")
Back to Journals » Drug Design, Development and Therapy » Volume 12
A study on screening and antitumor effect of CD55-specific ligand peptide in cervical cancer cells
Authors Liu G, Yin Q, Ji H, Wang Y, Liu H, Jiang L, Zhu F, Li B
Received 2 August 2018
Accepted for publication 10 October 2018
Published 13 November 2018 Volume 2018:12 Pages 3899—3912
DOI https://doi.org/10.2147/DDDT.S182337
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Guoxiang Liu, Qifeng Yin, Huanhuan Ji, Yujuan Wang, Huihui Liu, Liangqian Jiang, Feng Zhu, Bing Li
Department of Genetics and Cell Biology, Basic Medical College, Qingdao University, Qingdao 266071, People’s Republic of China
Background: To improve the targeting ability of antitumor drugs, we identified the antigens with high expression on the surface of tumor cells associated with tumor escape, such as the complement regulatory protein CD55 molecule, which is also known as the decay accelerating factor. In this study, phage display technology was used to screen and identify CD55-specific ligand peptide (CD55sp) bound to CD55 molecule on the surface of cervical cancer HeLa cells. We then explored the role of this peptide in inhibiting the growth of cervical cancer cells in vitro. Our characterization of CD55sp will provide implication for tumor target therapy.
Methods: The phage bound to the surface of HeLa cells were isolated by phage display technology. Positive phage clones were identified by ELISA. Phage was then amplified and determined by agarose gel electrophoresis after monoclonal DNA extraction. DNA sequencing and bioinformatical analysis were conducted to obtain specific ligand peptides. Flow cytometry and immunofluorescence were used to measure the expression of CD55 molecule on the surface of tumor and normal cells. Subsequently, the effects of CD55sp on the proliferation and apoptosis of HeLa and SiHa cells were determined by Cell Counting Kit-8 (CCK-8), flow cytometry, and TUNEL assay, respectively. The morphology of apoptotic cells was examined by electron microscope. The distribution of Cleaved caspase-3 was detected by immunofluorescence. The expression of bcl-2 and Cleaved caspase-3 were determined by Western blot.
Results: The results showed that the peptide (QVNGLGERSQQM) can bind to the CD55 molecule on the surface of cervical cancer HeLa and SiHa cells as a ligand peptide. It can also effectively inhibit the proliferation of cervical cancer cells and induce cell apoptosis.
Conclusion: This study demonstrates that CD55sp screened by phage display technology plays a strong antitumor role.
Keywords: phage display, cervical cancer, CD55sp, ligand peptide, apoptosis
Introduction
Great progress has been made in recent years in finding treatments for tumors. Chemotherapy has been widely used in tumor treatment.1,2 However, due to its limited ability to distinguish human tumor cells from normal ones, chemotherapy drugs also affect normal cells while killing tumor cells.3 To improve the therapeutic efficacy of antitumor drugs, it is essential to develop tumor-targeted drugs that can specifically accumulate in tumors and cause death of tumor cells without doing harm to normal ones.3–5 Previous studies have found that one or more complement regulatory proteins (CD46, CD55, CD59) are highly expressed on various surface of tumor cells such as uterine serous carcinomas.6 Studies showed that small interfering RNAs inhibiting CD55 and CD59 (but not CD46) expression in vitro could sensitize uterine serous carcinomas to complement-dependent cytotoxicity and antibodies-dependent cytotoxicity. In addition, they can significantly increase antibody-mediated therapeutic effects in vivo when specifically targeted to tumor cells.7 The newly identified sp22 peptide can, in a dose-dependent manner, inhibit CD59 expression. Meanwhile, sp22 can increase complement-mediated lysis and apoptosis signals.8 Other studies have shown that HPV-E6 protein expression enriched the CD55 (+) population, which contributes to the tumorigenicity and radio resistance of cervical cancer cells and targeting CD55 through CRISPR/Cas9 could also be a potential strategy for the treatment of cervical cancer.9
Phage display technology has been proven to be a powerful tool to screen useful ligand that specifically bind biomarkers on the surface of tumor cells.10 Ligand peptide identified by this technique has been successfully applied to early cancer diagnosis and chemotherapy. For example, a novel non-small-cell lung cancer targeting peptide (sequence RCPLSHSLICY) was screened in vivo using the Ph.D.-C7C (TM) phage display library.11 In this study, we screened the CD55sp from random 12-peptide phage display library, characterized the targeted combination of CD55sp to CD55 molecule on the surface of tumor cells, and performed mechanistic studies of the antitumor effects of CD55sp.
Cervical cancer is one of the common gynecological malignancies that threaten human health. According to epidemiology studies, globally there are approximately 500,000 new cases of cervical cancer each year.12 Studies have shown that TMC120 displayed potent cytotoxic effect on human cervical carcinoma through enhancing the polymerization of microtubules, thus can be used as a potential chemoprophylactic and therapeutic agent for cervical cancers in the similar manner as paclitaxel and TMC120 is also suitable for preventing HIV infection and cervical cancer in women.13 The sensitivity of primary tumor cell from patients with cervical cancer to eight chemotherapeutic combinations were as follows: gemcitabine < docetaxel < topotecan + cisplatin < cisplatin + cyclophosphamide + doxorubicin < gemcitabine + cisplatin < taxol < taxol + carboplatin < taxol + cisplatin.14 However, most existing anticancer drugs have strong side effects making them unable to reach tumor tissues effectively, which requires the development of effective anticancer drugs that can be specifically or selectively delivered to tumor tissues. These drugs can have specific distribution pattern across different physiological sites, which could reduce the side effects and toxicity of anticancer drugs. Studies reveal that an existing drug atovaquone can effectively inhibit cervical cancer cells through increased mitochondrial biogenesis, and atovaquone-targeting mitochondrial respiration can be used as a therapeutic strategy for cervical cancer.15 In addition, the Hippo/YAP pathway has been suggested to regulate cervical cancer progression through interaction with EGFR signaling and HPV oncoproteins, thus combined targeting of the Hippo and the ERBB signaling pathways represents a novel therapeutic strategy for prevention and treatment of cervical cancer.16
Our group has previously found that CD55 gene was highly expressed in a variety of solid tumors such as colorectal carcinoma, lung cancer, and cervical cancer but has a lower expression level in normal cells.17 CD55, also known as a decay factor, is a glycosylphosphatidylinositol-anchoring glycoprotein on the cell membrane.17 The main function of CD55 is to compete with C2 and C4b, inhibiting the formation of C3 convertase and promoting its decomposition.17 In addition, it can inhibit deposition and activation of complement C3 and suppress of classical complement and alternative pathways.17 Thus, tumors can evade from immune system defenses due to the inhibition of the complement system which attacks tumor cells. It was suggested that the CD55 molecule can be used as a novel target to study the escape of tumor cells from autoimmune surveillance and tumor immunotherapy. Therefore, it is important to find effective peptide that could specifically bind to CD55 molecule on the surface of tumor cells, which will help to achieve targeted therapy. In this study, flow cytometry was used to determine the expression of CD55 on the surface of cervical cancer HeLa cells, and the phage bound to HeLa cells was screened by phage display technology to obtain phage bound to CD55 molecule, and further obtain sequence of CD55sp.18,19
In the following experiments, we treated SiHa and HeLa cells with CD55sp and checked the indicators to verify the occurrence of tumor cell apoptosis and the inhibition of tumor cell proliferation, thereby prove the antitumor effects of CD55sp.
Materials and methods
Materials
Human cervical cancer SiHa cell line, human cervical cancer HeLa cell line, and mouse osteogenic MC3T3-E1 cell line were preserved by our laboratory. Random 12-peptide phage display library, −96 gIII Sequencing Primer 5′-HOCCC TCA TAG TTA GCG TAA CG-3′, and Escherichia coli ER2738 host strain were purchased from New England Biolabs (Ipswich, MA, USA). The short peptide (QVNGLGERSQQM) was purchased from Gill Biochem Co., Ltd., Shanghai, China. Anti-human CD55 monoclonal antibody was purchased from Santa Cruz Biotechnology Inc., Dallas, TX, USA. Flow cytometry kit was purchased from Ebioscience, Inc. (Thermo Fisher Scientific, Waltham, MA, USA). Cell Counting Kit-8 (CCK-8) was purchased from Biosharp, Hefei, China. TUNEL Apoptosis Detection Kit was purchased from YEASEN, Shanghai, China. Enzyme label analyzer (9,602A) was purchased from Shanghai Chuangxin Science & Education Equipment Co., Ltd, Shanghai, China.
