")
Back to Journals » Drug Design, Development and Therapy » Volume 14
A Triple Combination of Metformin, Acetylsalicylic Acid, and Oseltamivir Phosphate Impacts Tumour Spheroid Viability and Upends Chemoresistance in Triple-Negative Breast Cancer
Authors Sambi M, Samuel V, Qorri B , Haq S, Burov SV , Markvicheva E, Harless W, Szewczuk MR
Received 16 December 2019
Accepted for publication 27 April 2020
Published 25 May 2020 Volume 2020:14 Pages 1995—2019
DOI https://doi.org/10.2147/DDDT.S242514
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Qiongyu Guo
Manpreet Sambi,1 Vanessa Samuel,1,2 Bessi Qorri,1 Sabah Haq,1,3 Sergey V Burov,4 Elena Markvicheva,5 William Harless,6 Myron R Szewczuk1
1Department of Biomedical and Molecular Sciences, Queen’s University, Kingston, ON, Canada; 2Faculty of Medicine, University of British Columbia, Vancouver, BC, Canada; 3Department of Pathology and Molecular Medicine, McMaster University, Hamilton, ON, Canada; 4Laboratory of Novel Peptide Therapeutics, Cytomed J.S.Co., St. Petersburg, Russia; 5Biomedical Materials Laboratory, Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, Moscow, Russia; 6Encyt Technologies, Inc., Membertou, Nova Scotia, Canada
Correspondence: Myron R Szewczuk
Department of Biomedical and Molecular Sciences, Queen’s University, Kingston, ON K7L3N6, Canada
Tel +1 613 533 2457
Fax +1 613 533 6796
Email [email protected]
Introduction: Targeted multimodal approaches need to be strategically developed to control tumour growth and prevent metastatic burden successfully. Breast cancer presents a unique clinical problem because of the variety of cellular subtypes that arise. The tumour stage and cellular subtypes often dictate the appropriate clinical treatment regimen. Also, the development of chemoresistance is a common clinical challenge with breast cancer. Higher doses and additional drug agents can produce additional adverse effects leading to a more aggressive malignancy. Acetylsalicylic acid (ASA), metformin (Met), and oseltamivir phosphate (OP) were investigated for their efficacy to sensitize MDA-MB-231 triple-negative breast cancer and its tamoxifen (Tmx) resistant variant (MDA-MB-231-TmxR) together in combination with Tmx treatment.
Methods: Microscopic imaging, the formation of 3D multicellular tumour spheroids, immunocytochemistry, flow cytometry, Annexin V Assay, Caspase 3/7 Apoptosis Assay, tube formation assay and analysis, and WST-1 cell viability assay evaluated the formation of MCTS, morphologic changes, cell viability, apoptosis activity and the expression levels of ALDH1A1, CD44 and CD24 on the cell surface, MDA-MB231 triple-negative breast cancer, tamoxifen (Tmx) resistant variant (MDA-MB-231-TmxR).
Results: The results using a triple combination of ASA, Met and OP on MDA-MB-231 and MDA-MB-231-TmxR cells and their matrix-free 3D multicellular tumour spheroids (MCTS) formed by using the cyclic Arg-Gly-Asp-D-Phe-Lys peptide modified with 4-carboxybutyl-triphenylphosphonium bromide (cyclo-RGDfK(TPP)) peptide method demonstrate a consistent and significant decrease in cell and tumour spheroid viability and volume with increased apoptotic activity, and increased sensitivity to Tmx therapy. Tmx treatment of MDA-MB-231 cells in combination with ASA, Met and OP markedly reduced the CD44/CD24 ratio by 6.5-fold compared to the untreated control group. Tmx treatment of MDA-MB-231-TmxR cells in combination with ASA, Met and OP markedly reduced the ALDH1A1 by 134-fold compared to the same treatment for the parental cell line. Also, the triple combination treatment of ASA, Met, and OP inhibited vasculogenic endothelial cell tube formation and induced endothelial cell apoptosis.
Conclusion: For the first time, the findings demonstrate that repurposing ASA, Met, and OP provides a novel and promising targeted multimodal approach in the treatment of triple-negative breast cancer and its chemoresistant variant.
Keywords: MDA-MB-231, MDA-MB-231-TmxR, human umbilical vein endothelial cell lines, multicellular tumour spheroids, MCTS, endothelial cell tube formation, apoptosis
Introduction
Triple-negative breast cancer (TNBC) is classified as one of the more aggressive subtypes of breast cancer,1,2 accounting for 11% to 17% of all breast cancers.3 TNBC is commonly diagnosed at a premenopausal age, with a 20% greater chance of metastasizing to visceral organs.4 Additionally, TNBC is characterized by its poor prognosis, early relapse, and a shorter progression-free survival period following recurrence when compared with other forms of breast cancer.5 This can be attributed to its lack of targetable receptors as TNBC does not express the estrogen receptor (ER), progesterone receptor (PR), or the human epidermal growth factor receptor 2 (HER-2).1,6 The lack of expression of receptors that control proliferation contributes to the higher proliferation rate of TNBC when compared with other breast cancers.4 While other breast cancer treatment options include a combination of chemotherapy in addition to targeted therapy (targets including ER, PR, or HER-2),6 there is yet to be agold standard treatment for TNBC.7,8 Current treatment options for TNBC include surgery with neoadjuvant or adjuvant chemotherapy or very selective targeted therapies. Due to the highly aggressive state of TNBC, treatment options available to patients are limited to those that offer a balance between efficacy and toxicity, and these decisions are made with the patient’s consent.5
The therapeutic regimen for TNBC is aggressive and has a rapid progression time between relapse and subsequent death compared to other types of breast cancer.3 Furthermore, the heterogeneous nature of TNBC has resulted in the maintenance of a relatively stagnant survival outcome over the last 20 years, primarily due to metastases to the central nervous system and visceral organs, all of which limit treatment options.1 Due to this pathophysiology and the lack of therapeutic targets, the standard of care options is limited to surgery in combination with chemotherapy.9,10 There are consistently poor treatment outcomes because many proposed and conventional treatment options are unable to target tumour heterogeneity or overcome chemoresistance. Given these clinical challenges, investigations into repurposing medications such as metformin (Met) and acetylsalicylic acid (ASA) for the treatment of patients with various types of cancer are being considered.
The rationale for this repurposing drug approach began following the observation that diabetic patients on Met treatment appeared to have a lower incidence of cancer over their lifetime.11 Several reports have provided evidence that the anti-tumour effect of Met may be attributed to regulating aberrant metabolic activity typically observed in many cancers via increased uptake of glucose.11 This characteristic property of Met may be an essential target. The aberrant metabolic activity is one of the emerging hallmarks of cancer, contributing to replicative cancer immortality and sustained proliferative activity.12 From a molecular standpoint, Met inhibits 5ʹ adenosine monophosphate-activated protein kinase (AMPK)-mediated inflammation and mammalian target of rapamycin complex-1 (mTORC1)-mediated cancer cell proliferation.13 There has also been an anti-tumour effect of Met observed in TNBC.14 Mice with MDA-MB-231 TNBC tumour xenografts displayed significant reductions in tumour growth and cell proliferation following Met treatment.14 Recently, Ryabaya et al15 have demonstrated that Met can increase the antitumor activity of mitogen-activated protein kinase kinase (MAP2K) inhibitor binimetinib in 2D cell cultures and 3D spheroid models of human metastatic melanoma cells. The combination of Met and binimetinib reduced the number of melanoma-formed colonies, and inhibited cell invasion and migration, leading to G0/G1 cell cycle arrest through the cyclin D/CDK4/CDK6 pathway. The mechanism of Met and binimetinib synergy in melanoma cells was associated with increased activation of p-AMPKα and decreased phosphorylated extracellular signal-regulated kinase (p-ERK), with no effects on p-mTOR.
Acetylsalicylic acid (ASA), an anti-inflammatory drug, was initially used for its anti-inflammation, anti-platelet, and analgesic properties.16,17 ASA’s mechanism of action was limited to the inhibition of prostaglandin production but was later used to prevent cardiovascular and cerebrovascular diseases.18 Recently, its chemopreventative mechanism(s) of action has been the subject of intense research investigation. ASA’s potential as an anti-cancer agent was revealed from the evidence that ASA irreversibly inhibits cyclooxygenase (COX) enzyme.17,18 Indeed, pancreatic ductal adenocarcinoma (PDAC) expresses aberrations in COX genes that have been reported to be upregulated.19 Although the COX enzymes come in two different isoforms, COX-2 is the inducible isoform and is upregulated in 47% to 66% of pancreatic cancers.20 The COX enzyme is known as prostaglandin-endoperoxide synthase (PGHS), which exists in two isoforms, COX-1 and −2, and is an integral protein with fatty acid oxygenase and peroxidase activity.17,18 The irreversible mechanism of action of ASA results from the acetylation of the hydroxyl group of serine residues in COX-1 and COX-2, which limit accessibility to the catalytic site of the enzyme that is necessary for arachidonic acid to bind. Arachidonic acid is metabolized to produce prostaglandins. Additional research has indicated that the mutation of Ras contributes to the increased stability of COX-2 mRNA and thus participates in crosstalk amongst themselves and other signaling pathways to collectively contribute to the unique pathologic characteristic of pancreatic cancer.21
ASA, an anti-inflammatory cyclooxygenase (COX)-1 and COX-2 inhibitor,22 has been repurposed as an anti-cancer agent given its broad efficacy in regulating the hallmarks of cancer.23 Investigations on the anti-cancer efficacy of ASA have revealed that it inhibits not only inflammatory COX-1/2 pathways, but also inhibits AMPK-mediated inflammatory pathways, and mTORC1-mediated cancer cell growth and proliferation.24 Several preclinical studies have demonstrated improved efficacy of ASA given in combination with Met, rather than alone, with or without conventional chemotherapeutics improved treatment outcomes in patients with colorectal,25 pancreatic,24,26 and breast cancer.27 In vitro studies following treatment with ASA and Met, the MDA-MB-231 TNBC cells not only demonstrated a decrease in cell viability but a decrease in colony formation and migration through a synergistic mechanism(s) of action of ASA and Met. It is noteworthy that ASA and Met can affect not only aberrant metabolic pathways but also specific cells and features of the breast cancer tumour microenvironment.27,28
Recently, oseltamivir phosphate (OP) has been shown to inhibit mammalian neuraminidase-1 (Neu-1) because it has the structural analog of α-2,3-sialic acid residues located on the receptor tyrosine kinases (RTKs), many of which are overexpressed on cancer cells.29,30 OP downregulates several EGFR-mediated pathways such as JAK/STAT, PI3K/Akt, and MAPK involved in cancer cell proliferation, metastasis, and tumour vascularization.31 Previously, our group reported on the efficacy of OP monotherapy in mouse models of human ovarian,32 breast,33 and pancreatic cancers31,34 and the mechanism of action of OP in regulating multistage tumorigenesis.35 Using a mouse model of human MDA-MB-231 TNBC tumours, Haxho et al33 have demonstrated that OP treatment at 30 mg/kg daily intraperitoneally reduced tumour vascularization and growth rate as well as significantly reduced tumour weight and spread to the lungs compared with the untreated cohorts. OP treatment at 50 mg/kg completely ablated tumour vascularization, tumour growth and spread to the lungs, with significant long-term survival at day 180 post-implantation, tumour shrinking, and no relapses after 56 days off-drug. Also, OP 30 mg/kg treated cohort tumours expressed significantly reduced levels of human N-cadherins and host CD31+ endothelial cells with significant concomitant expression of E-cadherins compared to the untreated cohorts. We have shown that a combination of OP and gemcitabine reduced tumour volume, and decreased tumour vascularization. Most importantly, the combination prevented chemoresistance in mouse models of human pancreatic cancer.36 Here, OP targets pathways and components of multistage tumorigenesis that ASA and Met have not been shown to affect.
In this present study, the triple combination of ASA, Met, and OP administered in combination with tamoxifen (Tmx) reduced cell proliferation, upended Tmx chemoresistance, and increased apoptotic activity in monolayer and spheroid cultures of MDA-MB-231 triple-negative cancer cells and their Tmx resistant variant. Also, ASA, Met, and OP downregulated the acquisition of stem-like cell properties of MDA-MB-231 cells. For the first time, these findings demonstrate that ASA, Met, and OP provide a significant therapeutic approach in targeting multistage tumorigenesis in triple-negative breast cancer.
Materials and Methods
Cell Lines and Culture Procedures
MDA-MB-231 (ATCC® HTB-26™, American Type Culture Collection, Manassas, VA 20110, USA), a human triple-negative breast cancer cell line, was obtained from the metastatic pleural effusion site of a 51-year-old woman with metastatic breast cancer. MDA-MB-231 is an invasive ductal adenocarcinoma.
MDA-MB-231 cell lines resistant against 10 μM tamoxifen (Tmx) (MDA-MB-231-TmxR) were established in culture to gradual increases in Tmx concentration in 1×Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco, Rockville, MD, USA) conditioned medium. After removing dead cells, the viable cells were maintained in culture containing 10 μM Tmx. At ~80% confluence, cells were continuously passaged in the conditioned medium containing 10 μM Tmx for over one year.
The cell lines were grown in 1×DMEM conditioned media with 10% fetal bovine sera (FBS; HyClone, Logan, UT, USA) and 5 μg/mL plasmocin™ (InvivoGen, San Diego, CA, USA). All cells were stored in T25 cell culture flasks in a 5% CO2 incubator at 37°C. Cells were passaged as needed (approximately every 4–5 days) using TrypLE Express (Gibco, Rockville, MD, USA).
Human umbilical vein endothelial cells (HuVECs) were obtained from Dr. Chandrakant Tayade’s lab, Queen’s University, Kingston, Ontario. The cells were acquired initially from American Type Culture Collection (ATCC® catalogue number CRL-1730, Manassas, VA 20110 USA) and were passaged three times to confluency before use. HuVECs were grown in complete human endothelial cell media (ATCC Cat #211-500). HuVECs were subcultured until they were needed for experiments. Cells were fed with human endothelial media every other day. Cells washed with 1×phosphate-buffered saline (PBS) were followed by 4 mL of warmed Trypsin-EDTA and incubated for 5 to 10 minutes. Cells were observed under the microscope to determine if they had detached. Trypsin-EDTA was neutralized by complete human endothelial media, counted, and plated as needed.
Reagents
The cyclic Arg-Gly-Asp-D-Phe-Lys (cyclo-RGDfK) peptide was synthesized and modified with 4-carboxy-butyl-triphenylphosphonium bromide to produce the cyclo-RGDfK(TPP) peptide. Standard methods of solid-phase peptide synthesis37 were conducted in the laboratory of Prof. Dr. S. Burov, Saint-Petersburg, Russia.
Acetylsalicylic acid (ASA, >99% pure, Sigma-Aldrich, Steinheim, Germany) dissolved in dimethyl sulfoxide (DMSO) was prepared to make a 550 mM stock solution, and stored in aliquots at −20°C. Tamiflu® (oseltamivir phosphate-free base (OP), Hoffman-La-Roche Ltd, Mississauga, ON, Canada) was obtained from 75 mg capsules that were dissolved in sterile phosphate-buffered saline (1×PBS), vortexed for 3 minutes, and centrifuged at 900 × g for 7 minutes to remove insoluble materials. The supernatant was transferred to a 50 mL vial, frozen at −80°C, lyophilized for 24–48 hours, and stored at −80°C. The stock-extracted OP solution had a concentration of 20 mg/mL. Metformin hydrochloride (Met, Sigma-Aldrich Canada Co., Oakville, ON, Canada) was dissolved in ddH2O to prepare a 387 mM or 116.84 mM stock solution, which was then aliquoted and stored at −20°C. Tamoxifen citrate salt (Tmx, ≥99% pure, Sigma-Aldrich, Steinheim, Germany) was dissolved in methanol (99.8% pure, Sigma-Aldrich, Steinheim, Germany) at 50 mg/mL to make 1 mM stock solution, aliquoted, wrapped in aluminum foil (light-sensitive), and stored at 4°C. Cocktail therapy refers to ASA, Met, and OP. The combination therapy refers to ASA, Met, OP, and Tmx.
