")
Back to Journals » Drug Design, Development and Therapy » Volume 9
A novel small-molecule inhibitor of HIV-1 entry
Authors Heredia A, Latinovic O, Barbault F, de Leeuw E
Received 27 May 2015
Accepted for publication 10 July 2015
Published 1 October 2015 Volume 2015:9 Pages 5469—5478
DOI https://doi.org/10.2147/DDDT.S89338
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Shu-Feng Zhou
Alonso Heredia,1,3 Olga S Latinovic,2,3 Florent Barbault,4 Erik PH de Leeuw3,5
1Department of Medicine, 2Department of Microbiology and Immunology, 3Institute of Human Virology, University of Maryland Baltimore School of Medicine, Baltimore, MD, USA; 4Univ Paris Diderot, Sorbonne Paris Cité, ITODYS, UMRCNRS7086, Paris, France; 5Department of Biochemistry and Molecular Biology, University of Maryland Baltimore School of Medicine, Baltimore, MD, USA
Background: Antiretroviral therapy has transformed HIV-1 infection into a managed condition with near-normal life expectancy. However, a significant number of patients remain with limited therapeutic options due to HIV-1 resistance, side effects, or drug costs. Further, it is likely that current drugs will not retain efficacy, due to risks of side effects and transmitted resistance.
Results: We describe compound 5660386 (3-ethyl-2-[3-(1,3,3-trimethyl-1,3-dihydro-2H-indol-2-ylidene)-1-propen-1-yl]-1,3-benzothiazol-3-ium) as a novel inhibitor of HIV-1 entry. Compound 5660386 inhibits HIV-1 entry in cell lines and primary cells, binds to HIV-1 envelope protein, and inhibits the interaction of GP120 to CD4. Further, compound 5660386 showed a unique and broad-range activity against primary HIV-1 isolates from different subtypes and geographical areas.
Conclusion: Development of small-molecule entry inhibitors of HIV-1 such as 5660386 may lead to novel classes of anti-HIV-1 therapeutics. These inhibitors may be particularly effective against viruses resistant to current antiretroviral drugs and could have potential applications in both treatment and prevention.
Keywords: HIV-1, defensin, drug, entry, antiviral therapy, CD4
Background
Infection with HIV-1 necessitates lifelong treatment, and the availability of suppressive combination antiretroviral therapy has greatly impacted the epidemic. Improvements in drug safety and potency of therapeutic regimens have greatly ameliorated long-term toxicity and tolerability of treatment. However, the acquisition and transmission of HIV-1 drug resistance pose a major risk to therapy.1 Currently, there are 26 drugs licensed for treatment of infection, targeting viral entry, reverse transcription, integration, or maturation, with new compounds under development.2,3 Viral entry of HIV-1 is a multistep process.4 Attachment of the virus particle to the cell is facilitated by a variety of molecules, such as gangliosides, DC-SIGN (dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin), heparan sulfate proteoglycans, and α4β7 integrin.5–7 HIV-1 infection occurs when the viral envelope glycoprotein GP120 binds to cellular receptor CD4 and subsequently to cellular co-receptors.8,9 Major co-receptors in HIV-1 binding and subsequent HIV-1 entry are CCR5 and CXCR4, and infection can be blocked by the natural ligands of these G-protein-coupled receptors.10 Co-receptor binding triggers fusion of the viral and cell membranes involving the transmembrane protein GP41.11
Viral entry is relatively underused as a small-molecule drug target.12,13 Enfuvirtide, a 36-amino acid peptide derived from the C-terminal region of helical domain 2 of GP41, is currently the only clinically approved inhibitor of fusion.14 Two additional peptides with overlapping binding sites to enfuvirtide, tifuvirtide and sifuvirtide, are currently in preclinical development.15 VIR-576, a derivate of a peptide termed virus inhibitory peptide that inhibits fusion, shows efficacy in Phase I/II clinical trials.16 The CCR5 antagonist Maraviroc (MVC) is the only clinically approved compound that targets co-receptor binding.17 Development of two other CCR5 antagonists, Aplaviroc and Vicriviroc, was halted due to hepatotoxicity and failure to demonstrate superiority, respectively.18,19 Cenicriviroc, an antagonist of both CCR5 and CCR2, is currently in Phase IIB clinical trials.20 In addition, derivatives of the RANTES, the natural ligand of CCR5, have been explored as potential microbicides.21,22
