")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Adenoviral delivery of truncated MMP-8 fused with the hepatocyte growth factor mutant 1K1 ameliorates liver cirrhosis and promotes hepatocyte proliferation
Authors Liu J , Li J, Fu W, Tang J, Feng X, Chen J, Liang Y, Jin R, Xie A, Cai X, Meng F
Received 16 July 2015
Accepted for publication 19 August 2015
Published 16 October 2015 Volume 2015:9 Pages 5655—5667
DOI https://doi.org/10.2147/DDDT.S92481
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Jinghua Liu,1 Jianbo Li,1 Weiwei Fu,2 Jiacheng Tang,3 Xu Feng,1 Jiang Chen,1 Yuelong Liang,1 Ren’an Jin,1 Anyong Xie,4 Xiujun Cai1,3
1Department of General Surgery, Sir Run Run Shaw Hospital, Zhejiang University, Hangzhou, Zhejiang, 2Department of Pathology, The Affiliated Hospital of Medical College, Qingdao University, Qingdao, Shandong, 3Key Lab of Surgery of Zhejiang Province, Sir Run Run Shaw Hospital, Zhejiang University, Hangzhou, Zhejiang, 4Sir Run Run Shaw Hospital and Institute of Translational Medicine, Zhejiang University School of Medicine, Hangzhou, Zhejiang, People’s Republic of China
Abstract: Liver cirrhosis is a chronic liver disease caused by chronic liver injury, which activates hepatic stellate cells (HSCs) and the secretion of extracellular matrix (ECM). Cirrhosis accounts for an extensive level of morbidity and mortality worldwide, largely due to lack of effective treatment options. In this study, we have constructed a fusion protein containing matrix metalloproteinase 8 (MMP-8) and the human growth factor mutant 1K1 (designated cMMP8-1K1) and delivered it into hepatocytes and in vivo and in cell culture via intravenous injection of fusion protein-harboring adenovirus. In doing so, we found that the cMMP8-1K1 fusion protein promotes the proliferation of hepatocytes, likely resulting from the combined inhibition of type I collagen secretion and the degradation of the ECM in the HSCs. This fusion protein was also observed to ameliorate liver cirrhosis in our mouse model. These changes appear to be linked to changes in downstream gene expression. Taken together, these results suggest a possible strategy for the treatment of liver cirrhosis and additional work is warranted.
Keywords: liver cirrhosis, MMP-8, HGF, hepatocytes proliferation
Introduction
Cirrhosis can accompany virtually any chronic liver disease. In a normal, fully functioning liver, the homeostatic hepatic cell/extracellular matrix (ECM) ratio is maintained by the well-balanced synthesis and degradation of various ECM components.1 However, during chronic liver injury, hepatic stellate cells (HSCs) are activated, which subsequently lead to a morphology change to a more myofibroblast-like phenotype2 and an increase in the secretion of ECM components, especially collagen I. If this unbalanced level of ECM secretion remains unchecked, the excess ECM can deposit in the sinusoid space, eventually resulting in liver fibrosis. The end stage of fibrogenesis involves nodule formation in hepatic parenchyma, at which point cirrhosis can be officially diagnosed and altered hepatic function is often observed.3 Thus, while hepatic fibrogenesis may be induced by a variety of factors, the decompensated cirrhosis that ultimately occurs in many patients is considered to be the final, irreversible stage of the process and accounts for the high levels of morbidity and mortality associated with this disease worldwide.4
In fact, this traditional view that liver cirrhosis is not reversible has resulted in many conventional therapies solely focusing on preventing or reducing the biosynthesis of collagen and/or factors responsible for ECM degradation.5,6 However, recent reports suggest that liver cirrhosis may be partially reversed in experimental rats treated with specific matrix metalloproteinases (MMPs), including MMP-17 and MMP-8.8,9 Notably, research regarding the cirrhosis-reversing effects of these MMPs is largely limited.
1K1, encoding the n and K1 domains, in which the low affinity binding site for heparan sulfate proteoglycan (HSPG) of K1 is disrupted through reverse charge mutations of Lys132 and Arg134, is a mutant of the naturally occurring human hepatocyte growth factor (HGF) splice variant of NK1 (K132E:R134E).10 They are convenient agents to develop because they can be efficiently produced in yeast.
In this study, we have constructed a fusion protein using the ammo terminal catalytic domain of MMP-8 and 1K1, with a short linker sequence ((GGGGS)4). By utilizing adenoviral administration, we investigated the effects of this fusion protein on cirrhotic animals, focusing on changes in hepatocyte proliferation as well as inhibition of HSC function in vitro.
Materials and methods
Recombinant plasmid adenoviral vector construction
The full sequence of the cMMP8-1K1 vector was chemically synthesized with a 5′ BamHI and 3′ AgeI excise sites. pAdGV314-cMMP8-1K1, an adenovirus type 5 plasmid vector expressing cMMP8-1K1 under the control of the cytomegalovirus promoter (pCMV) and with a simian virus 40 polyadenylation EGFP signal was constructed using an in vitro ligation method. The expression cassette coding for cMMP8-1K1 was excised with BamHI/AgeI and inserted into the multiple cloning site of the shuttle plasmid pAdGV314, and the resulting plasmid, pAdcMMP8-1K1 was made.
Generation, amplification, and purification of adenovirus
The AdcMMP8-1K1 adenoviral vector was transfected with lipofectamine 2000 into human embryonic kidney 293 (HEK293) cells seeded in six-well plate. The culture medium was refreshed 8 hours later with DMEM containing 10% fetal bovine serum (FBS). The transfected cells were collected 2 days later, a large proportion of which had plaques and observed cytopathic effects. The collected cells were freeze–thawed three times at −80°C and 37°C, respectively, followed by centrifugation at 7,000 rpm for 5 minutes at 4°C. The supernatants were then stored at −80°C.
Viruses were amplified by transduction of HEK293 cells seeded in a T75 flask. Recombinant viruses were harvested and expanded in HEK293 cells to prepare the large-scale stocks. Stocks were stored at −80°C and thawed immediately before use. The purification was performed with an Adeno-X™ Virus Purification Kit (BD Biosciences, San Jose, CA, USA) according to the manufacturer’s instructions. Recombinant adenovirus was tittered with end-point dilutions.
Murine hepatectomy
All animal studies were performed on Balb/c mice in accordance with the animal experimentation guidelines of Sir Run Run Shaw Hospital School of Medicine, Zhejiang University. This study was approved by the ethics committee review board of Sir Run Run Shaw Hospital, School of Medicine, Zhejiang University. AdcMMP8-1K1 and Ad-GFP were intravenously injected into the tail vein of male Balb/c mice (weighing approximately 20–22 g) at a concentration of 109 PFU per mouse. A laparotomy was performed on each mouse 3 days later under anesthesia induced with 8% chloral hydrate (0.4 g/kg), followed by removal of 70% of the left and median lobes of the liver. All mice were killed 3 days after laparotomy. Blood and remnant liver samples were excised, blotted, and weighed, followed by formalin fixation (histology/immunohistochemistry) and snap freezing (total protein content analysis).
