")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Adjunctive therapies to reduce thrombotic events in patients with a history of myocardial infarction: role of vorapaxar
Authors Farag M, Patel H, Gorog DA
Received 16 February 2015
Accepted for publication 2 April 2015
Published 22 July 2015 Volume 2015:9 Pages 3801—3809
DOI https://doi.org/10.2147/DDDT.S68391
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Mohamed Farag,1,2 Hiten Patel,1 Diana A Gorog1–3
1Department of Cardiology, East and North Hertfordshire NHS Trust, Stevenage, 2Postgraduate Medical School, University of Hertfordshire, Hatfield, 3National Heart and Lung Institute, Imperial College, London, UK
Abstract: Acute myocardial infarction (AMI) is generally attributed to coronary atherothrombotic disease. Platelet activation is essential for thrombus formation and is thus an important target for pharmacological intervention to prevent and treat AMI. Despite contemporary treatment with dual antiplatelet therapy, including acetylsalicylic acid and adenosine diphosphate receptor antagonists, patients with prior AMI remain at increased risk of future thrombotic events. This has stimulated the search for more potent antithrombotic agents. Among these is the oral protease-activated receptor-1 antagonist vorapaxar, which represents a new oral antiplatelet agent to reduce thrombotic risk in patients with atherothrombotic disease. The TRACER and the TRA 2°P-TIMI 50 trials concluded that vorapaxar in addition to standard therapy reduced ischemic adverse cardiac events. A remarkable benefit was observed in patients with stable atherosclerotic disease, particularly those with a previous history of AMI. Although favorable effects were seen in reduction of adverse cardiac events, this was associated with excess major and intracranial bleeding, particularly in patients at high risk of bleeding and those with a history of stroke or transient ischemic attack. Currently, the lack of a reliable individualized risk stratification tool to assess patients for thrombotic and bleeding tendencies in order to identify those who might gain most net clinical benefit has led to limited use of vorapaxar in clinical practice. Vorapaxar may find a niche as an adjunct to standard care in patients at high risk of thrombotic events and who are at low risk of bleeding.
Keywords: myocardial infarction, thrombosis, antiplatelet agents, protease-activated receptor-1, vorapaxar
Introduction
The predominant cause of death from cardiovascular disease is believed to be coronary artery thrombosis.1–3 Thrombotic occlusion of a coronary artery in response to atherosclerotic plaque rupture is considered the ultimate and key step in the pathogenesis of acute myocardial infarction (AMI).4 The propensity to provoke thrombosis depends on a complex cascade of events involving inflammatory pathways, and more importantly, platelet activation with subsequent aggregation.5 Guidelines recommend dual antiplatelet therapy with acetylsalicylic acid and adenosine diphosphate (ADP) receptor antagonists for a period of up to 1 year following the qualifying AMI event, to reduce recurrent thrombosis.6–9 The introduction of more potent oral antiplatelet agents, such as the more recent ADP receptor antagonists ticagrelor and prasugrel, has further reduced the risk of recurrent thrombosis.10,11 However, despite modern treatments, many patients remain at increased risk of future thrombotic events. In recent studies, some 10%–15% of patients went on to have a major adverse cardiac event during the first 12 months after AMI, which was attributed predominantly to thrombotic complications.12–15 Additionally, there has been a growing concern over the safety profile of oral antiplatelet agents in terms of increased bleeding, which is now known to be a marker of an adverse prognosis and has negatively affected their use.10,16 In order to reduce thrombotic risk even further, oral anticoagulant agents were added to dual antiplatelet therapy, but this was found to be associated with increased bleeding.17,18 This has led to the search for novel antiplatelet agents with effects to reduce thrombotic risk, taking into consideration the potential for excess bleeding. Among these are the oral protease-activated receptor (PAR)-1 antagonists, which represent a new class of oral antiplatelet agents for patients with atherothrombotic disease. A key step in the process of thrombus formation is the role thrombin plays in the activation of platelets by binding to PARs, especially PAR-1.19 Targeting this thrombin signaling receptor has led to greater inhibition of platelet activation and inhibition, and in turn of thrombosis.
Several PAR-1 antagonists have been evaluated for clinical use. The clinical efficacy appeared to be superior with vorapaxar, compared with atopaxar, but this was associated with a higher risk of serious bleeding.20 Only vorapaxar has completed Phase III clinical trial investigation to assess its efficacy and safety in the clinical arena.21,22 The present review provides an overview of the role of adjunctive therapy with vorapaxar in the secondary prevention of atherothrombotic disease, particularly AMI, and the potential role for vorapaxar in modern practice.