Several compounds that inhibit the GP120–CXCR4 interaction are under development, although none have been approved. Clinical development of the potently active compound AMD-3100 was halted due to cardiac abnormalities and lack of efficacy.23 Inhibition of the interaction between GP120 and CD4 has been attempted using soluble forms of CD4 as molecular decoys; however, this approach was relatively ineffective in humans.24 The small-molecule GP120–CD4 inhibitor BMS-378806 was identified based on the crystal structure of the GP120–CD4 complex.25,26 An optimized compound, BMS-626529, is currently being explored in Phase II clinical trials.27,28 Here, we report on a new small-molecule antimicrobial compound with anti-HIV-1 activity derived from a class of innate immune peptides termed defensins.29,30
Materials and methods
Cell culture
TZM-bl cells were obtained from the American Type Culture Collection and maintained in Rosewell Park Memorial Institute (RPMI) medium supplemented with 10% fetal bovine serum, antibiotics, and l-glutamine. Peripheral blood lymphocytes (PBLs) were separated from purchased buffy coats (New York Blood Bank). Cells were cultured in RPMI medium containing 10% fetal bovine serum plus antibiotics and activated by PHA treatment (2.5 μg/mL) for 3 days. Activated cells were maintained in complete medium containing interleukin-2 (100 U/mL). No ethics statement was required from the institutional review board for the use of this cell line.
Viruses and drugs
The following isolates were tested to assay the potency and breadth of compounds: subtypes A, B, C, D, E, F, and G. The lab-adapted strains HIV-1 BaL and HIV-1 IIIB, and the primary isolates from subtypes A, B, D, E, F, and G were obtained from the NIH AIDS Repository (Germantown, MD, USA). Subtype C primary isolates were provided by Dr Mark Wainberg (McGill University AIDS Centre, Montreal, QC, Canada). MVC-resistant HIV-1 strain 6061 was provided by Dr Steven Deeks (San Francisco VA Medical Center and University of California, San Francisco, CA, USA). Two primary isolates of each subtype were tested initially in the TZM-bl assay. Anti-HIV-1 activity against these isolates was confirmed in HIV-1 infectivity assays as described using the appropriate viruses and compound BMS as control.31,32 For the BlaM assay, JRFL140T (CCR5) and HxB2 (CXCR4) pseudovirus were used. Compound 5660386 was obtained from Chembridge Corp. BMS-488043 (herein referred to as BMS) was provided by Mark Krystal (Brystol-Myers Squibb, Princeton, NJ, USA), and enfuvirtide (T20) was from Sigma Aldrich.
TZM-bl assays
Drug susceptibility assays of compounds were performed in TZM-bl cells infected with 250 TCID50 HIV-1 BaL (CCR5) or IIIB (CXCR4). Compounds were used at twofold serial dilutions starting at 2 μM, using MVC or AMD-3100 as control for CCR5-tropic (BaL) or CXCR4-tropic (IIIB) virus, respectively. Inhibition of viral growth was measured by luminescence using the BrightGlo kit from Promega.
HIV-1 infectivity assays
Drug susceptibility assays of compounds were performed in PHA-activated PBLs using a multiplicity of infection (MOI) of 0.001. Compounds were used at twofold serial dilutions starting at 1 μM. Viral growth was measured by standard p24 ELISA.
Cytotoxicity
The effect of compounds on cell viability was assessed by measuring the mitochondrial activity using MTS assays (Cell Titer 96 proliferation assay, Promega).
BlaM assay
TZM-bl cells (4×104 cells/well) were grown overnight in 96-well plates in a phenol red-free growth medium. Virus (CXCR4, IIIB; CCR5, BaL) was added to cells at MOI of 0.7–1.0 and subsequently centrifuged at 2,095× g for 30 minutes at 4°C. Cells were washed with cold phosphate-buffered saline to remove unbound viruses, and virus–cell fusion was initiated by shifting the samples to 37°C. Samples were maintained at 37°C for a total of 90 minutes, briefly chilled by placing on ice, loaded with the fluorescence substrate CCF2-AM from the BlaM kit, and incubated overnight at 18°C. BlaM activity was quantified using Synergy HT fluorescence plate reader. The extent of virus–cell fusion is determined from the ratio of blue (440–480 nm) and green (518–538 nm) emission upon exciting the cells at 405–415 nm.