Formation of 3D Multicellular Tumour Spheroids (MCTS)
MDA-MB-231 and MDA-MB-231-TmxR cells were grown in T25 flasks to ~90% confluence, plated in 96-well plates with 20,000 cells/well (100 µL/well), and incubated for 3 hours at 37°C to allow for cell adhesion. After 3 hours, the media were replaced with 33.3 µL of cyclo-RGDfK(TPP) peptide (50 µM), and 66.6 µL of 1x DMEM supplemented with 10% FBS. Cells were incubated for four days at 37°C to allow for MCTS formation. After MCTS formation, ASA, OP, Met, and/or Tmx were added, while some MCTS did not receive drugs and served as untreated controls. After treatment, all cells remained in culture for 72 hours (Day 7).
Phase-Contrast Microscopy and Measurement of MCTS Volume
The morphology of MDA-MB-231 and MDA-MB-231-TmxR cells was studied before and after the addition of the cyclo-RGDfK(TPP) peptide. The cellular morphology, aggregation, and MCTS formation were observed using phase-contrast microscopy. Images were acquired using a scope-mounted camera (Fisher Scientific) at 4× and 10× magnification throughout each experiment (7 days). An MCTS was defined as a compact rounded sphere of diameter >20 µm with a distinct border containing cells indistinguishable from one another. Some experiments yielded a mixture of defined MCTS and cell aggregates. Both MCTS and cell aggregate measurements were included in the results. ImageJ software (ImageJ, Bethesda, Maryland, USA) was used to measure two diameters from each MCTS or cell aggregate, with 4–10 MCTS/cell aggregates measured per image. All diameters were measured to the scale bar in the phase-contrast images. The two measured diameters were then averaged and divided to calculate the average radius. The following formulae were used to determine MCTS/cell aggregate volume, as previously described in detail:37–40
10× objective images: Volume = (4/3) πr3 where r = average radius (μm);
4× objective images: Volume = (2.5)(4/3) πr3 where r = average radius (μm)
The formula for the 4× objective images includes 2.5 factor to normalize values to the 10× objective images.
WST-1 Cell Proliferation Assay
The water-soluble tetrazolium salt-1 (WST-1) assay measures cell viability based on the reduction of a tetrazolium compound to a soluble derivative.41 The absorbance at 420 nm of the reaction correlates directly to the number of living cells in culture. For monolayer breast cancer cells, cells were plated at a density of 10,000 cells/well in a 96-well plate. The cells were incubated overnight, and they were then exposed to increasing concentrations of drug cocktail or left untreated as controls for 24, 48, and 72 hours. The treatment conditions were performed in triplicates, and the entire experiment was repeated. Absorbance readings at 0, 24, 48, and 72 hours were taken by adding 10% WST-1 reagent (Roche Diagnostics, Division de Hoffman La Roche Limitée, Laval-des-Rapides, QC, Canada) in Dulbecco’s Modified Eagle’s Medium to each well followed by incubation at 37°C for 2 hours before reading at the indicated time points. Cell viability is presented as a mean percent of control and illustrated as a bar graph using GraphPad Prism software (GraphPad Software, La Jolla, CA, USA). The following formula was used to determine cell viability as a percent of control for each time point and drug concentration:
[(Absorbance of cells in a drug concentration) – (Media absorbance)]/[(Absorbance of cells alone) – (Media absorbance)] × 100.
Concerning MCTS, the WST-1 assay has explicitly been recognized as a valid measure of cell viability in small (≤200 μm diameter) MCTS, as the WST-1 reagent can successfully permeate through thin 3D layers of cells.42 Cells were grown to 80–90% confluence in T25 flasks and seeded in a 96-well tissue culture plate at a density of 10,000 cells/well for 3 hours, followed by the addition of 50 µM cyclo-RGDfK(TPP) peptide directly to the cells. As described above, on Day 4, MCTS were treated with ASA, OP, Met, or Tmx at indicated concentrations or were left untreated as controls. On Day 7, 18 μL of 10% WST-1 reagent (Roche Diagnostics, Laval-des-Rapides, QC, Canada) in 1x PBS was added to each well. The 96-well plate at 37°C was then incubated for 2 hrs before an absorbance reading using the SpectraMax250 machine and SoftMax software. Cell viability as a percentage of control was illustrated as a bar graph using GraphPad Prism software (GraphPad Software, La Jolla, CA, USA).
The following formula was used to determine MCTS cell viability as a percentage of control (Day 4) after 72 hours of drug treatment (Day 7):
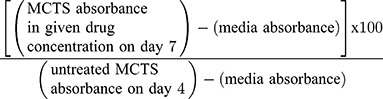
The fully-formed MCTS in 50 μM cyclo-RGDfK(TPP) peptide on Day 4 represents the untreated control group.
Immunocytochemistry
MDA-MB-231 cells and MDA-MB-231-TmxR variant cells were plated at a density of 100,000 to 200,000 cells/mL on glass coverslips in 24-well plates. Cells were treated with single and combination agents for 24, 48, and 72 hours. At the end of each time-point, cells were washed, fixed with 4% PFA for 30 minutes and blocked for 1 hour in 10% FBS + 0.1% Triton X-100 + 1×PBS (0.1% Triton X-100 was omitted from blocking buffer for membrane-only stains to block intracellular non-specific binding). Following blocking, cells were washed with 1×PBS, and the primary antibody diluted to 1:250 using a 1% FBS + 1×PBS + 0.1% Triton X-100 overnight at 4°C. Primary antibodies were obtained from Santa Cruz BioTechnology ALDH1A1 (sc-374149), CD44 (sc-DF1485), CD24 (sc-19585). Cells were then washed 3× for 10 minutes with 1×PBS and incubated for 1 hour with AlexaFluor 594 (for CD24, MDR1, and ALDH1A1) and AlexaFluor 488 (for CD44) secondary antibodies. Secondary antibody controls were included for unspecific staining. Cells were then washed 5× for 15 mins with 1×PBS (note: one wash included 0.1% Triton X-100 to permeabilize cells for DAPI). DAPI containing mounting media (Vector Laboratories H-1200-10) was added to slides, and coverslips were inverted on to mounting media droplet and sealed.
Relative fluorescence density readings were quantified using images captured at 20x to ensure a wide field of view was obtained. Two representative images were taken at 20x. Background means, image mean, and pixel measurements were obtained from Corel Photo-Paint X8. Red (Alexafluor 594) or green (Alexafluor 488) colour channel images quantified. The background density means represent an unstained section of the image, and the image means represented the total fluorescence of the image. These measurements were used to quantify the relative fluorescence density using the equation below:
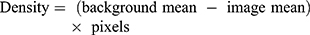
Flow Cytometry
MDA-MB-231 cells and MDA-MB-231-TmxR variant cells were exposed to combination therapy, Tmx only, or left untreated for 72 hours. They were harvested and counted for a final concentration of 1.0 x106 cells/mL. All subsequent steps were done on ice. Cells were washed twice in 2% FBS + 1×PBS before adding the primary antibody. Cells were treated with 100 μL of CD44, CD24, or ALDH1A1 (note: Cells stained with ALDH1A1 were permeablized with blocking and antibody buffer containing 0.1% Triton X-100) primary monoclonal mouse antibodies at 10 μg/mL, and incubated for 60 minutes. Secondary control cells were treated with 100 μL of 2% FBS + 1×PBS and incubated for 60 minutes. The cells were then washed twice with 2% FBS+ 1×PBS followed by incubation for 60 minutes with 100 μL of secondary anti-mouse antibody AlexaFluor 488 at a final concentration of 10 μg/mL. The cells were washed twice with 2% FBS + 1×PBS and fixed in 1 mL of 4% paraformaldehyde solution before flow cytometry analysis.
Annexin V-FITC Apoptosis Assay and Propidium Iodide
MDA-MB-231 cells and MDA-MB-231-TmxR variant cells were plated at a density of 2x106 cells in a T25 tissue culture flask. Cells were treated with individual or a combination of acetylsalicylic acid, metformin, oseltamivir phosphate, and tamoxifen for 48 hours or left untreated. The cells were then trypsinized with TrypLE express, and 100,000 cells were analyzed for apoptotic, necrotic, and viable cell populations using the Annexin V-FITC Apoptosis Kit (BioVision K101-25). Briefly, the collected cells were resuspended in 500 μL of 1×Binding Buffer followed by 5 μL of each Annexin V-FITC and propidium iodide. The cells were incubated for 5 minutes at room temperature in the dark, followed by quantification by flow cytometry analysis.
Tube Formation Assay and Analysis
Human umbilical vein endothelial cells (HuVECs) were grown to confluency and were washed with 1×PBS and trypsinized with 4mL of Trypsin-EDTA at room temperature. Cells were incubated for 5–10 minutes and observed under an inverted phase-contrast microscope to assess whether cells were detached. Trypsin-EDTA was neutralized with complete human endothelial cell media and cells were counted. The tube formation assay protocol was conducted using the Angiogenesis (Tube Formation) Assay Kit (BioVision K905) following the manufacturer’s manual. In brief, chilled 96-well plates were coated in 50 μL of Matrigel thaw to 4°C and incubated for 1 hour. HuVECs were plated at a density of 10,000 cells/well. Treatment conditions with Tmx alone, cocktail therapy, combination therapy were immediately added to the wells or left untreated as controls. Images were acquired using the Incucyte Zoom Imager every 2 hours with a 10× objective. Images were analyzed using ImageJ software with the Angiogenesis Analyzer Plugin.
Caspase 3/7 Apoptosis Assay
HuVECs were plated at a density of 10,000 cells/well and treated with cocktail therapy (ASA at 8 mM, Met 4 mM and OP 300 μg/mL), Tmx (20 μM) or combination therapy (ASA at 8 mM, Met 4 mM and OP 300 μg/mL, Tmx 20 μM) or left untreated for 3 days. Caspase 3/7 enzyme analysis was completed using the CellEvent™ Caspase 3/7 Green Detection Reagent ThermoFisher Scientific. The reagent was diluted to the manufacturer-recommended concentration of 5 μM in human endothelial cell media from the stock solution of 2 mM. Phase contrast and green fluorescent images were acquired using the Incucyte Zoom Imager every 2 hours at a 10× objective for 3 days. Images were analyzed using Incucyte analysis software for counting the number of cells expressing green representing Caspase 3/7 expression.
Statistical Analysis
Data as means ± standard error of the mean (SEM) are from two repeats of each experiment (WST-1 data represents two independent experiments done in triplicates). Statistical analyses were performed with GraphPad Prism software. Half-maximal inhibitory concentrations (IC50) were calculated using logarithmic regression on GraphPad Prism. Results were compared with one-way analysis of variance (ANOVA) and Fisher’s LSD Test, with p < 0.05 defined as statistically significant.
Results
Acetylsalicylic acid (ASA), metformin (Met), and oseltamivir phosphate (OP) and in combination dose-dependently reduce cell viability in MDA-MB-231 monolayer cell culture and 3D tumour spheroid volume
MDA-MB-231 cells were exposed to doses of ASA and Met ranging from 0.5 mM to 16 mM, while OP was administered ranging from 25 to 800 μg/mL. The selected dose ranges correspond with past in vitro cancer studies using ASA, OP, and Met33,43,44 and are representative of safe in vivo drug equivalents.45–47 As shown in Figure 1, significant decreases in cell viability by the individual drugs were observed at higher concentrations compared to the untreated controls. Following treatment with ASA (Figure 1A), significant decreases in cell viability were observed in doses ranging from 2 mM to 16 mM at the 48- and 72-hour treatments. Similarly, a significant decrease in cell viability was observed after 48 hours following treatment with 100 μg/mL OP (p < 0.01), and after 72 hours, 800 μg/mL OP (p < 0.001) (Figure 1B). Lastly, significant decreases in cell viability were observed at 48 hours when MDA-MB-231 cells were treated with 8 mM Met (p < 0.05) and following 72 hours, 16 mM Met (p < 0.001) (Figure 1C).
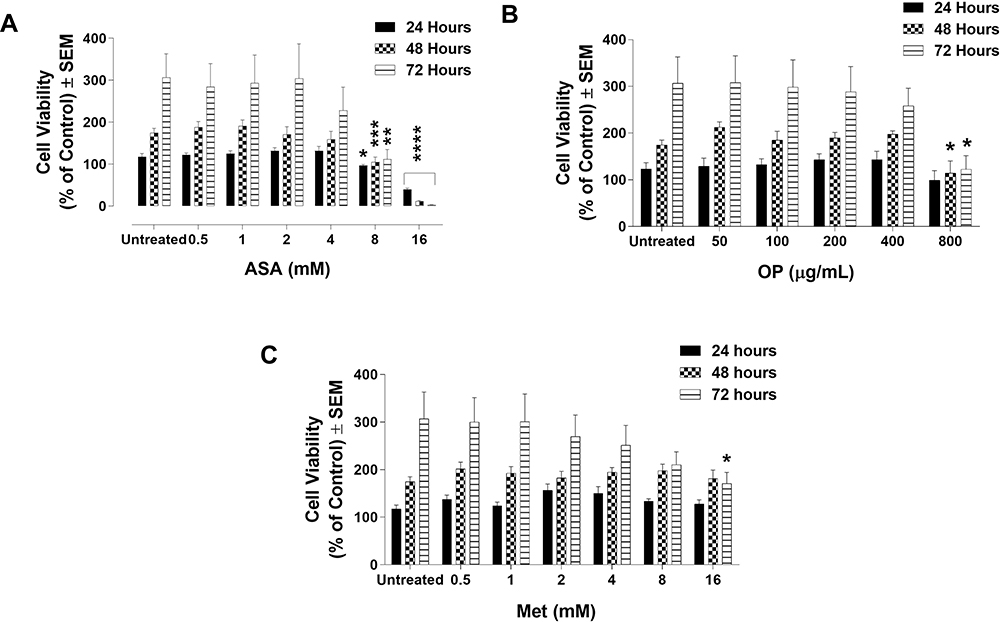 |
Figure 1 Cell viability of MDA-MB-231 cells following treatment with (A) acetylsalicylic acid (ASA), (B) oseltamivir phosphate (OP), and (C) metformin (Met) using the WST-1 cell proliferation assay. MDA-MB-231 cells were plated at 10,000 cells/mL per well in triplicates. The indicated drug concentrations were serially diluted, and the cells were exposed to the treatments for 24, 48, and 72 hours. At the end of each time point, the WST-1 reagent was added to each well. The cells were incubated for 1.5 hours, after which the plates were read on a colorimetric plate reader. The data are cell viability as a mean percent of control ± S.E.M. of three independent experiments performed in triplicates. The cell viabilities were compared to the untreated control for each time point by ANOVA using the uncorrected Fisher’s LSD multiple comparisons test with 95% confidence with indicated asterisks for statistical significance. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, n = 3. |
To assess the effects of the individual drugs on 3D multicellular tumour spheroids (MCTS), the percent decrease in spheroid volume was calculated, and the WST-1 cell proliferation assay was performed for cell viability following treatment with ASA, Met, and OP dose-dependently, and in their combination. In Figure 2, after MCTS were fully formed on Day 4, ASA, OP, and Met were independently added to MDA-MB-231 MCTS for 72 hours (Day 7). ASA and Met were administered in doses ranging from 0.5–16 mM, while OP was administered in doses ranging from 50–800 μg/mL. After 72 hours of drug treatment (Day 7), MCTS volume was measured using ImageJ software, and WST-1 assays were conducted to assess cell viability. It is noteworthy that with increasing concentrations of drugs, proper MCTS shape was not maintained as the spheroids would disassemble and/or shrink due to the effects of the drugs. However, we measured the volume of these cell aggregates to quantify how aggregate volume changed with drug treatment. ASA significantly decreased MCTS volume, starting at a low dose of ASA (4 mM) and displayed a dose-dependent decrease in MCTS volume (Figure 2A). Out of all three drugs, ASA displayed the earliest significant decreases in MCTS cell viability (Figure 2A). These MCTS findings are consistent with those observed in the monolayer cell culture experiments depicted in Figure 1. OP also produced a significant decrease in MCTS cell viability only at concentrations of 400 and 800 μg/mL (Figure 2B), a similar trend observed in Figure 1B. OP displayed significant decreases in MCTS volume at doses of 400 μg/mL and 800 μg/mL (Figure 2B). Met showed a significant decrease in MCTS volume at doses of 4, 8 and 16 mM (Figure 2C). Met also produced significant decreases in MCTS cell viability at doses of 4 mM and 16 mM (Figure 2C).