Mechanism of action of PAR-1 antagonism with vorapaxar
Hemostasis is considered a protective mechanism that maintains the integrity of blood vessels after vascular injury. Thrombin signaling in platelets contributes to hemostasis and thrombosis by converting circulating fibrinogen into fibrin, the fibrous matrix of blood clots. The cellular effects of thrombin are mainly mediated by PARs.23 The mechanism of PAR activation and signaling is complex. PARs are G protein-coupled receptors that are expressed in vascular endothelial cells and activated by cleavage of part of their extracellular domain, causing the physiological response.24 They play an important role in thrombosis, coagulation, hemostasis, atherosclerosis, and inflammation.25–27 There are four known types of PARs, numbered from PAR-1 to PAR-4. Thrombin triggers platelet activation with subsequent aggregation primarily by activating PAR-1 and PAR-4. PAR-1 is activated at a much smaller concentration, resulting in rapid platelet activation.28 Many of the downstream mediators of the PAR-1 pathway, such as thromboxane A2 and ADP, are involved in platelet activation. In an animal model, administration of the PAR-1 antagonist vorapaxar caused complete and dose-dependent inhibition of thrombin receptor activating peptide (TRAP)-induced platelet aggregation without affecting the coagulation cascade, including activated clotting time, prothrombin time, and activated partial thromboplastin time, a finding that is consistent with the fact that this agent interacts with specific platelet receptors,29 and suggested that it could inhibit thrombosis without undue bleeding risk.
Vorapaxar (formerly known as SCH 530348) is a synthetic tricyclic 3-phenylpyridine derived from the natural product himbacine. It is an oral competitive PAR-1 antagonist that exerts its action by inhibition of TRAP-induced platelet aggregation in a dose-dependent manner30 (Figure 1). The loading dose is 20 or 40 mg, with a higher dose achieving greater inhibition of platelet aggregation, and the maintenance dose is 2.5 mg daily.31 Vorapaxar is rapidly absorbed via the gastrointestinal tract, with high bioavailability. Its peak concentration is 1–2 hours after oral loading and it has a half-life of 159–310 hours, with no antidote available at present.32 Vorapaxar is predominantly metabolized via the cytochrome P450 3A4 pathway and is mainly excreted in bile with only minor renal excretion.32,33
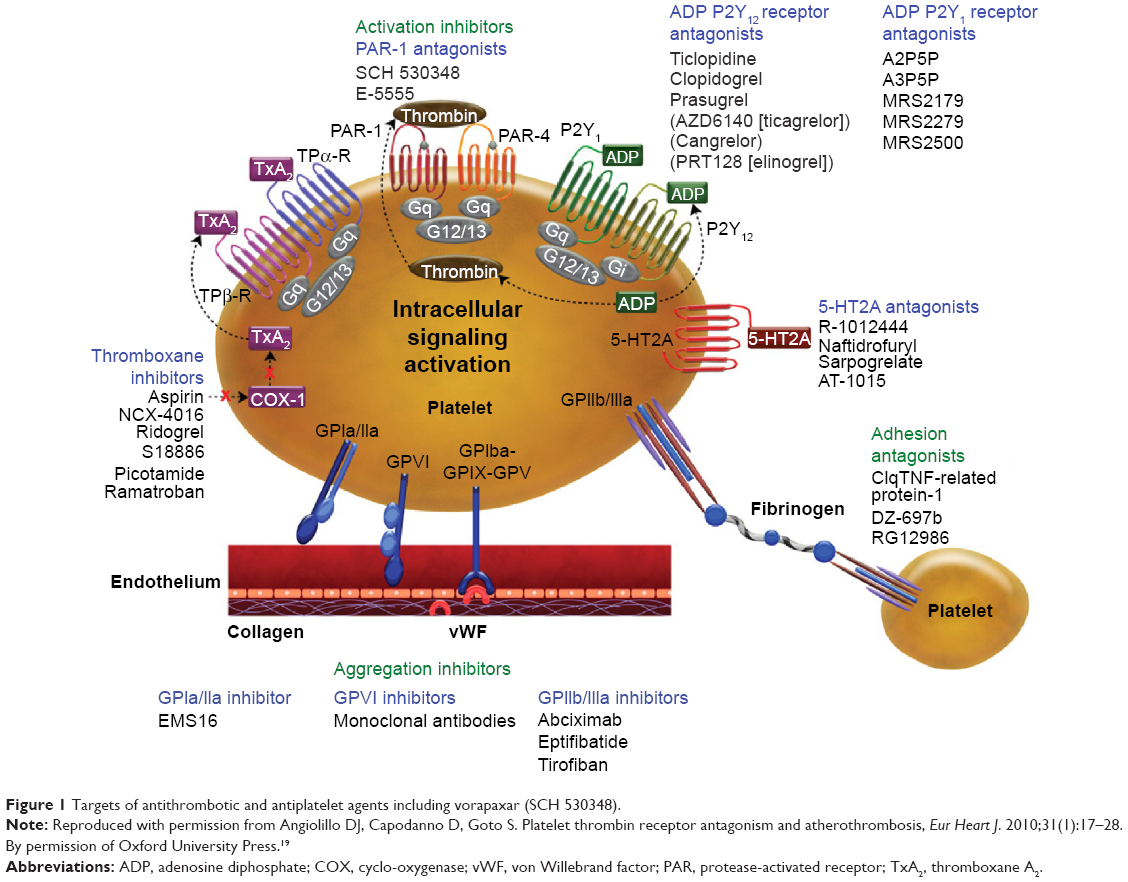 | Figure 1 Targets of antithrombotic and antiplatelet agents including vorapaxar (SCH 530348). |
A summary of key randomized trials evaluating the use of vorapaxar in the three phases of clinical trial investigation is shown in Table 1.