Biacore assays
Binding of active entry inhibitors to immobilized GP-120 (1,500 response units) was evaluated on an in-house Biacore T100 system in a 96-well format at 25°C essentially as described. The ability of active entry inhibitors that bind GP120 to inhibit the binding of CD4 to GP120 was evaluated by determining the binding of soluble CD4 (1 μg/mL) to GP120 in the absence or presence of increasing concentration of compound.
Molecular docking
The starting coordinates of the GP120 HIV-1 protein were extracted from the PDB under the accession code 4DKQ. This entry represents a crystal structure of the viral enzyme in complex with the OLK inhibitor at a resolution of 1.80 Å.33 We chose this reference structure because the molecular size of OLK is similar to our compounds and is also carrying one positive charge. Some amino-acid side chains are missing in the PDB 4DKQ; these are not located at the interacting site. However, to ensure the GP120 integrity, we added these side chains with the help of the xleap module of Amber according to the protein force field ff99SB.34 An optimization was then made with 2,000 steps of steepest descent followed by 2,000 steps of conjugated gradient with General Born water implicit solvation. The GP120 target interacting site was defined with a grid of 15 Å ×15 Å ×19.5 Å in the xyz direction on cavity where 5660386 is found. The compounds shown in Figure 1 were built with the help of the maestro interface of Schrodinger software package.35 Atomic partial charges were determined with the semiempirical AM1-BCC method.36,37 Molecular geometries were optimized through 5,000 steps of steepest descent followed by 5,000 steps of conjugated gradient with the gaff force field.38 The compounds were then prepared for molecular docking calculations with the help of Raccoon software.39 In this step, all rotatable dihedral angles were set free to move during the calculations. Molecular docking calculations were performed with the Autodock 4.2 software.40 The Lamarckian Genetic Algorithm method41 was employed for the global optimum binding position search. One hundred cycles of calculations were performed in order to get a final binding position as accurate as possible. The resulting docking structures were then clustered into conformation families according to a root mean square deviation lower than 2 Å. The conformation selected was the one which presented the lowest docking free energy of binding in the most populated cluster.42 Visualization and analysis of protein–ligand interactions were made with the help of the visual molecular dynamic software.43 A hydrogen bond was considered to be present when the donor–acceptor distance is smaller than 3.5 Å and the H-donor–acceptor angle is smaller than 45°. The visualization of protein hydrophobicity was made by coloring of its molecular surface according to the Eisenberg scale.44
 | Figure 1 Docking orientation of 5660386 inside the binding pocket of viral GP120 protein. |
Results and discussion
Human defensins act as effectors of innate immunity against invading microbes, including many viruses.29 The compounds identified in this work were derived from the human defensin Human Neutrophil Peptide-1 (HNP-1).45 In the case of HNP-1, several anti-HIV-1 entry mechanisms have been described, including binding to GP120 and CD4, and interfering with the GP120–CD4 interaction.46–48 More recently, the effects of HNP-1 on HIV-1 entry were further dissected. The defensin appeared to inhibit binding of envelope to CD4 and co-receptors as well as formation of the helical bundle structure of envelope, thus productively inhibiting HIV-1 uptake.49 We have recently identified critical residues of the human defensin HNP-1 involved in binding to bacterial Lipid II.50 In that study, compounds were identified that do not bind to Lipid II, yet have potent antimicrobial activity. Given the reported anti-HIV-1 activity of HNP-1,51 we reasoned that such compounds could have potential anti-HIV-1 activity. We therefore screened these compounds for their ability to inhibit infection of TZM-bl cells with the CCR5-dependent HIV-1 BaL strain in vitro. Compounds were preincubated with virus for 60 minutes, and the mixture of compound and virus was subsequently incubated with the cells for 48 hours. In this screen, we identified two compounds, 7165-0606 and 5660386 (3-ethyl-2-[3-(1,3,3-trimethyl-1,3-dihydro-2H-indol-2-ylidene)-1-propen-1-yl]-1,3-benzothiazol-3-ium), with anti-HIV-1 properties. The Lipid II-binding compounds BAS00127538, 4890-0291, and 2332-101450 did not show antiviral activity (Figure S1). Compound 5660386 appeared most potent against HIV-1 (IC50 ~2,000 nM) and was selected for further study. 5660386 inhibited HIV-1 infection in a dose-dependent fashion and did not negatively affect cell growth as determined by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assays (Figure 2A). Time-of-addition experiments indicated that compound was able to inhibit infection after preincubation with virus for only 5 minutes (not shown). We next tested the possibility of synergism between 5660386 and the CCR5-dependent drug MVC or the attachment inhibitor BMS.25 Virus was preincubated with BMS or MVC at concentrations ranging from 0 nM to 500 nM in the absence or presence of 500 nM 5660386. Compound 5660386 inhibited both CCR5-(BaL, Figure 2D) and CXCR4-dependent virus (IIIB, Figure 2C). Further, 5660386 enhanced the inhibition of infection by MVC but not BMS, suggesting that inhibition by compound is independent of CCR5 (Figure 2C and D).