Cell culture and bacterial culture
Cell lines were cultured in RPMI-1640 medium containing 10% newborn calf serum and incubated in a humid incubator containing 5% CO2 at 37°C. The cell morphology was observed using an inverted microscope. E. coli ER2738 were plated on Luria-Bertani-tetracycline (LB-Tet) plates, were incubated at 37°C overnight, and then were inoculated into LB medium to achieve log-phase growth. The human cell lines provided in this experiment have been approved by the ethics review committee of Qingdao University and all applicable institutional and governmental regulations concerning the ethical use of human cell lines were followed.
Phage display technology
A total of 10 μL of different dilutions of phage solution were mixed with E. coli ER2738 medium, added to the upper layer of agar containing IPTG/X-gal, and immediately poured into solid LB plates containing IPTG/X-gal to be coagulated. After incubation overnight at 37°C, blue plaques appeared and were counted to determine the titer. Adherent HeLa cells were washed with serum-free RPMI-1640 and blocked with 16% culture medium containing 0.1% BSA for 1 hour, and then added to the stock solution of Ph.D.-12 phage peptide library (titer: 1 × 1011 pfu/mL) for 1 hour. After washing on ice with a pre-cooled 0.1% PBST at 4°C to remove non-cell-bound phage, the phage bound to the cell surface was eluted on ice with a glycine buffer (pH 2.2) pre-cooled at 4°C immediately, and then added to a centrifuge tube pre-filled with 250 μL Tris buffer (pH 2.2). The second and third rounds of screening were performed using the amplification solution from the eluted phage in the previous round of screening and the recovery was calculated.
Selection, amplification, and confirmation of positive phage clones by ELISA
Fifteen ER2738 monoclones were selected, added to a 20 mL LB liquid culture shaker tube, and ordered 1–15, respectively, which were incubated at 37°C for 4.5 hours with vigorous shaking. Fourteen blue plaques with well-developed color development and well-isolated plaques were randomly selected from the screening plate in the fourth round for HeLa surface eluent and added to the shaker tube, followed by vigorous shaking at 37°C for 4.5 hours, and stored at 20°C for use. The titers of the 15 amplified monoclonal phage samples (No 1, 2, 3, ...15) were determined, and 1 × 1010 pfu of each clone was detected by ELISA20 and stored at 4°C. In a 96-well plate, HeLa cells were serum-free, fixed in 4% paraformaldehyde, blocked with 5% PBS-BSA, added with a monoclonal phage titer of approximately 1 × 1010 pfu, and incubated at 37°C for 1–2 hours with shaking. HRP/Anti-M13 (1:5,000 dilution) was added and incubated at 37°C for 1 hour. Finally, the color was developed with TMB and the reaction was terminated with HCL. The OD value at 450 nm was detected by a microplate reader. The affinity was calculated by the following formula: (OD 450 nm of random clone − OD 450 nm of blank control) ÷ (OD 450 nm of negative control − OD 450 nm of blank control). When the value of the affinity was higher than 2.1, the clone had high affinity.
Extraction and detection of single-stranded DNA for sequencing analysis
The high-affinity monoclonal phage amplification solution was sedimented with PEG/NaCl, resuspended in iodide buffer, and dissociated with ethanol. DNA was precipitated and finally resuspended in TE for detection by agarose gel electrophoresis. The −96 primers were provided by the kit and the DNA was submitted to Beijing Jinweizhi Biotechnology Co., Ltd, (Beijing, China) for sequencing.
CCK-8 assay
In a 96-well plate, cells were treated with different concentrations of CD55sp (0 μg/mL, 100 μg/mL, 200 μg/mL, 300 μg/mL) and placed in a 37°C, 5% CO2 incubator for 36 hours. The medium was aspirated, and CCK-8 mixture was added and incubated for 2 hours. The absorbance at 450 nm was measured with microplate reader.21
Flow cytometry
In a six-well plate, cells were treated with −CD55sp and +CD55sp, respectively. Then they are digested with pancreatin without EDTA, collected and centrifuged, and cells are washed repeatedly with PBS. After the cells were resuspended in 1× Binding Buffer, Annexin V/PE and 7-AAD were added, gently mixed and protected from light, and reacted at room temperature for 15 minutes. An appropriate amount of 1× Binding Buffer was added, mixed, and placed on ice and the sample was detected by flow cytometry within 1 hour.
TUNEL detection
In a 24-well plate, cells were treated with −CD55sp and +CD55sp, respectively. Subsequently, 4% paraformaldehyde was added for immobilization. Proteinase K solution (20 μg/mL) was added and incubated for 5 minutes at room temperature. After washing with PBS, 1× Equilibration Buffer was added and incubated for 30 minutes at room temperature. After suctioning excess 1× Equilibration Buffer, TDT incubation buffer was added and incubated at 37°C for 1 hour in the dark. After washing with PBS, DAPI solution was added for staining for 10 minutes. After washing with deionized water, PBS was added to keep wet, and the samples were immediately analyzed under a fluorescence microscope. Alexa Fluor 647 red fluorescence was detected at 620 nm and blue DAPI was observed at 460 nm.
Electron microscopy
In a six-well plate, the cells were treated with −CD55sp and +CD55sp, respectively. Cells were then scraped off, centrifuged, and the supernatant was aspirated. The precipitate was fixed by glutaraldehyde and observed under the transmission electron microscopy.22,23
Immunofluorescence assay
In a 24-well plate, cells were treated with −CD55sp and +CD55sp, respectively. Subsequently, 4% paraformaldehyde was added for immobilization. The normal serum blocking solution was used to seal cells for 10 minutes. The excessive fluid was shaken off and appropriate primary antibody was added at 4°C for overnight. After cells were washed with PBS, biotin-labeled secondary antibody was added at 37°C for 30 minutes. SABC-Cy3 was diluted by 0.01 M PBS with 1:100 dilution and treated at 37°C for 30 minutes. After washing with PBS, DAPI solution was added for staining for 10 minutes. After washing with PBS, the water-soluble encapsulated tablets were mounted and observed under the fluorescence microscopy.
Western blot analysis
In a six-well plate, cells were treated with −CD55sp and +CD55sp, respectively. The cells were collected after adding the lytic solution on ice. And after centrifugation, supernatants were collected, and the protein concentration was measured by BCA. The target protein was separated by SDS-PAGE and transferred to a PVDF membrane, which was immersed in 5% skimmed milk powder for 1 hour for blocking, and the target primary antibody was added to a well-diluted solution overnight at 4°C. Washed with PBST, PVDF membrane was treated with HRP-secondary antibody for 1 hour. Chemiluminescence visualization was performed, and the grayvalue analysis of the bands was performed using ImageJ.
Statistical analysis
Statistical analysis was conducted with Prism 5.0 (GraphPad Software, Inc., La Jolla, CA, USA). Results were expressed as the mean ± SEM of three or more observations (as indicated in each experiment). The mean values for biochemical data from two groups were compared by two-tailed Student’s t-test. The P-value was less than 0.05, which was considered statistically significant.
Results and discussion
Determination of phage titer and recovery
Before the first round of screening, phage stocks were plated to determine the titer, and a series of dilution concentration (10, 102, 103, ⋯1010 times) was set. When concentration was diluted to 109 fold, three replicate groups were cultured for phage blue spots in the plates (Figure 1). According to the calculation formula24: N = Y ÷ V × X (N: efficacy value is the required titer, Y: average plaque/dish, V: sample volume, X: dilution), the phage titer was 5.1 × 1012 pfu. To obtain CD55sp, we performed four rounds of affinity screening. The recovery rate was calculated for each round (Table 1). The phages with high affinity gradually enriched after several rounds of selection and became relatively stable in rounds 3 and 4. After being stabilized, the phage enriched on the cells can be considered saturated. On this basis, we continue to positively identify by ELISA to screen high-affinity phage.
 | Figure 1 Plaque of the culture plate. |
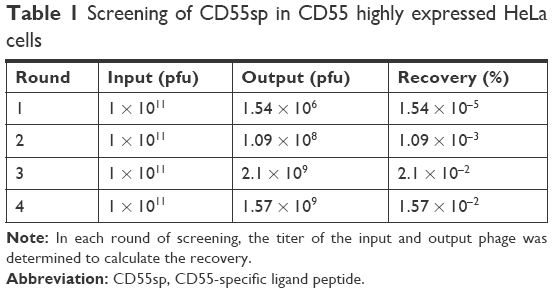 | Table 1 Screening of CD55sp in CD55 highly expressed HeLa cells |
Specific binding of monoclonal phage to HeLa cells with CD55 high expression
The CD55-specific binding ability of the amplified 15 phage clones was evaluated by ELISA assay. Optical density was determined (Figure 2). According to the value of the affinity >2.1, it suggests that the clone had high affinity with HeLa cells. Among the 15 phage clones, the clones numbered 1, 2, 4, 5, 6, 9, 10, 11, 13, 14, and 15 had high affinity to the HeLa cells. Since we determined that the expression of CD55 on the surface of HeLa cells was extremely high in the pre-experiment, we need to obtain highly repetitive sequences of the selected high-affinity phage by sequencing.