CCl4-induced liver fibrosis
Balb/c mouse (18–22 g) were treated with 0.125 mL/kg intraperitoneal (IP) doses of CCl4 in olive oil three times a week for 6 weeks. AdcMMP8-1K1 and Ad-GFP were then intravenously injected into the tail vein at a concentration of 109 PFU per mouse during week 5 and 6. Mice were killed at the end of the 6-week experiment. Liver and blood samples were obtained for each animal as described earlier.
In vitro protein isolation
HEK293 cells, human liver HL-7702 cells, and hepatic stellate LX2 cells were seeded in 100 mm dishes and infected with Ad-GFP or AdcMMP8-1K1 at a concentration of 1×107 PFU/mL in DMEM/10% FBS. The culture medium was replaced with serum-free medium 24 hours later. The cellular lysate and serum-free medium supernatant were collected 72 hours after replacement. The supernatant was subsequently concentrated 40-fold by ultrafiltration (Amicon Ultra-4 series, EMD Millipore, Billerica, MA, USA) at 4°C. The cellular lysate and supernatant were collected from each sample and stored at −80°C until use.
In vitro collagenase activity analysis
Type I collagen proteolytic activity was analyzed as described elsewhere.8 Briefly, HL-7702 cells were treated with AdcMMP8-1K1 or Ad-GFP. The supernatant was collected and ultrafiltrated. To activate the MMP-8 proenzyme, the concentrated media supernatants were incubated for 4 hours at 37°C with 1 mmol/L of activator of proenzyme metalloproteinases (APMA) in APMA buffer (50 mmol/L Tris, 0.2 mol/L NaCl, 10 mmol/L CaCl2, 1 mmol/L ZnCl2, 0.05% Brij35, and 100 mmol/L arginine, pH 7.5). The activated supernatants were then incubated at 37°C for 48 hours with 50 μg/mL of acid-soluble calf type I collagen (Sigma-Aldrich Co., St Louis, MO, USA) with/without 2 mM ethylenediaminetetraacetic acid (EDTA) (100 μg/mL). The type I collagen activity was measured at 4, 24, and 48 hours. The reaction products were analyzed on an 8% sodium dodecyl sulfate–polyacrylamide gel (SDS–PAGE). The gel was then stained with Coomassie Brilliant Blue R-250 (Bio-Rad Laboratories Inc., Hercules, CA, USA) and destained with 35% methanol (or ethanol) and 10% acetic acid.
Analysis of cellular proliferation
HL-7702 cells were seeded in T75 flasks and incubated with AdcMMP8-1K1 and Ad-GFP 24 hours later. These cells were then seeded at the following concentrations, depending on their intended proliferation-related experiments: (1) 15,000/8,000/5,000 cells per well in 96-well plates to be used in MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assays at 24, 48, and 72 hours, respectively, with and without 250 μg/mL 5-fluorouracil (5-FU); (2) 200,000 cells per well in six-well plates for use in the quantitative polymerase chain reaction (qPCR) experiments to analyze proliferating cell nuclear antigen (PCNA) expression, primer shown in Table 1; and (3) 400,000 cells per 60 mm dish to be used in a Western blot analysis for PCNA and phosphorylation of c-Met, Akt, Mek, and Erk.
 | Table 1 Gene primers used in this study |
Quantitative PCR
Likewise, total RNA was extracted from HL-7702 cells and LX2 cells treated with AdcMMP8-1K1 or Ad-GFP to examine transforming growth factor (TGF)-β1, TIMP-1, α-SMA1/2, collagen type I/III, and MMP-9 expression during liver cirrhosis. All primers are shown in Table 1.
Apoptosis detection with flow cytometry
LX2 and HL-7702 cells were seeded in 60 mm dish with 400,000 cells per dish, followed by treatment with AdcMMP-8-1K1 or Ad-GFP. Apoptosis was detected using flow cytometry at 48 hours with 1,000 μg/mL 5-FU with an apoptosis detection kit (BD Biosciences).
Western blot analysis
For the detection of the target proteins in the sample tissues, cells, and supernatants, FLAG (Sigma-Aldrich Co.) and GFP (Sino Biologial Inc., Beijing, People’s Republic of China) antibodies were used. To determine the changes in proliferation and apoptosis protection in the HL-7702 and LX2 cells, a PCNA antibody (Santa Cruz Biotechnology Inc., Dallas, TX, USA), PARP antibody (Cell Signaling Technology, Inc., Danvers, MA, USA), and CASP-3 antibody (Signalway Antibody LLC, College Park, MD, USA) were utilized. Antibodies for phosphorylated c-Met, Akt, Erk1/2, and Mek (all purchased from Cell Signaling Technology, Inc.) were also used to detect their expression in HL-7702 cells. Further, the expression of α-SMA and TGF-β1 in the LX2 was also evaluated using their respective antibodies (Abcam, Cambridge, UK).
Western blot was performed as described: transfected cells proteins were collected and stored at −80°C after a centrifugation at 12,000× g for 15 minutes. Protein amount was determined by bicinchoninic acid assay (BCA, Thermo Fisher Scientific, Waltham, MA, USA). For testing, after denaturation, the proteins were individually separated with gel electrophoresis using 8%–12% SDS–PAGE individually and transferred to polyvinylidene fluoride membrane for 2 hours of blocking in 5% skim milk. The membrane was washed once with TBST (a mixture of Tris-buffered saline and Tween 20) and incubated overnight at 4°C with the relevant aforementioned antibodies. The membrane was again washed three times with TBST, and incubated with secondary antibody (goat anti-rabbit/mouse IgG 1:1,000) for 2 hours at room temperature. The membrane was washed the third time with TBST and then electrochemiluminescence liquid was added, and placed to react in a darkroom. β-Actin was used as a positive control.
Statistical analysis
Statistical analysis was performed using SPSS 16.0, and the results were expressed as values of mean ± SD. Analysis of variance was used to analyze variance among all groups. We performed independent samples t-test, and the statistical significance was set at P<0.05.
Results
Construction and purification of recombinant adenoviral AdcMMP8-1K1
An adenovirus type 5 plasmid vector, pAdGV314-cMMP8-1K1, expressing cMMP8-1K1, was made under the control of the cytomegalovirus promoter (pCMV). The recombinant adenoviral vector AdcMMP8-1K1 was generated after transfection of pAdGV314-cMMP8-1K1 into HEK293 cells, and plaques from the first and second rounds of amplification and from the final amplification production were identified by the appearance of various cytopathic effects and by PCR sequence analysis of the fusion cMMP8-1K1 gene (data not shown).
Expression and localization of AdcMMP8-1K1 and the fusion protein following infection
After injecting Balb/c mice with AdcMMP8-1K1, we sought to evaluate the localization of adenoviral infection using GFP and FLAG antibodies (Figure 1A). It appears that infection of the fusion protein is not homogeneous throughout the animal, and most of the adenovirus was localized to liver, with very little being detected in other tissues, such as heart, spleen, lung, kidney, bowel, or muscle.
