")
Back to Journals » Drug Design, Development and Therapy » Volume 14
ACE2 Attenuates Epithelial-Mesenchymal Transition in MLE-12 Cells Induced by Silica
Authors Li S, Li Y , Xu H , Wei Z, Yang Y, Jin F , Zhang M, Wang C, Song W, Huo J, Zhao J, Yang X, Yang F
Received 3 March 2020
Accepted for publication 2 April 2020
Published 21 April 2020 Volume 2020:14 Pages 1547—1559
DOI https://doi.org/10.2147/DDDT.S252351
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Tuo Deng
Shumin Li,1,2,* Yaqian Li,2,3,* Hong Xu,3 Zhongqiu Wei,2 Yi Yang,4 Fuyu Jin,2,3 Min Zhang,3 Chen Wang,3 Wenxiong Song,2 Jingchen Huo,2 Jingyuan Zhao,2 Xiuhong Yang,2 Fang Yang1,3
1School of Public Health, North China University of Science and Technology, Tangshan, Hebei 063210, People’s Republic of China; 2School of Basic Medical Sciences, North China University of Science and Technology, Tangshan, Hebei 063210, People’s Republic of China; 3Hebei Key Laboratory for Organ Fibrosis, North China University of Science and Technology, Tangshan, Hebei 063210, People’s Republic of China; 4Academic Affairs Office, North China University of Science and Technology, Tangshan, Hebei 063210, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Fang Yang
School of Public Health, North China University of Science and Technology, No. 21 Bohai Road, Caofeidian Eco-City, Tangshan, Hebei 063210, People’s Republic of China
Tel +86 18633309386
Fax +86-315-8805522
Email [email protected]
Xiuhong Yang
School of Basic Medical Sciences, North China University of Science and Technology, No. 21 Bohai Road, Caofeidian Eco-City, Tangshan, Hebei 063210, People’s Republic of China
Email [email protected]
Purpose: The role of angiotensin-converting enzyme 2 (ACE2) in silicosis remains unknown, although previous studies have suggested that ACE2 may be beneficial. We, therefore, investigated the effect of ACE2 on silicosis, particularly with regard to its role in regulating the epithelial-mesenchymal transition (EMT) induced by silica, with the aim to uncover a new potential target for the treatment of pulmonary fibrosis.
Materials and Methods: We employed wild-type mice treated with diminazene aceturate (DIZE, an ACE2 activator, 15 mg/kg/day for 4 weeks), hACE2-transgenic mice (overexpress the ACE2 gene), and the mouse lung type II epithelial cell line treated with DIZE (10− 7 M for 48 h) or angiotensin-(1– 7) [Ang-(1– 7)] (10− 4 M for 48 h), following induced fibrotic responses to determine the protective potential of ACE2. Silicosis models were established by orotracheal instillation of SiO2 (2.5 mg/mouse). Immunostaining was used to determine α-smooth muscle actin (α-SMA) expression. The activities of angiotensin-converting enzyme (ACE) and ACE2 and the levels of angiotensin II (Ang II) and Ang-(1-7) were detected by enzyme-linked immunosorbent assay. The mRNA expression of ACE and ACE2, and protein expression of the renin-angiotensin system (RAS) components and EMT indicators were studied by qRT-PCR and Western blot, respectively.
Results: DIZE treatment and overexpression of ACE2 markedly inhibited the formation of silica-induced lung fibrosis and increased the level of E-cadherin, with concomitant downregulation of pro-collagen, vimentin, and α-SMA via RAS signaling. Furthermore, DIZE and Ang-(1– 7) attenuated the EMT and collagen deposition induced by silica in MLE-12 cells. Moreover, these effects were abrogated by MLN-4760 (a specific ACE2 inhibitor) and A779 (a specific Mas receptor blocker).
Conclusion: The overexpression of ACE2 and treatment with DIZE can ameliorate EMT in silicotic mice via activation of the ACE2-Ang-(1– 7)-Mas receptor axis, and these changes are accompanied by suppression of the ACE–Ang II–AT1 receptor axis.
Keywords: angiotensin-converting enzyme 2, angiotensin-converting enzyme, silicosis fibrosis, diminazene aceturate, renin–angiotensin system, transgenic mice
Introduction
Silicosis is a major occupational health hazard throughout the world.1 Previous studies by our group found that macrophage activation, myofibroblast differentiation, and epithelial-mesenchymal transition (EMT) are important factors in the pathogenesis of silicosis,2,3 while several other studies have confirmed that activation of the local renin–angiotensin system (RAS) in lung tissue is involved in the development of pulmonary fibrosis.4–6 However, the pathomechanisms of silicosis remain to be fully elucidated.
Inhibition of the upstream angiotensin-converting enzyme (ACE)/Ang II/angiotensin receptor type 1 (AT1R) pathway and activation of the downstream angiotensin-converting enzyme 2 (ACE2)/Ang-(1-7)/Mas receptor pathway are two feasible treatment strategies with demonstrated efficacy in various pulmonary disease models.7 Our recent studies also showed that Ac-SDKP (N-acetyl-seryl-aspartyl-lysyl-proline) exerts an anti-silicotic effect through activation of the ACE2-Ang-(1–7)-Mas axis.8,9
Our previous study found that the expression of ACE, Ang II and AT1 increased while that of ACE2 decreased in local lung tissue in mice with acute lung injury induced by limb ischemia-reperfusion. After preventive intervention with DIZE for 4 weeks, the lesions of the lung injury were attenuated, and the production of Ang-(1–7) and Mas receptor protein were up-regulated in the lung, whereas the opposite trend was observed for Ang II and the AT1 receptor protein. Compared with wild-type mice, the application of DIZE in hACE2 (human ACE2) transgenic mice resulted in similar expression changes but to a greater magnitude.10 Moreover, we observed high ACE2 expression in the epithelial cells of the lung, and ACE2 inhibited myofibroblast differentiation induced by Ang II in fibroblasts.8 We, therefore, hypothesized that increased ACE2 expression may have a beneficial effect on alveolar epithelial cells. Several studies have shown that diminazene aceturate (DIZE) is an activator of ACE2 and that treatment with DIZE is beneficial for myocardial, liver, and biliary fibrosis in mice.11–13 In addition, DIZE combined with physical exercise potentiates the reduction of bleomycin-induced pulmonary fibrosis.14
Therefore, in the present study, we focused on the effect of ACE2 on silicosis, particularly with regards to its role in regulating EMT induced by silica. For this purpose, we used silica-treated hACE2 (human ACE2)-transgenic mice and MLE-12 cells to explore the respective effects of endogenous overexpression of ACE2 and exogenous activation (DIZE intervention) on silicosis, with the aim to uncover a new potential target for the treatment of pulmonary fibrosis.
Materials and Methods
Animals and Experimental Procedures
Ninety-eight- to twelve-week-old male ICR mice, weighing 30 ± 3 g, were used in the experiments. The wild-type and F1 generation of the human ACE2 (hACE2)-transgenic ICR mice were kindly supplied by the Institute of Laboratory Animal Sciences, CAMS & PUMC (Beijing, China), and fed in the Laboratory Animal Center of North China University of Science and Technology. All mice were housed in a specific pathogen-free facility and were maintained under conditions of constant temperature (23–25°C), humidity (40–50%), and a 12-h light/dark cycle. Animals were cared for in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 8023, revised 1978), and the experimental procedures were subject to the approval of the Ethics Committee of the North China University of Science and Technology (approval number 2013–038 and 2017–025).
Wild-type mice were randomly divided into five groups (n = 10 each) as follows: 1) wild-type control group, 2) wild-type silicosis model group, 3) DIZE (15 mg/kg/day, i.p., D7770; Sigma-Aldrich, St. Louis, MO, USA)15 treatment group, 4) DIZE + A779 (5 mg/kg/day, s.c., 4030357; Bachem, Bubendorf, Switzerland)16 treatment group, and 5) DIZE + MLN-4760 (1 mg/kg/day, s.c. 530,616; Millipore, Darmstadt, Germany)17 treatment group. The hACE2-transgenic ICR mice were randomly divided into four groups (n = 10 each) as follows: 1) hACE2-transgenic control group; 2) hACE2-transgenic silicosis model group, 3) A779 treatment group, and 4) MLN-4760 treatment group.