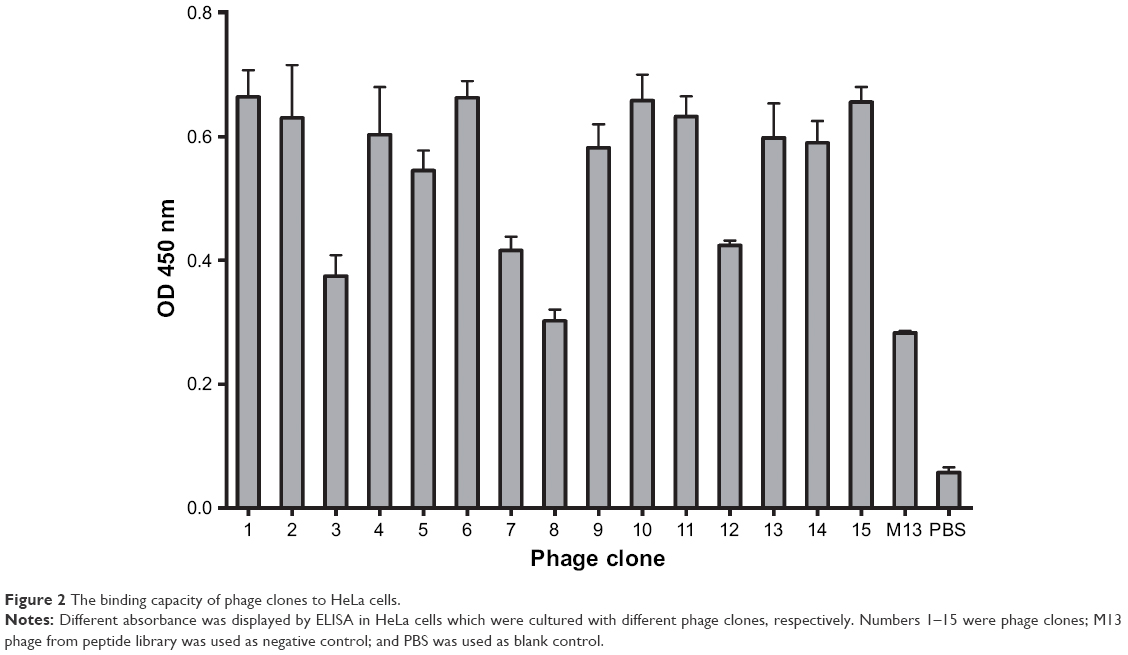 | Figure 2 The binding capacity of phage clones to HeLa cells. |
Detection and sequencing of monoclonal phage ssDNA
The ssDNA of 11 phage clones were extracted and analyzed by agarose gel electrophoresis. The extracted ssDNA bands show the expected size of 7,249 bp (Figure 3). The concentration of ssDNA was relatively high and accorded with the sequencing requirements. The 11 ssDNA samples were then sequenced (Figure 4). According to the sequencing results, the DNA sequence of the inserted 12-mer peptide was analyzed by combining the N-terminal sequence of the random dodecapeptide-gIII fusion protein. The amino acid sequence of the short peptide was read using the condensed genetic code table and three short peptides were obtained based on the analysis result (Table 2). The highly repetitive sequence QVNGLGERSQQM was submitted to the PIR International Protein Database41–43 (http://pir.georgetown.edu/) as a ligand peptide that specifically binds to the CD55 molecule on HeLa cells. No matching sequence was found. The peptide was synthesized by GL Biochem, with FITC fluorescence attached to the N-terminus for cellular experiments.
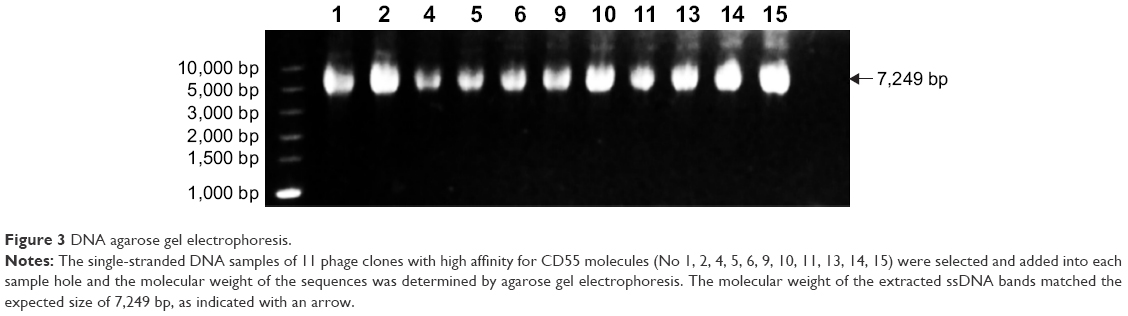 | Figure 3 DNA agarose gel electrophoresis. |
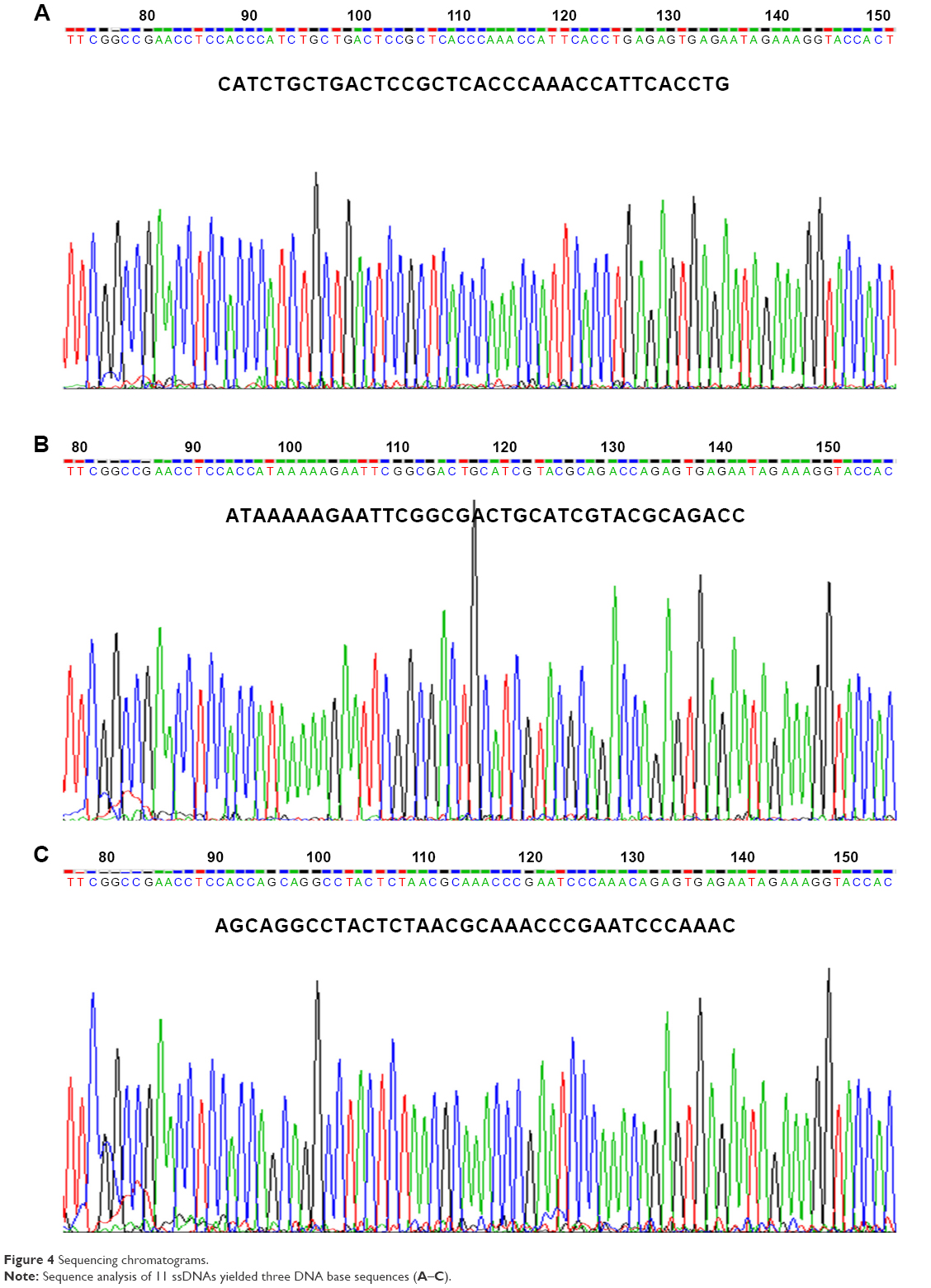 | Figure 4 Sequencing chromatograms. |
 | Table 2 Amino acid sequences deduced from DNA sequences |
Detection of the expression of CD55 on the cell surface
Cervical cancer HeLa cells, cervical cancer SiHa cells, and normal osteoblast MC3T3-E1 cells suspension were added 1:100 dilution of CD55 monoclonal antibody labeled by FITC, respectively, incubated for 1 hour at room temperature in the dark. The negative cell control was set by replacing CD55 monoclonal antibody with PBS, and the expression of CD55 proteins was detected by flow cytometry.18 Flow cytometry is a powerful tool for detailed analysis of complex populations in a short period of time, and its working depends on the light scattering features of the cells under investigation, which can be derived from dyes or monoclonal antibodies targeting either extracellular molecules located on the surface or intracellular molecules inside the cell.25 Compared with osteoblast MC3T3-E1 cells (13.5% ± 1.5%), the detected positive rate of SiHa (93.3%±1.5%) and HeLa cells (93.4% ± 0.8%) with high expression of CD55 proteins was significantly higher (**P < 0.01, ***P < 0.001) (Figure 5).