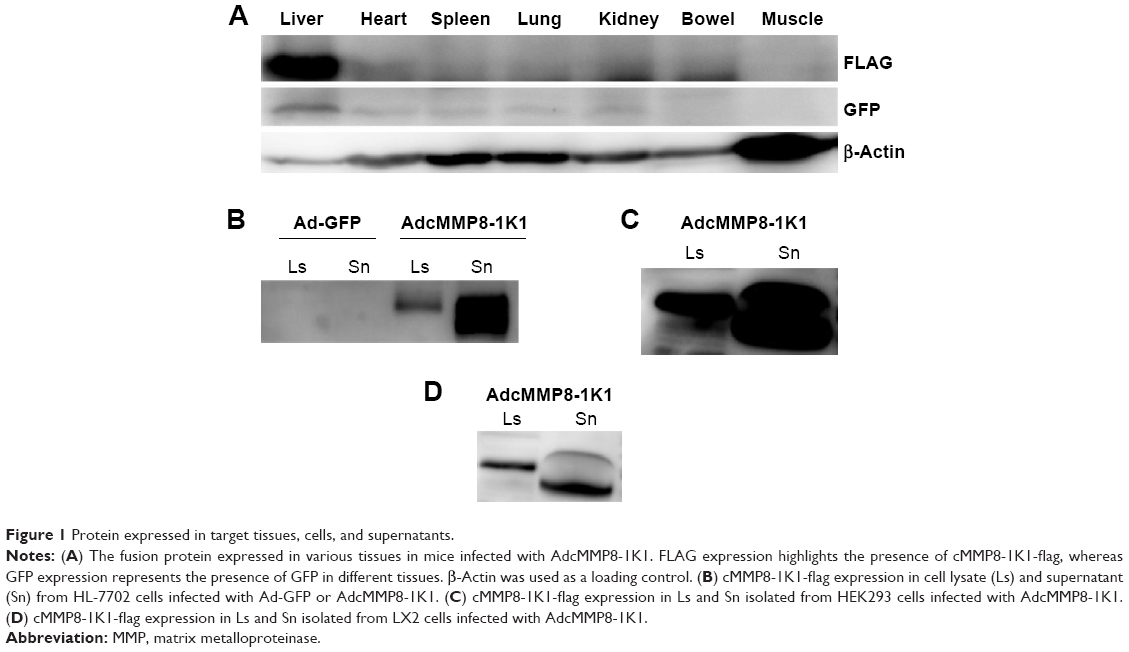 | Figure 1 Protein expressed in target tissues, cells, and supernatants. |
Transduced human liver HL-7702 cells were also incubated with AdcMMP8-1K1 for 48 hours, and the level of fusion protein cMMP8-1K1 (with FLAG-tag) expression in the cell lysate and supernatant was detected by Western blotting with an anti-FLAG antibody. The cells incubated with AdcMMP8-1K1 do, in fact, express cMMP8-1K1, which was observed to be higher in the culture supernatants compared with the cellular lysate (Figure 1B). Similar results were also noted for HEK293 cells (Figure 1C) and hepatic stellate LX2 cells (Figure 1D). Thus, it appears that cells infected with AdcMMP8-1K1 can express the fusion protein cMMP8-1K1 and also secrete it into the supernatant. Notably, the size of the fusion protein in the supernatant is slightly smaller than that found in the lysate, likely due to a structural changing during its activation from proenzyme to enzyme.11
Effects of AdcMMP8-1K1 following 70% hepatectomy
Furthermore, 3 days after injection with either AdcMMP8-1K1 or Ad-GFP, we performed laparotomy and 70% hepatectomy on the animals to investigate the possible function of this adenovirus during the wound-healing response in the liver. The proliferative rate of the hepatocytes, highlighted by the expression of PCNA (Figure 2G) and serum-TGF-β1 (Figure 2F; n=5,4 for AdcMMP8-1K1 and Ad-GFP, individually), was significantly greater in the animals receiving AdcMMP8-1K1 after surgery (P<0.01). Notably, liver function, as determined by the levels of ALT (Figure 2A), AST (Figure 2B), and TBIL (Figure 2D), was also significantly better in the AdcMMP8-1K1-treated mice compared with the Ad-GFP-treated group (P<0.01). However, ALB expression was not significantly altered (Figure 2C, P=0.73). We also observed the weight of the liver in GFP group to be significantly higher (Figure 2E, P<0.01), indicating that AdcMMP8-1K1 may also prevent this postsurgical issue after hepatectomy.
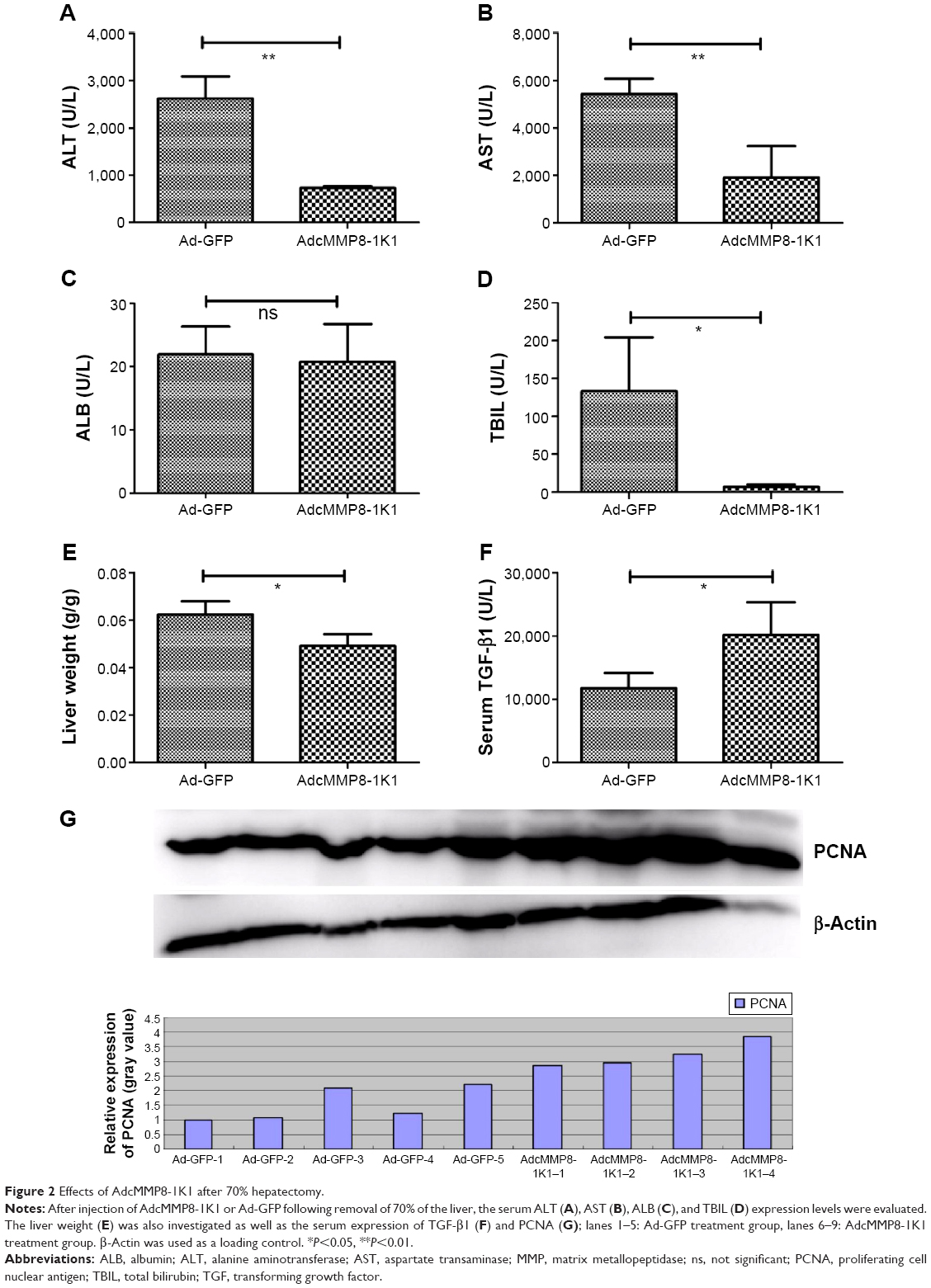 | Figure 2 Effects of AdcMMP8-1K1 after 70% hepatectomy. |
Prevention of CCl4-induced liver fibrosis
Liver fibrosis was induced in Balb/c mice using CCL4 for 6 weeks. In the final 2 weeks of treatment, AdcMMP8-1K1 or Ad-GFP was injected once per week (n=6,4 alive for AdcMMP8-1K1 and Ad-GFP, individually). Following this treatment, we observed the serum ALT (Figure 3A), AST (Figure 3B) to be significantly lower in the AdcMMP8-1K1-treated group (P<0.01). On the other hand, no significant differences were observed in ALB (Figure 3C; P=0.65) or TBIL (Figure 3D; P=0.44) expression. Serum TGF-β1 (Figure 3E) and MMP-9 (Figure 3F) were significantly lower and higher in the AdcMMP8-1K1 group, respectively (P<0.05 and P<0.01, respectively).
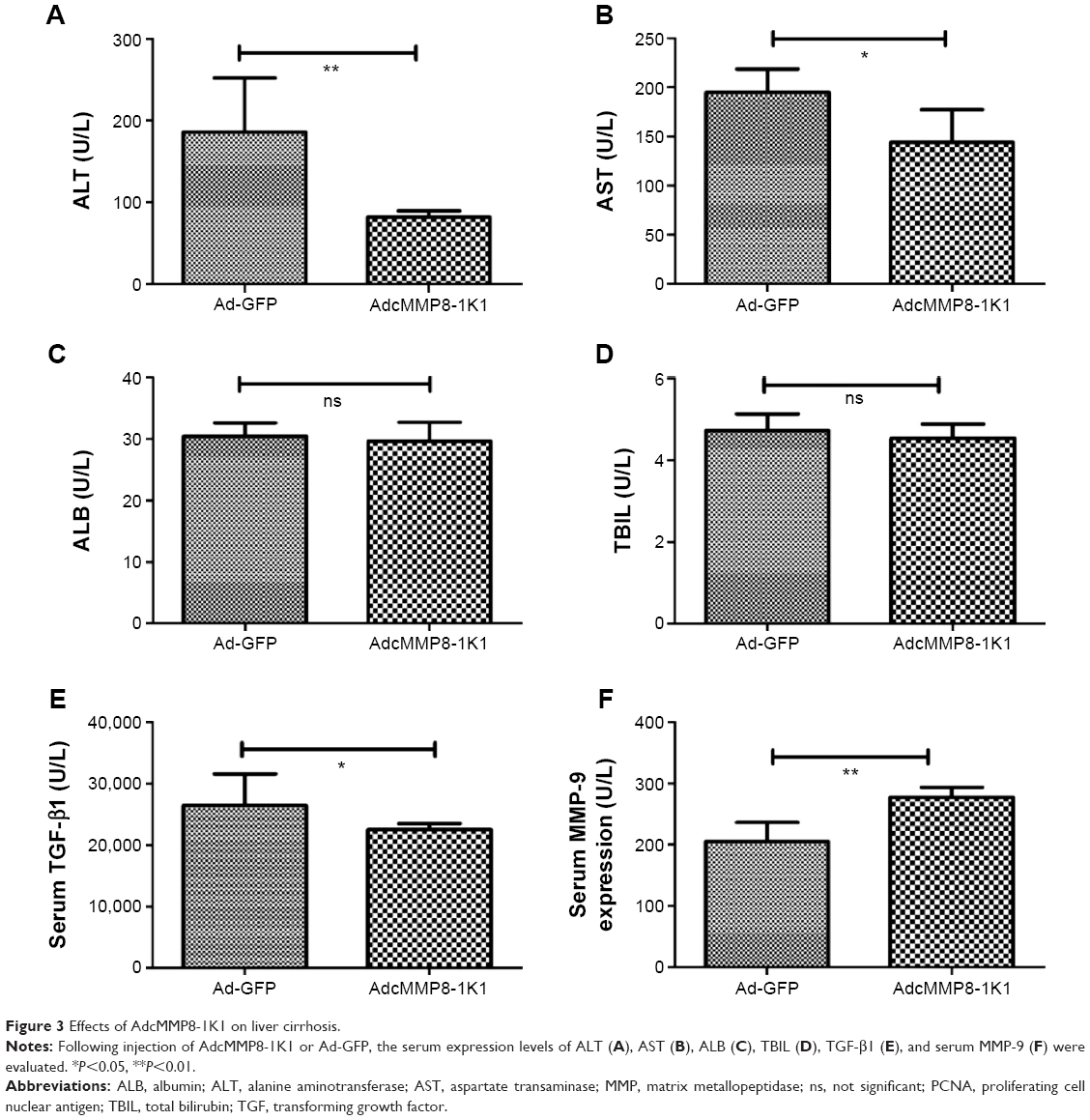 | Figure 3 Effects of AdcMMP8-1K1 on liver cirrhosis. |
Furthermore, our Masson Dyeing analysis showed that collagen, indicating fibrosis, was significantly more abundant in the control Ad-GFP group compared with the AdcMMP8-1K1 group (Figure 4A and B; P<0.05). It also appears that PCNA (Figure 4C and D; P<0.01) is abundantly expressed in the experiment group, whereas the reverse is observed for α-SMA (Figure 4E).