Mice were anesthetized by 1% pentobarbital sodium (50 mg/kg, i.p.),18 except for the wild-type and hACE2-transgenic control groups (intratracheal administration of 50 μL saline), all mice were subjected to once-off intratracheal administration of 50 μL SiO2 suspension (50 mg/mL, equal to 2.5 mg/mouse, Cat. No. 274739, mean particle size <5 μm; Sigma-Aldrich)18 at the same time as receiving the treatment specific to each experimental group. The mice were sacrificed after 4 wk of treatment, and their lungs were isolated. Left lung tissues were fixed with 4% paraformaldehyde solution, and the rest of the lungs were stored at −80°C until analysis.
Cell Culture and Experimental Procedures
MLE-12 cells (mouse lung type II epithelial cell line) were obtained from the American Type Culture Collection (CRL-2110; ATCC, Manassas, VA, USA). MLE-12 cells were grown as monolayer cultures routinely in DMEM/F-12 50/50 medium (10-092-CVR; Mediatech, VA, USA) supplemented with 10% fetal bovine serum (10099141; Gibco, Thermo Fisher Scientific, Madison, WI, USA) and 1% penicillin and streptomycin (P06-07001; Pan Biotech, Aidenbach, Germany) and maintained at 5% CO2 and 37°C.
After serum starvation for 24 h, the cells were cultured with various combinations of the following: SiO2 (50 μg/cm3), 10−7 mol/L Ang-(1–7) (4084113; Bachem),19 10−5 mol/L A779, 10−4 mol/L DIZE,20 and 10−5 mol/L MLN-4760.21 Cells were harvested for total RNA and protein extraction after treatment for 48 h.
Immunohistochemistry (IHC)
Immunostaining for α-smooth muscle actin (α-SMA, EPR5368; Epitomics, Burlingame, CA, USA) was performed on lung sections as described previously.8,9 Brown staining indicated samples positive for α-SMA expression.
Enzyme-Linked Immunosorbent Assay (ELISA)
The ELISA method was used to determine ACE/ACE2 activity and Ang II/Ang-(1–7) levels in the lung tissue and serum samples. For this purpose, 100 mg of lung tissue was rinsed with phosphate buffer saline (PBS), homogenized in 1 mL of PBS, and stored overnight at −20°C. Two freeze-thaw cycles were then performed to disrupt the cell membranes, and the homogenates were centrifuged at 5,000 ×g, 4°C for 5 min. The resulting supernatants were removed and stored at −80°C. The samples were then centrifuged again after thawing before the assay. Serum samples were allowed to clot overnight at 4°C before centrifugation at 1,000 ×g, 4°C for 20 min. The supernatant was harvested immediately and stored at −80°C for later analysis. Then, the ACE (MB-0005A; Jiangsu Meibiao Biological Technology, Yancheng, China)/ACE2 (MB-3569A; Jiangsu Meibiao Biological Technology) activity and Ang II (CSB-E04495m; Cusabio, Baltimore, MD, USA)/Ang-(1–7) (CSB-E13763m; Cusabio) concentration in the lung tissue and serum were detected with the ELISA kits according to the manufacturer’s instructions. Enzyme activity was expressed in U/L and peptide concentration in pg/mL.
Total RNA Extraction, Reverse Transcription, and Quantitative Real-Time PCR (qRT-PCR)
Lung tissue was ground in liquid nitrogen and total RNA was extracted with TRIzol reagent (15,596–018; Invitrogen, Carlsbad, CA, USA) according to a standard method. RNA samples (2 μg each) were reverse-transcribed to synthesize cDNA (ZR102-2; Beijing Zoman Biotechnology, Beijing, China). Lung mRNA expression of ACE and ACE2 was evaluated by qRT-PCR (2× SYBR qPCR Mix, ZF102-2; Beijing Zoman Biotechnology) according to the manufacturer’s instructions on an ABI 7500 Real-Time PCR System (Thermo Fisher Scientific).
The primer sequences (synthesized by Thermo Fisher Scientific) were: ACE sense, 5′-CAGTGTCTACCCCCAAGCAT-3′; ACE antisense, 5′-CTTCCATCAAAGACCCTCCA-3′;22 ACE2 sense, 5′-CCTTCTCAGCCTTGTTGCTGTTAC-3′; ACE2 antisense, 5′-TGCCCAGAGCCTAGAGTTGTAGTC-3′;23 β-actin sense, 5′-GTCGTACCACAGGCATTGTGATGG-3′; and β-actin antisense, 5′-GCAATGCCTGGGTACATGGTGG-3′.22 Expression levels were normalized with the 2−ΔΔCt formula based on the Ct values of the respective RNA samples relative to the housekeeping gene, β-actin.
Protein Isolation and Western Blot
Lung tissues and cells were homogenized in ice-cold RIPA lysis buffer containing 1% protease inhibitor for 30 min. The lysates were centrifuged at 13,800 ×g, 4°C for 20 min and the supernatants were then recovered. Protein concentrations were measured with a bicinchoninic acid protein assay kit [PQ0012; Multisciences (Lianke) Biotech, Hangzhou, China]. The supernatants were boiled in 5× bromophenol blue sample buffer for 5 min, and 10 μL (20 μg) of each sample was applied to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The separated proteins were transferred to a polyvinylidene fluoride membrane. The membranes were then blocked with 5% non-fat milk and hybridized with a primary antibody against each of α-SMA (EPR5368; Epitomics), collagen I (pro-Col, AF7001; Affinity Biosciences, OH, USA), collagen III (pro-Col III, AF0136; Affinity Biosciences), vimentin (Vim, ARG57462; Arigo Biolaboratories, Hsinchu, Taiwan), E-cadherin (E-cad, A11453; ABclonal Technology, Boston, MA, USA), ACE (EPR2757; Epitomics), ACE2 [EPR4435(2); Epitomics], AT1 (EPR3873; Epitomics), Mas (sc-390453; Santa Cruz Biotechnology, Dallas, TX, USA), Ang II (GTX37789; GeneTex Inc., San Antonio, TX, USA) and Ang-(1-7) (P1787; EIAab Science Co., Wuhan, China) at 4°C overnight. ECL and horseradish peroxidase-conjugated secondary antibodies toward rabbit (5220–0336; SeraCare, Samuel, CA, USA) and mouse (5220–0341; SeraCare) IgG were used for detection. Protein expression was normalized to that of GAPDH (AC036; ABclonal) or tubulin (AF7010; Affinity Biosciences).
Data Presentation and Statistical Analysis
SPSS 17.0 statistical software (SPSS Inc, Chicago, IL, USA) was used to analyze the data. All data were expressed as the mean ± standard deviation (SD). One-way analysis of variance (ANOVA) and the least significant difference (LSD) test were used to determine the significance of differences between groups. Statistical significance was defined as p < 0.05.
Results
DIZE Inhibits SiO2-Induced EMT and Attenuates Silicotic Fibrosis
Based on histology using immunohistochemistry staining for α-SMA, we observed nodule formation with extensive α-SMA expression in the nodular lesions and pulmonary interstitial region (Figure 1A), and large regions of silicosis were observed, indicating fibrotic response in the lung tissue of silicotic mice (Figure 1B). Treatment with DIZE overcame the fibrotic remodeling and reduced the silicotic area in silica-exposed lungs. Western blotting (Figure 1C) indicated that treatment with DIZE dramatically decreased the protein expression of α-SMA, Vim, pro-Col I, and pro-Col III in the lungs (Figure 1D–G) while augmenting the expression of E-cad (Figure 1H). Furthermore, we observed that treatment with MLN-4760 (an ACE2 inhibitor) or A779 (a Mas receptor blocker) reversed the suppression of silicosis by DIZE, with significantly increased expression of α-SMA, Vim, pro-Col I, and pro-Col III expression in the lungs along with markedly reduced expression of E-cad (Figure 1D–H).