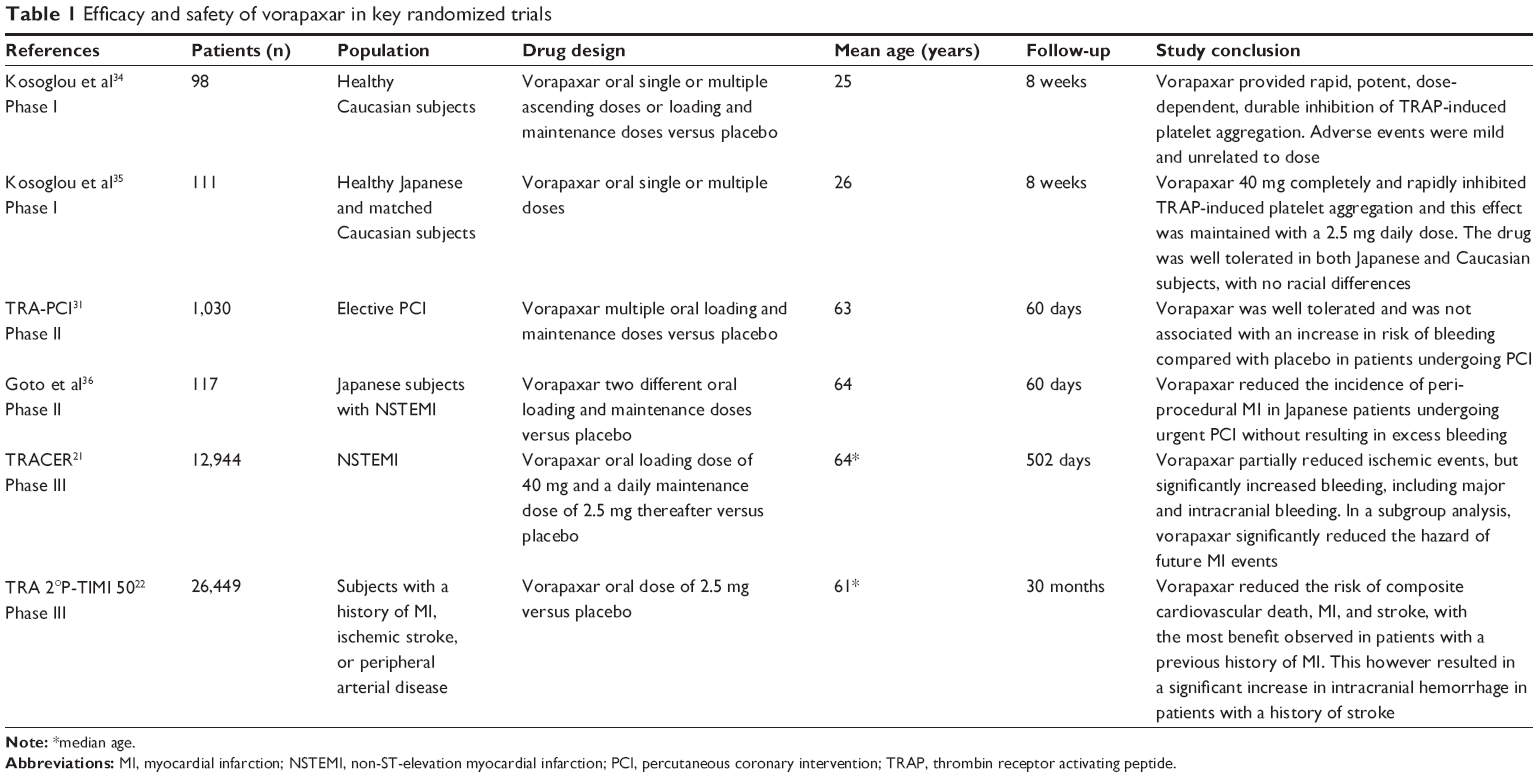 | Table 1 Efficacy and safety of vorapaxar in key randomized trials |
Phase I clinical trial data
In healthy Caucasian subjects, single 20 and 40 mg doses of vorapaxar administered in a randomized, double-blind placebo-controlled fashion inhibited TRAP-induced platelet aggregation (>80% inhibition) at 1 hour, and this level of inhibition was sustained for up to 72 hours.34 Multiple ascending doses for 28 days (1, 3, or 5 mg/day) resulted in complete inhibition of platelet aggregation on day 1 (5 mg/day) and day 7 (1 and 3 mg/day). Adverse events were generally mild and unrelated to dose. In another randomized open-label trial in healthy Japanese and matched Caucasian subjects, complete inhibition of TRAP-induced platelet aggregation was achieved most rapidly with vorapaxar 40 mg and was sustained with a maintenance dose of 2.5 mg daily.35 No racial difference as regard to the safety, pharmacokinetics, or pharmacodynamics of vorapaxar was found. These findings supported further investigation of vorapaxar in Phase II clinical trials.