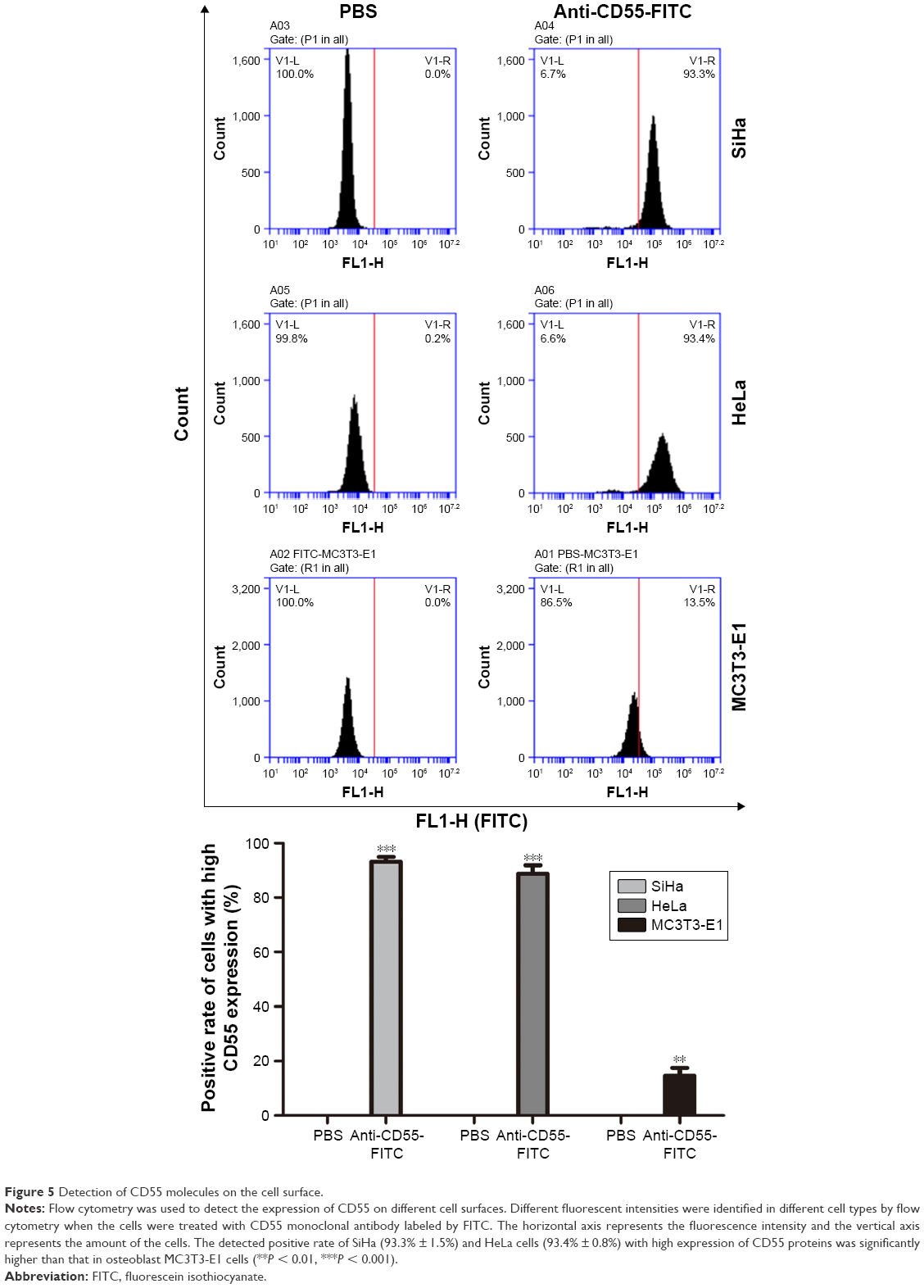 | Figure 5 Detection of CD55 molecules on the cell surface. |
Identification of CD55sp
Based on the detection of the expression of CD55 on the cell surface, we demonstrated the specificity of CD55sp by fluorescence experiments. Immunofluorescence assay is one of the commonly used methods for the identification of ligand peptides.19,20 After being fixed in 4% paraformaldehyde, three types of cells were incubated in the CD55sp for 2 hours. The levels of CD55sp bound to the cell surface of cervical cancer SiHa and HeLa cells were higher than that in osteoblast MC3T3-E1 cells, which was in accordance with the CD55 levels presented in the cell surface, as measured by the fluorescence intensity.19,20 The results suggested that cells with high CD55 expression could specifically bind to more CD55sp identified in our screen. Compared with normal cells, CD55sp can specifically bind more to the surface of cervical cancer SiHa and HeLa cells and then be ingested by tumor cells (Figure 6A). Three types of cells were incubated in the medium containing the CD55 ligand peptide for 36 hours. The level of CD55sp ingested into cervical cancer SiHa and HeLa cells was higher than that in osteoblast MC3T3-E1 cells, as measured by the fluorescence intensity. CD55sp entered tumor cells to achieve the effect of inhibiting proliferation (Figure 6B).