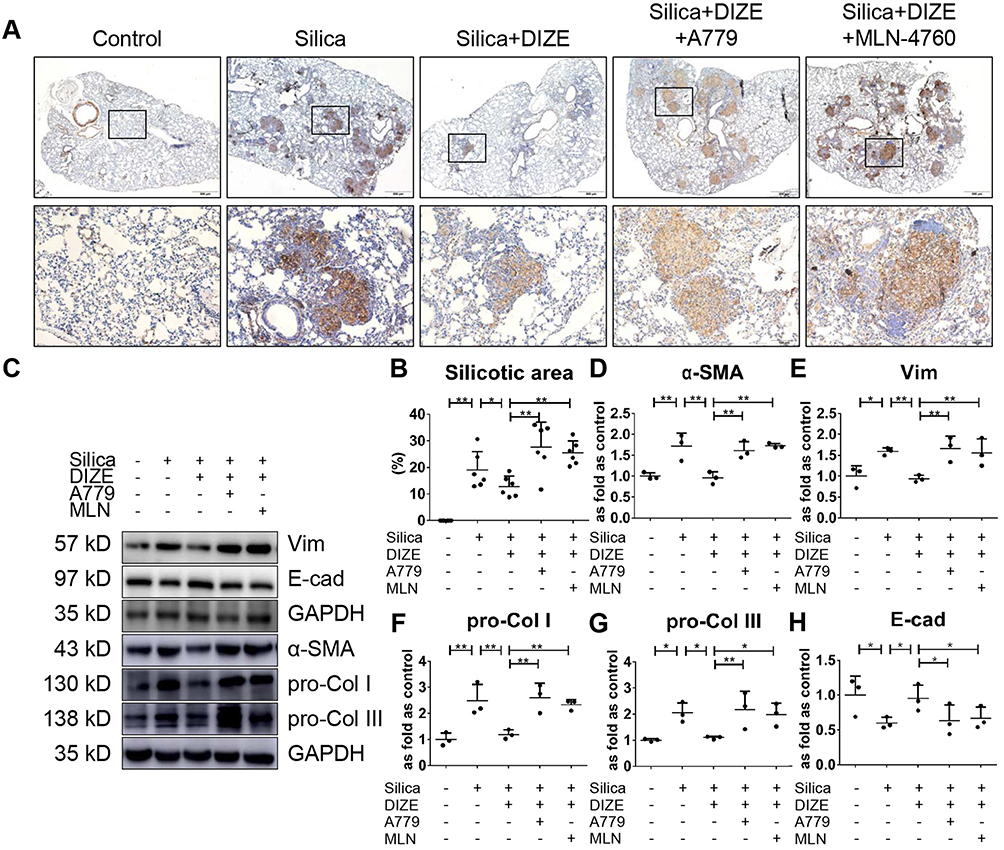 |
Figure 1 DIZE inhibits SiO2-induced EMT and collagen deposition in wild-type silicotic mice. (A) Immunochemistry staining of α-SMA expression in the lungs of wild-type mice, n = 6 in each group. Magnification, ×40 (top panel) and ×200 (bottom panel). Silicotic mice underwent various treatment combinations with DIZE (ACE2 activator), A779 (Mas receptor blocker), and MLN-4760 (ACE2 inhibitor). (B) The proportion of silicotic areas in the lung samples in (A). (C) Western blot showing the protein expression of α-SMA (D), Vim (E), pro-Col I (F), pro-Col III (G), and E-cad (H) in the lungs of mice from the various treatment groups. Values represent the mean ± SD, n = 3 independent experiments, fold change is expressed relative to the control (no treatments), *P < 0.05 vs corresponding group, **P < 0.01 vs corresponding group. |
We also measured RAS activity in the lung tissue and serum samples to analyze the role of circulating and local RAS in silicosis. Serum ACE2 activity was decreased in silicotic mice and this effect was reversed by DIZE treatment, while the level of ACE activity and the concentration of Ang II, and Ang-(1–7) in the serum were unchanged (Figure 2A–D). In the lung tissue, treatment with DIZE decreased the activity of ACE but increased the activity of ACE2, while the concentration of Ang II decreased and that of Ang-(1–7) increased (Figure 2E–H). Furthermore, the beneficial effects of DIZE on the lung tissue were blocked by MLN-4760 and A779. These results implied that circulatory and local RAS may perform distinct physiological and pathological roles in mice exposed to silica.
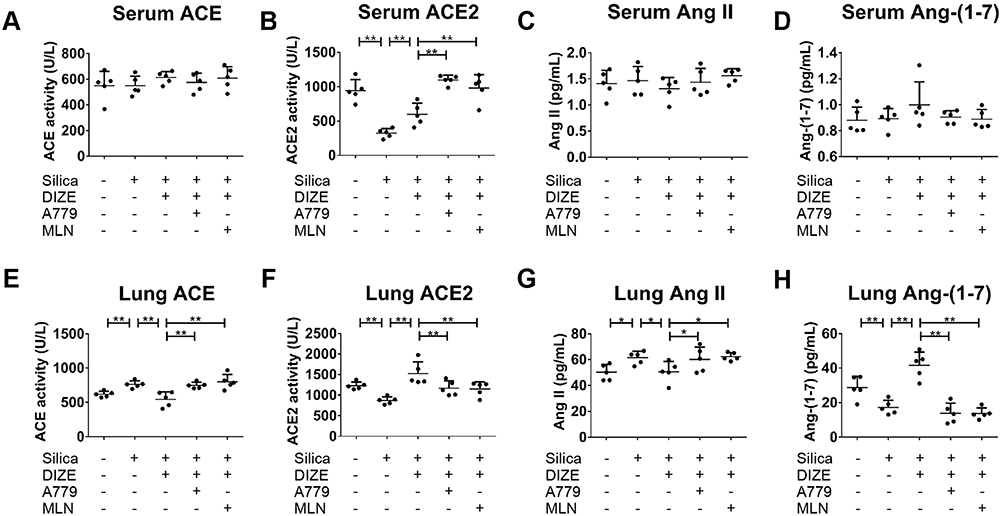 |
Figure 2 DIZE regulates the activity of ACE/ACE2 and the concentration of Ang II/Ang-(1–7) in wild-type silicotic mice. (A, B) The activity of ACE/ACE2 was measured in the serum samples of silicotic mice, along with (C, D) the concentration of Ang II/Ang-(1–7), following various treatment combinations with DIZE, A779, and MLN-4760. (E, F) The activity of ACE/ACE2 was measured in lung tissue samples from silicotic mice, along with (G, H) the concentration of Ang II/Ang-(1–7). ELISA was used for all experiments. Values represent the mean ± SD, n = 5 independent experiments, *P < 0.05 vs corresponding group, **P < 0.01 vs corresponding group. |
Additionally, Western blotting results (Figure 3A) indicated that treatment with DIZE resulted in increased protein and mRNA levels of ACE2 (Figure 3B and C) and elevated protein levels of Mas (Figure 3D), accompanied by reduced protein and mRNA expression of ACE (Figure 3E and F) and reduced protein expression of AT1R (Figure 3G). All of these changes were reversed by A779 and MLN-4760 treatment. These results indicated that the activation of ACE2 by DIZE suppressed the lung fibrotic response toward exposure to silica.