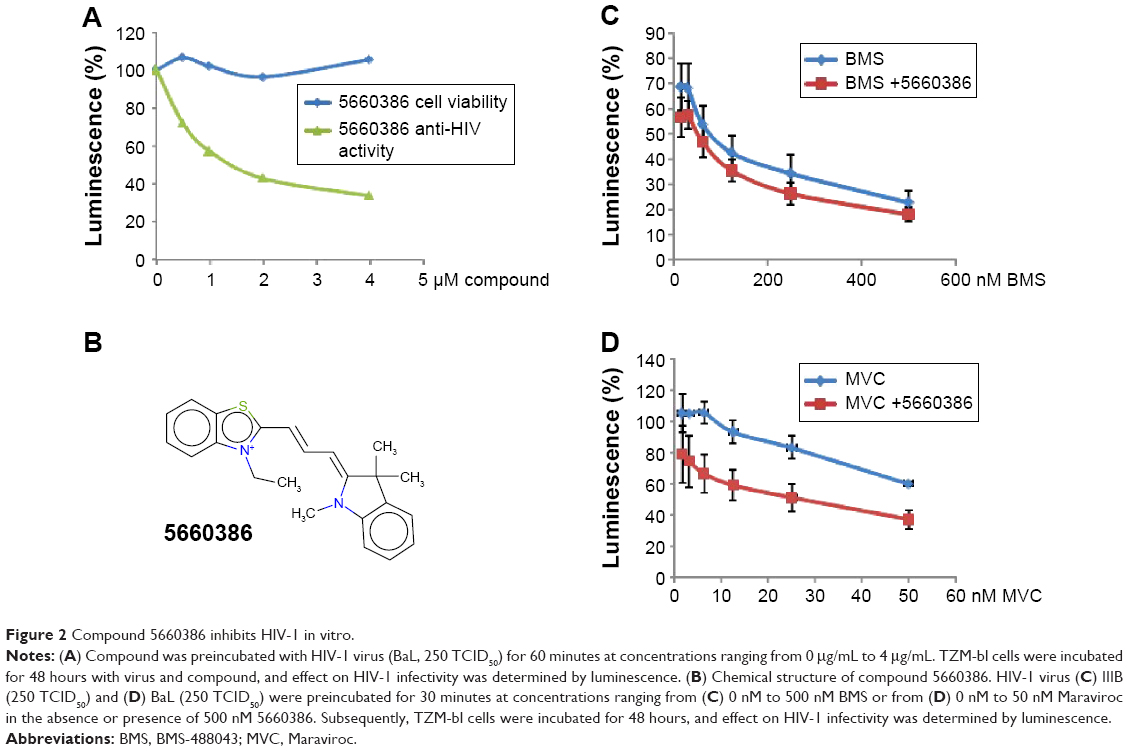 | Figure 2 Compound 5660386 inhibits HIV-1 in vitro. |
We next examined the potency and breadth of the inhibition of HIV-1 infection by our compound in primary cells. In a first set of experiments, compound and virus were preincubated for 30 minutes, added to phytohemagglutinin (PHA)-stimulated peripheral blood mononuclear cells (PBMCs) for 2 hours, and then removed from the medium by extensive washing. Infected cells were cultured in the absence of compound for 7 days, after which virus production was measured in supernatants by p24 enzyme-linked immunosorbent assay (ELISA) (Figure 3A). In these experiments, we used both CCR5-dependent and CXCR4-dependent strains. In a second set of experiments, compound was added immediately after removal of the virus from the cells by washing, and it was kept in the cultures for the 7-day duration of the experiment (Figure 3B). Compound 5660386 was not toxic to cells under these conditions and inhibited replication of HIV-1 reference strains BaL (CCR5 tropic) and IIIB (CXCR4 tropic) in both experimental conditions. We examined the ability of 5660386 to inhibit primary isolates of HIV-1 from different subtypes, using the attachment inhibitor BMS as control in the TZM-bl assay. BMS antiviral activity was highest against subtype B, followed by clades C and A, with little or no activity against clade F and G, in accordance with previous findings.25 In contrast, 5660386 potently inhibited clade B and C primary isolates, and to a lesser extent, clade F, G, and A isolates. Subtype G was not inhibited by BMS but was inhibited