Phase II clinical trial data
TRA-PCI (Thrombin Receptor Antagonist Percutaneous Coronary Intervention) was a multicenter, randomized, double-blind, placebo-controlled trial of patients undergoing non-urgent or elective percutaneous coronary intervention (PCI).31 This was a Phase II trial involving 1,030 patients comparing different oral loading doses of 10, 20, and 40 mg vorapaxar followed by maintenance doses of 0.5, 1, and 2 mg daily against matched placebo in a 3:1 ratio. After the loading doses, the vorapaxar group continued receiving vorapaxar maintenance doses and the placebo group continued placebo for 60 days after PCI. Patients were continued on standard dual antiplatelet therapy with acetylsalicylic acid and ADP receptor antagonists during the study. The primary endpoint was the incidence of TIMI (Thrombolysis In Myocardial Infarction) major bleeding (defined as intracranial hemorrhage, overt bleeding associated with a fall in hemoglobin >5 g/dL), or TIMI minor bleeding (defined as overt clinical signs of bleeding associated with a hemoglobin reduction of 3–5 g/dL) in the PCI cohort. The secondary endpoints were overt bleeding that did not meet TIMI criteria (defined as clinically overt signs of bleeding with a reduction in hemoglobin <3 g/dL), or ischemic events (defined as composite of death, myocardial infarction [MI], and stroke). The study showed no increased risk of TIMI major or minor bleeding or non-TIMI bleeding with vorapaxar using any of the dosing regimens compared with placebo. As the study was underpowered, there was a non-significant reduction in ischemic events among PCI-treated patients with vorapaxar using any of the dosing regimens compared with placebo (odds ratio 0.67; 95% confidence interval [CI] 0.33–1.34). The conclusion of the TRA-PCI study was that, in addition to standard dual antiplatelet therapy, vorapaxar was well tolerated and not associated with an increased risk of bleeding compared with placebo in patients undergoing PCI.
Another Phase II trial was reported by Goto et al.36 This was a multicenter, randomized, double-blind, placebo-controlled trial performed to assess the efficacy and safety of vorapaxar in Japanese patients with non-ST-segment elevation MI planned for PCI. The study involved 117 patients to compare two different vorapaxar oral loading doses of 20 mg and 40 mg followed by maintenance doses of 1 mg and 2.5 mg daily against matched placebo in a 4:1 ratio. The patients received loading doses of standard-of-care medication at the time of the study (acetylsalicylic acid, ticlopidine, and heparin) as well as a loading dose of the study treatment; the vorapaxar group continued receiving vorapaxar maintenance doses and the placebo group continued placebo for 60 days after PCI. Patients were continued on standard dual antiplatelet therapy with acetylsalicylic acid and ADP receptor antagonists during the study. The efficacy endpoint was major adverse cardiac event or all-cause death and the safety endpoint was TIMI major and minor bleeding or non-TIMI bleeding. The study showed a significant reduction in peri-procedural AMI in the group treated with vorapaxar compared with the placebo group (16.9% versus 42.9%, respectively; P=0.013). Peri-procedural AMI was defined as elevation of cardiac enzymes above three times the upper limit of normal, with at least a 50% increase from the value prior to the procedure. The incidence of combined TIMI major and minor bleeding was 14% in the vorapaxar group and 10% in the placebo group. TIMI major bleeding occurred in five patients (7%) in the vorapaxar group versus none in the placebo group. The rates of non-TIMI bleeding or bleeding of any severity were comparable between the two groups. The authors concluded that in addition to standard dual antiplatelet therapy, vorapaxar significantly reduced the incidence of peri-procedural MI in Japanese patients undergoing urgent PCI without resulting in excess bleeding.
Phase III clinical trial data
To date, two large randomized clinical trials have been conducted, ie, TRACER (Thrombin Receptor Antagonist for Clinical Event Reduction in Acute Coronary Syndrome) and TRA 2°P-TIMI 50 (Thrombin Receptor Antagonist in Secondary Prevention of Atherothrombotic Ischemic Events-Thrombolysis in Myocardial Infarction 50).21,22 Several hypothesis-generating subgroup analyses were derived from these trials. In this review, we highlight the key findings and important data from these trials.
TRACER
TRACER was the first large study to evaluate the efficacy and safety of vorapaxar.21 It was a multicenter, randomized, double-blind, placebo-controlled, Phase III trial, in which patients with non-ST-segment elevation MI (12,944 patients) received vorapaxar at a loading dose of 40 mg and a daily maintenance dose of 2.5 mg thereafter, or matched placebo. Management of the patients included medical therapy (32.2%), PCI (57.7%), and coronary artery bypass grafting (CABG, 10.1%), according to usual clinical care. Patients were continued on standard dual antiplatelet therapy with acetylsalicylic acid and an ADP receptor antagonist during the study. The primary efficacy endpoint was a composite of cardiovascular death, MI, stroke, recurrent ischemia with rehospitalization, and urgent coronary revascularization. The primary safety endpoints were a composite of moderate or severe bleeding according to the GUSTO (Global Use of Strategies to Open Occluded Coronary Arteries) classification and clinically significant bleeding according to the TIMI classification. The trial was terminated early owing to safety concerns. At a median follow-up of 502 days, the study primary composite ischemic endpoint was non-significantly reduced in patients randomized to vorapaxar when compared with placebo (18.5% versus 19.9%; hazard ratio [HR] 0.92; 95% CI 0.85–1.01; P=0.07). However, a secondary non-prespecified ischemic endpoint (defined as composite of cardiovascular death, MI, or stroke) was significantly reduced with vorapaxar (14.7% versus 16.4%; HR 0.89; 95% CI 0.81–0.98; P=0.02). The rate of moderate or severe bleeding (GUSTO classification) was higher with vorapaxar compared with placebo (7.2% versus 5.2%; HR 1.35; 95% CI 1.16–1.58; P<0.001). Also, the rate of intracranial hemorrhage was higher with vorapaxar (1.1% versus 0.2%; HR 3.39; 95% CI 1.78–6.45; P<0.001) compared with placebo. The data and safety monitoring board recommended premature termination of the trial as a result of increased bleeding risk with vorapaxar. The conclusion of the study was that in addition to standard dual antiplatelet therapy, although vorapaxar partially reduced ischemic events, its use significantly increased bleeding, including major and intracranial bleeding.