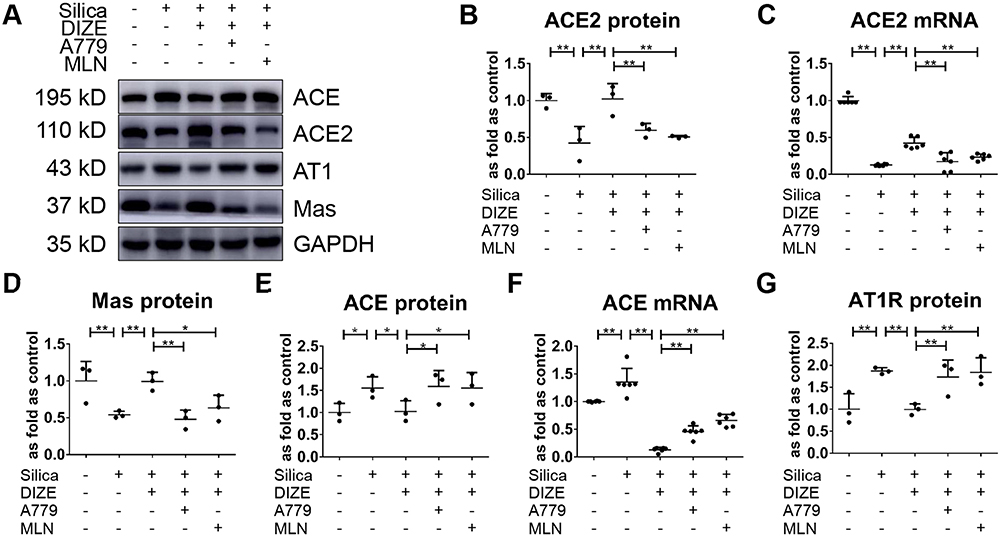 |
Figure 3 DIZE regulates lung RAS in wild-type silicotic mice. (A) Western blot showing the protein expression of various components of lung RAS in the lungs of wild-type silicotic mice. (B, C) The protein and mRNA expression levels of ACE2, respectively. (D) The protein expression levels of Mas. (E, F) The protein and mRNA expression levels of ACE, respectively. (G) The protein expression level of AT1R. Protein and mRNA expression levels were quantified based on the results of the Western blot in (A) and RT-qPCR, respectively. Values represent the mean ± SD, n = 3 and n = 6 independent experiments for protein and mRNA detection, respectively, fold change is expressed relative to the control (no treatments), *P < 0.05 vs corresponding group, **P < 0.01 vs corresponding group. |
Overexpression of hACE2 Exerts an Anti-Fibrotic Effect in Mice Exposed to Silica
To assess whether augmenting endogenous ACE2 has anti-fibrotic effects in vivo, murine overexpression of hACE2 was employed. We detected fewer nodules, lower α-SMA positive expression, and a lower proportion of silicotic area in the silicotic hACE2-transgenic mice compared with silicotic wild-type mice (Figure 4A and B), as well as sustained decreases in α-SMA, Vim, pro-Col I, and pro-Col III (Figure 4C–G), and a slight increase of E-cad (Figure 4H) in hACE2 mice exposed to silica in comparison with wild-type silicotic mice.
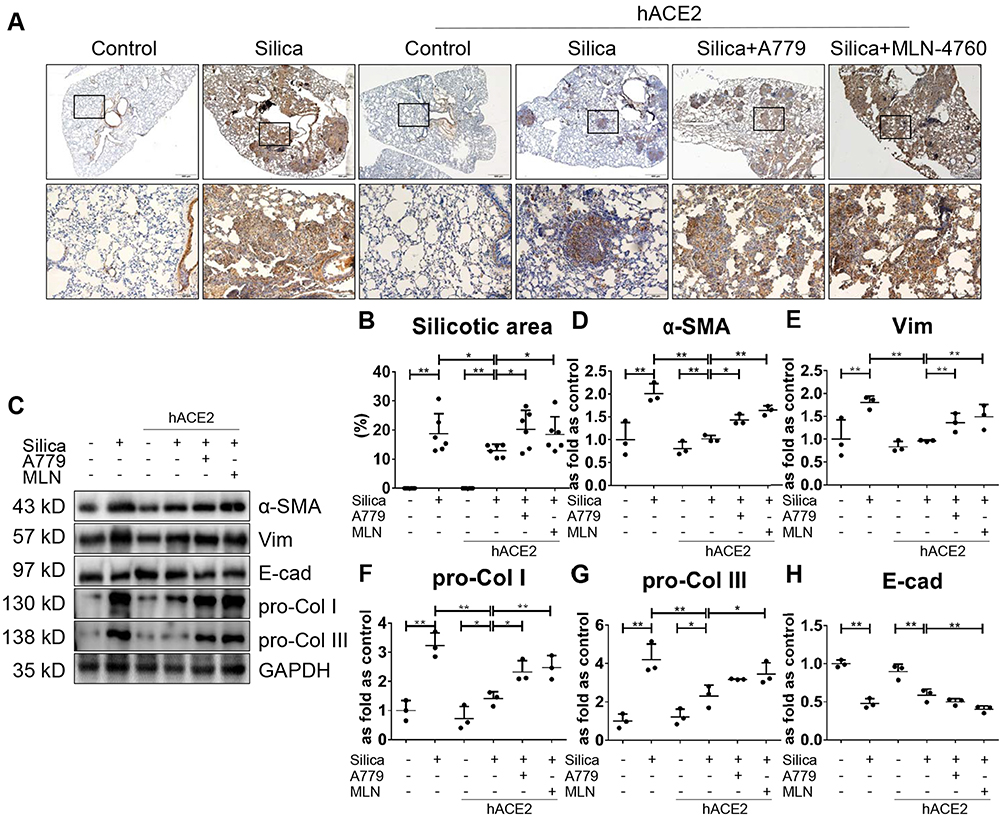 |
Figure 4 Overexpression of hACE2 inhibits SiO2-induced EMT and collagen deposition in hACE2-transgenic silicotic mice. (A) Immunochemistry staining of α-SMA expression in the lungs of hACE2-transgenic mice, n = 6 in each group. Magnification, ×40 (top panel) and ×200 (bottom panel). Silicotic mice underwent various treatment combinations with DIZE (ACE2 activator), A779 (Mas receptor blocker), and MLN-4760 (ACE2 inhibitor). (B) The proportion of silicotic areas in the lung samples in (A). (C) Western blot showing the protein expression of α-SMA (D), Vim (E), pro-Col I (F), pro-Col III (G), and E-cad (H) in the lungs of mice from the various treatment groups. Values represent the mean ± SD, n = 3 independent experiments, fold change is expressed relative to the control (wild-type mice with no treatments), *P < 0.05 vs corresponding group, **P < 0.01 vs corresponding group. |
The ACE2 activity in the lung tissue and serum (Figure 5A and B), as well as the protein and mRNA expression levels of ACE2 in the lung tissue (Figure 6A-C), were higher in the silicotic hACE2-transgenic mice compared with the silicotic wild-type mice. Similarly, the concentration of Ang-(1–7) in the lung tissue and serum was higher in the hACE2-transgenic group (Figure 5C and D). This was accompanied by decreased ACE activity in the lung tissue (unchanged in the serum) (Figure 5E and F) and decreased ACE protein and mRNA expression levels in the lungs of the hACE2-transgenic group (Figure 6D and E). Ang II concentration was also lower in the lung tissue (unchanged in the serum) in the hACE2-transgenic group compared with the wild-type group (Figure 5G and H), with increased Mas and lower AT1R protein levels present in the lung tissue from the transgenic group (Figure 6F and G). Thus, the overexpression of ACE2 was shown in vivo to have a beneficial effect on silicosis by upregulating the ACE2-Ang-(1–7)-Mas receptor axis and suppressing the ACE-Ang II-AT1 receptor axis.
