J, Sullivan-Brown J, Burdine RD. Zebrafish curly up encodes a Pkd2 ortholog that restricts left-side-specific expression of southpaw. Development. 2007;134(8):1605–1615. | |
Praetorius HA, Frokiaer J, Nielsen S, Spring KR. Bending the primary cilium opens Ca2+-sensitive intermediate-conductance K+ channels in MDCK cells. J Membr Biol. 2003;191(3):193–200. | |
AbouAlaiwi WA, Takahashi M, Mell BR, et al. Ciliary polycystin-2 is a mechanosensitive calcium channel involved in nitric oxide signaling cascades. Circ Res. 2009;104(7):860–869. | |
Nonaka S, Shiratori H, Saijoh Y, Hamada H. Determination of left-right patterning of the mouse embryo by artificial nodal flow. Nature. 2002; 418(6893):96–99. | |
Sarmah B, Latimer AJ, Appel B, Wente SR. Inositol polyphosphates regulate zebrafish left-right asymmetry. Dev Cell. 2005;9(1):133–145. | |
Sammels E, Devogelaere B, Mekahli D, et al. Polycystin-2 activation by inositol 1,4,5-trisphosphate-induced Ca2+ release requires its direct association with the inositol 1,4,5-trisphosphate receptor in a signaling microdomain. J Biol Chem. 2010;285(24):18794–18805. | |
Li Y, Santoso NG, Yu S, Woodward OM, Qian F, Guggino WB. Polycystin-1 interacts with inositol 1,4,5-trisphosphate receptor to modulate intracellular Ca2+ signaling with implications for polycystic kidney disease. J Biol Chem. 2009;284(52):36431–36441. | |
Cowley BD Jr. Calcium, cyclic AMP, and MAP kinases: dysregulation in polycystic kidney disease. Kidney Int. 2008;73(3):251–253. | |
Wang X, Ward CJ, Harris PC, Torres VE. Cyclic nucleotide signaling in polycystic kidney disease. Kidney Int. 2010;77(2):129–140. | |
Veleri S, Bishop K, Dalle Nogare DE, et al. Knockdown of Bardet-Biedl syndrome gene BBS9/PTHB1 leads to cilia defects. PLoS One. 2012;7(3):e34389. | |
May-Simera HL, Kai M, Hernandez V, Osborn DP, Tada M, Beales PL. Bbs8, together with the planar cell polarity protein Vangl2, is required to establish left-right asymmetry in zebrafish. Dev Biol. 2010;345(2):215–225. | |
Davis RE, Swiderski RE, Rahmouni K, et al. A knockin mouse model of the Bardet-Biedl syndrome 1 M390R mutation has cilia defects, ventriculomegaly, retinopathy, and obesity. Proc Natl Acad Sci U S A. 2007;104(49):19422–19427. | |
Zhang Q, Nishimura D, Vogel T, et al. BBS7 is required for BBSome formation and its absence in mice results in Bardet-Biedl syndrome phenotypes and selective abnormalities in membrane protein trafficking. J Cell Sci. 2013;126(pt 11):2372–2380. | |
Nishimura DY, Fath M, Mullins RF, et al. Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc Natl Acad Sci U S A. 2004;101(47):16588–16593. | |
Mykytyn K, Mullins RF, Andrews M, et al. Bardet-Biedl syndrome type 4 (BBS4)-null mice implicate Bbs4 in flagella formation but not global cilia assembly. Proc Natl Acad Sci U S A. 2004;101(23):8664–8669. | |
Pospisilik JA, Schramek D, Schnidar H, et al. Drosophila genome-wide obesity screen reveals hedgehog as a determinant of brown versus white adipose cell fate. Cell. 2010;140(1):148–160. | |
Teperino R, Amann S, Bayer M, et al. Hedgehog partial agonism drives Warburg-like metabolism in muscle and brown fat. Cell. 2012; 151(2):414–426. | |
Loktev AV, Jackson PK. Neuropeptide Y family receptors traffic via the Bardet-Biedl syndrome pathway to signal in neuronal primary cilia. Cell Rep. 2013;5(5):1316–1329. | |
Chavali PL, Putz M, Gergely F. Small organelle, big responsibility: the role of centrosomes in development and disease. Philos Trans R Soc Lond B Biol Sci. 2014;369(1650). | |
Chavali PL, Gergely F. Cilia born out of shock and stress. EMBO J. 2013;32(23):3011–3013. | |
Sung CH, Leroux MR. The roles of evolutionarily conserved functional modules in cilia-related trafficking. Nat Cell Biol. 2013;15(12):1387–1397. | |
Sang L, Miller JJ, Corbit KC, et al. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell. 2011;145(4):513–528. | |
Garcia-Gonzalo FR, Corbit KC, Sirerol-Piquer MS, et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet. 2011;43(8):776–784. | |
Chih B, Liu P, Chinn Y, et al. A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat Cell Biol. 2012;14(1):61–72. | |
Leitch CC, Zaghloul NA, Davis EE, et al. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat Genet. 2008;40(4):443–448. | |
Muller J, Stoetzel C, Vincent MC, et al. Identification of 28 novel mutations in the Bardet-Biedl syndrome genes: the burden of private mutations in an extensively heterogeneous disease. Hum Genet. 2010;127(5):583–593. | |
Xing DJ, Zhang HX, Huang N, et al. Comprehensive molecular diagnosis of Bardet-Biedl syndrome by high-throughput targeted exome sequencing. PLoS One. 2014;9(3):e90599. | |
Forsythe E, Beales PL. Bardet-Biedl Syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al. editors. GeneReviews(R). Seattle, WA; 1993. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.