Subgroup analyses play an important role in the interpretation of clinical trials. The net clinical benefit, defined as the difference in ischemic and bleeding event rates, was evaluated in a post hoc analysis of the TRACER trial.37 The analysis was performed by application of multivariate risk stratification strategies, which were unique to this analysis and not widely validated. The results showed that vorapaxar was associated with an improved net benefit in a large group of patients with acute coronary syndrome (ACS) at high risk of recurrent ischemic events and at low risk of bleeding (26% of patients; net benefit +2.8%). However, among patients with a high risk of bleeding and irrespective of ischemic risk (low or high), vorapaxar was associated with worse net benefit (11% of patients; net benefit −3%). Vorapaxar exerted a neutral effect on those with a low risk of bleeding and a low risk of ischemic events (63% of patients; net benefit −0.1%). The results of this analysis highlight the potentially beneficial role of vorapaxar in patients at high risk of ischemic events and low risk of bleeding, but similarly, the potential for harm using vorapaxar in patients at high risk of bleeding.
Although the TRACER trial did not meet its primary efficacy endpoint, a significant reduction in the rate of MI was observed with vorapaxar compared with placebo. Therefore, the effect of vorapaxar on MI was further explored in a post hoc analysis.38 A blinded, independent central endpoint adjudication committee prospectively defined and classified MI according to the universal MI definition.39 During a median follow-up of 502 days, 1,580 MI events occurred in 1,319 patients. Compared with placebo, vorapaxar reduced the hazard of a first MI of any type by 12% (HR 0.88; 95% CI 0.79–0.98; P=0.021) and the hazard of total numbers of MI (first and subsequent) by 14% (HR 0.86; 95% CI 0.77–0.97; P=0.014). Also, vorapaxar reduced type 1 MI (the most common type) by 17% (HR 0.83; 95% CI 0.73–0.95; P=0.007), but not type 4a MI (PCI-related, HR 0.90; 95% CI 0.73–1.12; P=0.35) compared with placebo. These findings support the potential role of vorapaxar in the management of ACS patients at high-risk of future MI events.
Platelet activation and thrombosis play an important role in acute bypass graft occlusion. A post hoc analysis was performed for the subgroup of 1,312 patients who underwent CABG in the TRACER trial.40 Of these, 78% were on vorapaxar at the time of surgery. Compared with placebo, the vorapaxar group had a 45% significant reduction in incidence of the primary composite ischemic endpoint, ie, a composite of cardiovascular death, MI, stroke, recurrent ischemia with rehospitalization, and urgent coronary revascularization (HR 0.55; 95% CI 0.36–0.83; P=0.005). These findings differed significantly from the non-CABG group, with a significant interaction (P=0.012). CABG-related major bleeding was similar with vorapaxar and placebo (9.7% versus 7.3%; HR 1.36; 95% CI 0.92–2.02; P=0.12). Although derived from subgroup analysis, these results show promise for use of vorapaxar in ACS patients undergoing CABG.
Another post hoc analysis was performed according to stent type in the 7,479 patients who underwent PCI in the TRACER trial (n=7,479).41 The efficacy and safety of vorapaxar among PCI patients were largely consistent with the overall TRACER trial results. A trend toward reduction in ischemic events and less bleeding was noted in patients who had bare metal stents compared with drug-eluting stents. In another post hoc analysis of ACS patients who were initially managed with medical treatment (n=4,194), the efficacy and safety of vorapaxar appeared consistent with the overall TRACER trial results.42
TRA 2°P-TIMI 50
TRA 2°P-TIMI 50 was a secondary prevention study.22 This was a multicenter, randomized, double-blind, placebo-controlled, Phase III trial. Patients with a history of MI, ischemic stroke, or peripheral arterial disease (26,449 patients) received vorapaxar (2.5 mg daily) or matching placebo. Patients were continued on standard-of-care therapy with acetylsalicylic acid or ADP receptor antagonists during the study. The study excluded patients with a high risk of bleeding, including those who had a history of bleeding diathesis or recent active bleeding, were receiving concurrent anticoagulation therapy, or had active hepatobiliary disease. The primary efficacy endpoint was the composite of cardiovascular death, MI, or stroke. The primary safety endpoints were a composite of moderate or severe bleeding according to the GUSTO classification and clinically significant bleeding according to the TIMI classification. The composite of cardiovascular death, MI, stroke, or recurrent ischemia leading to urgent coronary revascularization was the major secondary efficacy endpoint. The trial was terminated early owing to safety concerns. At a median follow-up of 30 months, the primary efficacy endpoint occurred in 9.3% of the vorapaxar group and in 10.5% of the placebo group (HR 0.87; 95% CI 0.80–0.94; P<0.001). The secondary efficacy endpoint occurred in 11.2% of the vorapaxar group and in 12.4% of the placebo group (HR 0.88; 95% CI 0.82–0.95; P=0.001). The ischemic benefit observed with vorapaxar was driven mainly by a reduction in MI (5.2% versus 6.1%; HR 0.83; 95% CI 0.74–0.93; P=0.001). Also, the incidence of cardiovascular death or MI was less with vorapaxar (7.3% versus 8.2%; P=0.002). No treatment difference was found in the incidence of stroke or death from any cause. The rate of moderate or severe bleeding (GUSTO classification) was higher with vorapaxar than with placebo (4.2% versus 2.5%; HR 1.66; 95% CI 1.43–1.93; P=0.001). The rate of intracranial hemorrhage was also notably higher with vorapaxar (1.0% versus 0.5%; HR 1.94; 95% CI 1.39–2.70; P<0.001). The data and safety monitoring board recommended premature termination of the trial in patients with a history of stroke or new stroke as a result of an increased risk of intracranial hemorrhage with vorapaxar. The conclusion of the study was that vorapaxar in addition to standard therapy reduced the risk of composite cardiovascular death, MI, and stroke, with the most benefit observed in patients with a stable atherosclerotic disease, particularly those with a previous history of MI. However, this resulted in a significant increase in bleeding risk, particularly intracranial hemorrhage in patients with a history of stroke.