by 5660386 by up to ~50% (Figure S2). To confirm these results, we tested HIV-1 inhibition by compound using primary cells and isolates of different subtypes (Figure 4). In these experiments, virus and compound were preincubated for 60 minutes, and the mixture was added to PBMCs for 120 minutes. Compound and virus were subsequently removed by gentle washing, and inhibition of HIV-1 infection was determined by p24 ELISA after 7 days. Compound 5660386 inhibited clade B (Figure 4A), C (Figure 4B), and G (Figure 4C) viruses at concentrations >100 nM, in agreement with our findings in the TZM-bl assays (Figure S2). Compound negatively affected growth of PBMCs only at the highest concentration tested (2 μM) under these experimental conditions. Combined, these observations indicate that 5660386 exhibits inhibitory activity against a panel of primary HIV-1 isolates infecting primary cells. Further, 5660386 inhibits CCR5- and CXCR4-dependent viruses and potentiates the CCR5 inhibitor MVC.
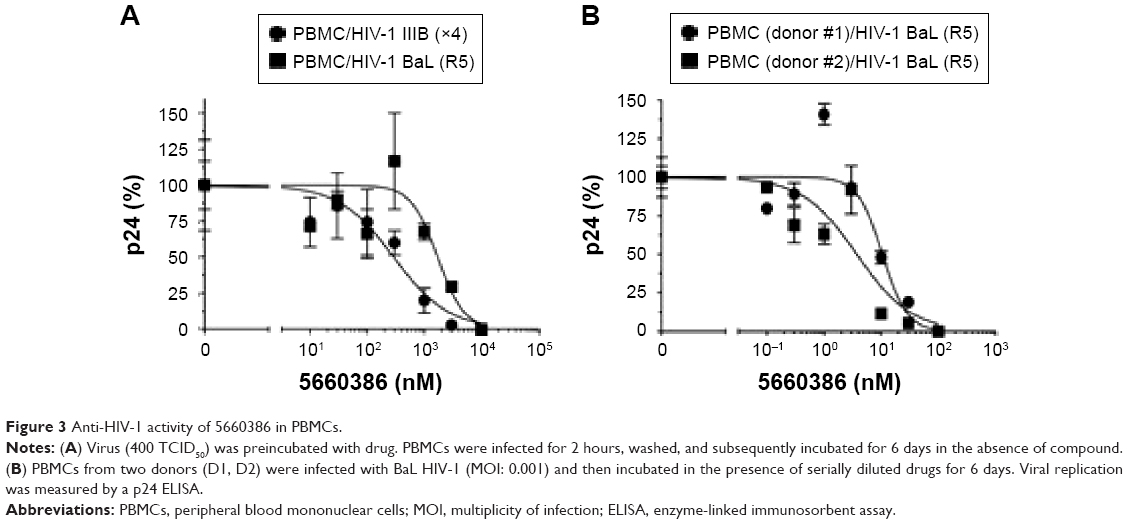 | Figure 3 Anti-HIV-1 activity of 5660386 in PBMCs. |
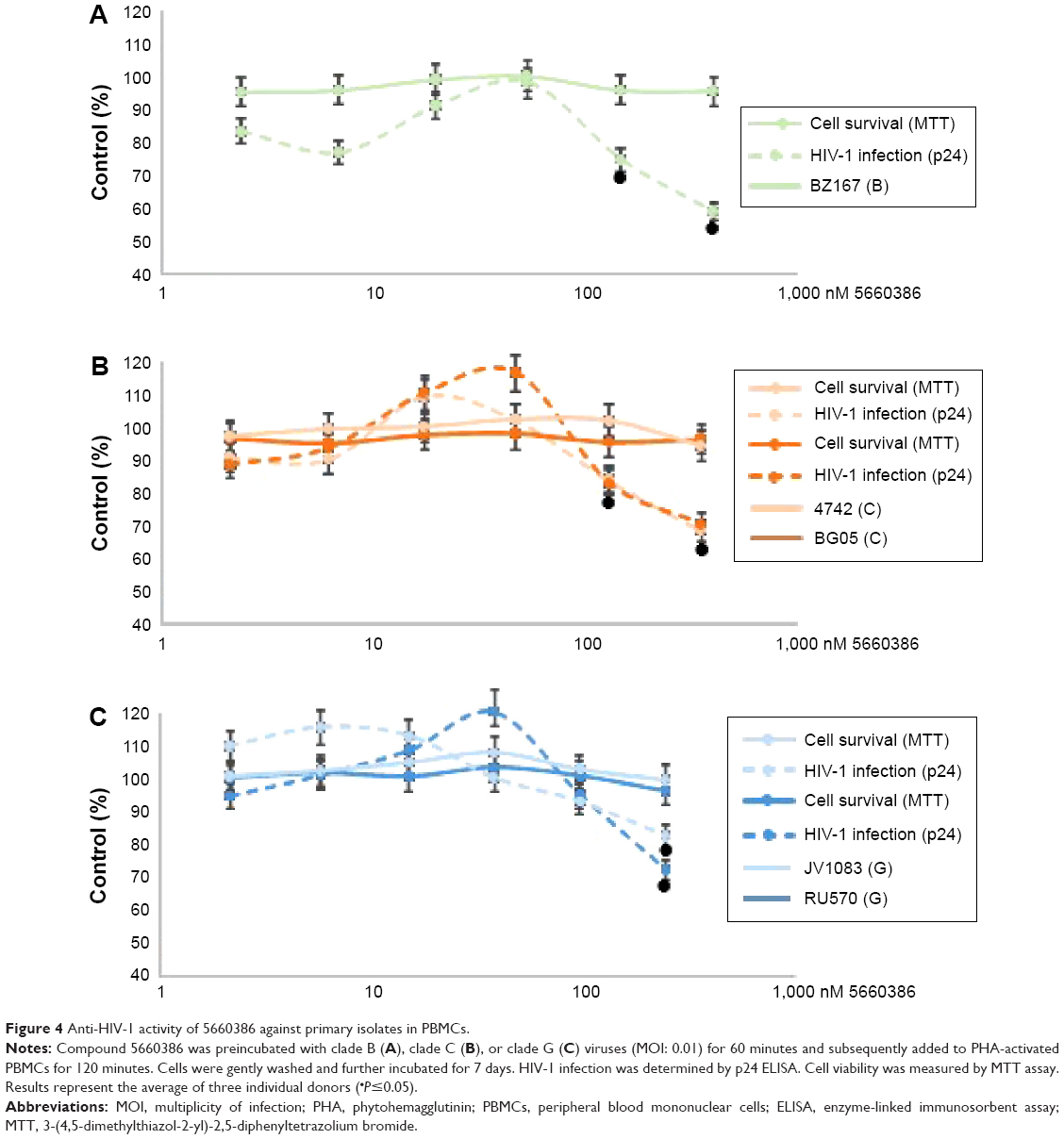 | Figure 4 Anti-HIV-1 activity of 5660386 against primary isolates in PBMCs. |
Based on the aforementioned studies, and the observation that HIV-1 replication in the virus/compound preincubation experiments was inhibited, 5660386 may interfere with HIV-1 attachment and/or entry. To examine if 5660386 inhibits viral entry directly, we measured inhibition of replication-incompetent CCR5- and CXCR4-dependent viruses by BlaM fusion assay52 (Figure 5). Compound was preincubated with virus