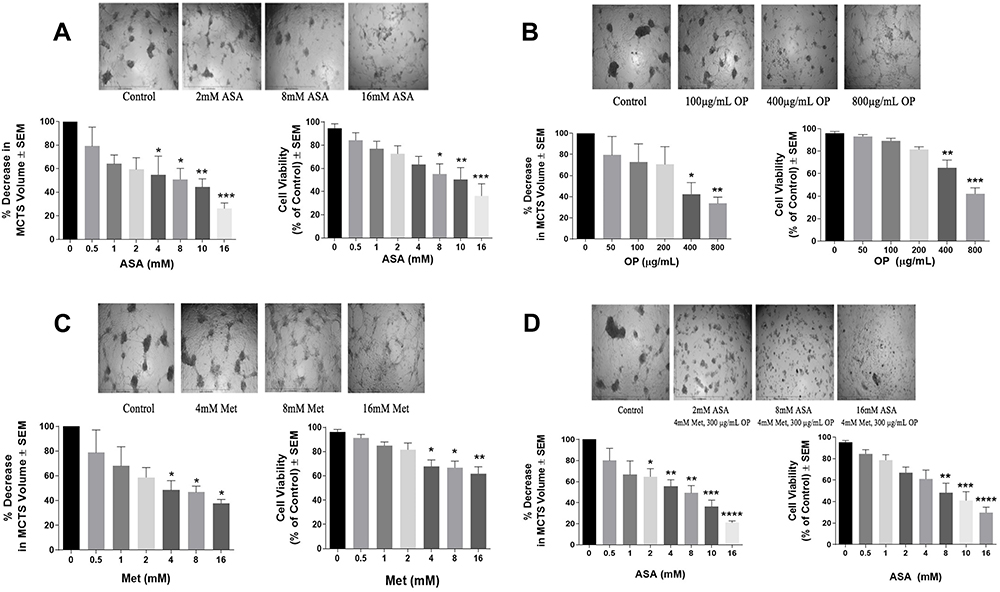 |
Figure 2 Effect of acetylsalicylic acid (ASA), oseltamivir phosphate (OP), and metformin (Met) on MDA-MB-231 multicellular tumour spheroid (MCTS) volume and viability for 72 hours. Cells were grown in culture media containing 50 μM cyclo-RGDfK(TPP) peptide for 4 days in forming MCTS at which time they were treated with (A) ASA, (B) OP, (C) Met, and (D) ASA plus 300 µg/mL OP and 4 mM Met, at their indicated concentrations. On Day 7, MCTS volumes were measured using V = (4/3) πr3, where π = 3.1415, r = average radius (μm). The images were taken with a 4× objective, and the radius was measured using the scale bar in the phase-contrast image. Percent decrease in MCTS volumes was calculated compared to the untreated control. MCTS viability was assessed after 72 hrs of drug treatment using the WST-1 cell viability assay on day 7. At the end of 72 hrs, the WST-1 reagent was added to each well. The MCTS were incubated for 1 to 3 hours, after which the plates were read on a colorimetric plate reader. The data are MCTS viability as a percent of control ± S.E.M. of two independent experiments performed in triplicates, n = 6. Each bar for the MCTS volume represents n=30–60 spheroids from two replicates of each experiment. The spheroid volumes and viabilities were compared by ANOVA using the Tukey’s multiple comparisons test with 95% confidence with indicated asterisks for statistical significance. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001. Abbreviation: MCTS, multicellular tumour spheroids. |
To test the additive effects of all three drugs in combination, we performed a dose-response of ASA (0.5–16 mM) in combination with OP (300 µg/mL) and Met (4 mM) constant concentration (Figure 2D). The cocktail combination also produced significant (p ≤ 0.01) decreases in MCTS volume and cell viability at doses of 8 mM to 16 mM ASA (p ≤ 0.0001) (Figure 2D). Compared to the individual administration of ASA, OP, and Met, the cocktail combination (Figure 2D) displayed an additive anti-cancer effect by producing the earliest and most significant decreases in MCTS volume and cell viability. Hereafter, the cocktail refers to the combination of ASA, Met, and OP.
ASA, Met, and OP Sensitize MDA-MB-231 Cancer Cells to Tamoxifen (Tmx)
Although Tmx acts primarily by antagonizing the estrogen receptor (ER) and thus would be an ineffective treatment option for TNBC,48 Tmx might have a modest anti-tumour effect through ER-independent mechanism(s) in TNBC.49 ASA, Met, and OP may prove to be an effective treatment of TNBC, especially with the chemoresistant TNBC tumours.50 The data in Figure 3A indicate a marked and significant (p < 0.0001) reduction in viability of MDA-MB-231 cells at 40 and 80 μM Tmx. As shown in Figure 3B, the cocktail of ASA (8 mM), Met (4 mM), and OP (300 μg/mL) produced and maintained a significant (p < 0.0001) decrease in viability of MDA-MB-231 cells together with Tmx dose-dependently. These data provide strong supporting evidence for the efficacy of the drug cocktail, not only to increase the MDA-MB-231 cancer cell sensitivity to Tmx but also provide an immediate and significant inhibition on cell proliferation.
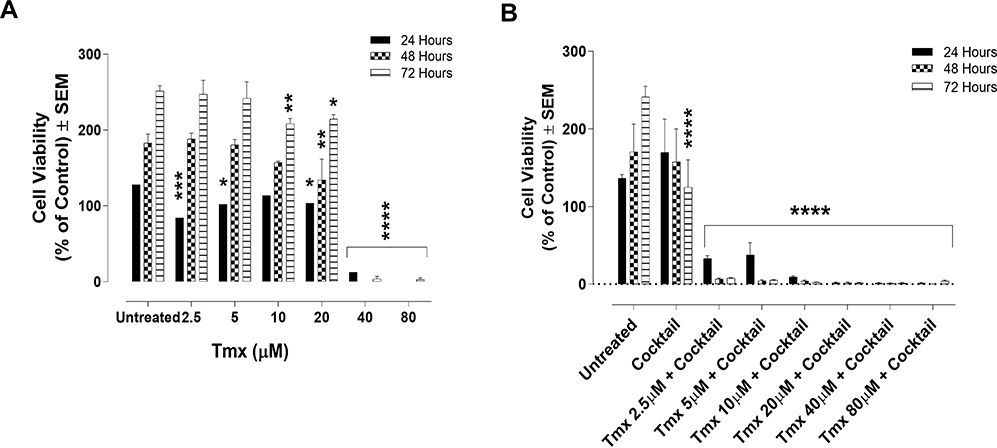 |
Figure 3 Cell viability of MDA-MB-231 cells treated with tamoxifen (Tmx) in combination with the drug cocktail of ASA, Met, and OP using the WST-1 cell proliferation assay. MDA-MB-231 cells were plated at 10,000 cells/mL in triplicates. The Tmx alone (A) or (B) in combination with drug cocktail containing ASA (8 mM), Met (4 mM), and OP (300 µg/mL) was serially diluted at the indicated concentrations, and the cells were exposed to the treatments for 24, 48, and 72 hours. At the end of each time point, the WST-1 reagent was added to each well. The cells were incubated for 1.5 hours, after which the plates were read on a colorimetric plate reader. The data are cell viability as a mean percent of control ± S.E.M. of three independent experiments performed in triplicates. The cell viabilities were compared to the untreated control for each time point by ANOVA using the uncorrected Fisher’s LSD multiple comparisons test with 95% confidence with indicated asterisks for statistical significance. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, n = 3.Abbreviations: Tmx, tamoxifen; ASA, acetylsalicylic acid; Met, metformin; OP, oseltamivir phosphate. |
ASA, Met, and OP Sensitize MDA-MB-231-TmxR Variant Cell Culture and Spheroids to Tmx Treatment
To test whether the drug cocktail would sensitize Tmx resistant breast cancer cells, the MDA-MB-231-TmxR cell line was grown in conditioned medium containing 10 µM Tmx for over one year. In Figure 4A–C, MDA-MB-231-TmxR cells were treated with ASA and Met ranging from 0.5 mM to 16 mM, and OP ranged from 25 to 800 μg/mL. Consistent with the results observed in the parental MDA-MB-231 cell line, higher concentrations of Met and OP significantly reduced the viability of MDA-MB-231-TmxR cells (Figure 4B and C). ASA had a significant effect on cell viability at low concentrations ranging from 2 to 8 mM (Figure 4A).
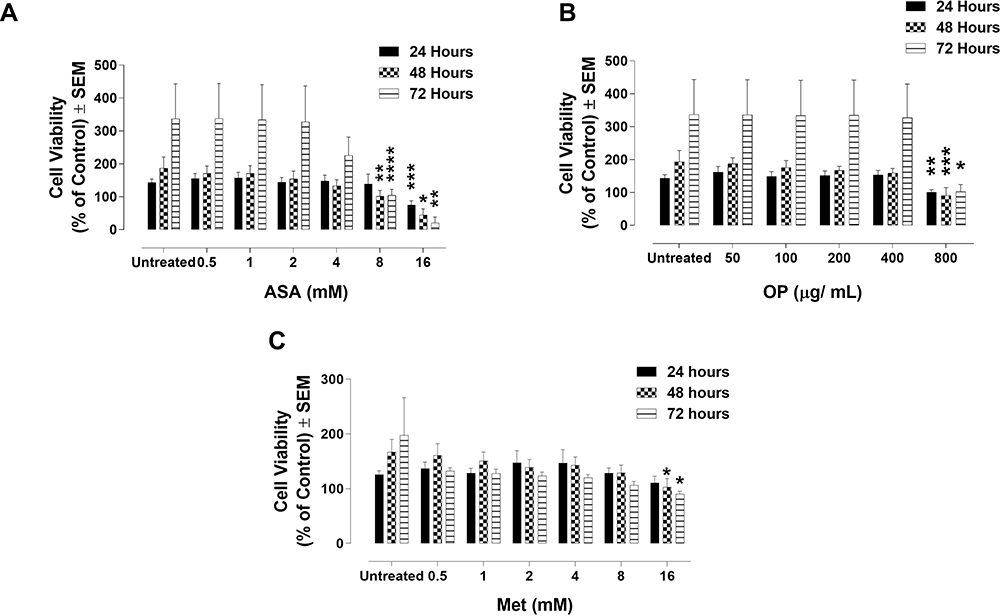 |
Figure 4 Cell viability of tamoxifen (Tmx) resistant MDA-MB-231-TmxR cells following treatment with (A) acetylsalicylic acid (ASA), (B) oseltamivir phosphate (OP), and (C) metformin (Met) using the WST-1 cell proliferation assay. MDA-MB-231-TmxR cells were plated at a density of 10,000 cells/mL in triplicates. The indicated drug concentrations were serially diluted, and the cells were exposed to the treatments for 24, 48, and 72 hours. At the end of each time point, the WST-1 reagent was added to each well. The cells were incubated for 1.5 hours, after which the plates were read on a colorimetric plate reader. The data are cell viability as a mean percent of control ± S.E.M. of three independent experiments performed in triplicates. The cell viabilities were compared to the untreated control for each time point by ANOVA using the uncorrected Fisher’s LSD multiple comparisons test with 95% confidence with indicated asterisks for statistical significance. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, n = 3. |
Furthermore, we treated MDA-MB-231-TmxR cells with Tmx concentrations ranging from 2.5 μM to 80 μM to establish the sensitivity of the Tmx resistant cells to Tmx alone. In Figure 5A, the MDA-MB-231-TmxR cells continued to be resistant using Tmx ranging from 2.5 μM to 20 μM. To test whether the drug cocktail would sensitize the MDA-MB-231-TmxR cells to Tmx treatment, Figure 5B showed a significant (p < 0.0001) decrease in cell viability observed at all concentrations of Tmx. These data are consistent with those observed with the parental MDA-MB-231 cell line in Figure 3B. Collectively, these results strongly support the efficacy of our drug cocktail of ASA, Met, and OP to sensitize both MDA-MB-231 and MDA-MB-231-TmxR cancer cells to Tmx.
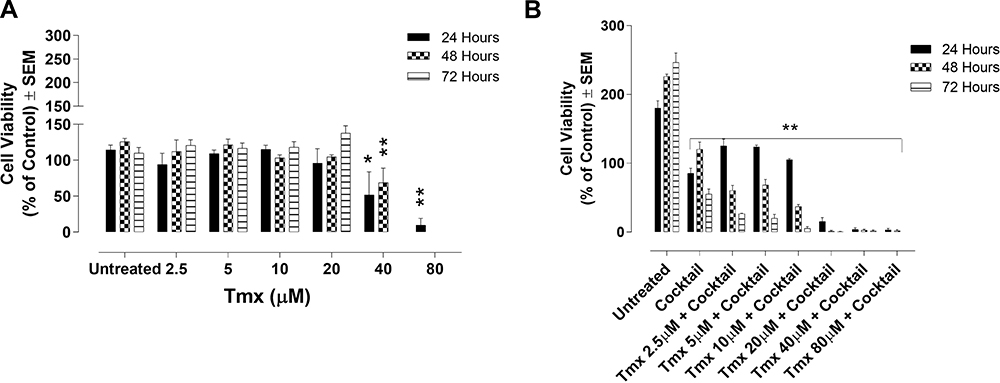 |
Figure 5 Cell viability of MDA-MB-231-TmxR cells treated with tamoxifen (Tmx) in combination with the drug cocktail of ASA, Met, and OP using the WST-1 cell proliferation assay. MDA-MB-231-TmxR cells were plated at a cell density of 10,000 cells/mL per well in triplicates. The Tmx alone (A) or (B) Tmx in combination with a drug cocktail containing ASA (8 mM), Met (4 mM), and OP (300 µg/mL) was serially diluted at the indicated concentrations. The cells were exposed to the treatments for 24, 48, and 72 hours. At the end of each time point, the WST-1 reagent was added to each well. The cells were incubated for 1.5 hours, after which the plates were read on a colorimetric plate reader. The presented data are cell viability as a percent of control ± S.E.M. of three independent experiments performed in triplicates. The cell viabilities were compared to the untreated control for each time point by ANOVA using the uncorrected Fisher’s LSD multiple comparisons test with 95% confidence with indicated asterisks for statistical significance. *p ≤ 0.05, **p ≤ 0.01, n = 3.Abbreviations: Tmx, tamoxifen; TmxR, tamoxifen resistance; ASA, acetylsalicylic acid; Met, metformin; OP, oseltamivir phosphate. |
We also tested whether ASA, OP, and Met would sensitize MDA-MB-231-TmxR 3D tumour spheroids (MCTS) to Tmx treatment (Figure 6). The cell viability of MDA-MB-231-TmxR tumour spheroids with increasing Tmx concentrations (5–25 μM) was measured in the presence or absence of ASA, OP, or Met. The IC50 values to inhibit 50% of the cell viability were calculated after 72 hours with the drug concentrations. ASA was able to decrease the IC50 of 22.92 μM Tmx alone to 20.83 μM Tmx with 8mM ASA in MDA-MB-231-TmxR MCTS (Figure 6A). At 5 μM Tmx, 8 mM ASA was able to produce a significant (p ≤ 0.01) decrease in cell viability when compared to Tmx treatment alone (Figure 6A). OP was able to decrease the IC50 of 22.92 μM Tmx alone to 21.80 μM Tmx with 300 μg/mL OP in MDA-MB-231-TmxR MCTS (Figure 6B). At 5 μM Tmx, 300 μg/mL OP was also able to produce a significant (p ≤ 0.05) decrease in cell viability when compared to Tmx alone (Figure 6B). Met was able to decrease the IC50 of 22.92 μM Tmx alone to 21.58 μM Tmx with 4 mM Met in MDA-MB-231-TmxR MCTS (Figure 6C). Unlike ASA and OP, 10 mM Met produce a significant difference in cell viability when compared to Tmx treatment alone at 5 μM (Figure 6C).