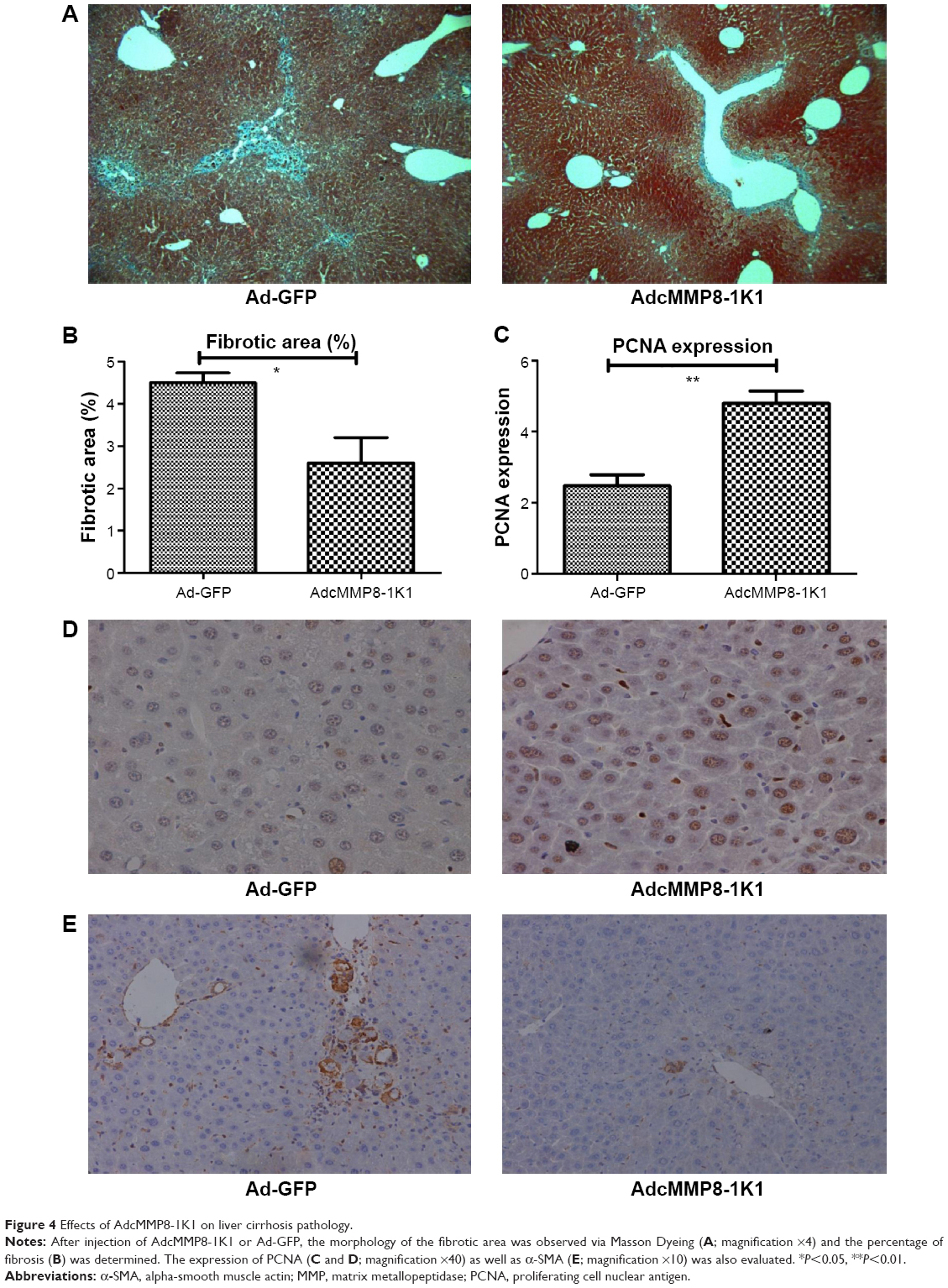 | Figure 4 Effects of AdcMMP8-1K1 on liver cirrhosis pathology. |
In vitro type I collagen activity after AdcMMP8-1K1 infection
The supernatants from human liver HL-7702 cells incubated with Ad-GFP or AdcMMP8-1K1 were used to analyze the proteolytic activity of this fusion protein against type I collagen. To do so, activated culture medium was incubated with type I collagen at room temperature for 48 hours. Indeed, the culture media from cells incubated with Ad-GFP were observed to contain native type I collagen at 120 kDa, whereas the media isolated from cells cultured with AdcMMP8-1K1 appear to contain the characteristic 1A (3/4) and 2A (1/4) cleavage products of type I collagen (Figure 5A), indicating that the MMP8-1K1 fusion protein can cleave this specific fibrosis-related protein.
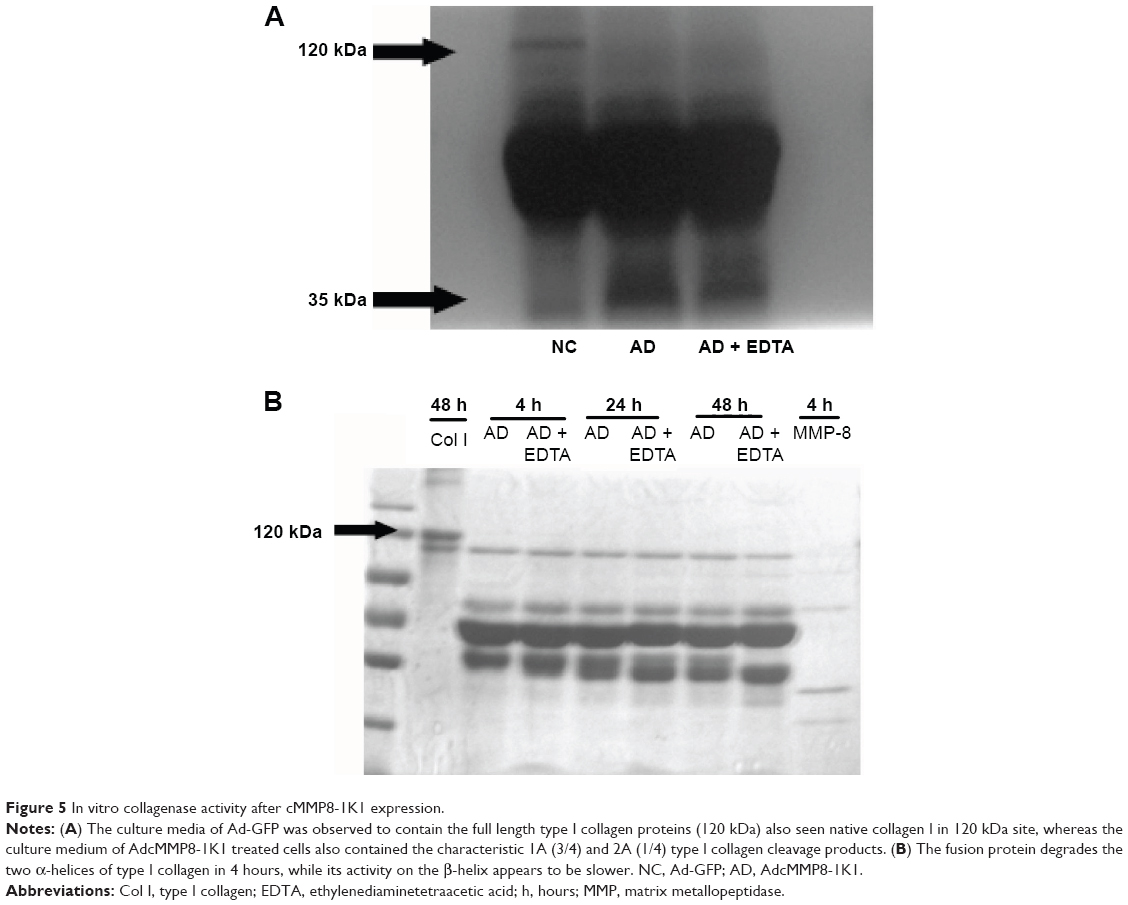 | Figure 5 In vitro collagenase activity after cMMP8-1K1 expression. |
To further test this, we used a time-dependent degradation test, whereby 100 μg/mL of type I collagen was degraded by the fusion protein over a set time course. It appears that the fusion protein can degrade the two α-helices of type I collagen in as little as 4 hours, whereas it takes much longer to degrade the β-helix (Figure 5B). In fact, even after 48 hours, MMP8-1K1 was unable to completely degrade it, indicating that the fusion protein may function on the α-helices rather than on the β-helix.