for 60 minutes, after which cells were spinoculated to promote virus binding and shifted to 37°C to initiate fusion. HIV-1 entry was determined after 48 hours by determination of the β-lactamase activity. Under these conditions, 5660386 clearly inhibited entry of both CCR5- and CXCR4-dependent viruses, at comparable concentrations to the entry inhibitor T20.15 In the TZM-bl assays, we observed that 5660386 was able to potentiate the CCR5-antagonist MVC but not BMS (Figure 2), suggesting that 5660386 does not compete with MVC for binding to CCR5. In addition, 5660386 did not inhibit infection by 6061, a CCR5-dependent virus that can use CCR5 bound to MVC as a receptor, further suggesting that the compound does not act primarily on this co-receptor (not shown). Based on these observations, we next evaluated binding of 5660386 to GP120. Surface plasmon resonance studies demonstrated that compound 5660386 binds to GP120 (Figure 6A). Since 5660386 binds to GP120, we examined the possibility that 5660386 affects the interaction between GP120 and CD4 as a possible mechanism of action. Binding of soluble CD4 to GP120 was found to be inhibited by increasing concentrations of 5660386 (Figure 6B). Combined, these findings suggest that the 5660386 compound targets the HIV-1 envelope and inhibits CD4 binding. Attempts to generate resistance mutants against compound 5660386 were unsuccessful, due to induction of cellular toxicity during extended culture at increased concentrations. At sub-EC50 concentrations (100 nM), no breakthrough viral replication could be detected by p24 ELISA after 2 weeks (not shown).