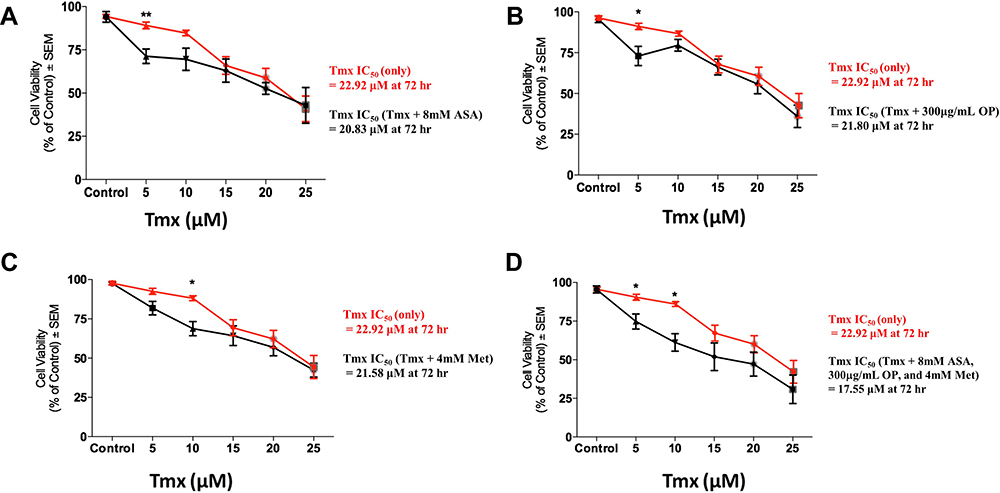 |
Figure 6 Cell viability of tamoxifen (Tmx) resistant MDA-MB-231-TmxR tumour MCTS following treatment with Tmx in a dose-response or in combination with (A) acetylsalicylic acid (ASA), (B) oseltamivir phosphate (OP), (C) metformin (Met) and (D) cocktail of ASA, OP, and Met using the WST-1 cell proliferation assay. The cell viability of MDA-MB-231-TmxR MCTS with increasing Tmx concentrations in the presence or absence of 8 mM ASA (A), 300 µg/mL OP (B), 4mM Met (C), and a cocktail of 8 mM ASA, 300 µg/mL OP, and 4 mM Met (D). The IC50 values represent the individual drug concentration to inhibit 50% of the cell viability. The half-maximal inhibitory concentration (IC50) was calculated using logarithmic regression with GraphPad Prism. The data are representative of two replicates of two independent experiments performed in triplicate with n=30–60 MCTS analyzed at each Tmx concentrations. Unpaired t-test was used to determine significant differences in cell viability between MDA-MB-231-TmxR MCTS with Tmx alone or in combination with ASA, OP, and Met at each Tmx concentration with asterisks for statistical significance *p ≤ 0.05, **p ≤ 0.001. Abbreviations: Tmx, tamoxifen; TmxR, tamoxifen resistance; MCTS, multicellular tumour spheroids; ASA, acetylsalicylic acid; Met, metformin; OP, oseltamivir phosphate. |
The combination of ASA (8 mM), Met (4 mM), and OP (300 μg/mL) was able to decrease the IC50 of 22.92 μM Tmx alone to 17.55 μM Tmx with 8mM ASA in MDA-MB-231-TmxR MCTS (Figure 6D). These latter data demonstrate a more marked decrease in IC50 of Tmx compared to the independent effects of ASA (Figure 6A), OP (Figure 6B), and Met (Figure 6C). At 5 μM and 10 μM of Tmx, the drug cocktail produced a significant (p ≤ 0.05) decrease in MDA-MB-231-TmxR MCTS cell viability when compared to Tmx alone (Figure 6D).
Cocktail of ASA, Met, and OP Activates Early and Late Apoptosis in Both MDA-MB-231 and MDA-MB-231-TmxR Breast Cancer Cells
To assess the effects of the ASA, Met, and OP inducing apoptotic activity in MDA-MB-231 breast cancer cells, cells were treated with individual drugs, and in combination or left untreated for 48 hours (Figure 7). The apoptotic activity of the cell treatments was assessed using Annexin-V and flow cytometry. Here, an early time point was taken after treatment to ensure that a viable number of cells could be analyzed. As shown in Figure 7A–C, the cocktail of ASA, Met, and OP did not significantly increase early apoptotic and late apoptotic events when compared to the untreated cells; however, when MDA-MB-231 cells were treated with a combination of ASA, Met, OP, and Tmx, a significant percentage (36.9%) of viable cells compared to early apoptotic (55.5%) ones entering the early phase was observed (Figure 7B). Furthermore, the percentage of viable cells when compared to the untreated cells was significantly decreased following Tmx and cocktail treatment, with a more significant reduction of viable cells being observed following combination treatment. When compared to the percentage of cells in the early apoptotic phase, there was a significantly increased number of cells in this phase following treatment with Tmx and the drug cocktail, with a more increased percentage of early apoptotic cells observed post-combination treatment.
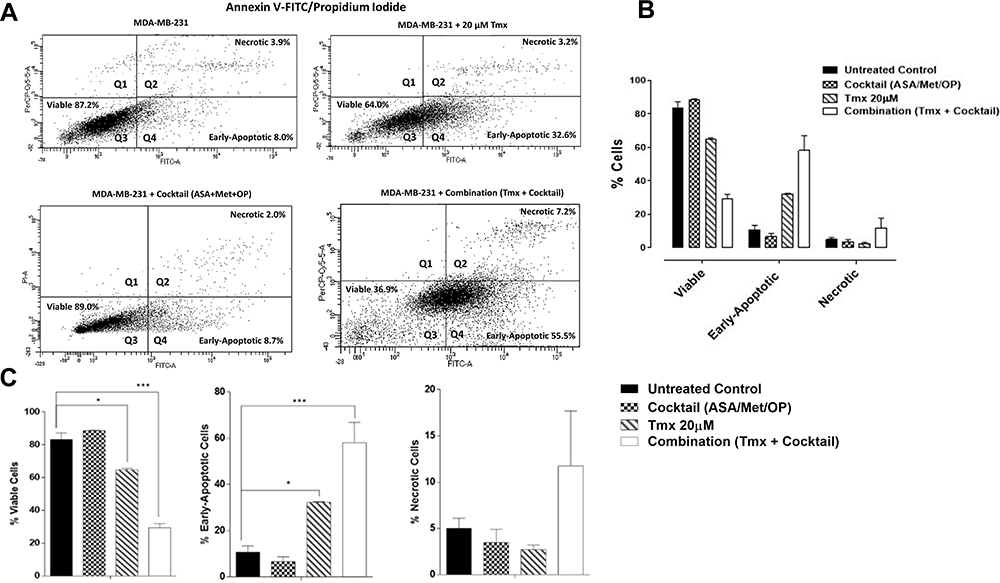 |
Figure 7 Viability, early apoptosis and necrosis of MDA-MB-231 cells 48 hours after treatment with Tmx (20μM), cocktail (ASA 8mM, Met 4mM, OP 300μg/mL) and combination (Tmx plus cocktail) using the Annexin V-FITC apoptosis and propidium iodide assay. (A) 100,000 cells were collected and stained with Annexin V-FITC/Propidium Iodide to assess early apoptosis and necrosis. (B) Comparisons were made within treatment groups to assess the shift of percent of cells that are viable, early apoptotic and necrotic stages of cell death following 48 hours of indicated treatments. (C) Comparisons were made between untreated cohorts and treatment groups for percent viable, early apoptotic, and necrotic cells. The data are cell viability as a percent of control ± S.E.M. of two independent experiments performed. The cell viabilities were compared to the untreated control for each treatment by ANOVA using the uncorrected Fisher’s LSD multiple comparisons test with 95% confidence with indicated asterisks for statistical significance. *p ≤ 0.05, ***p ≤ 0.001, n = 2.Abbreviations: Tmx, tamoxifen; ASA, acetylsalicylic acid; Met, metformin; OP, oseltamivir phosphate; Annexin V-FITC, Annexin V conjugated with fluorescein isothiocyanate; FITC-A; fluorescein isothiocyanate axis; PI-A, propidium iodide axis. |
To assess the effects of the combination treatment on MDA-MB-231-TmxR cells, a consistent early apoptotic trend was observed compared to that of the parental cells (Figure 8A–C). Interestingly, although there is a definite increase in early apoptotic activity post-Tmx and combination cocktail treatment, these increases in apoptotic activity were not statistically significant when compared to the untreated cohort. Surprisingly, the percentage of cells entering the necrotic cell phase was statistically significant compared with the untreated control cells and was only observed with the combination drug treatment.
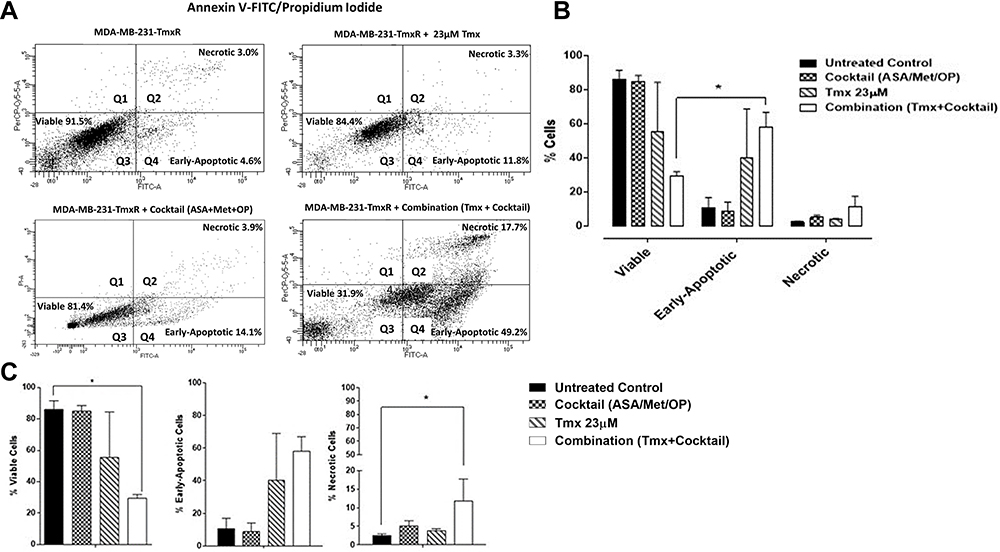 |
Figure 8 Viability, early apoptosis and necrosis of tamoxifen (Tmx) resistant MDA-MB-231-TmxR cells 48 hours after treatment with Tmx (23μM), cocktail (ASA 8mM, Met 4mM, OP 300μg/mL) and combination (Tmx plus cocktail) using the Annexin V-FITC apoptosis and propidium iodide assay. (A) 100,000 cells were collected and stained with Annexin V-FITC/Propidium Iodide to assess early apoptosis and necrosis. (B) Comparisons were made within treatment groups to assess the shift of percent of cells that are viable, early apoptotic and necrotic stages of cell death following 48 hours of indicated treatments. (C) Comparisons were made between untreated cohorts and treatment groups for percent viable, early apoptotic, and necrotic cells. The presented data are cell viability as a percent of control ± S.E.M. of two independent experiments. The cell viabilities were compared to the untreated control for treatments by ANOVA using the uncorrected Fisher’s LSD multiple comparisons test with 95% confidence with indicated asterisks for statistical significance. *p ≤ 0.05, n = 2.Abbreviations: Tmx, tamoxifen; TmxR, tamoxifen resistance; ASA, acetylsalicylic acid; Met, metformin; OP, oseltamivir phosphate; Annexin V-FITC, Annexin V conjugated with fluorescein isothiocyanate; FITC-A; fluorescein isothiocyanate axis; PI-A, propidium iodide axis. |
Cocktail of ASA, Met, and OP with Tmx Combination Disrupts Vasculogenic Endothelial Cell Tube Formation of Human Umbilical Vein Endothelial Cells (HuVECs)
About 23% of patients with triple-negative breast cancer (TNBC) present with lymphovascular invasion, where both the lymphatic and blood vessel invasion are lying within an endothelial lined space in the area surrounding the invasive tumour.51 Additionally, the lymphovascular invasion has been correlated with an increased metastatic burden, typical of patients with this subtype of breast cancer.52 Currently, the anti-angiogenic therapy using VEGF inhibitor bevacizumab was approved for use in combination with paclitaxel as a treatment option for this metastatic TNBC disease.53 However, the patient response rates to this anti-angiogenic therapy have been minimal.
To this end, we investigated if the drug cocktail had anti-angiogenic properties. Human umbilical vein endothelial cells (HuVECs) were used to assess vasculogenic endothelial cell tube formation following ASA (8 mM), Met (4 mM), OP (300 μg/mL) and Tmx (20 μM) alone and in a combination of drug cocktail and Tmx. As shown in Figure 9A, after 10 hours of endothelial cell tube formation in the conditioned culture medium, HuVEC cells formed vasculogenic endothelial networks with a distinct network of vessels. In contrast, the drug cocktail and its combination with Tmx disrupted the morphology and architecture of these networks into abnormal and segmented networks. Further data analysis on the number of nodes, mean mesh size, and the number of isolated segments demonstrated no significant differences between the treatment groups (Figure 9B). However, the cocktail treatment alone showed more significant numbers of isolated segments as well as reduced mesh sizes. It is noteworthy that the integrity of the vascular endothelial cell network was disrupted by the cocktail of ASA, Met, and OP.
 |
Figure 9 Tube formation assay of HuVEC cells 10 hours after treatment with tamoxifen (Tmx 20 µM), cocktail (ASA 8 mM, Met 4 mM OP 300 μg/mL) and Tmx plus cocktail. (A) HuVECs were plated at 10,000 cells/well on 50 μL of Matrigel followed by treatment with Tmx (20 μM), cocktail, Tmx plus the cocktail or left untreated. Phase-contrast images were acquired by the Incucyte Zoom Imager every 2 hours at 10x magnification. Scale bar represents 300 μm. (B) Analyses of tube formation were done with ImageJ Software using the Angiogenesis Analyzer Plugin. The number of nodes, mean mesh size and the number of isolated segments were analyzed for each treatment condition. The data are mean ± SEM of two independent experiments. Abbreviations: ASA, acetylsalicylic acid; Met, metformin; OP, oseltamivir phosphate; Tmx, tamoxifen; A/M/OP, acetylsalicylic acid/metformin/oseltamivir phosphate. |
Cocktail of ASA, Met, and OP Alone and in Combination with Tmx Induces Apoptosis in Human Umbilical Vein Endothelial Cells (HuVECs)
To further assess whether the drug cocktail would induce anti-angiogenic effects, HuVECs were treated with ASA at 8 mM, Met at 4 mM and OP at 300 μg/mL, Tmx at 20 μM and in combination (cocktail + Tmx) supplemented with the CellEvent™ Caspase 3/7 Green Detection Reagent for 72 hours. As shown in Figure 10A, the number of cells expressing the caspase 3/7 enzyme increased gradually for all treatment conditions as well as with the untreated cohort. However, as shown in Figure 10B, the most significant increase in caspase 3/7 activity was observed after 24 hours in the Tmx treatment group and in the combination (cocktail and Tmx) compared to the untreated HuVECs. These results suggest that the combination of drug cocktail and Tmx induces apoptosis in human umbilical vein endothelial cells. Further investigations are required to determine which components of the cocktail components possess anti-angiogenic properties.