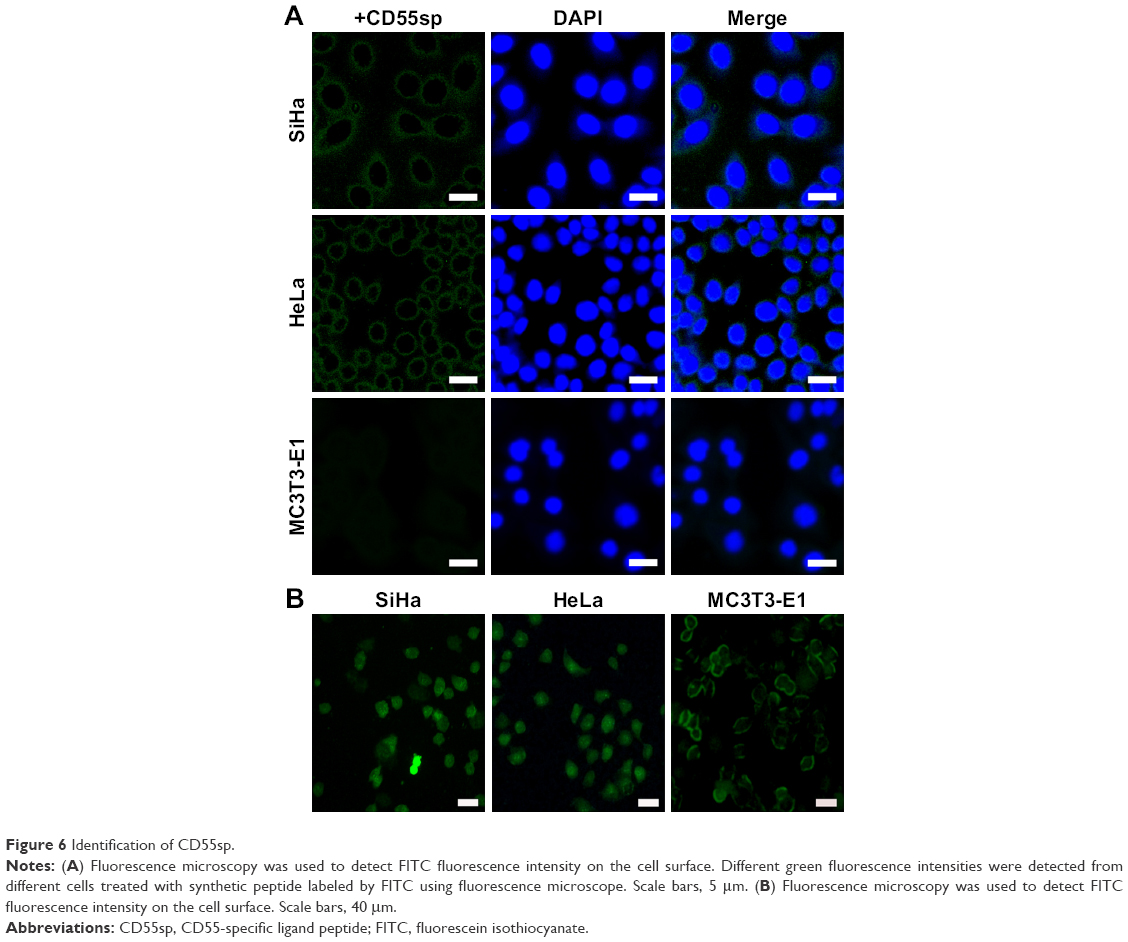 | Figure 6 Identification of CD55sp. |
Inhibitory effects of CD55sp on cell proliferation
Cell viability is defined as the number of healthy cells in a sample, and proliferation of cells is a vital indicator for understanding the mechanisms of action of certain genes, proteins, and pathways involved in cell survival or death after exposing to toxic agents.26 Generally, methods used to determine viability are also common for the detection of cell proliferation.26 Cell proliferation assays are generally used for drug screening to detect whether the test molecules have effects on cell proliferation.26 After CD55sp treatment, we found that with increased CD55sp concentration, the activity of SiHa , HeLa, and Mc3T3-E1 cells gradually declined. The results showed that the growth status of SiHa, HeLa, and Mc3T3-E1 cells depend on the concentration of CD55sp. The results also revealed that CD55sp could strongly inhibit the proliferation of tumor cells with high expression of CD55 and could weakly inhibit the proliferation of normal cells with low expression of CD55. The half-maximal inhibitory concentration (IC50) of the CD55sp on SiHa and HeLa cells was 208.4 ± 13.5 μg/mL and 230.3 ± 20.1 μg/mL, respectively. The IC50 values were used as the drug dose in the following groups: −CD55sp groups (0 μg/mL) as control groups, and +CD55sp groups (IC50 values) as drug treatment groups (Figure 7).
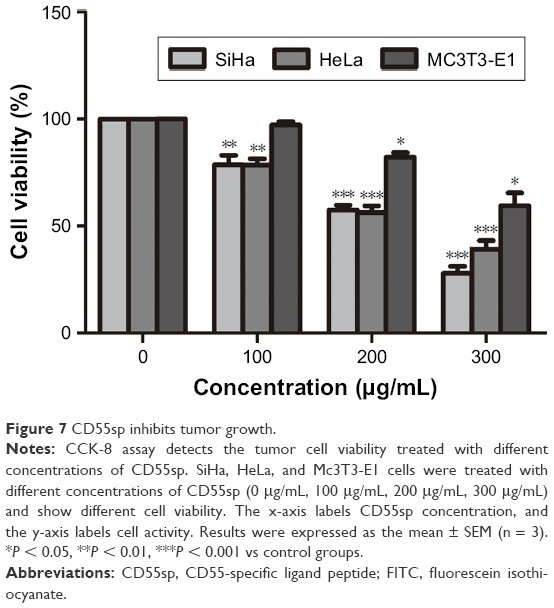 | Figure 7 CD55sp inhibits tumor growth. |
CD55sp induces apoptosis of tumor cells
After cervical cancer SiHa and HeLa cells were treated with CD55sp, the numbers of apoptotic SiHa and HeLa cells increased (Figure 8A). When the cells are apoptotic, they activate some DNase, which cleaves the genomic DNA between the nucleosomes.27 TUNEL can be used to detect cell nuclear DNA during the late stage of apoptosis to demonstrate cell apoptosis. After cervical cancer SiHa and HeLa cells were treated with CD55sp, the apoptosis rate of both SiHa and HeLa cells increased (Figure 8B). In normal living cells, phosphatidylserine is located inside the cell, but in early apoptosis, phosphatidylserine is flipped from inside of the cell membrane to the surface and exposed to the extracellular environment.28,29 By flow cytometry, the dye can bind to the membrane of the early apoptotic cells by binding to the phosphatidylserine exposed on the outside of the cell and can also penetrate and bind to the DNA of the late apoptotic cells or necrotic cells. This result proved that CD55sp can promote apoptosis of tumor cells.