Effect of cMMP8-1K1 on hepatocyte proliferation in vitro
When we cultured HL-7702 cells with either AdcMMP8-1K1 or Ad-GFP and analyzed the effects on proliferation using an MTT assay, it was apparent that AdcMMP8-1K1 can promote the proliferation of hepatocytes, whereby the ratio of viable HL-7702 cells infected with AdcMMP8-1K1 to Ad-GFP at 24, 48, and 72 hours was increasing. Further, these data were supported by our analysis performed after treatment with 5-FU, with the most obvious proliferative effects being observed at 48 and 72 hours (Figure 6A, P<0.01). Finally, the expression of PCNA detected by qPCR and Western blotting was also shown to increase (Figure 6B, P<0.05), accompanied by increases in the phosphorylation of c-Met, Mek, Erk, and Akt, as shown in Figure 6C.
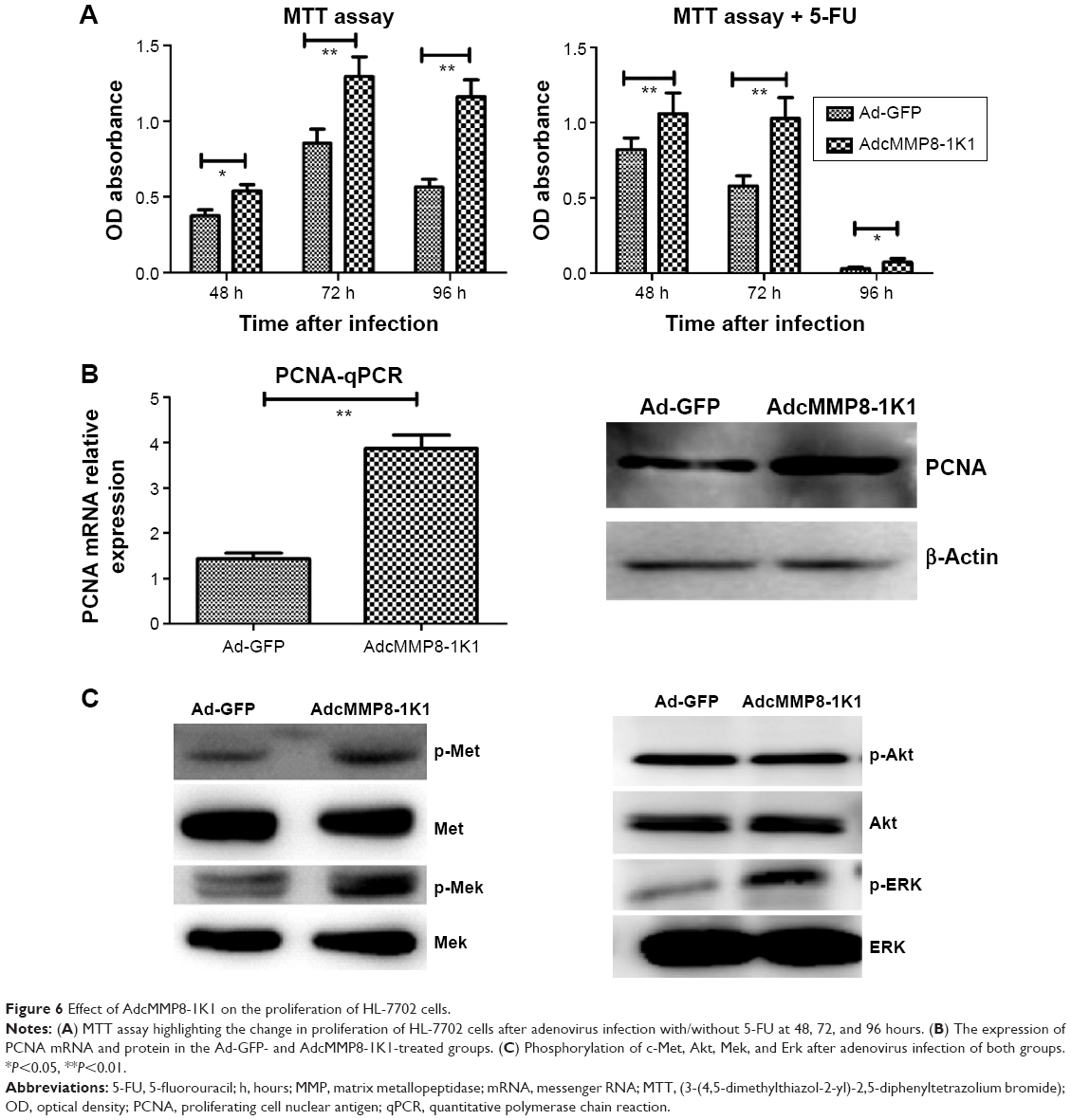 | Figure 6 Effect of AdcMMP8-1K1 on the proliferation of HL-7702 cells. |
Inhibition of liver cirrhosis-related gene expression
Total RNA was extracted from HL-7702 cells and LX2 cells that had been treated with either AdcMMP8-1K1 or Ad-GFP, and the expression of TGF-β1, α-SMA, TIMP-1, MMP-2, MMP-9, Col I, and III was measured by qPCR. Notably, almost all of these cirrhosis-related genes were downregulated in the AdcMMP8-1K1 group compared with the Ad-GFP group (Figure 7A and B, P<0.01).
 | Figure 7 Inhibition of liver cirrhosis-related gene expression and induced apoptosis of HSCs after 5-FU treatment. |
Inducing apoptosis in HSCs after 5-FU treatment
In addition to proliferation- and cirrhosis-related gene expression, we also utilized flow cytometry to analyze the effect the MMP8-1K1 fusion protein on apoptosis using Annexin-PE and 7-AAD. These data indicate that a higher rate of apoptosis was present after 5-FU treatment accompanied by treatment with AdcMMP8-1K1 (Figure 7C, P<0.01). The decreasing protein expression of both TGF-β1 and α-SMA after AdcMMP8-1K1 treatment, determined via Western bolting (Figure 7D), also supports the decrease in fibrosis. Furthermore, the increase in PARP and CASP-3 cleavage, known markers of apoptosis, also highlights these AdcMMP8-1K1-mediated changes after treatment with 5-FU (Figure 7E).