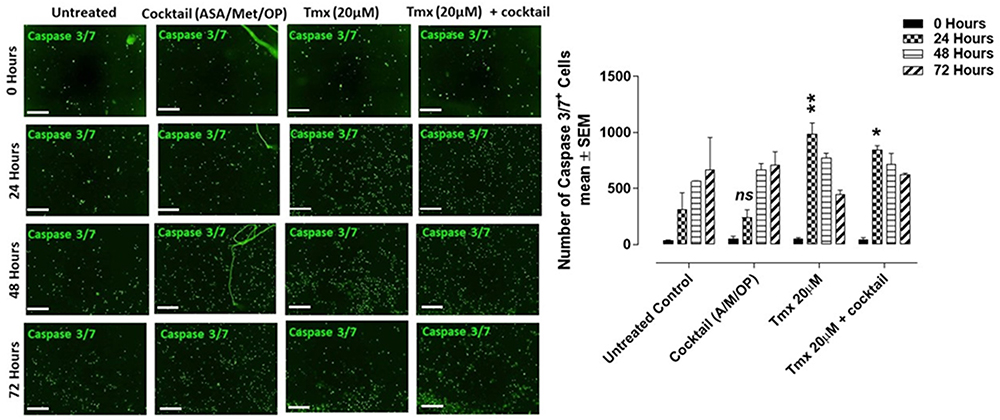 |
Figure 10 CellEvent Caspase 3/7 Green Assay of HuVEC cells at 0, 24, 48, and 72 hours after treatment with a cocktail (ASA 8 mM, Met 4 mM, OP 300 μg/mL), tamoxifen (Tmx 20 µM), Tmx 20 µM + Cocktail, or left untreated. (A) HuVECs were plated at 10,000 cells/well. Cells adhered to the well were treated with Tmx (20 μM), cocktail (OP 300 μg/mL, Met 4 mM, ASA 8 mM), and Tmx 20 μM plus the cocktail or left untreated. CellEvent™ Caspase 3/7 Green Detection Reagent was added at a concentration of 5 μM in human endothelial cell media. Green fluorescent images were acquired by the Incucyte Zoom Imager every 2 hours at 10x magnification. Scale bar represents 300 μm. (B) Analyses were done using the Incucyte Zoom Imager Software Analyzer to count the number of expressing Caspase 3/7 (green) cells. The images are representative of two independent experiments showing similar results. The graph represents the mean ± SEM combined results from two independent experiments compared to the untreated control for each treatment by ANOVA using the uncorrected Fisher’s LSD multiple comparisons test with 95% confidence with indicated asterisks for statistical significance. ns, not significant, *p ≤ 0.01, **p ≤ 0.001, n = 2.Abbreviations: ASA, acetylsalicylic acid; Met, metformin; OP, oseltamivir phosphate; Tmx, tamoxifen; A/M/OP, acetylsalicylic acid/metformin/oseltamivir phosphate; T, tamoxifen; A/M/OP, acetylsalicylic acid/metformin/oseltamivir phosphate. |
Tamoxifen (Tmx) in Combination with the Cocktail of ASA, Met, and OP Prevents the Acquisition of Stem-Like and Invasive Phenotype Markers and Inhibits MDA-MB-231 Cell Growth
The accumulation of somatic mutation and factors of tumour microenvironment such as fibroblasts, extracellular matrix (ECM), inflammatory cells, and blood vasculature are known to be responsible for invasion and metastasis of cancer cells. Breast cancer stem cells can also induce the formation of new blood vessels at the site of metastasis and enhance a stromal cell response similar to that of their primary breast cancer.51 Also, the acquisition of stem-like characteristics of cancer cells is typically an advent of the survival response following chemotherapeutic treatments.54 Here, we investigated whether the drug cocktail, in combination with Tmx, would affect the acquisition of cancer stem-like biomarkers in MDA-MB-231 cells.
Markers of chemoresistance and cancer stem-like (CSC) properties included alcohol dehydrogenase-1 family member A1 (ALDH1A1), CD24, and CD44. The standard way to identify CSCs is the expression of these characteristic markers on the cell surface. High CD44 expression and low expression of CD24 (CD44+/CD24−/low) is one of such marker characteristics. Breast tumours with an expression of CD44+/CD24−/low have been shown to exhibit enhanced invasion and metastasis.55,56 Aldehyde dehydrogenase-1 (ALDH1) is also used to characterize cancer cell stemness. ALDH1 is an enzyme responsible for the oxidation of retinol to retinoic acid, which is essential for the early differentiation of stem cells.57 As shown in Figure 11A, cells treated alone with Tmx (20 µM), ASA (8 mM), Met (4 mM), or OP (300 μg/mL) revealed a significant increase in the expression of CD44 when compared to the untreated group. Surprisingly, the cells treated with Tmx in combination with the drug cocktail showed a significant decrease in CD44 compared to the untreated control.
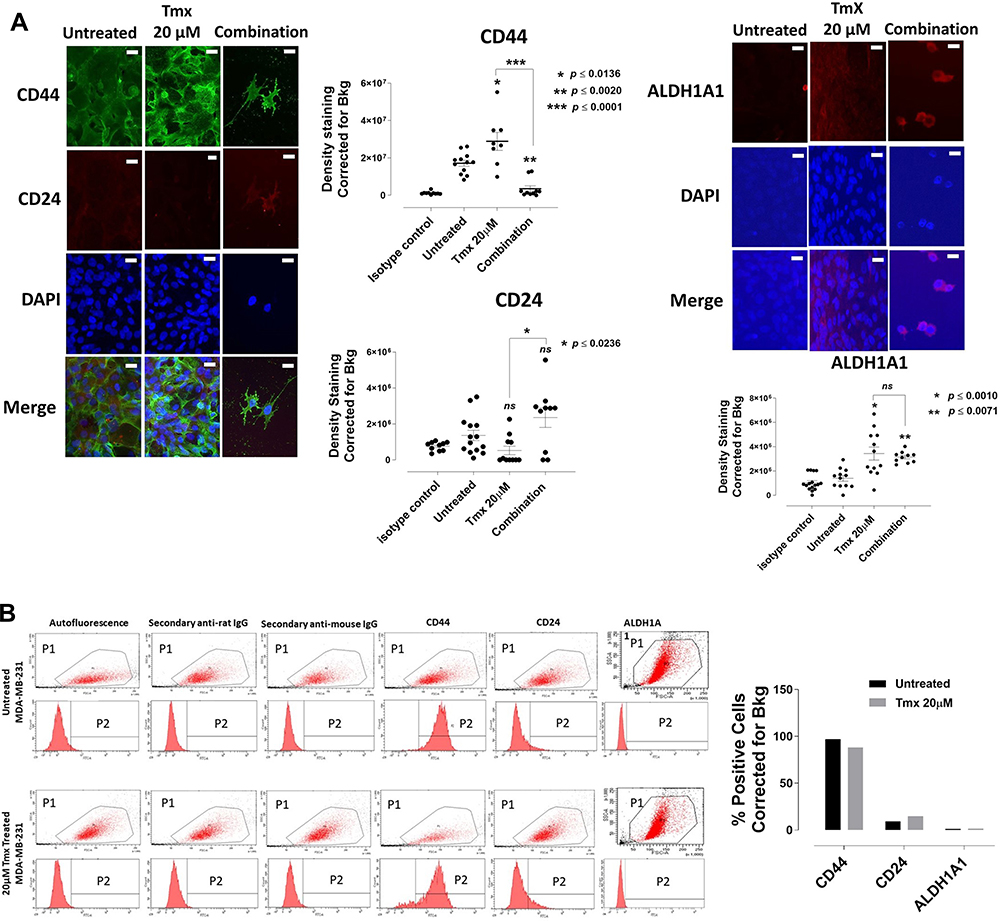 |
Figure 11 Analyses of MDA-MB-231 cells for CD44, CD24 and ALDH1A1 stem-like markers following 72-hour exposure to Tmx, ASA, Met, OP and Tmx in a combination of ASA, Met and OP cocktail using immunocytochemistry and flow cytometry. (A) MDA-MB-231 cells were plated at 150,000 cells/mL and left untreated, treated with Tmx (20 μM), ASA (8 mM), Met (4 mM), OP (300 μg/mL) and Tmx (20 μM) in combination with ASA, Met and OP for 72 hours. Cells were immunostained for markers of CD44, CD24 and ALDH1A1. The results are depicted as a scatter plot of data visualization using dots to represent the indicated marker values obtained from 2 independent experiments of multiple images (n = 4–12). The mean density staining corrected for background (Bkg) ± S.E.M. for indicated marker values is indicated for each group. The density staining values of each group were compared to the untreated control as well as Tmx in combination with drug cocktail with Tmx alone by ANOVA using the uncorrected Fisher’s LSD multiple comparisons test with 95% confidence with indicated asterisks for statistical significance. (B) Flow cytometry of untreated and Tmx (20µM) treated MDA-MB-231 cells for 72 hrs. One million cells were immunostained for CD44, CD24, and ALDH1A1. Data are presented as the percentage of cells expressing the markers corrected for background autofluorescence. ns, not significant, *p ≤ 0.01, **p ≤ 0.001, ***p ≤ 0.0001. P1, all viable cells; P2, antibody of interest. Abbreviations: Tmx, tamoxifen; ASA, acetylsalicylic acid; Met, metformin; OP, oseltamivir phosphate; DAPI, 4′,6-diamidino-2-phenylindole, IgG, immunoglobulin G; Bkg, background;ALDH1A1, aldehyde dehydrogenase 1 family, member A1; CD44, cancer stem cell marker participates in cell-cell interaction, cell adhesion and migration; CD24, a cell adhesion molecule and a glycophosphatidylinositol-linked sialoprotein that serves as a ligand for P-selectin. |
Interestingly, the MDA-MB-231 cells treated with Tmx (20 µM), ASA (8 mM), or OP (300 μg/mL) alone or Tmx in the combination of ASA, Met and OP showed no differences in their expression levels of CD24 compared to the untreated control group, except for a significant CD24 increase with Met (4 mM) treatment. The ratio of CD44/CD24 expressed on MDA-MB-231 cells was 55 for the Tmx treatment compared with 13 for the untreated control group (Table 1). These data strongly support the evidence for Tmx treatment of MDA-MB-231 cells for 72 hrs induces an invasive and metastatic cancer stem cell characteristic property, which is dramatically reversed with Tmx in a combination of ASA, Met and OP (CD44/CD24 ratio of 2). Surprisingly, the ALDH1A1 expression was significantly increased following Tmx treatment alone or Tmx in combination with the drug cocktail. Consistent with our earlier results, a minimal number of cells may remain following the combination of drug cocktail and Tmx treatment, as indicated by the reduced number of nuclei observed in the DAPI panel (supplementary Figure S1 and S2).
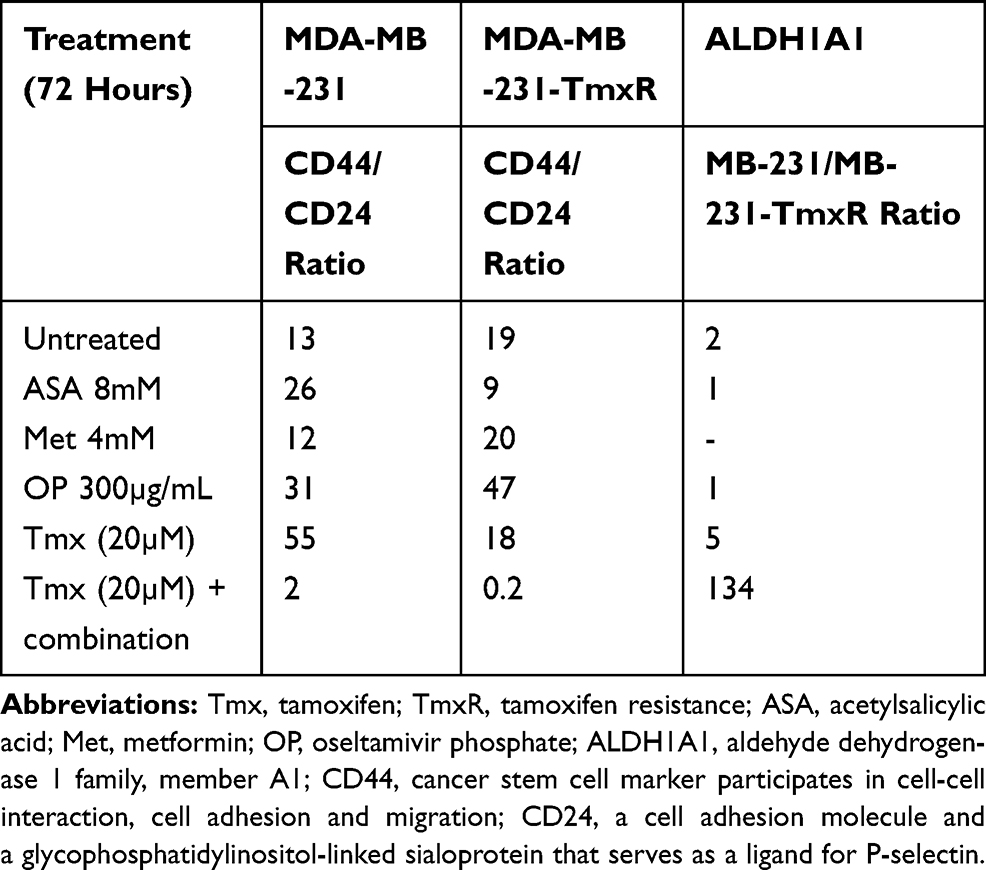 |
Table 1 The Ratio of CD44/CD24 Expression on MDA-MB-231 and MDA-MB-2321-TmxR Cells After Treatment with Tmx, ASA, Met, OP Alone and Tmx in Combination with ASA, Met and OP |
We further confirmed these staining patterns with flow cytometry to quantify the number of cells expressing the stem-like markers in MDA-MB-231 cells treated with Tmx (20µM) for 72 hrs compared with the untreated control group (Figure 11B). It is noteworthy that the majority of cells treated with the triple combination were ablated following 72 hours of treatment (data not shown), leaving only the untreated and Tmx treated cells available for flow cytometry analysis. The flow cytometry data are consistent with the immunofluorescence data for the Tmx and untreated cells where the percentage of cells expressing CD44, CD24, and ALDH1A1 are relatively comparable between the Tmx treated group and the untreated group. These data are consistent with reported findings with MDA-MB-231 cells, where the CD44/CD24 ratio was the highest in the basal mesenchymal cell line MDA-MB-231, compared to the basal epithelial cell lines MDA-MB-468, HER2-OE and SK-BR-3, and the luminal A cell line MCF-7.58 ALDH1 is highly expressed in the most malignant cell line, MDA-MB-231. Surprisingly, the Tmx treatment of MDA-MB-231 cells in combination with the drug cocktail markedly reduced the CD44/CD24 ratio by 6.5-fold compared to the untreated control group (Table 1). Tmx treatment of MDA-MB-231-TmxR cells in combination with the drug cocktail markedly reduced the ALDH1A1 by 134-fold compared to the same treatment for the parental cell line. The Tmx treatment of MDA-MB-231-TmxR variants in combination with ASA, Met and OP markedly suppressed the CD44 and ALDH1A1, leading to a decreased tumorigenicity and cell invasion. Li et al have demonstrated that suppression of CD44 and ALDH1 by siRNA knockdown led to decreased tumorigenicity and cell migration capacity.58 Moreover, they also demonstrated that both high CD44/CD24 ratio and ALDH1+ markers in TNBC cells were conserved during metastasis, from the primary tumours to the circulating tumour cells (CTCs) and the distant metastases.