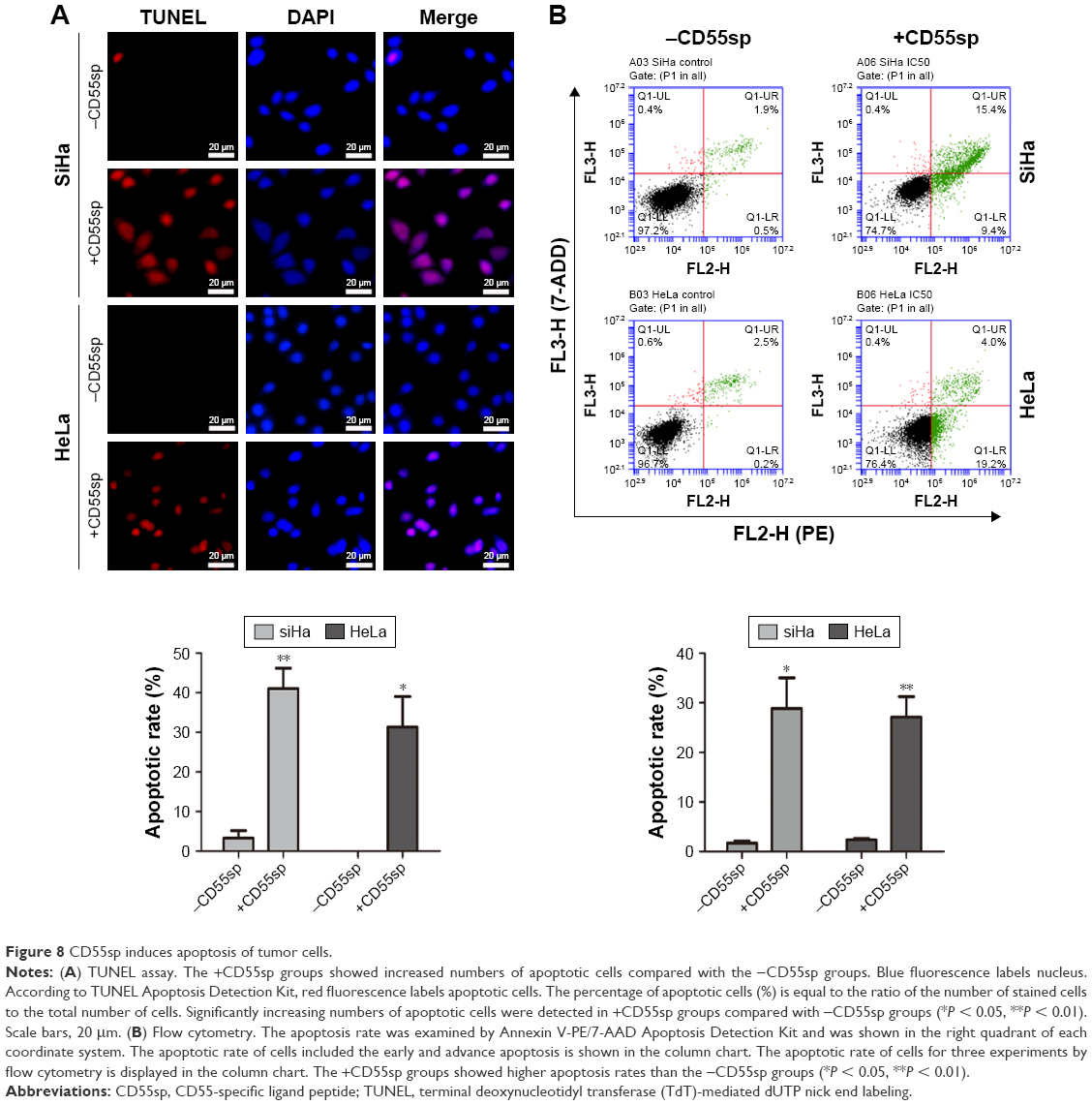 | Figure 8 CD55sp induces apoptosis of tumor cells. |
Ultrastructural analysis of apoptotic cells using electron microscopy
Ultrastructural analysis of untreated SiHa and HeLa cells revealed normal morphology of the organelles within cells in −CD55sp groups, such as nucleus and mitochondria. However, cells treated with CD55sp showed the apoptotic morphology, including condensed nucleus and the appearance of apoptotic bodies (Figure 9).30 Dynamic changes in the compaction of nuclear chromatin are one of the characteristic phenomena of apoptotic execution.31 During apoptotic execution, chromatin undergoes a phase change from a heterogeneous, genetically active network to an inert highly condensed form that is fragmented and packaged into apoptotic bodies.22,23 These observations suggested that apoptosis of tumor cells appeared after CD55sp treatment.
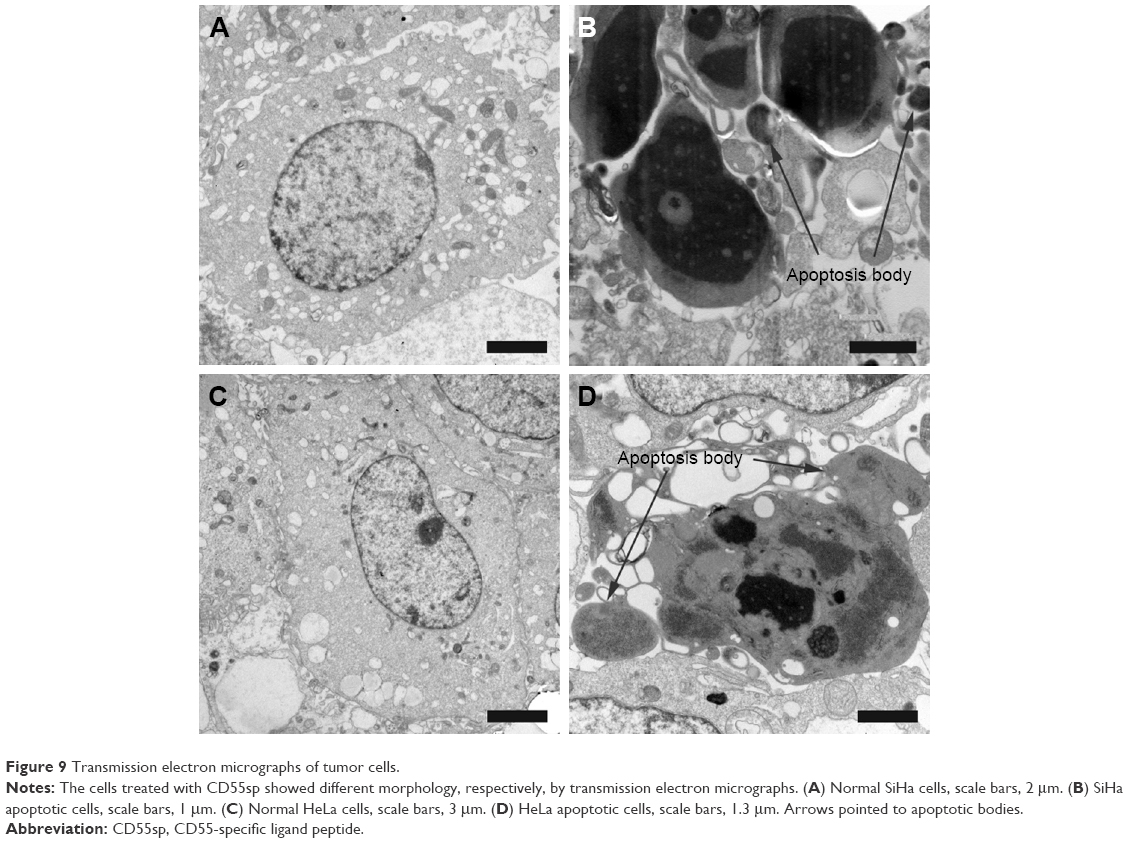 | Figure 9 Transmission electron micrographs of tumor cells. |
CD55sp treatment increases Cleaved caspase-3 levels in tumor cells
After cervical cancer SiHa and HeLa cells were treated with CD55sp, Cleaved caspase-3 levels of both SiHa and HeLa cells increased (Figure 10). Apoptosis-related proteins can be localized and quantified by immunofluorescence.32,33 Cleaved caspase-3 has been used as a marker for cell apoptosis. Increased expression of the proapoptotic protein Cleaved caspase-3 means apoptosis of tumor cells.32 The results suggested that CD55sp could promote the expression apoptosis-related proteins in tumor cells to induce tumor cell apoptosis.