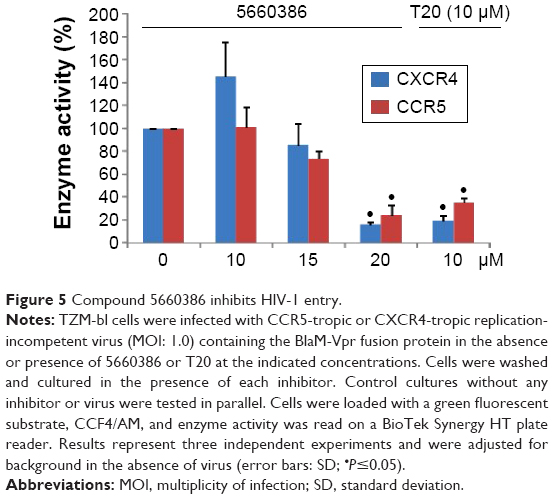 | Figure 5 Compound 5660386 inhibits HIV-1 entry. |
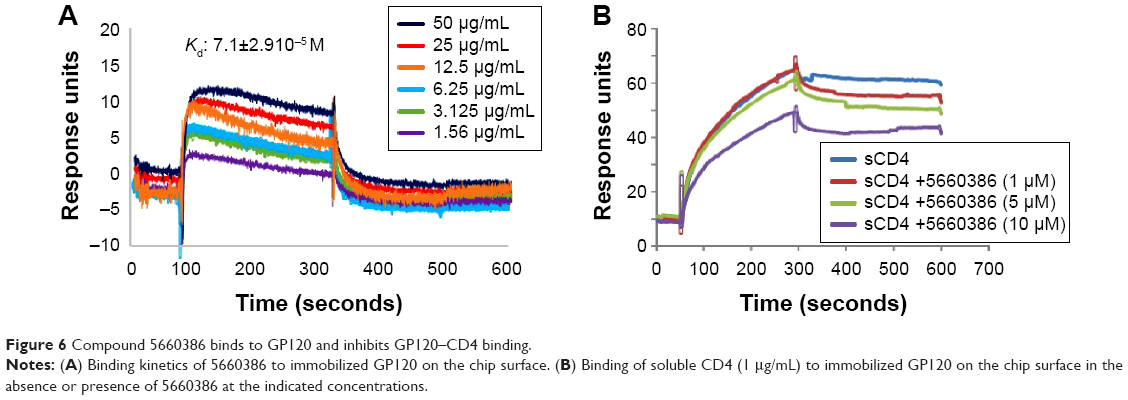 | Figure 6 Compound 5660386 binds to GP120 and inhibits GP120–CD4 binding. |
Molecular docking studies were conducted to find the optimal position of compound 5660386 (Figure 1). The starting coordinates of the GP120 HIV-1 protein were extracted from the PDB under the accession code 4DKQ. This entry represents a crystal structure of the viral enzyme in complex with the N-[(1S,2S)-2-carbamimidamido-2,3-dihydro-1H-inden-1-yl]-N′-(4-chloro-3-fluorophenyl)ethanediamide inhibitor (hereafter named OLK) at a resolution of 1.80 Å.33 First, we performed a docking test to ensure the validity of our computational protocol. The compound OLK (Figure 7), for which a crystal structure exists, was docked into the GP120 target. The predicted position of the OLK ligand closely resembles its position in the crystal structure, preserving the interacting pattern toward GP120 and validating our computational approach (Figure S3). For 5660386, the calculation yielded only two clusters with a lowest energy of −9.4 kcal/mol and occupancy of 86%. As expected, an electrostatic interaction with the ionic moiety of the ligand and the Asp carboxylate moiety is observed with a distance of 6.5 Å. Besides, another electrostatic interaction is also recorded between the ammonium of the carboxyl atom of the glycine 473 with a distance of 2.5 Å. Viral GP120 protein recognizes the CD4 human protein, and this event triggers the viral entry process. The association between these two proteins involves mainly two CD4 residues: Phe34 and Arg59. The phenyl-alkaline moiety is inserted into a GP120 cavity, whereas the positive charge of the guanidinium part of arginine performs an electrostatic interaction with the carboxylate part of the viral residue Asp368.53 This cavity represents the interacting site of the Phe43 residue from CD4 protein. The 5660386 compound makes strong van der Waals interaction into the Phe34 cavity of GP120. This cavity is delineated by the hydrophobic residues Trp427, Ile475, Val255, Phe382, Met426, and Ile424. The filling of this