Discussion
In the current literature, the primary focus concerning cirrhosis is on prevention and the basic underlying mechanisms of the disease. Although some studies have investigated treatments to promote hepatocyte proliferation, inhibit HSC activation and/or function, and degrade ECM during liver-related wound-healing responses, some of which are used clinically or are under clinical trial, a cure for liver cirrhosis has yet to be revealed. Liver fibrosis results from chronic damage to the liver in conjunction with the accumulation of ECM proteins, which is characteristic of most types of chronic liver disease.12 This disease also involves a series of activated HSCs, production of type I and type III collagens, hepatocyte dysfunction, and various other clinic syndromes. Thus, understanding the changes occurring in the hepatocyte proliferation rates and degradation of the ECM in vivo and in vitro is important in order to develop an effective cure for liver cirrhosis. To study this, we constructed a fusion protein by connecting cMMP-8 and 1K1 with a short linker and tested its function during liver cirrhosis.
MMP-8 is a collagenase in the MMP family, and some reports have shown that the catalytic domain has enzymatic activity against native type I collagen,13 and tMMP-8 can ameliorate rat liver cirrhosis in vivo.14 Furthermore, MMP-8 is also known to promote liver proliferation in vivo.8,9 We believe that it is unlikely that the effects of MMP-8 on liver cell proliferation and collagen degradation are related; however, until now, there was no previously published information concerning this phenomenon in vitro. The other region of our fusion protein, 1K1 (K132E:R134E), is a mutant of NK1, a naturally occurring splice variant of HGF that encodes the n and k1 domains.10 The mutations at Lys132 and Arg134 cause a charge reversal, which results in disrupted/lower binding affinity for HSPGs.15 The HGF ligand and its receptor, Met receptor tyrosine kinase, play an important role in mediating tissue regeneration.16 HGF is an approximately 80 kDa multidomain protein composed of an N-terminal hairpin domain, four Kringle domains, and a C-terminal serine protease homology domain.17 Notably, HGF has long been considered a potential therapeutic agent. However, there are many limitations for its use: its complex structure, the need for mammalian expression systems, and its rapid sequestration HSPGs.18 Thus, protein engineering was necessary in order to study the effects of this protein in our analysis.
Structural protein engineering is primarily aimed at changing the innate structure of the protein to improve its natural function for therapeutic application or scientific research. In this engineering process, the domains and amino acids essential to the protein functions are determined in order to analyze what changes may or may not alter function, which changes may not be suitable, and what changes may make the protein smaller and easier to process. In this study, protein engineering was used to focus on the function and possible therapeutic effects of MMP-8 and 1K1 because while their function during cirrhosis has been widely investigated, their use as therapeutic agents in clinical trials is limited due to structural/functional restrictions. To this end, we chose the truncated forms of these proteins, which are known to be more stable, and constructed a fusion protein with allowing the combined expression of MMP-8 and 1K1.
After constructing our fusion protein, it was essential to develop a way to express the proteins in vivo as well as in cell culture. Gene therapy using viral gene carriers has been shown to be efficient and safe, especially during treatment for severe liver diseases.19 Adenovirus is an ideal candidate in liver disease treatment as it has a highly efficient infection rate and is distributed in the liver at a high concentration in vivo. Here, we have used Ad-5 as the carrier of our fusion protein and incubated it with HL-7702 and LX-2 cells. The efficiency of infection was above 80%–95%. We also injected this adenoviral carrier into the caudal veins of mice and tested its tissue distribution. In doing so, we found that the vast majority of the infection occurs in the liver, and little of the virus was distributed in other tissue, such as the heart, spleen, lung, kidney, and muscle.8 Furthermore, in a mouse model of liver cirrhosis, 1011 PTU/kg of AdcMMP8-1K1 was enough to induce changes in morphology. Notably, this dose, administered multiple times, did not appear to influence the behavior of the mice. The three cell types we investigated in our in vitro analysis, HEK293, HL-7702, and LX2 cells, were incubated with AdcMMP8-1K1 and appear to not only express the cMMP8-1K1 fusion protein, but also secrete it into the culture supernatant at a high concentration. Further, it is also likely that this secretion mediates activation of the fusion protein from proenzyme to enzyme as this allows other secreted factors to come into contact with the protein in the ECM and culture medium.
Moreover, in our experiments, the fusion protein cMMP8-1K1 was shown to ameliorate liver cirrhosis and promote liver proliferation in our Balb/c mouse model. In fact, in our 70% hepatectomy model, our results show that the fusion protein can protect liver function and aids hepatocyte proliferation. Furthermore, the level of TGF-β1 was also shown to increase in the cMMP8-1K1-treated cell culture group, and we proved that overexpression of the fusion protein in vivo would also lead to an increase of MMPs, such as MMP-9 and a decrease of TIMP-1.20 It is has been shown that an increase in serum TGF-β1 levels results from increased growth of hepatocytes and ECM secretion.21,22 Moreover, in our liver cirrhosis mouse model, the area and density of the fibrous bands in the Masson trichrome staining were thin in animals treated with the cMMP8-1K1 fusion protein compared with those treated with GFP. Liver function, as highlighted by the serum levels of AST and ALT, also supports the function of this fusion protein to amelioration the effects of cirrhosis. These data were not altogether surprising as the effect on hepatocyte proliferation and cirrhosis for MMP-8 and 1K1 have been previously identified in vivo. Thus, we also sought to test the mechanisms underlying these phenomena in vitro.
Using cultures of HL-7702 cells treated with cMMP8-1K1- or GFP-containing adenovirus, we sought to determine if a similar protective/curative effect was observed. Regardless of the time point (48, 72, or 96 hours) and treatment (with/without 5-FU), the effect of cMMP8-1K1 on the proliferation rate of cultured HL-7702 cells was obvious. In this cellular context, cMMP8-1K1 also appears to activate the classic downstream signaling pathways, involving the phosphorylation of c-Met, Erk, Merk, and Akt as well as increased expression of PCNA mRNA and protein.
Notably, many MMPs, including MMP-1, -8, and -13, utilize type I collagen as their main substrate, either the α- or β-helix, and can degrade it into 90 and 30 kDa fragments. In order to determine if this cleavage mechanism functions in our culture model, we analyzed the supernatant of cultured HL-7702 cells infected with AdcMMP8-1K1 or Ad-GFP. In the control, only the full length 120 kDa type I collagen was observed, whereas the 30 kDa degradation product was also found along with the full-length protein when the cells were infected with AdcMMP8-1K1. These data indicate that the fusion protein has the ability to degrade type I collagen, with the main substrate being the α-helices, rather than the β-helix. In addition to promoting hepatocyte proliferation and degrading type I collagen, we also found that the fusion protein can inhibit the expression of TIMP-1, TGF-β1, and α-SMA in the HSC cell line LX2. Notably, these genes are related to the product of ECM, indicating that cMMP8-1K1 may inhibit ECM production by altering the expression of TGF-β1 and α-SMA in the HSC cells in vivo and in vitro.