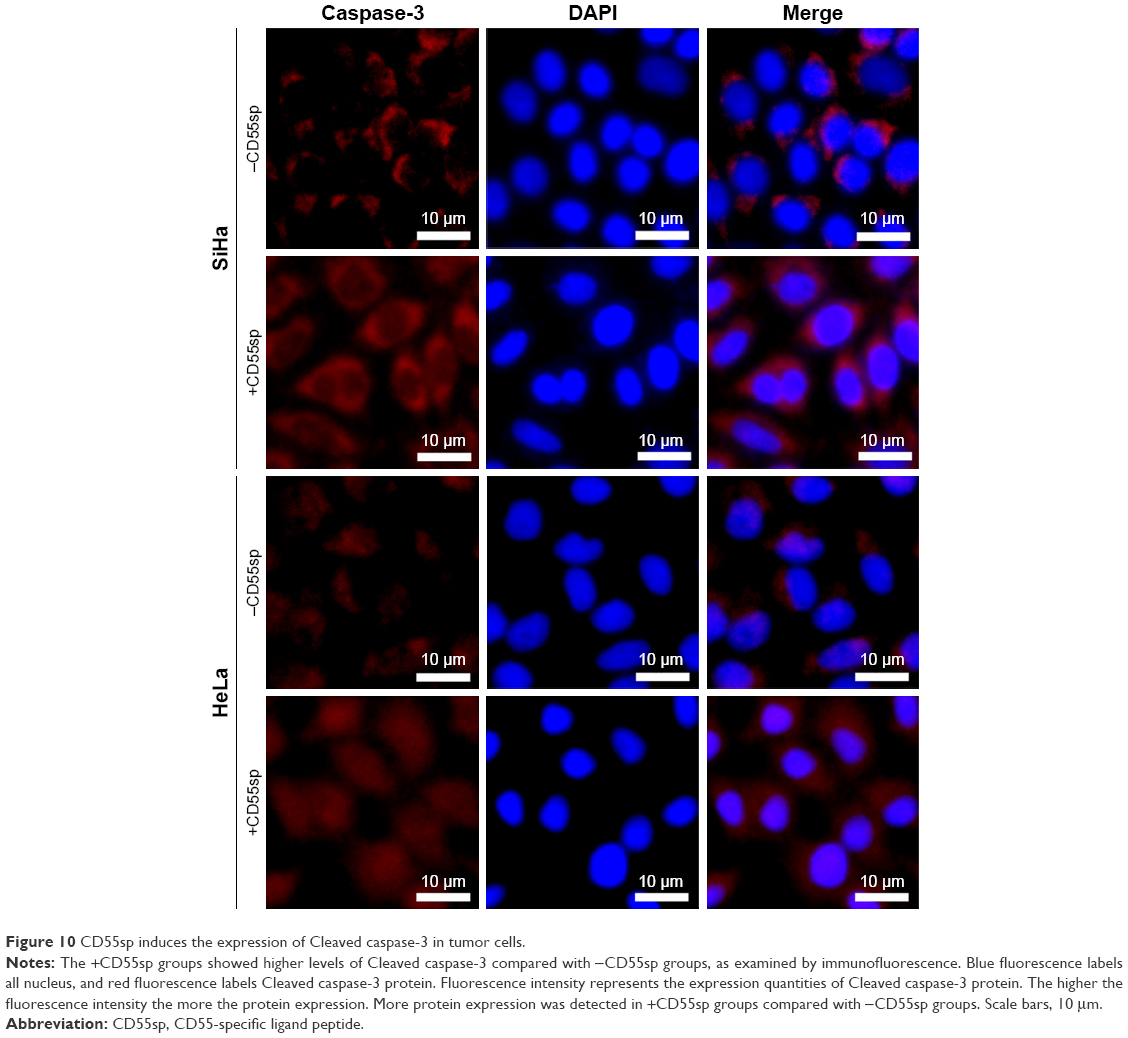 | Figure 10 CD55sp induces the expression of Cleaved caspase-3 in tumor cells. |
Reduced levels of bcl-2 proteins and increased levels of Cleaved caspase-3 proteins detected in tumor cells with CD55sp treatment
After cervical cancer, SiHa and HeLa cells were treated with CD55sp, bcl-2 protein levels of both SiHa and HeLa cells reduced, and Cleaved caspase-3 proteins increased (Figure 11). Tumorigenesis is due to the disruption of the balance between cell proliferation and apoptosis, and apoptosis signal transduction is the key to cell apoptosis.34 Among them, apoptosis-related proteins such as the caspase 3 protease family35,36 and the anti-apoptotic protein bcl-237,38 play a key role in apoptosis. By Western blot,39,40 we detected the expression of Cleaved caspase-3 and bcl-2 to verify the apoptosis in tumor cells. These results illuminated that CD55sp could induce apoptosis of tumor cells by altering the expression of apoptosis-related proteins.
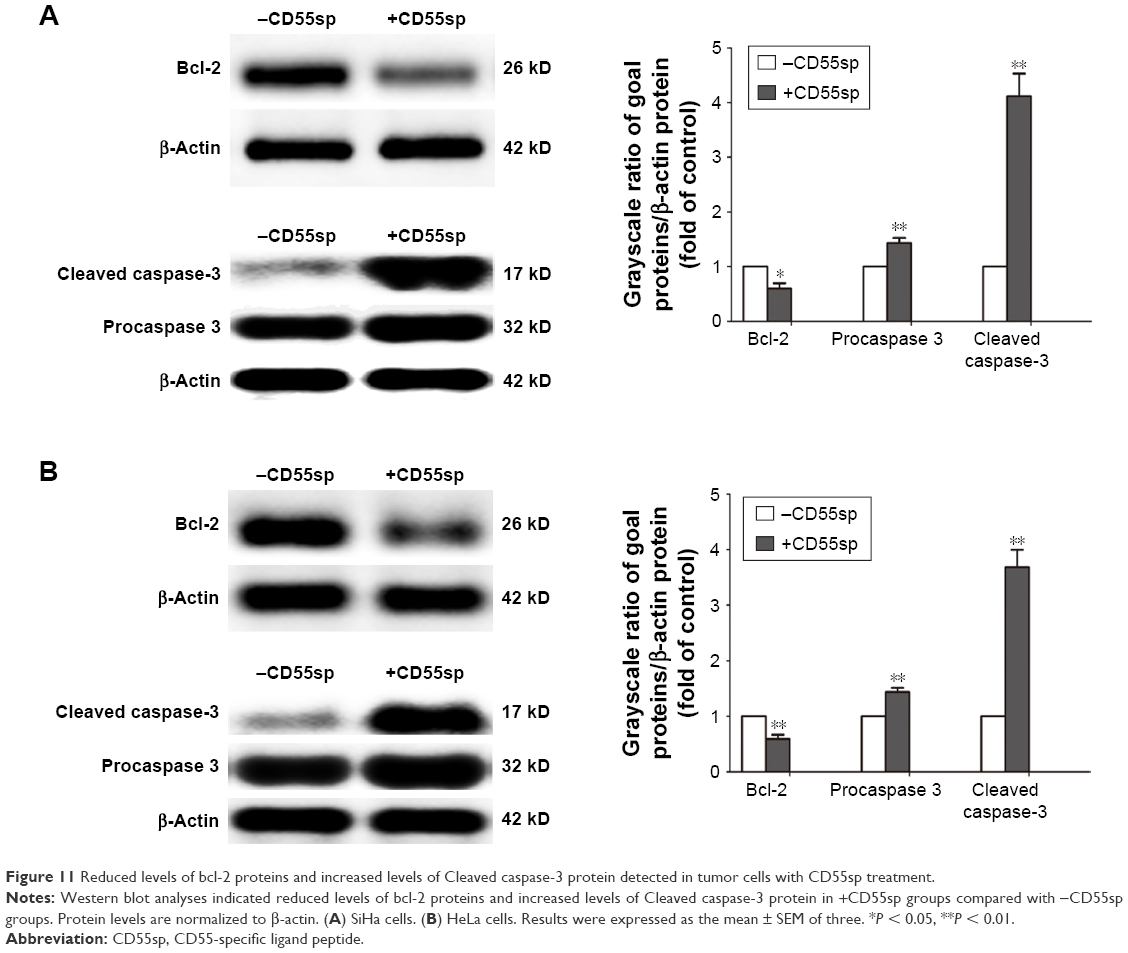 | Figure 11 Reduced levels of bcl-2 proteins and increased levels of Cleaved caspase-3 protein detected in tumor cells with CD55sp treatment. |
Conclusion
The peptides specifically bind to CD55 on the cell surface were successfully identified from the screen using phage display technology. The antitumor effects of this ligand peptide were further characterized in vitro. In the future, in view of CD55sp specifically binding to CD55, our research will focus on the construction of the targeted drug delivery system to improve clinical efficacy in cervical cancer.
Acknowledgment
This work was supported by grants from the National Natural Science Foundation of China (Nos 81471546 and 81871231) and Qingdao industrial incubation project (No 18-6-1-91-nsh).
Disclosure
The authors report no conflicts of interest in this work.