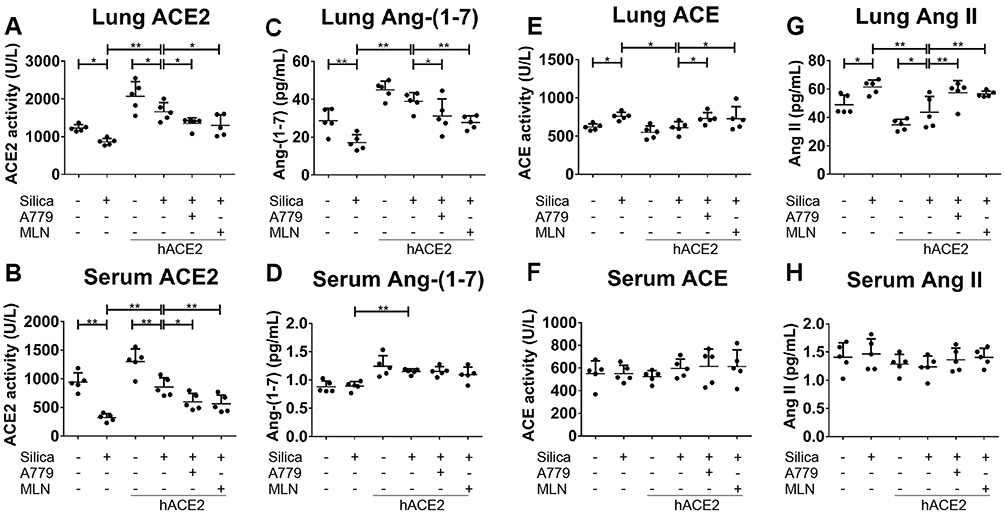 |
Figure 5 Overexpression of hACE2 regulates the activity of ACE/ACE2 and the concentration of Ang II/Ang-(1–7) in hACE2-transgenic silicotic mice. (A, B) The activity of ACE2 in the lung tissue and serum, respectively. (C, D) The concentration of Ang-(1–7) in the lung tissue and serum, respectively. (E, F) The activity of ACE in the lung tissue and serum, respectively. (G, H) The concentration of Ang II in the lung tissue and serum, respectively. ELISA was used for all experiments. Values represent the mean ± SD, n = 5 independent experiments, *P < 0.05 vs corresponding group, **P < 0.01 vs corresponding group. |
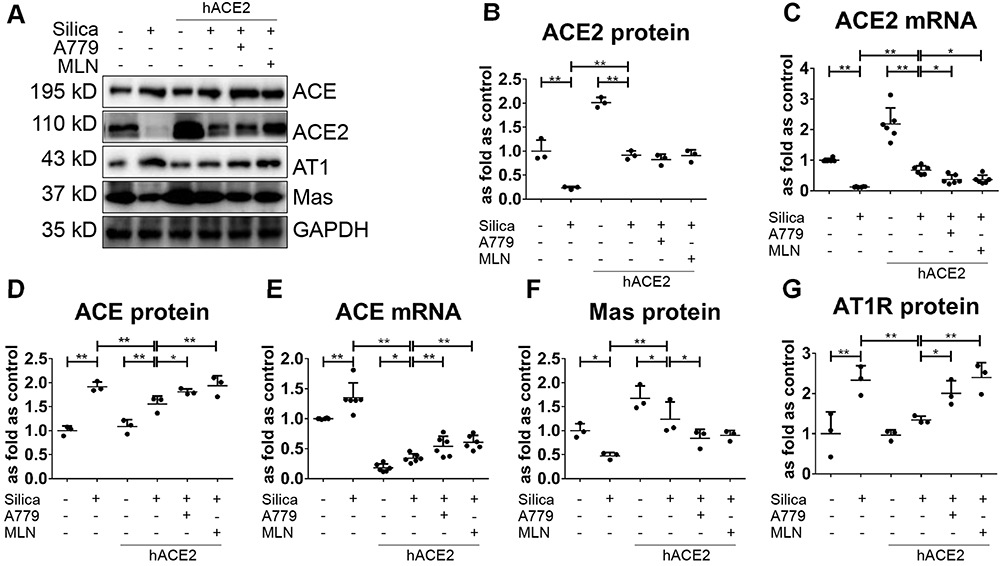 |
Figure 6 Overexpression of hACE2 regulates lung RAS in hACE2-transgenic silicotic mice. (A) Western blot showing the protein expression of various components of lung RAS in the hACE2-transgenic silicotic mice. (B, C) The protein and mRNA expression levels of ACE2, respectively. (D, E) The protein and mRNA expression levels of ACE, respectively. (F) The protein expression levels of Mas. (G) The protein expression level of AT1R. Protein and mRNA expression levels were quantified based on the results of the Western blot in (A) and RT-qPCR, respectively. Values represent the mean ± SD, n = 3 and n = 6 independent experiments for protein and mRNA detection, respectively, fold change is expressed relative to the control (wild-type mice with no treatments), *P < 0.05 vs corresponding group, **P < 0.01 vs corresponding group. |
Ang-(1–7) Inhibits SiO2-Induced EMT in MLE-12 Cells
We treated MLE-12 alveolar type II epithelial cells with SiO2. Immunohistochemistry staining (Figure 7A) and Western blotting (Figure 7B) indicated that silica treatment resulted in increased protein expression of α-SMA, Vim, pro-Col I, and pro-Col III (Figure 7C–F), along with decreased protein expression of E-cad (Figure 7A and G). Treatment with exogenous Ang-(1–7) was able to reverse the effects of silica, while A779 treatment abrogated the beneficial effects of Ang-(1–7).
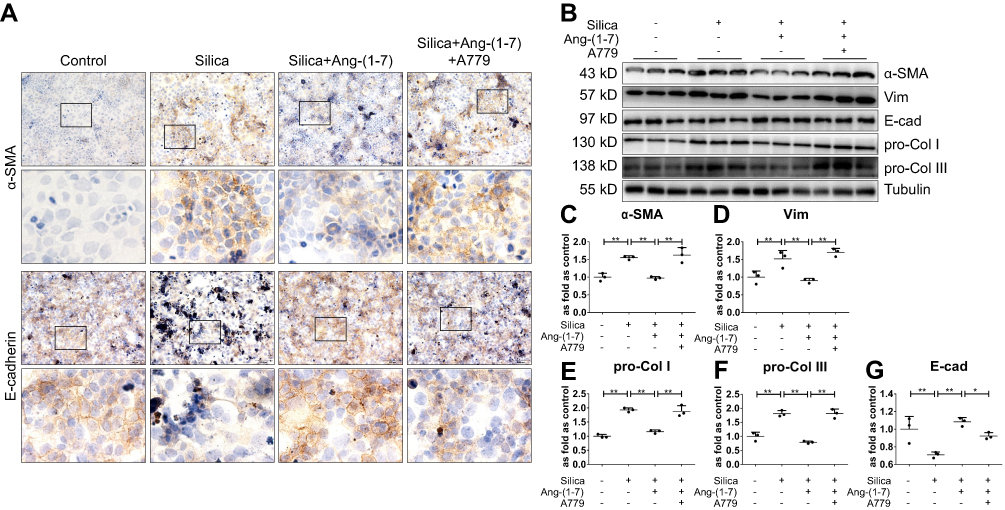 |
Figure 7 Ang-(1–7) inhibits SiO2-induced EMT in MLE-12 alveolar type II epithelial cells. (A) Immunohistochemistry staining of α-SMA and E-cad expression in silica-treated MLE-12 cells. Magnification, ×40 (top panels) and ×200 (bottom panels). (B) Western blot showing the protein expression of α-SMA (C), Vim (D), pro-Col I (E), pro-Col III (F), and E-cad (G) in these cells. Silica-treated MLE-12 underwent various treatment combinations with Ang-(1–7), and A779 (Mas receptor blocker). Values represent the mean ± SD, n = 3 independent experiments, fold change is expressed relative to the control (no treatments), *P < 0.05 vs corresponding group, **P < 0.01 vs corresponding group. |
In addition, Western blotting (Figure 8A) indicated that silica treatment of the MLE-12 cells resulted in increased protein levels of ACE, Ang II, and AT1R (Figure 8B–D), while that of ACE2, Ang-(1–7), and Mas decreased (Figure 8E–G). Treatment with exogenous Ang-(1–7) reversed these changes in the RAS markers, while the beneficial effects of Ang-(1–7) were abrogated by treatment with A779.