Finally, induction of HSC apoptosis has also been identified as a kind of method to alleviate liver cirrhosis or retard its advancement. Thus, it was essential to also evaluate changes in apoptosis in our cell culture model. Using flow cytometry, it appears that cMMP8-1K1 can, in fact, induce apoptosis of LX2 cells, and this process involves an increase in the cleavage of PARP and activation of CASP-3 expression. Taken together, it appears that our cMMP8-1K1 fusion proteins could be used as a clinical means to ameliorate liver cirrhosis.
Conclusion
Our results show that cMMP8-1K1 can promote hepatocyte proliferation and ameliorate liver cirrhosis in vivo. This fusion protein can also protect liver function following 70% hepatectomy. The mechanism underlying this function involves the proliferation of hepatocytes, degradation of the ECM, induction of HSC apoptosis, and inhibition of ECM production/secretion. It is important to note that although substantial improvements in function and structure may occur in advanced fibrosis, it is uncertain whether the architectural distortion and vascular derangements in advanced cirrhosis can return to their normal structure.23 However, at the very least, an incomplete resolution of micronodular cirrhosis can occur in experimental animals as well as in cell culture, highlighted in this study. Further, tissue transglutaminase-mediated cross-linking of fibrillar collagens may be a control point for matrix resorption, rendering the hepatic scar resistant to degradation.24 This study provides a foundation for future research investigating the basic function of cMMP8-1K1, and additional studies are warranted in order to further understand the curative abilities of this fusion protein during cirrhosis.
Acknowledgments
The authors thank Dr Lv Zhen of the First Affiliated Hospital of Zhejiang University for providing the HL-7702 cells. This study was supported by grants from the Fundamental Research Funds for the Central Universities (number 2014FZA7005); International Science and Technological Cooperation Projects (number 2012DFA30410); Key Science and Technology Innovation Team Project of Zhejiang Province (number 2009R50040); and the National Science and Technology Support Plan Projects (number 2012BAI14B06). The authors report this research did not involve use of human tissues, and therefore did not require approval by a formally constituted review board for the use of human tissues.
Disclosure
The authors report no conflicts of interest in this work.
References
Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;275:2247–2250. | ||
Geerts A. History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin Liver Dis. 2001;21:311–335. | ||
Friedman SL. Liver fibrosis – from bench to bedside. J Hepatol. 2003;38(Suppl 1):S38–S53. | ||
Guo J, Friedman SL. Hepatic fibrogenesis. Semin Liver Dis. 2007;27:413–426. | ||
Arthur MJ. Reversibility of liver fibrosis and cirrhosis following treatment for hepatitis C. Gastroenterology. 2002;122:1525–1528. | ||
Wu J, Danielsson A. Inhibition of hepatic fibrogenesis: a review of pharmacological candidates. Scand J Gastroenterol. 1994;29:385–391. | ||
Iimuro Y, Nishio T, Morimoto T, et al. Delivery of matrix metalloproteinase-1 attenuates established liver fibrosis in the rat. Gastroenterology. 2003;124:445–458. | ||
Siller-López F, Sandoval A, Salgado S, et al. Treatment with human metalloproteinase-8 gene delivery ameliorates experimental rat liver cirrhosis. Gastroenterology. 2004;126:1122–1133. | ||
Garcia-Bañuelos J, Siller-Lopez F, Miranda A, et al. Cirrhotic rat livers with extensive fibrosis can be safely transduced with clinical-grade adenoviral vectors. Evidence of cirrhosis reversion. Gene Ther. 2002;9:127–134. | ||
Ross J, Gherardi E, Mallorqui-Fernandez N, et al. Protein engineered variants of hepatocyte growth factor/scatter factor promote proliferation of primary human hepatocytes and in rodent liver. Gastroenterology. 2012;142:897–906. | ||
Bode W, Reinemer P, Huber R, Kleine T, Schnierer S, Tschesche H. The X-ray crystal structure of the catalytic domain of human neutrophil collagenase inhibited by a substrate analogue reveals the essentials for catalysis and specificity. EMBO J. 1994;13:1263–1269. | ||
Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. | ||
Siller-López F, García-Bañuelos J, et al. Truncated active matrix metalloproteinase-8 gene expression in HepG2 cells is active against native type I collagen. J Hepatol. 2000;33:758–763. | ||
Liu J, Cheng X, Guo Z, et al. Truncated active human matrix metalloproteinase-8 delivered by a chimeric adenovirus-hepatitis B virus vector ameliorates rat liver cirrhosis. PLoS One. 2013;8:e53392. | ||
Schwall RH, Chang LY, Godowski PJ, et al. Heparin induces dimerization and confers proliferative activity onto the hepatocyte growth factor antagonists NK1 and NK2. J Cell Biol. 1996;133:709–718. | ||
Jones DS 2nd, Tsai PC, Cochran JR. Engineering hepatocyte growth factor fragments with high stability and activity as Met receptor agonists and antagonists. Proc Natl Acad Sci U S A. 2011;108:13035–13040. | ||
Donate LE, Gherardi E, Srinivasan N, Sowdhamini R, Aparicio S, Blundell TL. Molecular evolution and domain structure of plasminogen-related growth factors (HGF/SF and HGF1/MSP). Protein Sci. 1994;3:2378–2394. | ||
Catlow KR, Deakin JA, Wei Z, et al. Interactions of hepatocyte growth factor/scatter factor with various glycosaminoglycans reveal an important interplay between the presence of iduronate and sulfate density. J Biol Chem. 2008;283:5235–5248. | ||
Nakatani T, Kuriyama S, Tominaga K, et al. Assessment of efficiency and safety of adenovirus mediated gene transfer into normal and damaged murine livers. Gut. 2000;47:563–570. | ||
Yoshiji H, Kuriyama S, Yoshii J, et al. Tissue inhibitor of metalloproteinases-1 attenuates spontaneous liver fibrosis resolution in the transgenic mouse. Hepatology. 2002;36:850–860. | ||
Evanko SP, Potter-Perigo S, Petty LJ, Workman GA, Wight TN. Hyaluronan controls the deposition of fibronectin and collagen and modulates TGF-β1 induction of lung myofibroblasts. Matrix Biol. 2014;42:74–92. | ||
Tan J, Tong BD, Wu YJ, Xiong W. MicroRNA-29 mediates TGFβ1-induced extracellular matrix synthesis by targeting wnt/β-catenin pathway in human orbital fibroblasts. Int J Clin Exp Pathol. 2014;7:7571–7577. | ||
Desmet VJ, Roskams T. Cirrhosis reversal: a duel between dogma and myth. J Hepatol. 2004;40:860–867. | ||
Issa R, Zhou X, Constandinou CM, et al. Spontaneous recovery from micronodular cirrhosis: evidence for incomplete resolution associated with matrix cross-linking. Gastroenterology. 2004;126:1795–1808. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.