In a subgroup analysis of patients with a history of ischemic stroke (n=4,883),43 vorapaxar increased the risk of intracranial hemorrhage compared with placebo (2.5% versus 1.0%; HR 2.52; 95% CI 1.46–4.36; P<0.001). Also, vorapaxar increased the risk of moderate and severe bleeding (4.2% versus 2.4%; HR 1.93; 95% CI 1.33–2.79; P<0.001), without any significant effect on the primary ischemic endpoint (13.0% versus 11.7%; P=0.75). This analysis highlighted the potential harm of vorapaxar in patients with a history of stroke.
Another large subgroup analysis was performed of patients with a prior MI within the previous 2 weeks to 12 months (17,779 patients).44 Vorapaxar significantly reduced the primary ischemic endpoint compared with placebo (8.1% versus 9.7%; HR 0.80; 95% CI 0.72–0.89; P<0.0001). This benefit was consistent in all key subgroups, including subgroup analysis based on timing between the qualifying MI events and randomization; <3 months (HR 0.82; 95% CI 0.70–0.95; P=0.011), 3–6 months (HR 0.79; 95% CI 0.65–0.97; P=0.023), and >6 months (HR 0.78; 95% CI 0.62–0.97; P=0.026). However, the observed benefit occurred at a cost of an excess of moderate or severe bleeding (vorapaxar group 3.4% versus placebo group 2.1%; HR 1.61; 95% CI 1.31–1.97; P<0.0001), and clinically significant bleeding (vorapaxar group 15.1% versus placebo group 10.4%; HR 1.49; 95% CI 1.36–1.63; P<0.0001). In this subgroup analysis, there was no significant risk of intracranial hemorrhage associated with vorapaxar compared with placebo (0.6% versus 0.4%; HR 1.54; 95% CI 0.96–2.48; P=0.076). In a further analysis of patients at low bleeding risk, defined as those <75 years of age and without a history of stroke or transient ischemic attack, similar results were obtained, with still significantly higher rates of moderate or severe bleeding with vorapaxar than with placebo (2.7% versus 1.8%), although with fewer bleeds overall. The conclusion of this analysis was that prolonged treatment with vorapaxar when added to standard antiplatelet therapy may be beneficial for long-term secondary prevention in patients with prior MI.
The efficacy of vorapaxar in terms of the occurrence of stent thrombosis (defined using Academic Research Consortium criteria), was recently investigated.45,46 During a median follow-up of 30 months, there were 152 definite stent thrombosis events, with the majority (92%) occurring late (30 days to 1 year) or very late (>1 year). Vorapaxar consistently reduced stent thrombosis including very late stent thrombosis (1.1% versus 1.4%; HR 0.71; 95% CI 0.51–0.98; P=0.037), regardless of dual antiplatelet use, stent type, history of diabetes, or time from PCI.
Conclusion
Patients with atherosclerosis and those with prior thrombotic events such as MI are at increased risk of future thrombotic events. Since platelet activation is an essential key step in thrombus formation, aggressive secondary preventive measures with antiplatelet agents have been devised to reduce thrombotic risk. PARs are an important target to reduce platelet activation. Vorapaxar, a PAR-1 antagonist, has been tested in addition to dual antiplatelet therapy for prevention of thrombotic events in patients mostly with a history of MI. Although Phase II trials showed a significant reduction in recurrent ischemic events with no increase in bleeding, Phase III trials showed similar reductions in ischemic events, but unveiled an increase in major bleeding, including intracranial bleeding, associated with vorapaxar. There are currently no ongoing trials further assessing the efficacy and safety of vorapaxar in the setting of MI.
Based on these findings, vorapaxar was approved for use only in patients at high risk of thrombosis and low risk of bleeding. Subsequently, the US Food and Drug Administration has recommended vorapaxar as an addition to dual antiplatelet therapy for treatment of patients with a history of MI, a low risk of bleeding, and no prior stroke or transient ischemic attack.47 However, given the concerns around bleeding associated with the drug, its use is still restricted due to the challenges of safely identifying patients at high risk of thrombosis and low risk of bleeding who may gain the most from vorapaxar.