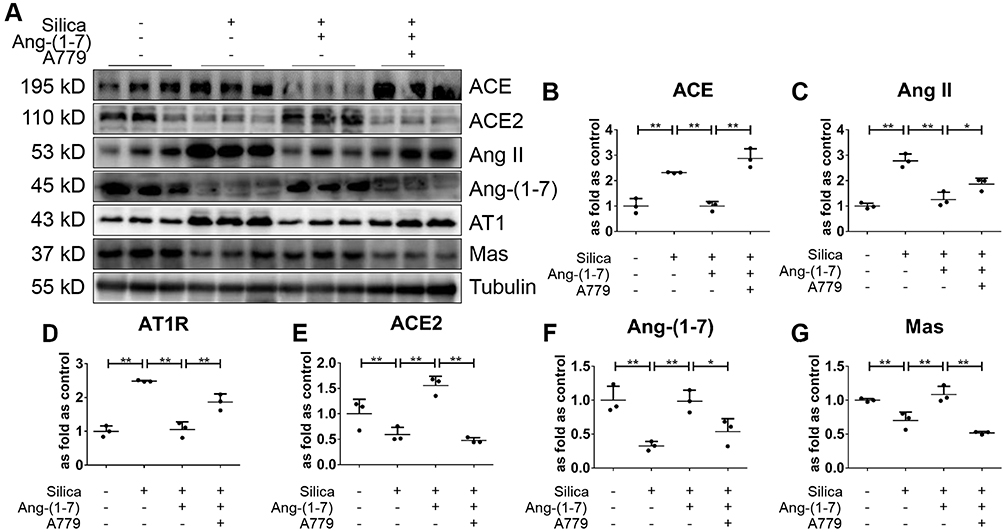 |
Figure 8 Ang-(1–7) regulates RAS in SiO2-treated MLE-12 alveolar type II epithelial cells. (A) Western blot showing the protein expression of ACE (B), Ang II (C), AT1R (D), ACE2 (E), Ang-(1–7) (F), and Mas (G) in silica-treated MLE-12 cells subjected to various treatment combinations with Ang-(1–7), and A779. Values represent the mean ± SD, n = 3 independent experiments, fold change is expressed relative to the control (no treatments), *P < 0.05 vs corresponding group, **P < 0.01 vs corresponding group. |
DIZE Attenuates SiO2-Induced EMT in MLE-12 Cells
We further treated MLE-12 alveolar type II epithelial cells induced by SiO2 with DIZE to investigate its anti-fibrotic effect. Immunohistochemistry staining (Figure 9A) and Western blotting (Figure 9B) showed that treatment with DIZE reversed the increased protein expression of α-SMA, Vim, pro-Col I, and pro-Col III (Figure 9C–F) and reversed the reduced protein expression of E-cad (Figure 9A and G), thereby mitigating the pro-fibrotic effect of SiO2 in MLE-12 cells. Treatment with either A779 or MLN-4760 abrogated the beneficial effects of DIZE in silica-treated MLE-12 cells.
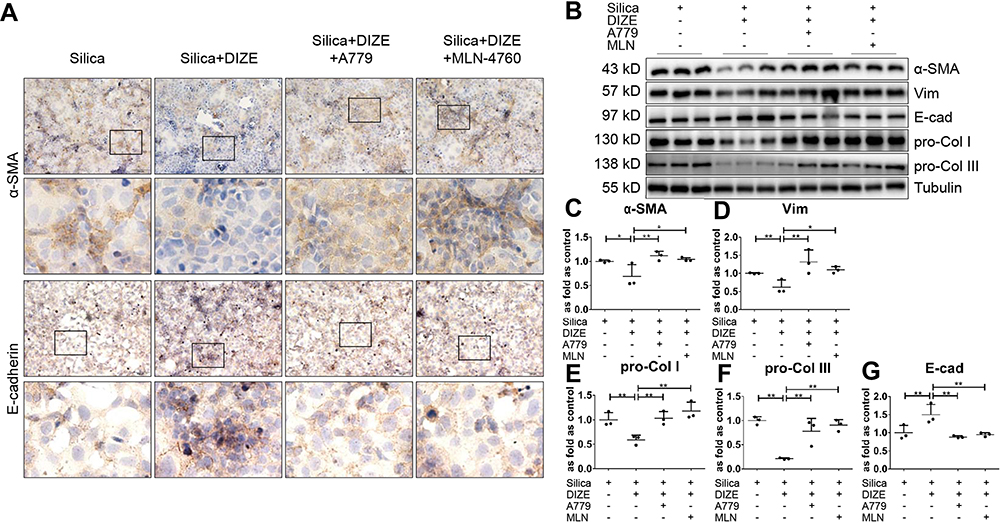 |
Figure 9 DIZE inhibits SiO2-induced EMT in MLE-12 alveolar type II epithelial cells. (A) Immunohistochemistry staining of α-SMA and E-cad expression in silica-treated MLE-12 cells subjected to various treatment combinations with DIZE, A779, and MLN-4760. Magnification, ×40 (top panels) and ×200 (bottom panels). (B) Western blot showing the protein expression of α-SMA (C), Vim (D), pro-Col I (E), pro-Col III (F), and E-cad (G) in these cells. Values represent the mean ± SD, n = 3 independent experiments, fold change is expressed relative to the control (silica only), *P < 0.05 vs corresponding group, **P < 0.01 vs corresponding group. |
Western blotting results (Figure 10A) further indicated that DIZE treatment of MLE-12 cells exposed to silica reversed the elevated protein expression of ACE, Ang II, and AT1R (Figure 10B–D) while reversing the suppressed protein expression of ACE2, Ang-(1–7), and Mas (Figure 10E–G), which are all changes associated with silica treatment. Treatment with MLN-4760 and A779 abrogated the beneficial effects of DIZE. In short, these observations indicated that DIZE treatment of MLE-12 cells promoted the expression of ACE2, thereby inhibiting the EMT induced by silica.
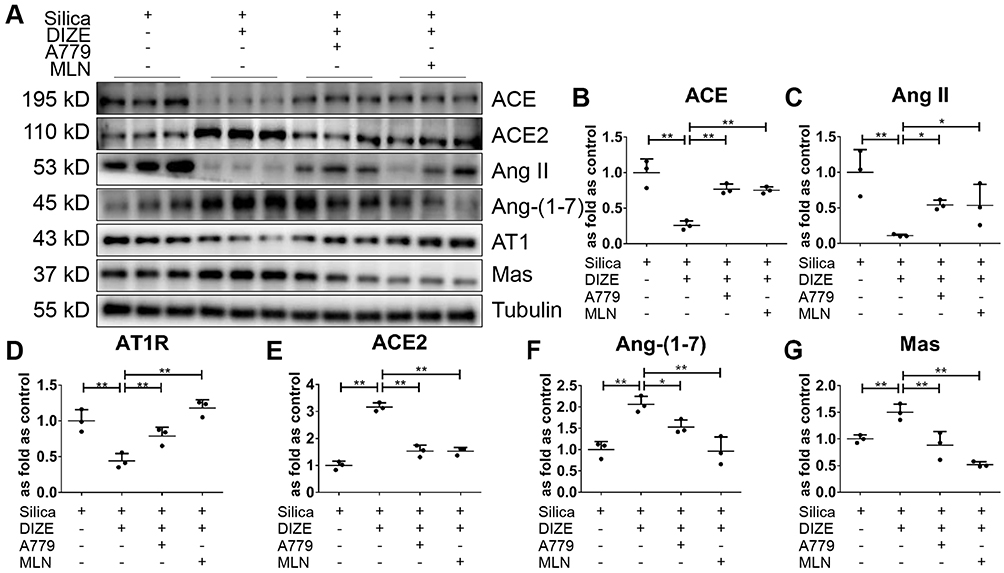 |
Figure 10 DIZE regulates RAS in SiO2-treated MLE-12 alveolar type II epithelial cells. (A) Western blot showing the protein expression of ACE (B), Ang II (C), AT1R (D), ACE2 (E), Ang-(1–7) (F), and Mas (G) in silica-treated MLE-12 cells subjected to various treatment combinations with DIZE, A779, and MLN-4760. Values represent the mean ± SD, n = 3 independent experiments, fold change is expressed relative to the control (silica only), *P< 0.05 vs corresponding group, **P<0.01 vs corresponding group. |
Discussion
Studies have shown that the overexpression of ACE2 inhibits inflammatory injury and cell proliferation, which protects the lung, heart, and kidney from the effects of injury.24–27 Knocking out ACE2 in mice leads to an imbalance between ACE/ACE2 and Ang II/Ang-(1–7), which aggravates lung injury,22 whereas exogenous ACE2 significantly reduces the injury lesions.28 The dysregulation of RAS may trigger this pathological pulmonary process.