To further examine the changes in CD44, CD24 and ALDH1A1 expression levels in Tmx-resistant MDA-MB-231-TmxR cells, the cells were treated with Tmx (23 μM), ASA (8 mM), Met (4 mM), or OP (300 μg/mL) alone, and Tmx in combination with ASA, Met, OP or left untreated for 72 hours as shown in Figure 12A and B. These findings further confirm the ability of the Tmx in combination with ASA, Met and OP to potentially target cells expressing markers typically associated with a malignant and cancer stem-like phenotype. MDA-MB-231-TmxR cells treated with this combination of drug therapy were too few to be analyzed by the flow cytometry (no data). The ratio of CD44/CD24 expressed on MDA-MB-231-TmxR cells was 18 for the Tmx treatment compared to 0.2 for the Tmx in the combination group (Table 1). These data strongly support the evidence for Tmx treatment of MDA-MB-231-TmxR cells for 72 hrs induces an invasive and metastatic potential characteristic property, which can be significantly reduced with Tmx in combination with ASA, Met and OP.
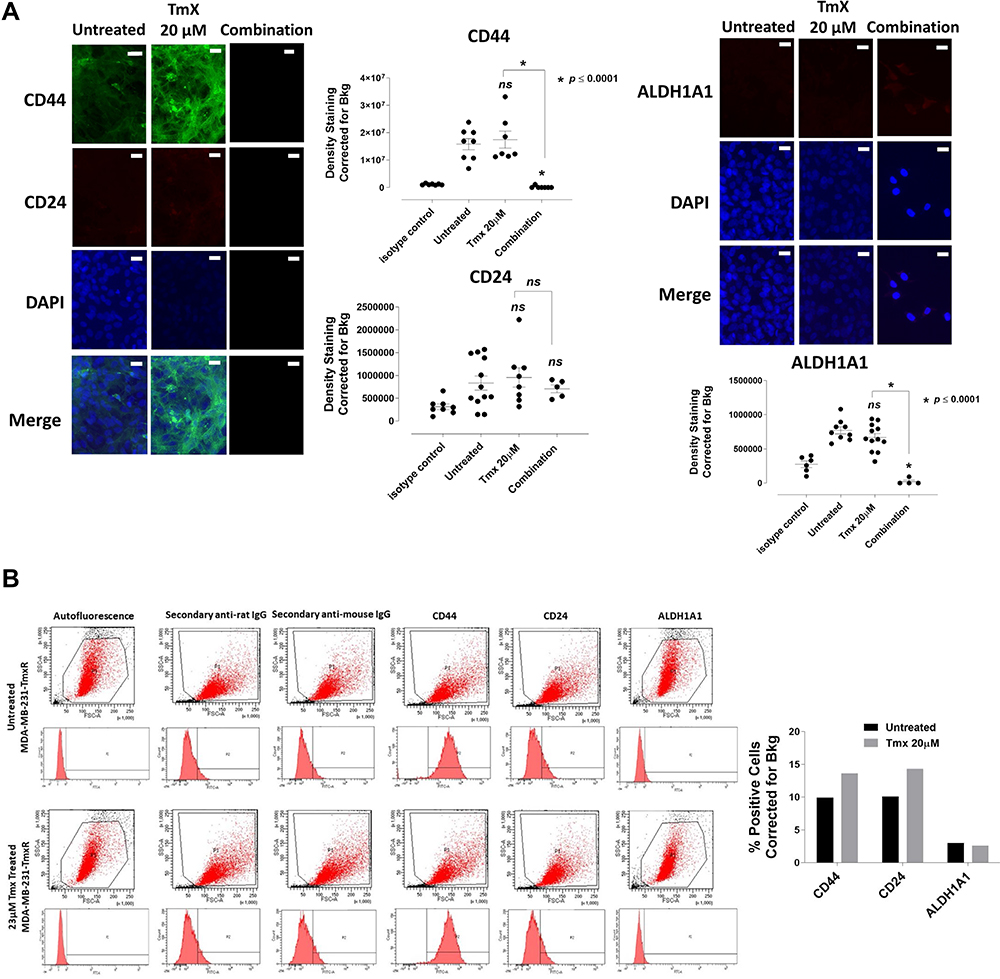 |
Figure 12 Analyses of MDA-MB-231-TmxR cells for CD44, CD24 and ALDH1A1 stem-like markers following 72-hour exposure to Tmx, ASA, Met, OP and Tmx in a combination of ASA, Met and OP cocktail using immunocytochemistry and flow cytometry. (A) MDA-MB-231-TmxR cells were plated at 150,000 cells/mL and left untreated, treated with Tmx (20 μM), ASA (8 mM), Met (4 mM), OP (300 μg/mL) and Tmx (20 μM) in combination with ASA, Met and OP for 72 hours. Cells were immunostained for markers of CD44, CD24 and ALDH1A1. The results are depicted as a scatter plot of data visualization using dots to represent the indicated marker values obtained from 2 independent experiments of multiple images (n = 4–12). The mean density staining corrected for background (Bkg) ± S.E.M. for indicated marker values is indicated for each group. The density staining values of each group were compared to the untreated control as well as Tmx in combination with drug cocktail with Tmx alone by ANOVA using the uncorrected Fisher’s LSD multiple comparisons test with 95% confidence with indicated asterisks for statistical significance. (B) Flow cytometry of untreated and Tmx (20 µM) treated MDA-MB-231-TmxR cells for 72 hrs. One million cells were immunostained for CD44, CD24, and ALDH1A1. Data are presented as the percentage of cells expressing the markers corrected for background autofluorescence. ns, not significant, *p ≤ 0.0001, P1, all viable cells; P2, antibody of interest. Abbreviations: Tmx, tamoxifen; ASA, acetylsalicylic acid; Met, metformin; OP, oseltamivir phosphate; DAPI, 4′,6-diamidino-2-phenylindole, IgG, immunoglobulin G; Bkg, background; ALDH1A1, aldehyde dehydrogenase 1 family, member A1; CD44, cancer stem cell marker participates in cell-cell interaction, cell adhesion and migration; CD24, a cell adhesion molecule and a glycophosphatidylinositol-linked sialoprotein that serves as a ligand for P-selectin. |
Discussion
Conventional 2D cell culture systems have served as the predominant in vitro screening platform for anti-cancer drug development, but 3D MCTS has recently emerged as an improved tumour model by more accurately representing the in vivo tumour microenvironment.59 As a result of their oncogene expression profiles,8 3D MCTS models are particularly useful in the study of aggressive or chemoresistant cancer cell subtypes, including triple-negative breast cancer (TNBC). Here, the effects of a novel treatment regimen on 2D monolayer TNBC cultures and 3D TNBC MCTS models were investigated to mimic in vivo tumours. ASA, Met and OP independently decreased in a dose-dependent manner 2D cell culture and tumour MCTS viability as well as MCTS volume in both MDA-MB-231 and MDA-MB-231-TmxR variant cells. Interestingly, ASA produced the most profound anti-cancer effects in both models at the earliest with significant decreases in both cell viability and MCTS volume. With increasing doses of ASA (0.5–16 mM), and constant OP (300 μg/mL) and Met (4 mM), an additive decrease in both viability of 2D cell culture and tumour MCTS as well as in MCTS volume were observed.
The rationale for the use of tamoxifen in our in vitro studies is based on reports that tamoxifen-resistance of breast cancer cell models may originate from cell populations to ER-related effects to exert stem-like properties60 or to induce a phenotype during Tmx treatment with the emergence of Tmx-resistant clones. However, the mechanism for tamoxifen to achieve this cell transformation remains unclear. Also, the induction of pluripotency by tamoxifen may be mediated through ER-unrelated mechanisms. Notas et al61 have reported that early/direct tamoxifen transcriptional actions related to the induction of pluripotency were mediated through specific ERα-dependent or ER-unrelated transcription. They performed a whole transcriptome analysis of an ERα-positive T47D cell line and TNBC MDA-MB-231 cells exposed to tamoxifen for 3-hours. They found that a subset of ERα-dependent genes associated with developmental processes and pluripotency are induced and this is accompanied by specific phenotypic changes (expression of pluripotency-related proteins). They also reported an association between the increased expression of pluripotency-related genes in ERα-positive breast cancer tissues samples and disease relapse after tamoxifen therapy. Collectively, we were interested to determine whether CD44, CD24 and ALDH1A1 stem-like pluripotency-related proteins following 72-hour exposure to Tmx and Tmx in a combination of ASA, Met and OP cocktail had an effect on MDA-MB-231-TmxR cells survival. Tamoxifen, in combination with the cocktail of ASA, Met, and OP, prevented the acquisition of stem-like and invasive phenotype markers and inhibited MDA-MB-231 and MDA-MB-231-TmxR cell growth. In our previously reported studies, the data indicated that both TNBC MDA-MB-231 and non-TNBC MCF-7 cells developed resistance against 5 μM and 10 μM of tamoxifen for over 1 year.33 At least for the MCF-7 cells, it has been reported that crosstalk between ER, HER-2, p38, and ERK may contribute to tamoxifen resistance.62
In the present studies, ASA, Met and OP may act through complementary mechanisms to produce anti-cancer effects in TNBC. Both ASA and Met have been proposed to act through anti-inflammatory (ie, anti-NF-κB) and anti-proliferative (ie, anti-mTORC1) pathways.24 OP has been shown to act through both anti-proliferative and anti-metastatic pathways (ie, anti-PI3K/Akt).34 Furthermore, the Warburg effect defines the process by which tumours produce the necessary energy required to drive their aberrant metabolic activities.63 Although this theory has been the source of controversy, maintained metabolic activity from aerobic glycolysis, even in the presence of fluctuating oxygen availability, allows rapidly proliferating cancer cells to maintain their growth.11 Given this conserved hallmark observed in all cancers, Hanahan and Weinberg proposed its inclusion as an emerging hallmark of cancer.12 In this novel combination treatment regimen, metformin is capable of targeting this aberrant aerobic activity in TNBC.
Additionally, owing to its rapidly proliferating and highly mutagenic profile, TNBC can effectively overcome the effects of current therapeutic options and present an additional challenge. One therapeutic mode is to target the epigenetic machinery to compensate for the cytotoxic effects of therapy. Therapeutically, targeting several conserved pathways were a critical aspect of this treatment regimen, designed with the highly adaptive nature of cancer in mind.
Here, the combination drug cocktail of ASA, Met and OP sensitized parental and MDA-MB-231-TmxR cells as well as MCTS to Tmx chemotherapy. The cell culture studies revealed a consistent and reproducible trend where the cocktail drug therapy was able to reduce cell viability of MDA-MB-231 significantly and its Tmx resistant MDA-MB-231-TmxR variant at the low dose of 2.5 μM of Tmx.
Additionally, ASA, OP, and Met independently decreased Tmx IC50 in MDA-MB-231-TmxR MCTS, suggesting that each of the drugs can sensitize MDA-MB-231-TmxR cells to Tmx treatment. In particular, ASA and OP produced significant decreases in MCTS cell viability at a low dose of 5 μM of Tmx. Met was able to produce significant decreases in MCTS cell viability at 10μM Met, but still at a low dose of Tmx treatment. In general, 3D tumour models are less sensitive to drug treatment than 2D cell cultures. For metformin, the significant dose of Met to reduce the cell viability of MDA-MB-231 2D culture was 16 mM (Figure 1C) after 72 hrs treatment compared to the low dose of 4 mM for MDA-MB-231 3D MCTS (Figure 2C). The question is what is the possible mechanism(s) of Met on our MDA-MB-231 MCTS 3D model system. Bizjak et al64 reported on the effects of nutrients on metformin-stimulated AMP-activated protein kinase activation using 3D tumour spheroids. The non-essential amino acids suppressed the effects of metformin on MDA-MB-231 cells in a 2D culture and in 3D tumour spheroids. Glutamine and pyruvate slightly diminished the effects of metformin in 2D culture. Furthermore, glucose protected tumour spheroids against metformin-induced disintegration.
The formation of 3D MDA-MB-231 MCTS in our studies uses a one-step, highly reproducible technique of matrix-free 3D multicellular tumour spheroid (MCTS) formation by using a synthetic cyclo-RGDfK modified with 4-carboxybutyl-triphenylphosphonium bromide (cyclo-RGDfK(TPP)) peptide under normal high glucose media conditions.39 The cyclo-RGDfK(TPP) peptide promotes the self-assembly of cancer cells by avoiding cell plate surface adhesion.37 The interactions of the cyclo-RGDfK(TPP) peptide with α5β1 integrin induce cadherin expressions on the cell membrane. Cadherin–cadherin interactions among cancer cells result in cell compaction and spheroid formation.65 The self-assembly formation of 2D monolayer cells into MCTS by the application of the cyclo-RGDfK(TPP) peptide involves cell aggregation and tight compaction of the cells. The fibronectin (FN) receptor α5β1 integrin plays an essential role in tumour growth, invasion, and malignant behaviour.66 Although α5β1 integrin-mediated adhesion is dependent on the binding of its subunits to a defined tripeptide RGD sequence of FN, the strength of this binding is modulated by various factors, including the glycosylation status of the integrin. The cyclo-RGDfK(TPP) peptide used in the formation of the MCTS in our study closely mimics the natural ECM FN protein binding to α5β1 integrins on the cell membrane. It is noteworthy, and interestingly that fibronectin is one of two major constituents of the extracellular matrix in the kidney. Yi et al67 showed that fibronectin deposition and staining in the glomeruli and the tubulointerstitial compartment in kidneys from sham-operated mice was significantly increased in obstructed kidneys. Treatment with metformin reduced the accumulation of fibronectin deposition in the obstructed kidneys. Collectively, metformin treatment of MDA-MB-231 MCTS by using cyclo-RGDfK(TPP) peptide may directly target the tripeptide RGD sequence of the peptide that mimics the same RGD sequence of fibronectin. Although α5β1 integrin-mediated adhesion is dependent on the binding of its subunits to a defined tripeptide RGD sequence of FN, the strength of this binding is also modulated by various factors including the glycosylation status of the integrin.
Although the individual Tmx-sensitizing effects of each drug were promising on their own, a combination of drug therapy of 8 mM ASA, 300 μg/mL OP, and 4 mM Met was designed to determine if these drugs could additively sensitize MDA-MB-231-TmxR MCTS to Tmx treatment. The triple combination treatment of MDA-MB-231-TmxR MCTS produced the most significant decrease in Tmx of IC50 (22.92 μM to 17.55 μM) and produced significant decreases in cell viability at both 5 μM and 10 μM doses of Tmx. The anti-cancer effects of all three drugs with low doses of Tmx suggest that an ASA/OP/Met combination treatment could serve as a potential adjuvant to Tmx chemotherapy in TNBC patients. With an ASA/OP/Met adjuvant therapy, TNBC patients would be able to receive lower doses of Tmx. They would thus experience fewer toxicities associated with moderate-to-high doses of Tmx chemotherapy.68 With the lower doses of Tmx, TNBC patients would also have a reduced likelihood of developing Tmx resistance and cancer relapse over time.69
Meta-analyses of randomized clinical trials have provided evidence for the efficacy of aspirin in cancer treatment. Daily aspirin doses of 75 mg and more reduced cancer incidence and mortality where colorectal cancer patients revealed a 30–40% reduction in incidence and mortality.70 Here, in the trials of aspirin versus control for a mean duration of scheduled treatment of 6 years, 391 (2·8%) of 14 033 patients had colorectal cancer (CRC) during a median follow-up of 18·3 years. Aspirin reduced the risk of colon cancer but not rectal cancer. The clinical trial concluded no benefits at doses of aspirin higher than 75 mg daily, with an absolute reduction of 1·76% after 5-years scheduled treatment with 75–300 mg daily.70 In another clinical trial, the risk of developing distant metastasis in patients with CRC was reduced using aspirin (HR 0.45; 95% CI 0.28–0.72), with a further lowering risk for these patients (HR 0.26, 95% CI, 0.11–0.57).71 Here, aspirin reduced the risk of adenocarcinoma with metastasis at initial diagnosis and on subsequent follow-up in patients without metastasis initially, particularly in patients with colorectal cancer and those who remained on the trial treatment up to or after diagnosis.