Author contributions
All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Davies MJ, Thomas A. Thrombosis and acute coronary-artery lesions in sudden cardiac ischemic death. N Engl J Med. 1984;310(18):1137–1140. | ||
Fuster V, Steele PM, Chesebro JH. Role of platelets and thrombosis in coronary atherosclerotic disease and sudden death. J Am Coll Cardiol. 1985;5(6 Suppl):175B–184B. | ||
Schwartz RS, Burke A, Farb A, et al. Microemboli and microvascular obstruction in acute coronary thrombosis and sudden coronary death: relation to epicardial plaque histopathology. J Am Coll Cardiol. 2009;54(23):2167–2173. | ||
Naghavi M, Libby P, Falk E, et al. From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies: Part II. Circulation. 2003;108(15):1772–1778. | ||
Libby P. Molecular and cellular mechanisms of the thrombotic complications of atherosclerosis. J Lipid Res. 2009;50 Suppl:S352–S357. | ||
Bassand JP, Hamm CW, Ardissino D, et al. Guidelines for the diagnosis and treatment of non-ST-segment elevation acute coronary syndromes: the task force for diagnosis and treatment of non-ST-segment elevation acute coronary syndromes of European Society of Cardiology. Eur Heart J. 2007;28(13):1598–1660. | ||
Hamm CW, Bassand JP, Agewall S, et al. ESC guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation: the task force for the management of acute coronary syndromes (ACS) in patients presenting without persistent ST-segment elevation of the European Society of Cardiology (ESC). Eur Heart J. 2011;32(23):2999–3054. | ||
O’Gara PT, Kushner FG, Ascheim DD, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association task force on practice guidelines. J Am Coll Cardiol. 2013;61(4):e78–e140. | ||
Amsterdam EA, Wenger NK, Brindis RG, et al. 2014 AHA/ACC guideline for the management of patients with non-ST-elevation acute coronary syndromes: a report of the American College of Cardiology/American Heart Association task force on practice guidelines. J Am Coll Cardiol. 2014;64(24):e139–e228. | ||
Wiviott SD, Braunwald E, McCabe CH, et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357(20):2001–2015. | ||
Wallentin L, Becker RC, Budaj A, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361(11):1045–1057. | ||
Kastrati A, Neumann FJ, Schulz S, et al. Abciximab and heparin versus bivalirudin for non-ST-elevation myocardial infarction. N Engl J Med. 2011;365(21):1980–1989. | ||
Patti G, Pasceri V, D’Antonio L, et al. Comparison of safety and efficacy of bivalirudin versus unfractionated heparin in high-risk patients undergoing percutaneous coronary intervention (from the Anti-Thrombotic Strategy for Reduction of Myocardial Damage During Angioplasty-Bivalirudin vs Heparin study). Am J Cardiol. 2012;110(4):478–484. | ||
Steg PG, van ‘t Hof A, Hamm CW, et al. Bivalirudin started during emergency transport for primary PCI. N Engl J Med. 2013;369(23):2207–2217. | ||
Shahzad A, Kemp I, Mars C, et al. Unfractionated heparin versus bivalirudin in primary percutaneous coronary intervention (HEAT-PPCI): an open-label, single centre, randomised controlled trial. Lancet. 2014;384(9957):1849–1858. | ||
Roe MT, Armstrong PW, Fox KA, et al. Prasugrel versus clopidogrel for acute coronary syndromes without revascularization. N Engl J Med. 2012;367(14):1297–1309. | ||
Oldgren J, Budaj A, Granger CB, et al. Dabigatran versus placebo in patients with acute coronary syndromes on dual antiplatelet therapy: a randomized, double-blind, phase II trial. Eur Heart J. 2011;32(22):2781–2789. | ||
Alexander JH, Lopes RD, James S, et al. Apixaban with antiplatelet therapy after acute coronary syndrome. N Engl J Med. 2011;365(8):699–708. | ||
Angiolillo DJ, Capodanno D, Goto S. Platelet thrombin receptor antagonism and atherothrombosis. Eur Heart J. 2010;31(1):17–28. | ||
Chatterjee S, Sharma A, Mukherjee D. PAR-1 antagonists: current state of evidence. J Thromb Thrombolysis. 2013;35(1):1–9. | ||
Tricoci P, Huang Z, Held C, et al. Thrombin-receptor antagonist vorapaxar in acute coronary syndromes. N Engl J Med. 2012;366(1):20–33. | ||
Morrow DA, Braunwald E, Bonaca MP, et al. Vorapaxar in the secondary prevention of atherothrombotic events. N Engl J Med. 2012;366(15):1404–1413. | ||
Martorell L, Martínez-González J, Rodríguez C, Gentile M, Calvayrac O, Badimon L. Thrombin and protease-activated receptors (PARs) in atherothrombosis. Thromb Haemost. 2008;99(2):305–315. | ||
Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407(6801):258–264. | ||
Vu TK, Hung DT, Wheaton VI, Coughlin SR. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64(6):1057–1068. | ||
Coughlin SR, Vu TK, Hung DT, Wheaton VI. Expression cloning and characterization of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Semin Thromb Hemost. 1992;18(2):161–166. | ||