References
Eifel PJ, Winter K, Morris M, et al. Pelvic irradiation with concurrent chemotherapy versus pelvic and para-aortic irradiation for high-risk cervical cancer: an update of radiation therapy oncology group trial (RTOG) 90-01. J Clin Oncol. 2004;22(5):872–880. | ||
Pereira E, Cooper HH, Zelaya PG, et al. Concurrent chemoradiation versus radiotherapy alone for the treatment of locally advanced cervical cancer in a low-resource setting. Gynecol Oncol Rep. 2016;19:50–52. | ||
Cao J, Chen Z, Chi J, Sun Y, Sun Y. Recent progress in synergistic chemotherapy and phototherapy by targeted drug delivery systems for cancer treatment. Artif Cells Nanomed Biotechnol. 2018:1–14. | ||
Gaertner FC, Kessler H, Wester HJ, Schwaiger M, Beer AJ. Radiolabelled RGD peptides for imaging and therapy. Eur J Nucl Med Mol Imaging. 2012;39(Suppl 1):S126–S138. | ||
Arosio D, Manzoni L, Corno C, Perego P. Integrin-targeted peptide- and peptidomimetic-drug conjugates for the treatment of tumors. Recent Pat Anticancer Drug Discov. 2017;12(2):148–168. | ||
Thielen AJF, van Baarsen IM, Jongsma ML, Zeerleder S, Spaapen RM, Wouters D. CRISPR/Cas9 generated human CD46, CD55 and CD59 knockout cell lines as a tool for complement research. J Immunol Methods. 2018;456:15–22. | ||
Bellone S, Roque D, Cocco E, et al. Downregulation of membrane complement inhibitors CD55 and CD59 by siRNA sensitises uterine serous carcinoma overexpressing Her2/neu to complement and antibody-dependent cell cytotoxicity in vitro: implications for trastuzumab-based immunotherapy. Br J Cancer. 2012;106(9):1543–1550. | ||
Li B, Gao MH, Chu XM, Xu YJ, Yang F. Identification of a novel short peptide seal specific to CD59 and its effect on HeLa cell growth and apoptosis. Cell Oncol (Dordr). 2012;35(5):355–365. | ||
Leung TH, Tang HW, Siu MK, et al. Human papillomavirus E6 protein enriches the CD55(+) population in cervical cancer cells, promoting radioresistance and cancer aggressiveness. J Pathol. 2018;244(2):151–163. | ||
Wu CH, Liu IJ, Lu RM, Wu HC. Advancement and applications of peptide phage display technology in biomedical science. J Biomed Sci. 2016;23:8. | ||
Liu J, Liu J, Chu L, et al. Novel peptide-dendrimer conjugates as drug carriers for targeting nonsmall cell lung cancer. Int J Nanomedicine. 2010;6:59–69. | ||
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Shi L, Yu L, Zhong D, et al. TMC120 displayed potent cytotoxic effect on human cervical carcinoma through enhancing the polymerization of microtubules. AIDS. 2018;32(9):1107–1114. | ||
Qiu F, Zhao X. Study on the sensitivity of primary tumor cells of patients with cervical cancer to chemotherapeutic combinations. J Buon. 2018;23(1):117–123. | ||
Tian S, Chen H, Tan W. Targeting mitochondrial respiration as a therapeutic strategy for cervical cancer. Biochem Biophys Res Commun. 2018;499(4):1019–1024. | ||
He C, Mao D, Hua G, et al. The Hippo/YAP pathway interacts with EGFR signaling and HPV oncoproteins to regulate cervical cancer progression. EMBO Mol Med. 2015;7(11):1426–1449. | ||
Mamidi S, Cinci M, Hasmann M, Fehring V, Kirschfink M. Lipoplex mediated silencing of membrane regulators (CD46, CD55 and CD59) enhances complement-dependent anti-tumor activity of trastuzumab and pertuzumab. Mol Oncol. 2013;7(3):580–594. | ||
Mazumdar B, Kim H, Meyer K, et al. Hepatitis C virus infection upregulates CD55 expression on the hepatocyte surface and promotes association with virus particles. J Virol. 2013;87(14):7902–7910. | ||
Zhou C, Kang J, Wang X, Wei W, Jiang W. Phage display screening identifies a novel peptide to suppress ovarian cancer cells in vitro and in vivo in mouse models. BMC Cancer. 2015;15:889. | ||
Zhang ZF, Shan X, Wang YX, Wang W, Feng SY, Cui YB. Screening and selection of peptides specific for esophageal cancer cells from a phage display peptide library. J Cardiothorac Surg. 2014;9:76. | ||
Liu Q, Song YJ, Meng LJ, et al. Role of LM23 in cell proliferation and apoptosis and its expression during the testis development. Asian J Androl. 2013;15(4):539–544. | ||
Toné S, Sugimoto K, Tanda K, et al. Three distinct stages of apoptotic nuclear condensation revealed by time-lapse imaging, biochemical and electron microscopy analysis of cell-free apoptosis. Exp Cell Res. 2007;313(16):3635–3644. | ||
Maruyama R, Takemura G, Aoyama T, et al. Dynamic process of apoptosis in adult rat cardiomyocytes analyzed using 48-hour videomicroscopy and electron microscopy: beating and rate are associated with the apoptotic process. Am J Pathol. 2001;159(2):683–691. | ||
Herrmann S, Leshem B, Lobel L, et al. T7 phage display of Ep15 peptide for the detection of WNV IgG. J Virol Methods. 2007;141(2):133–140. | ||
Adan A, Alizada G, Kiraz Y, Baran Y, Nalbant A. Flow cytometry: basic principles and applications. Crit Rev Biotechnol. 2017;37(2):163–176. | ||
Adan A, Kiraz Y, Baran Y. Cell proliferation and cytotoxicity assays. Curr Pharm Biotechnol. 2016;17(14):1213–1221. | ||
Fayzullina S, Martin LJ. Detection and analysis of DNA damage in mouse skeletal muscle in situ using the TUNEL method. J Vis Exp. 2014;(94). | ||
Wlodkowic D, Telford W, Skommer J, Darzynkiewicz Z. Apoptosis and beyond: cytometry in studies of programmed cell death. Methods Cell Biol. 2011;103:55–98. | ||
Jiang L, Paone S, Caruso S, et al. Determining the contents and cell origins of apoptotic bodies by flow cytometry. Sci Rep. 2017;7(1):14444. | ||
Taatjes DJ, Sobel BE, Budd RC. Morphological and cytochemical determination of cell death by apoptosis. Histochem Cell Biol. 2008;129(1):33–43. | ||
Wyllie AH, Morris RG, Smith AL, Dunlop D. Chromatin cleavage in apoptosis: association with condensed chromatin morphology and dependence on macromolecular synthesis. J Pathol. 1984;142(1):67–77. | ||
Sorrells S, Toruno C, Stewart RA, Jette C. Analysis of apoptosis in zebrafish embryos by whole-mount immunofluorescence to detect activated Caspase 3. J Vis Exp. 2013;(82):e51060. | ||
Dong QM, Ling C, Zhao L. Immunofluorescence analysis of cytokeratin 8/18 staining is a sensitive assay for the detection of cell apoptosis. Oncol Lett. 2015;9(3):1227–1230. | ||
Williams GT, Smith CA. Molecular regulation of apoptosis: genetic controls on cell death. Cell. 1993;74(5):777–779. | ||
Safavi M, Shakeri R, Ardestani SK, Davoodi J, Ajdary S, Foroumadi A. Caspase-dependent apoptosis induced by two synthetic halogenated flavanones, 3′,7-dichloroflavanone and 3′,6-dichloroflavanone, on human breast and prostate cancer cells. In Vitro Cell Dev Biol Anim. 2018;54(2):136–146. | ||
Hsu HY, Lin TY, Hu CH, Shu DTF, Lu MK. Fucoidan upregulates TLR4/CHOP-mediated caspase-3 and PARP activation to enhance cisplatin-induced cytotoxicity in human lung cancer cells. Cancer Lett. 2018;432:112–120. | ||
Beberok A, Wrześniok D, Rok J, Rzepka Z, Respondek M, Buszman E. Ciprofloxacin triggers the apoptosis of human triple-negative breast cancer MDA-MB-231 cells via the p53/Bax/Bcl-2 signaling pathway. Int J Oncol. 2018. | ||
Pihán P, Carreras-Sureda A, Hetz C. BCL-2 family: integrating stress responses at the ER to control cell demise. Cell Death Differ. 2017;24(9):1478–1487. | ||
Lin WW, Chen IJ, Cheng TC, et al. A secondary antibody-detecting molecular weight marker with mouse and rabbit IgG Fc linear epitopes for western blot analysis. PLoS One. 2016;11(8):e0160418. | ||
Saleh AM, Aljada A, Rizvi SA, Nasr A, Alaskar AS, Williams JD. In vitro cytotoxicity of Artemisia vulgaris L. Essential oil is mediated by a mitochondria-dependent apoptosis in HL-60 leukemic cell line. BMC Complement Altern Med. 2014;14:226. | ||
George DG, Dodson RJ, Garavelli JS, et al. The Protein Information Resource (PIR) and the PIR-International Protein Sequence Database. Nucleic Acids Res. 1997;25(1):24–28. | ||
Barker WC, Garavelli JS, McGarvey PB, et al. The PIR-International Protein Sequence Database. Nucleic Acids Res. 1999;27(1):39–43. | ||
Barker WC, George DG, Mewes HW, Pfeiffer F, Tsugita A. The PIR-International databases. Nucleic Acids Res. 1993;21(13):3089–3092. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.