In a clinical trial of individuals not taking non-steroidal anti-inflammatory drugs (NSAIDs), a 250 mg/day dose of metformin was sufficient to decrease aberrant crypt foci, suggesting that a lower dose of metformin could play a role in colon cancer prevention in this population.72 In another randomized, placebo-controlled, multicenter trial, patients who underwent resection of single or multiple colorectal adenoma polyps, received metformin (250 mg/day) for 1 year revealed a significant reduction in the risk of total polyps (RR = 0.67; 95% CI: 0.47–0.97) and adenomas (RR = 0.60, 0.39–0.92) compared with placebo.73 In the study, a low-dose of 250 mg/day of metformin for one year to these patients without diabetes was found to be safe.73 In another randomized, Phase II, double-blind, placebo-controlled, multicenter, ASAMET trial, the combination of aspirin and metformin was assessed for their potential synergistic interaction on a set of biomarkers associated with colon carcinogenesis.74 Here, the patient trial administered metformin at 850 mg BID (twice a day) and aspirin at 100mg QD (once a day) for one year.
We found that metformin at a dose of 16 mM (3.2 mmoles per day) was a sufficient dose to reduce cell proliferation of TNBC cells compared to 1700 mg Met (13.16 mmoles per day) or 250 mg Met (1.9 mmoles per day) in clinical trials. For aspirin, we found that ASA at a dose of 8 mM (1.6 µmoles per day) was a sufficient dose to reduce cell proliferation of TNBC cells compared to 100 mg ASA (560 µmoles per day) in clinical trials. Chen and Holmes recently reported a review of the current epidemiologic and clinical trial evidence for aspirin and breast cancer survival.75 The report concluded that the results of the three meta-analyses are remarkably similar, reporting an approximate 30% reduced risk of breast cancer death associated with aspirin use. Detailed information on the dosage of aspirin was not available for most of the clinical studies analyzed.75
We also asked the question if the combined effects of Tmx, ASA, OP, and Met would lead to an irreversible activation of apoptotic activity in MDA-MB-231 and MDA-MB-231-TmxR cells. Here, AnnexinV is a member of the annexin family of intracellular proteins that binds to phosphatidylserine (PS) in a calcium-dependent manner.75 PS is commonly only found on the intracellular leaflet of the plasma membrane in healthy cells. However, during early apoptosis, membrane asymmetry is lost, and PS translocates to the external leaflet. Fluorochrome-labeled Annexin V was used to specifically target and identify apoptotic cells expressing PS residues on the outer membrane, indicating early cell death. Propidium iodide, on the other hand, is impermeable for viable cells but enters necrotic cells to bind DNA. Both of these events are irreversible and thus are indicative of apoptotic and necrotic activity in cells following drug treatments. Significant decreases in viable cells were observed with Tmx and Tmx in combination with ASA, Met and OP with the highest significant percentage of early apoptotic and necrotic cells observed following the Tmx in combination with ASA, Met and OP. These observations support the efficacy potential that the combination of ASA, Met, and OP sensitizes TNBC and Tmx resistant TNBC cells to Tmx in an additive therapeutic effect in reducing cell viability and cell proliferation, and inducing early apoptosis and necrosis.
CD44, CD24, and ALDH1A1 markers have routinely been used to characterize the presence of cancer stem cells in tumorigenesis and metastasis.58 In breast cancer, a cancer stem-like population is typically characterized as being CD44+/CD24−/low and ALDH1A1 positive. Collectively, the expression of these markers is associated with greater tumorigenicity. CD44 is generally overexpressed in cancers and binds to hyaluronic acid, a component of the extracellular matrix.76 This interaction leads to the activation of several signaling pathways involved in multistage tumorigenesis, including cell proliferation, invasion, migration, and epithelial to mesenchymal transition (EMT). CD24 expression, however, is a source of controversy given the phenotypic variations that result in its varied expression levels.77 In triple-negative breast cancer, increased CD24 expression is associated with poor prognosis and a significant increase in metastatic burden.78 ALDH1A1 is another marker used to identify cancer stem cells in breast cancer.79 Aldehyde dehydrogenases are enzymes that detoxify aldehydes that are required for cellular processes, with the ALDH1A1 isoform being an essential regulator of cancer stem cell activity.80 Specifically, an increase in this marker has been associated with chemoresistance in several cancers.81
Although the expression and activity of these markers can be studied individually or relative to other markers associated with stemness, here, we investigated the changes in these markers individually post-treatment with Tmx treatment. Following treatment with Tmx, a significant increase in CD44 and ALDH1A1 expression is observed in MDA-MB-231 cells with a concomitant decrease in CD24 compared to the untreated group. This observation is consistent with studies demonstrating an increase in stemness and chemoresistance following conventional treatments.54 Using Tmx in combination with ASA, Met and OP, CD44 is significantly reduced compared to Tmx treatment alone and untreated control group. Using Tmx in combination with ASA, Met and OP, ALDH1A1 expression was similar to the Tmx treatment, both of which treatments showed significant increases in ALDH1A1 compared to the untreated control. It is noteworthy that the ratio of CD44/CD24 for MDA-MB-231 cells was 13 for the untreated group, 55 for Tmx alone and 2 for Tmx in combination with ASA, Met and OP. These data support the evidence that the triple combination of ASA, Met and OP treatment protocol can significantly reduce the acquisition of a cancer stem-like phenotype following Tmx therapy for TNBC by 36.6-fold.
For the Tmx resistant MDA-MB-231-TmxR cell variant, ALDH1A1 expression for Tmx treatment was not significantly different from the untreated control group. However, the triple combination with Tmx demonstrated a significant reduction in ALDH1A1 expression. The ratio of CD44/CD24 for MDA-MB-231-TmxR cells was 19 for the untreated group, 18 for Tmx alone and 0.2 for Tmx in combination with ASA, Met and OP. These data also support the evidence that the triple combination of ASA, Met and OP treatment protocol can significantly reduce the acquisition of a cancer stem-like phenotype following Tmx therapy for the Tmx resistant TNBC variant by 79.1-fold. These results suggest that the MDA-MB-231-TmxR breast cancer cells have a more cancer stem-like phenotype before treatment compared to the parental cells, but the MDA-MB-231-cells are highly induced post-treatment with Tmx. The triple combination of ASA, Met, and OP with Tmx can reduce the expression markers associated with cancer cell stemness. The exact mechanism of action of this reduction is unknown; however, studies on the effects of Met and ASA have consistently shown that the cancer stem cell/chemoresistant phenotype can be ablated by these agents individually82,83 or when administered as a dual therapy.27 Our group has also demonstrated the ablation of EMT and chemoresistance through the application of OP.31 Collectively, this triple-drug combination appears to regulate the acquisition of a stem-like phenotype in both parental and Tmx-resistant cell lines.
To determine whether the triple combination of ASA, Met and OP treatment had secondary anti-angiogenic effects, we tested the effectiveness of 8 mM ASA, 4 mM Met and 300 μg/mL OP on human umbilical vein endothelial cells (HuVECs). This cell line has routinely been used to study the physiological process of vasculogenic endothelial networks with a distinct network of vessels and vessel tube formation. Here, an anti-angiogenic effect of this triple-drug combination on healthy, normal HuVEC cells was investigated. Tmx, in combination with ASA, Met and OP, disrupted the morphology and architecture of these networks into abnormal and segmented networks. Further data analyses on the number of nodes, mean mesh size, and the number of isolated segments demonstrated no significant differences between the treatment groups. However, the cocktail treatment alone showed significant numbers of isolated segments as well as reduced mesh sizes. These findings suggest that the integrity of the vascular endothelial cell network was disrupted by the cocktail of ASA, Met, and OP. Surprisingly, the anti-angiogenic effect of the triple combination cocktail was dampened when administered with Tmx. Also, we assessed the caspase 3/7 apoptotic activity on HuVECs treated with the triple combination cocktail, Tmx, and in combination with Tmx treatments. Caspases are enzymes that are typically inactive zymogens, which are then activated to undergo catalysis when cells initiate apoptosis.84 Here, the caspases 3/7 were measured to assess the effects of the triple-drug combination treatments on HuVEC cell death. A steady increase in caspase 3/7 activity was observed within the first 24 hours in all treatment conditions followed by a decline in caspase 3/7 activity compared to an increase in caspase 3/7 activity in the untreated cells. The explanation of these observations may be that HuVEC cells are a primary large cell line requiring constant replenishment of nutrients and a larger surface area for long-term growth. Therefore, the first 24 hours post-treatment offer more reliable insight into the effects of the treatments investigated here rather than the later time points.
Due to the potential therapeutic implications of this study, it is worth acknowledging the clinical relevance of the doses that were used in the in vitro experiments and the specificity of the drugs as anti-cancer treatments. Firstly, the in vitro dose of ASA, OP, and Met are consistent with in vitro doses used by other groups studying the anti-cancer effects of these drugs.33,43,44 Collectively, the significance of the results of this study suggests that this novel combination of ASA, OP, and Met may be able to target several aspects of multistage tumorigenesis, including replicative immortality, cell proliferation, development of blood vessels as well as activating apoptotic pathways.
Conclusion
Although treatment options for triple-negative breast cancer are currently limited, additional biomarker discovery, as well as patient stratification based on cellular and molecular subtypes, may enhance the effectiveness of current treatment options. Several clinical trials are investigating the importance of patient stratification before treating patients and those that are taking a more personalized approach. Given the heterogeneity of TNBC, as well as the compensatory and highly mutagenic nature of this malignancy, a combination of therapeutic agents is necessary for consideration. Here, a combinatorial approach is used not only to sensitize TNBC cells to Tmx therapy but also to overcome resistance in the Tmx-resistant cell line. This triple combination of drug therapy not only can reduce the tumour spheroid volume and spheroid viability, but also induce apoptotic activity and dampen the cancer stem-like phenotype induced by chemotherapeutic Tmx. Collectively, these results show the efficacy of this triple combination of ASA, Met and OP therapy, which merits further investigation in preclinical animal studies and clinical trials.
Abbreviations
ALDH1A1, aldehyde dehydrogenase-1 family member A1; ASA, acetylsalicylic Acid; CD, cluster of differentiation; MCTS, multicellular tumour spheroid; MDA-MB-231, human triple-negative breast cancer cell line; MDA-MB-231-R, chemoresistant human triple-negative breast cancer cell line; Met, metformin; OP, oseltamivir phosphate; RGDfK, Arg-Gly-Asp-D-Phe-Lys; Tmx, Tamoxifen; TNBC, triple-negative breast cancer; TPP, 4-carboxybutyl-triphenylphosphonium bromide.
Acknowledgments
This work was supported in part by grants to M. R. Szewczuk from the Natural Sciences and Engineering Research Council of Canada (NSERC) and by a grant (N 18-04-01087) from the Russian Foundation for Basic Research (RFBR) to S. Burov and E. Markvicheva. M. Sambi is the recipient of the Queen’s University Graduate Award (QGA) and the R. Samuel McLaughlin Fellowship. B. Qorri is a recipient of the QGA, Terry Fox Research Institute Transdisciplinary Training Program in Cancer Research Scholarship, Dean’s Doctoral Award, and the Franklin Bracken Fellowship.
Disclosure
Dr William Harless reports funding from ACOA/NRC and is employed by Encyt Technologies, Inc.. Sergey V Burov is employed by the Laboratory of Novel Peptide Therapeutics, Cytomed J.S.Co.. The authors have no other possible conflicts of interest in this work.
References
1. Zeichner SB, Terawaki H, Gogineni K. A review of systemic treatment in metastatic triple-negative breast cancer. Breast Cancer (Auckl). 2016;10:25–36. doi:10.4137/BCBCR.S32783
2. Sambi M, Qorri B, Harless W, Szewczuk MR. Therapeutic options for metastatic breast cancer. In: Ahmad A, editor. Breast Cancer Metastasis and Drug Resistance: Challenges and Progress. Vol. 2.
3. Baselga J, Gomez P, Greil R, et al. Randomized Phase II study of the anti-epidermal growth factor receptor monoclonal antibody cetuximab with cisplatin versus cisplatin alone in patients with metastatic triple-negative breast cancer. J Clin Oncol. 2013;31:2586–2592. doi:10.1200/JCO.2012.46.2408
4. Liedtke C, Mazouni C, Hess KR, et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol. 2008;26:1275–1281. doi:10.1200/JCO.2007.14.4147
5. Rodler E, Korde L, Gralow J. Current treatment options in triple negative breast cancer. Breast Dis. 2010;32:99–122. doi:10.3233/BD-2010-0304
6. Costa RLB, Gradishar WJ. Triple-negative breast cancer: current practice and future directions. J Oncol Pract. 2017;13:301–303. doi:10.1200/JOP.2017.023333
7. Goldvaser H, Ribnikar D, Majeed H, Ocana A, Amir E. Absolute benefit from adjuvant chemotherapy in contemporary clinical trials: a systemic review and meta-analysis. Cancer Treat Rev. 2018;71:68–75. doi:10.1016/j.ctrv.2018.10.010
8. Sambi M, Haq S, Samuel V, et al. Alternative therapies for metastatic breast cancer: multimodal approach targeting tumor cell heterogeneity. Breast Cancer (Dove Med Press). 2017;9:85–93. doi:10.2147/bctt.s130838
9. Lehmann BD, Pietenpol JA. Clinical implications of molecular heterogeneity in triple negative breast cancer. Breast. 2015;24(Suppl 2):S36–S40. doi:10.1016/j.breast.2015.07.009
10. Wang J, Shi M, Ling R, et al. Adjuvant chemotherapy and radiotherapy in triple-negative breast carcinoma: a prospective randomized controlled multi-center trial. Radiother Oncol. 2011;100:200–204. doi:10.1016/j.radonc.2011.07.007
11. Leone A, Di Gennaro E, Bruzzese F, Avallone A, Budillon A. New perspective for an old antidiabetic drug: metformin as anticancer agent. Cancer Treat Res. 2014;159:355–376. doi:10.1007/978-3-642-38007-5_21
12. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi:10.1016/j.cell.2011.02.013
13. Kasznicki J, Sliwinska A, Drzewoski J. Metformin in cancer prevention and therapy. Ann Transl Med. 2014;2. doi:10.3978/j.issn.2305-5839.2014.06.01.
14. Liu B, Fan Z, Edgerton SM, et al. Metformin induces unique biological and molecular responses in triple negative breast cancer cells. Cell Cycle. 2009;8:2031–2040. doi:10.4161/cc.8.13.8814
15. Ryabaya O, Prokofieva A, Akasov R, et al. Metformin increases antitumor activity of MEK inhibitor binimetinib in 2D and 3D models of human metastatic melanoma cells. Biomed Pharmacother. 2019;109:2548–2560. doi:10.1016/j.biopha.2018.11.109
16. Jiang MJ, Dai JJ, Gu DN, Huang Q, Tian L. Aspirin in pancreatic cancer: chemopreventive effects and therapeutic potentials. Biochim Biophys Acta. 2016;1866:163–176. doi:10.1016/j.bbcan.2016.08.002
17. Vane JR, Botting RM. The mechanism of action of aspirin. Thromb Res. 2003;110:255–258. doi:10.1016/s0049-3848(03)00379-7
18. Montinari MR, Minelli S, De Caterina R. The first 3500years of aspirin history from its roots - A concise summary. Vascul Pharmacol. 2019;113:1–8. doi:10.1016/j.vph.2018.10.008
19. Mimeault M, Brand RE, Sasson AA, Batra SK. Recent advances on the molecular mechanisms involved in pancreatic cancer progression and therapies. Pancreas. 2005;31:301–316. doi:10.1097/01.mpa.0000175893.04660.1b
20. Okami J, Yamamoto H, Fujiwara Y, et al. Overexpression of cyclooxygenase-2 in carcinoma of the pancreas. Clin Cancer Res. 1999;5:2018–2024.