As previously reported, we found that EMT induced by silica has an important role in the progression of silicosis.29 The epithelial to mesenchymal or myofibroblast transition results in extracellular matrix (ECM) deposition and decreasing lung compliance. In the present study, we found that exogenous activation (DIZE intervention) and endogenous overexpression of ACE2 (hACE2-transgenic mice) could increase the level of E-cadherin and inhibit the markers of EMT and myofibroblast. We also observed that DIZE and Ang (1–7) could attenuate EMT induced by silica in MLE-12 cells. These results provide evidence that ACE2 exerts a protective effect against pulmonary fibrosis through the inhibition of EMT induced by silica.
In recent years, studies have found that DIZE, a traditional anti-trypanosomiasis drug without major toxicity30 and approved by the US Food and Drug Administration,11 can increase ACE2 activity, thereby promoting Ang-(1–7) production through the degradation of Ang II. DIZE intervention for 4 weeks improves cardiac remodeling in myocardial infarction rats, and it is associated with increased mRNA expression and enzyme activity of ACE2. Moreover, these effects are blocked by the inhibition of either ACE2 with MLN-476031 or the Mas receptor with A779.32 Recently, researchers have shed light on the applications of DIZE in the treatment of fibrotic diseases, indicating that it exerts protective effects against lung, myocardial, liver, and biliary fibrosis.11–14 As expected, DIZE effectively mitigated pathological changes in the lung tissue of silicotic mice and restored the RAS imbalance in the present study. Thus, the mechanism underlying the effect of DIZE on ACE2 activity is probably based, at least in part, on the upregulation of ACE2 mRNA and protein expression.
We employed transgenic mice overexpressing the human ACE2 gene as a model for our investigations. The mouse Ace2 gene has high homology with human ACE2. The model was prepared using the endogenous mouse Ace2 promoter; under the control of this promoter, the ACE2 gene is widely expressed in the lung, kidney, heart, and small intestine of mice.33 Previous studies have confirmed that ACE2 mRNA and protein levels in the lung tissue of hACE2-transgenic mice are higher than those in wild-type mice.22 In the present study, we also found that exogenous and endogenous overexpression of ACE2 protects the lung from exposure to silica, in particular by reversing the reductions in ACE2/Ang-(1-7)/Mas observed in silicotic mice.
In the current study, serum ACE/ACE2 activity and Ang II/Ang-(1−7) levels were also measured, but the observed changes were not completely consistent with those in the lung tissue. This suggests that the activation mechanism and the role of circulatory and tissue RAS are different. Circulatory Ang II mainly originates in the lung tissue, while Ang-(1−7) is a derivative of Ang II, formed when ACE2 cleaves the amino acid, phenylalanine, from the C-terminus.34 In short, circulatory and tissue RAS responds differently in the mouse silicosis model. Circulatory RAS plays an important role in maintaining blood pressure and body fluid balance, which may ensure blood supply to critical organs. Tissue RAS, however, may be necessary for maintaining local blood perfusion and for its roles in such processes as antioxidation and antiproliferation.22
Conclusion
In summary, unbalanced RAS homeostasis in the lung tissue plays an important role in the occurrence and development of lung fibrosis in silicotic mice. The current work provided evidence that both the overexpression of ACE2 and treatment with DIZE could activate the ACE2/Ang-(1–7)/Mas receptor axis to inhibit EMT, thereby exerting anti-fibrotic effects in silicotic mice, and this was accompanied by suppression of the ACE/Ang II/AT1 receptor axis. The regulation of RAS homeostasis, which is focused on ACE2, may provide a promising treatment approach for pulmonary fibrosis; however, the specific underlying mechanisms remain to be determined.
Abbreviations
SiO2, silicon dioxide; RAS, renin–angiotensin system; ACE, angiotensin-converting enzyme; ACE2, angiotensin-converting enzyme 2; Ang II, angiotensin II; Ang-(1–7), angiotensin-(1-7); AT1, angiotensin II receptor 1; DIZE, diminazene aceturate; EMT, epithelial-mesenchymal transition; ECM, extracellular matrix.
Acknowledgments
We thank Natasha Beeton-Kempen, PhD., from Liwen Bianji, Edanz Editing China for editing the English text of a draft of this manuscript.
Author Contributions
Shumin Li and Hong Xu designed the study. Shumin Li, Yaqian Li, Zhongqiu Wei, Yi Yang, Fuyu Jin, Min Zhang, Chen Wang, Wenxiong Song, Jingchen Huo and Jingyuan Zhao carried out the experimental work, analyzed the data, and drafted the manuscript. Fang Yang and Xiuhong Yang participated in the design of the study, critically reviewed the manuscript, and provided intellectual input. All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
This study was supported by the National Natural Science Foundation of China (grant no. 81972988); National Natural Science Foundation of China (grant no. 81472953); Natural Science Foundation of Hebei Province (grant no. H2016209170); Postgraduate Innovation Funding Program of Hebei Province (No. CXZZBS2017127); Postgraduate Student Innovation Fund of North China University of Science and Technology (grant no. 2017B10).
Disclosure
The authors report no conflicts of interest in this work.
References
1. The Lancet Respiratory M. The world is failing on silicosis. Lancet Respir Med. 2019;7(4):283. doi:10.1016/s2213-2600(19)30078-5.
2. Xu H, Yang F, Sun Y, et al. A new antifibrotic target of Ac-SDKP: inhibition of myofibroblast differentiation in rat lung with silicosis. PLoS One. 2012;7(7):e40301. doi:10.1371/journal.pone.0040301
3. Shifeng L, Hong X, Xue Y, et al. Ac-SDKP increases alpha-TAT 1 and promotes the apoptosis in lung fibroblasts and epithelial cells double-stimulated with TGF-beta1 and silica. Toxicol Appl Pharmacol. 2019;369:17–29. doi:10.1016/j.taap.2019.02.015
4. Meng Y, Li T, Zhou GS, et al. The angiotensin-converting enzyme 2/angiotensin (1-7)/Mas axis protects against lung fibroblast migration and lung fibrosis by inhibiting the NOX4-derived ROS-mediated RhoA/Rho kinase pathway. Antioxid Redox Signal. 2015;22(3):241–258. doi:10.1089/ars.2013.5818
5. Rago F, Melo EM, Kraemer L, et al. Effect of preventive or therapeutic treatment with angiotensin 1-7 in a model of bleomycin-induced lung fibrosis in mice. J Leukoc Biol. 2019. doi:10.1002/jlb.Ma1218-490rr
6. Wang J, Chen L, Chen B, et al. Chronic activation of the renin-angiotensin system induces lung fibrosis. Sci Rep. 2015;5:15561. doi:10.1038/srep15561
7. Tan WSD, Liao W, Zhou S, Mei D, Wong WF. Targeting the renin-angiotensin system as novel therapeutic strategy for pulmonary diseases. Curr Opin Pharmacol. 2018;40:9–17. doi:10.1016/j.coph.2017.12.002
8. Gao X, Xu H, Zhang B, et al. Interaction of N-acetyl-seryl-aspartyl-lysyl-proline with the angiotensin-converting enzyme 2-angiotensin-(1-7)-Mas axis attenuates pulmonary fibrosis in silicotic rats. Exp Physiol. 2019;104(10):1562–1574. doi:10.1113/EP087515
9. Zhang B, Xu H, Zhang Y, et al. Targeting the RAS axis alleviates silicotic fibrosis and Ang II-induced myofibroblast differentiation via inhibition of the hedgehog signaling pathway. Toxicol Lett. 2019;313:30–41. doi:10.1016/j.toxlet.2019.05.023
10. Li SM, Wang XY, Liu F, Yang XH. ACE2 agonist DIZE alleviates lung injury induced by limb ischemia-reperfusion in mice. Acta Physiologica Sinica. 2018;70(02):175–183.