Vergnolle N, Wallace JL, Bunnett NW, Hollenberg MD. Protease-activated receptors in inflammation, neuronal signaling and pain. Trends Pharmacol Sci. 2001;22(3):146–152. | ||
Kahn ML, Nakanishi-Matsui M, Shapiro MJ, Ishihara H, Coughlin SR. Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J Clin Invest. 1999;103(6):879–887. | ||
Chintala M, Strony J, Yang B, Kurowski S, Li Q. SCH 602539, a protease-activated receptor-1 antagonist, inhibits thrombosis alone and in combination with cangrelor in a Folts model of arterial thrombosis in cynomolgus monkeys. Arterioscler Thromb Vasc Biol. 2010;30(11):2143–2149. | ||
Doller D, Chackalamannil S, Czarniecki M, McQuade R, Ruperto V. Design, synthesis, and structure-activity relationship studies of himbacine derived muscarinic receptor antagonists. Bioorg Med Chem Lett. 1999;9(6):901–906. | ||
Becker RC, Moliterno DJ, Jennings LK, et al. Safety and tolerability of SCH 530348 in patients undergoing non-urgent percutaneous coronary intervention: a randomised, double-blind, placebo-controlled phase II study. Lancet. 2009;373(9667):919–928. | ||
TRA*CER Executive and Steering Committees. The Thrombin Receptor Antagonist for Clinical Event Reduction in Acute Coronary Syndrome (TRA*CER) trial: study design and rationale. Am Heart J. 2009;158(3):327–334.e4. | ||
Xia Y, Chackalamannil S, Greenlee WJ, et al. Discovery of a vorapaxar analog with increased aqueous solubility. Bioorg Med Chem Lett. 2010;20(22):6676–6679. | ||
Kosoglou T, Reyderman L, Tiessen RG, et al. Pharmacodynamics and pharmacokinetics of the novel PAR-1 antagonist vorapaxar (formerly SCH 530348) in healthy subjects. Eur J Clin Pharmacol. 2012;68(3):249–258. | ||
Kosoglou T, Reyderman L, Kasserra C, et al. No differences in the pharmacodynamics and pharmacokinetics of the thrombin receptor antagonist vorapaxar between healthy Japanese and Caucasian subjects. Eur J Clin Pharmacol. 2012;68(3):291–300. | ||
Goto S, Yamaguchi T, Ikeda Y, Kato K, Yamaguchi H, Jensen P. Safety and exploratory efficacy of the novel thrombin receptor (PAR-1) antagonist SCH530348 for non-ST-segment elevation acute coronary syndrome. J Atheroscler Thromb. 2010;17(2):156–164. | ||
Tricoci P, Huang Z, Van de Werf F, et al. Net clinical benefit of vorapaxar in NSTE ACS: role of ischemic and bleeding risk stratification. Circulation. 2012;126:A19049. | ||
Leonardi S, Tricoci P, White HD, et al. Effect of vorapaxar on myocardial infarction in the thrombin receptor antagonist for clinical event reduction in acute coronary syndrome (TRACER) trial. Eur Heart J. 2013;34(23):1723–1731. | ||
Thygesen K, Alpert JS, Jaffe AS, et al. Third universal definition of myocardial infarction. Eur Heart J. 2012;33(20):2551–2567. | ||
Whellan DJ, Tricoci P, Chen E, et al. Vorapaxar in acute coronary syndrome patients undergoing coronary artery bypass graft surgery: subgroup analysis from the TRACER trial (Thrombin Receptor Antagonist for Clinical Event Reduction in Acute Coronary Syndrome). J Am Coll Cardiol. 2014;63(11):1048–1057. | ||
Valgimigli M, Tricoci P, Huang Z, et al. Usefulness and safety of vorapaxar in patients with non-ST-segment elevation acute coronary syndrome undergoing percutaneous coronary intervention (from the TRACER Trial). Am J Cardiol. 2014;114(5):665–673. | ||
Held C, Tricoci P, Huang Z, et al. Vorapaxar, a platelet thrombin-receptor antagonist, in medically managed patients with non-ST-segment elevation acute coronary syndrome: results from the TRACER trial. Eur Heart J Acute Cardiovasc Care. 2014;3(3):246–256. | ||
Morrow DA, Alberts MJ, Mohr JP, et al. Efficacy and safety of vorapaxar in patients with prior ischemic stroke. Stroke. 2013;44(3):691–698. | ||
Scirica BM, Bonaca MP, Braunwald E, et al. Vorapaxar for secondary prevention of thrombotic events for patients with previous myocardial infarction: a prespecified subgroup analysis of the TRA 2°P-TIMI 50 trial. Lancet. 2012;380(9850):1317–1324. | ||
Mauri L, Hsieh WH, Massaro JM, Ho KK, D’Agostino R, Cutlip DE. Stent thrombosis in randomized clinical trials of drug-eluting stents. N Engl J Med. 2007;356(10):1020–1029. | ||
Bonaca MP, Scirica BM, Braunwald E, et al. Coronary stent thrombosis with vorapaxar versus placebo: results from the TRA 2° P-TIMI 50 trial. J Am Coll Cardiol. 2014;64(22):2309–2317. | ||
French SL, Arthur JF, Tran HA, Hamilton JR. Approval of the first protease-activated receptor antagonist: Rationale, development, significance, and considerations of a novel anti-platelet agent. Blood Rev. November 6, 2014. [Epub ahead of print]. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.