21. Sarkar FH, Banerjee S, Li Y. Pancreatic cancer: pathogenesis, prevention and treatment. Toxicol Appl Pharmacol. 2007;224:326–336. doi:10.1016/j.taap.2006.11.007
22. Mahdi EJ. Aspirin and its related non-steroidal anti-inflammatory drugs. Libyan J Med. 2013;8:21569. doi:10.3402/ljm.v8i0.21569
23. Zhang X, Feng Y, Liu X, et al. Beyond a chemopreventive reagent, aspirin is a master regulator of the hallmarks of cancer. J Cancer Res Clin Oncol. 2019;145:1387–1403. doi:10.1007/s00432-019-02902-6
24. Yue W, Yang CS, DiPaola RS, Tan XL. Repurposing of metformin and aspirin by targeting AMPK-mTOR and inflammation for pancreatic cancer prevention and treatment. Cancer Prev Res (Phila). 2014;7:388–397. doi:10.1158/1940-6207.CAPR-13-0337
25. De Monte A, Brunetti D, Cattin L, et al. Metformin and aspirin treatment could lead to an improved survival rate for Type 2 diabetic patients with stage II and III colorectal adenocarcinoma relative to non-diabetic patients. Mol Clin Oncol. 2018;8:504–512. doi:10.3892/mco.2018.1554
26. Yue W, Zheng X, Lin Y, et al. Metformin combined with aspirin significantly inhibit pancreatic cancer cell growth in vitro and in vivo by suppressing anti-apoptotic proteins Mcl-1 and Bcl-2. Oncotarget. 2015;6:21208–21224. doi:10.18632/oncotarget.4126
27. Amaral MEA, Nery LR, Leite CE, de Azevedo Junior WF, Campos MM. Pre-clinical effects of metformin and aspirin on the cell lines of different breast cancer subtypes. Invest New Drugs. 2018;36:782–796. doi:10.1007/s10637-018-0568-y
28. Orecchioni S, Reggiani F, Talarico G, et al. The biguanides metformin and phenformin inhibit angiogenesis, local and metastatic growth of breast cancer by targeting both neoplastic and microenvironment cells. Int J Cancer. 2015;136:E534–E544. doi:10.1002/ijc.29193
29. Amith SR, Jayanth P, Franchuk S, et al. Neu1 desialylation of sialyl alpha-2,3-linked beta-galactosyl residues of TOLL-like receptor 4 is essential for receptor activation and cellular signaling. Cell Signal. 2010;22:314–324. doi:10.1016/j.cellsig.2009.09.038
30. Abdulkhalek S, Szewczuk MR. Neu1 sialidase and matrix metalloproteinase-9 cross-talk regulates nucleic acid-induced endosomal TOLL-like receptor-7 and −9 activation, cellular signaling and pro-inflammatory responses. Cell Signal. 2013;25:2093–2105. doi:10.1016/j.cellsig.2013.06.010
31. O’Shea LK, Abdulkhalek S, Allison S, Neufeld RJ, Szewczuk MR. Therapeutic targeting of Neu1 sialidase with oseltamivir phosphate (Tamiflu(R)) disables cancer cell survival in human pancreatic cancer with acquired chemoresistance. Onco Targets Ther. 2014;7:117–134. doi:10.2147/ott.s55344
32. Abdulkhalek S, Geen OD, Brodhagen L, et al. Transcriptional factor snail controls tumor neovascularization, growth and metastasis in mouse model of human ovarian carcinoma. Clin Transl Med. 2014;3:28. doi:10.1186/s40169-014-0028-z
33. Haxho F, Allison S, Alghamdi F, et al. Oseltamivir phosphate monotherapy ablates tumor neovascularization, growth, and metastasis in mouse model of human triple-negative breast adenocarcinoma. Breast Cancer (Dove Med Press). 2014;6:191–203. doi:10.2147/BCTT.S74663
34. Gilmour AM, Abdulkhalek S, Cheng TS, et al. A novel epidermal growth factor receptor-signaling platform and its targeted translation in pancreatic cancer. Cell Signal. 2013;25:2587–2603. doi:10.1016/j.cellsig.2013.08.008
35. Haxho F, Neufeld RJ, Szewczuk MR. Neuraminidase-1: a novel therapeutic target in multistage tumorigenesis. Oncotarget. 2016;7:40860–40881. doi:10.18632/oncotarget.8396
36. Allison Logan S, Brissenden AJ, Szewczuk MR, Neufeld RJ. Combinatorial and sequential delivery of gemcitabine and oseltamivir phosphate from implantable poly(d,l-lactic-co-glycolic acid) cylinders disables human pancreatic cancer cell survival. Drug Des Devel Ther. 2017;11:2239–2250. doi:10.2147/DDDT.S137934
37. Akasov R, Gileva A, Zaytseva-Zotova D, et al. 3D in vitro co-culture models based on normal cells and tumor spheroids formed by cyclic RGD-peptide induced cell self-assembly. Biotechnol Lett. 2017;39:45–53. doi:10.1007/s10529-016-2218-9
38. Mikhail AS, Eetezadi S, Allen C. Multicellular tumor spheroids for evaluation of cytotoxicity and tumor growth inhibitory effects of nanomedicines in vitro: a comparison of docetaxel-loaded block copolymer micelles and Taxotere®. PLoS One. 2013;8:e62630. doi:10.1371/journal.pone.0062630
39. Akasov R, Zaytseva-Zotova D, Burov S, et al. Formation of multicellular tumor spheroids induced by cyclic RGD-peptides and use for anticancer drug testing in vitro. Int J Pharm. 2016;506:148–157. doi:10.1016/j.ijpharm.2016.04.005
40. Akasov R, Haq S, Haxho F, et al. Sialylation transmogrifies human breast and pancreatic cancer cells into 3D multicellular tumor spheroids using cyclic RGD-peptide induced self-assembly. Oncotarget. 2016;7:66119–66134. doi:10.18632/oncotarget.11868
41. Guertler A, Kraemer A, Roessler U, et al. The WST survival assay: an easy and reliable method to screen radiation-sensitive individuals. Radiat Prot Dosimetry. 2011;143:487–490. doi:10.1093/rpd/ncq515
42. Menyhart O, Harami-Papp H, Sukumar S, et al. Guidelines for the selection of functional assays to evaluate the hallmarks of cancer. Biochim Biophys Acta. 2016;1866:300–319. doi:10.1016/j.bbcan.2016.10.002
43. Zhang Y, Liu L, Fan P, et al. Aspirin counteracts cancer stem cell features, desmoplasia and gemcitabine resistance in pancreatic cancer. Oncotarget. 2015;6:9999–10015. doi:10.18632/oncotarget.3171
44. Luo Q, Hu D, Hu S, Yan M, Sun Z, Chen F. In vitro and in vivo anti-tumor effect of metformin as a novel therapeutic agent in human oral squamous cell carcinoma. BMC Cancer. 2012;12:517. doi:10.1186/1471-2407-12-517
45. Brennan BJ, Davies B, Cirrincione-Dall G, et al. Safety, tolerability, and pharmacokinetics of intravenous oseltamivir: single- and multiple-dose Phase I studies with healthy volunteers. Antimicrob Agents Chemother. 2012;56:4729–4737. doi:10.1128/aac.00200-12
46. Yin MJ, Yamamoto Y, Gaynor RB. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature. 1998;396:77–80. doi:10.1038/23948
47. Wilcock C, Bailey CJ. Accumulation of metformin by tissues of the normal and diabetic mouse. Xenobiotica. 1994;24:49–57. doi:10.3109/00498259409043220
48. Goodsell DS. The molecular perspective: tamoxifen and the estrogen receptor. Stem Cells. 2002;20:267–268. doi:10.1634/stemcells.20-3-267
49. Manna S, Holz MK. Tamoxifen action in ER-negative breast cancer. Sign Transduct Insights. 2016;5:1–7. doi:10.4137/STI.S29901
50. O’Reilly EA, Gubbins L, Sharma S, et al. The fate of chemoresistance in triple negative breast cancer (TNBC). BBA Clin. 2015;3:257–275. doi:10.1016/j.bbacli.2015.03.003
51. Al-Mahmood S, Sapiezynski J, Garbuzenko OB, Minko T. Metastatic and triple-negative breast cancer: challenges and treatment options. Drug Deliv Transl Res. 2018;8:1483–1507. doi:10.1007/s13346-018-0551-3
52. Ribatti D, Nico B, Ruggieri S, Tamma R, Simone G, Mangia A. Angiogenesis and antiangiogenesis in triple-negative breast cancer. Transl Oncol. 2016;9:453–457. doi:10.1016/j.tranon.2016.07.002
53. Robert NJ, Diéras V, Glaspy J, et al. RIBBON-1: randomized, double-blind, placebo-controlled, Phase III trial of chemotherapy with or without bevacizumab for first-line treatment of human epidermal growth factor receptor 2–negative, locally recurrent or metastatic breast cancer. J Clin Oncol. 2011;29:1252–1260. doi:10.1200/JCO.2010.28.0982
54. Creighton CJ, Li X, Landis M, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci U S A. 2009;106:13820–13825. doi:10.1073/pnas.0905718106
55. Sheridan C, Kishimoto H, Fuchs RK, et al. CD44+/CD24- breast cancer cells exhibit enhanced invasive properties: an early step necessary for metastasis. Breast Cancer Res. 2006;8:R59. doi:10.1186/bcr1610
56. Abraham BK, Fritz P, McClellan M, Hauptvogel P, Athelogou M, Brauch H. Prevalence of CD44+/CD24-/low cells in breast cancer may not be associated with clinical outcome but may favor distant metastasis. Clin Cancer Res. 2005;11:1154–1159.
57. Chute JP, Muramoto GG, Whitesides J, et al. Inhibition of aldehyde dehydrogenase and retinoid signaling induces the expansion of human hematopoietic stem cells. Proc Natl Acad Sci. 2006;103:11707. doi:10.1073/pnas.0603806103
58. Li W, Ma H, Zhang J, Zhu L, Wang C, Yang Y. Unraveling the roles of CD44/CD24 and ALDH1 as cancer stem cell markers in tumorigenesis and metastasis. Sci Rep. 2017;7:13856. doi:10.1038/s41598-017-14364-2
59. Bielecka ZF, Maliszewska-Olejniczak K, Safir IJ, Szczylik C, Czarnecka AM. Three-dimensional cell culture model utilization in cancer stem cell research. Biol Rev. 2016. doi:10.1111/brv.12293
60. Lin X, Li J, Yin G, et al. Integrative analyses of gene expression and DNA methylation profiles in breast cancer cell line models of tamoxifen-resistance indicate a potential role of cells with stem-like properties. Breast Cancer Res. 2013;15:R119. doi:10.1186/bcr3588
61. Notas G, Pelekanou V, Kampa M, et al. Tamoxifen induces a pluripotency signature in breast cancer cells and human tumors. Mol Oncol. 2015;9:1744–1759. doi:10.1016/j.molonc.2015.05.008
62. Gutierrez MC, Detre S, Johnston S, et al. Molecular changes in tamoxifen-resistant breast cancer: relationship between estrogen receptor, HER-2, and p38 mitogen-activated protein kinase. J Clin Oncol. 2005;23:2469–2476. doi:10.1200/jco.2005.01.172
63. Seyfried TN, Flores RE, Poff AM, D’Agostino DP. Cancer as a metabolic disease: implications for novel therapeutics. Carcinogenesis. 2014;35:515–527. doi:10.1093/carcin/bgt480
64. Bizjak M, Malavasic P, Pirkmajer S, Pavlin M. Comparison of the effects of metformin on MDA-MB-231 breast cancer cells in a monolayer culture and in tumor spheroids as a function of nutrient concentrations. Biochem Biophys Res Commun. 2019;515:296–302. doi:10.1016/j.bbrc.2019.05.090
65. Lin R-Z, Chang H-Y. Recent advances in three-dimensional multicellular spheroid culture for biomedical research. Biotechnol J. 2008;3:1172–1184. doi:10.1002/biot.200700228
66. Varner JA, Cheresh DA. Integrins and cancer. Curr Opin Cell Biol. 1996;8:724–730. doi:10.1016/S0955-0674(96)80115-3
67. Yi Y, Chen D, Ao J, et al. Metformin promotes AMP-activated protein kinase-independent suppression of DeltaNp63alpha protein expression and inhibits cancer cell viability. J Biol Chem. 2017;292:5253–5261. doi:10.1074/jbc.M116.769141
68. Lorizio W, Wu AHB, Beattie MS, et al. Clinical and biomarker predictors of side effects from tamoxifen. Breast Cancer Res Treat. 2012;132:1107–1118. doi:10.1007/s10549-011-1893-4
69. Baneshi MR, Warner P, Anderson N, Edwards J, Cooke TG, Bartlett JMS. Tamoxifen resistance in early breast cancer: statistical modelling of tissue markers to improve risk prediction. Br J Cancer. 2010;102:1503–1510. doi:10.1038/sj.bjc.6605627
70. Rothwell PM, Wilson M, Elwin CE, et al. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet. 2010;376:1741–1750. doi:10.1016/s0140-6736(10)61543-7
71. Rothwell PM, Wilson M, Price JF, Belch JF, Meade TW, Mehta Z. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet. 2012;379:1591–1601. doi:10.1016/s0140-6736(12)60209-8
72. Hosono K, Endo H, Takahashi H, et al. Metformin suppresses colorectal aberrant crypt foci in a short-term clinical trial. Cancer Prev Res (Phila). 2010;3:1077–1083. doi:10.1158/1940-6207.Capr-10-0186
73. Higurashi T, Hosono K, Takahashi H, et al. Metformin for chemoprevention of metachronous colorectal adenoma or polyps in post-polypectomy patients without diabetes: a multicentre double-blind, placebo-controlled, randomised Phase 3 trial. Lancet Oncol. 2016;17:475–483. doi:10.1016/s1470-2045(15)00565-3
74. Petrera M, Paleari L, Clavarezza M, et al. The ASAMET trial: a randomized, phase II, double-blind, placebo-controlled, multicenter, 2 × 2 factorial biomarker study of tertiary prevention with low-dose aspirin and metformin in stage I-III colorectal cancer patients. BMC Cancer. 2018;18:1210. doi:10.1186/s12885-018-5126-7
75. Chen WY, Holmes MD. Role of aspirin in breast cancer survival. Curr Oncol Rep. 2017;19:48. doi:10.1007/s11912-017-0605-6
76. Chen C, Zhao S, Karnad A, Freeman JW. The biology and role of CD44 in cancer progression: therapeutic implications. J Hematol Oncol. 2018;11:64. doi:10.1186/s13045-018-0605-5
77. Ortiz-Montero P, Liu-Bordes WY, Londono-Vallejo A, Vernot JP. CD24 expression and stem-associated features define tumor cell heterogeneity and tumorigenic capacities in a model of carcinogenesis. Cancer Manag Res. 2018;10:5767–5784. doi:10.2147/CMAR.S176654
78. Kwon MJ, Han J, Seo JH, et al. CD24 overexpression is associated with poor prognosis in luminal A and triple-negative breast cancer. PLoS One. 2015;10:e0139112. doi:10.1371/journal.pone.0139112
79. Ginestier C, Hur MH, Charafe-Jauffret E, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–567. doi:10.1016/j.stem.2007.08.014
80. Vassalli G. Aldehyde dehydrogenases: not just markers, but functional regulators of stem cells. Stem Cells Int. 2019;2019:3904645. doi:10.1155/2019/3904645
81. Kozovska Z, Patsalias A, Bajzik V, et al. ALDH1A inhibition sensitizes colon cancer cells to chemotherapy. BMC Cancer. 2018;18:656. doi:10.1186/s12885-018-4572-6
82. Shi P, Liu W, Wang H, et al. Metformin suppresses triple-negative breast cancer stem cells by targeting KLF5 for degradation. Cell Discov. 2017;3:17010. doi:10.1038/celldisc.2017.10
83. Saha S, Mukherjee S, Khan P, et al. Aspirin suppresses the acquisition of chemoresistance in breast cancer by disrupting an NFkappaB-IL6 signaling axis responsible for the generation of cancer stem cells. Cancer Res. 2016;76:2000–2012. doi:10.1158/0008-5472.CAN-15-1360
84. Shi Y. Caspase activation, inhibition, and reactivation: a mechanistic view. Protein Sci. 2004;13:1979–1987. doi:10.1110/ps.04789804
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.