11. Rajapaksha IG, Mak KY, Huang P, Burrell LM, Angus PW, Herath CB. The small molecule drug diminazene aceturate inhibits liver injury and biliary fibrosis in mice. Sci Rep. 2018;8(1):10175. doi:10.1038/s41598-018-28490-y
12. Velkoska E, Patel SK, Griggs K, Burrell LM. Diminazene aceturate improves cardiac fibrosis and diastolic dysfunction in rats with kidney disease. PLoS One. 2016;11(8):e0161760. doi:10.1371/journal.pone.0161760
13. Wang LP, Fan SJ, Li SM, Wang XJ, Gao JL, Yang XH. Protective role of ACE2-Ang-(1-7)-Mas in myocardial fibrosis by downregulating KCa3.1 channel via ERK1/2 pathway. Pflugers Archiv. 2016;468(11–12):2041–2051. doi:10.1007/s00424-016-1875-9
14. Prata LO, Rodrigues CR, Martins JM, et al. ACE2 activator associated with physical exercise potentiates the reduction of pulmonary fibrosis. Exp Biol Med. 2017;242(1):8–21. doi:10.1177/1535370216665174
15. Goru SK, Kadakol A, Malek V, Pandey A, Sharma N, Gaikwad AB. Diminazene aceturate prevents nephropathy by increasing glomerular ACE2 and AT2 receptor expression in a rat model of type1 diabetes. Br J Pharmacol. 2017;174(18):3118–3130. doi:10.1111/bph.13946
16. Souza LK, Nicolau LA, Sousa NA, et al. Diminazene aceturate, an angiotensin-converting enzyme II activator, prevents gastric mucosal damage in mice: role of the angiotensin-(1-7)/Mas receptor axis. Biochem Pharmacol. 2016;112:50–59. doi:10.1016/j.bcp.2016.05.010
17. Li Y, Zeng Z, Cao Y, et al. Angiotensin-converting enzyme 2 prevents lipopolysaccharide-induced rat acute lung injury via suppressing the ERK1/2 and NF-kappaB signaling pathways. Sci Rep. 2016;6:27911. doi:10.1038/srep27911
18. Li W, Xie L, Ma J, et al. Genetic loss of Gas6/Mer pathway attenuates silica-induced lung inflammation and fibrosis in mice. Toxicol Lett. 2019;313:178–187. doi:10.1016/j.toxlet.2019.07.008
19. Uhal BD, Li X, Xue A, Gao X, Abdul-Hafez A. Regulation of alveolar epithelial cell survival by the ACE-2/angiotensin 1-7/Mas axis. Am J Physiol Lung Cell Mol Physiol. 2011;301(3):L269–274. doi:10.1152/ajplung.00222.2010
20. De Maria ML, Araujo LD, Fraga-Silva RA, et al. Anti-hypertensive effects of diminazene aceturate: an angiotensin- converting enzyme 2 activator in rats. Protein Pept Lett. 2016;23(1):9–16. doi:10.2174/0929866522666151013130550
21. Garabelli PJ, Modrall JG, Penninger JM, Ferrario CM, Chappell MC. Distinct roles for angiotensin-converting enzyme 2 and carboxypeptidase A in the processing of angiotensins within the murine heart. Exp Physiol. 2008;93(5):613–621. doi:10.1113/expphysiol.2007.040246
22. Chen L-N, Yang X-H, Nissen DH, et al. Dysregulated renin-angiotensin system contributes to acute lung injury caused by hind-limb ischemia-reperfusion in mice. Shock. 2013;40(5):420–429. doi:10.1097/SHK.0b013e3182a6953e
23. Wiener RS, Cao YX, Hinds A, Ramirez MI, Williams MC. Angiotensin converting enzyme 2 is primarily epithelial and is developmentally regulated in the mouse lung. J Cell Biochem. 2007;101(5):1278–1291. doi:10.1002/jcb.21248
24. Kong E-L, Zhang J-M, An N, Tao Y, Yu W-F, Wu F-X. Spironolactone rescues renal dysfunction in obstructive jaundice rats by upregulating ACE2 expression. J Cell Commun Signal. 2019;13(1):17–26. doi:10.1007/s12079-018-0466-2
25. Li X-Y, Peng Y, Bu X-W, Yao J, Yao L. Balancing effect of biejiajian oral liquid () on ACE-Ang II-AT1R axis and ACE2-Ang-(1-7)-Mas axis in rats with CCl4-induced hepatic fibrosis. Chin J Integr Med. 2018;24(11):853–859. doi:10.1007/s11655-017-2909-7
26. Williams VR, Scholey JW. Angiotensin-converting enzyme 2 and renal disease. Curr Opin Nephrol Hypertens. 2018;27(1):35–41. doi:10.1097/mnh.0000000000000378
27. Zhao S, Ghosh A, Lo C-S, et al. Nrf2 deficiency upregulates intrarenal angiotensin-converting enzyme-2 and angiotensin 1-7 receptor expression and attenuates hypertension and nephropathy in diabetic mice. Endocrinology. 2018;159(2):836–852. doi:10.1210/en.2017-00752
28. Imai Y, Kuba K, Rao S, et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436(7047):112–116. doi:10.1038/nature03712
29. Deng H, Xu H, Zhang X, et al. Protective effect of Ac-SDKP on alveolar epithelial cells through inhibition of EMT via TGF-beta1/ROCK1 pathway in silicosis in rat. Toxicol Appl Pharmacol. 2016;294:1–10. doi:10.1016/j.taap.2016.01.010
30. Peregrine AS, Mamman M. Pharmacology of diminazene: a review. Acta Trop. 1993;54(3–4):185–203. doi:10.1016/0001-706X(93)90092-P
31. Qi Y, Zhang J, Cole-Jeffrey CT, et al. Diminazene aceturate enhances angiotensin-converting enzyme 2 activity and attenuates ischemia-induced cardiac pathophysiology. Hypertension. 2013;62(4):746–752. doi:10.1161/HYPERTENSIONAHA.113.01337
32. Shenoy V, Gjymishka A, Jarajapu YP, et al. Diminazene attenuates pulmonary hypertension and improves angiogenic progenitor cell functions in experimental models. Am J Respir Crit Care Med. 2013;187(6):648–657. doi:10.1164/rccm.201205-0880OC
33. XH Y. Susceptibility of hACE2 Transgenic Mice to SARS-Cov and Experimental Study of SARS Pathogenesis. Peking Union Medical College; 2007.
34. Zisman LS, Keller RS, Weaver B, et al. Increased angiotensin-(1-7)-forming activity in failing human heart ventricles: evidence for upregulation of the angiotensin-converting enzyme Homologue ACE2. Circulation. 2003;108(14):1707–1712. doi:10.1161/01.CIR.0000094734.67990.99
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.