cavity engenders more van der Waals interactions and also gives favorable entropic variations, since we expected that more water molecules will be displaced when 5660386 will act on GP120. Therefore, we predict that an inhibitor perturbs the GP120–CD4 association through these residues. As expected with the molecular formula of 5660386, hydrogen bonding is not observed. Indeed, there is no polar hydrogen, and neither sulfur nor aromatic tertiary amines make good acceptors for hydrogen bonds.
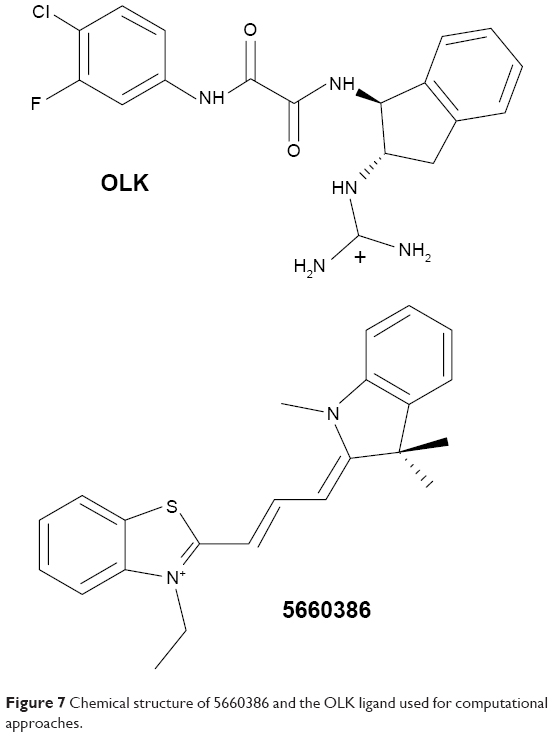 | Figure 7 Chemical structure of 5660386 and the OLK ligand used for computational approaches. |
Conclusion
Overall, the data show that 5660386 is a new inhibitor of HIV-1. The mechanistic studies demonstrate that it is working at the level of entry, targeting HIV-1 envelope. Compound 5660386 shows antiviral activity against primary virus isolates of varying subtypes in the TZM-bl cell line and showed reduced, but significant, antiviral activity in primary cells. These findings indicate that early lead small-molecule entry inhibitors of HIV-1 such as 5660386 can be developed into a second-generation of anti-HIV-1 therapeutics with a unique spectrum of activity.
Acknowledgments
We thank Mark Krystal (Brystol-Myers Squibb, Princeton, NJ, USA) for providing compound BMS-488043. We thank Mark Wainberg (McGill University AIDS Centre, Montreal, QC, Canada) for subtype C viruses, and Steven Deeks (San Francisco VA Medical Center and University of California, San Francisco, CA, USA) for MVC-resistant HIV-1.
Disclosure
The authors declare no competing financial interests. US provisional patent application No 62/054,137 “Small Molecule HIV Entry Inhibitors” has been filed. This does not alter our adherence to all Drug Design, Development and Therapy policies on sharing data and materials.
References
Menendez-Arias L. Molecular basis of human immunodeficiency virus type 1 drug resistance: overview and recent developments. Antiviral Res. 2013;98:93–120. | ||
De Clercq E. Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV. Int J Antimicrob Ag. 2009;33:307–320. | ||
Flexner C, Saag M. The antiretroviral drug pipeline: prospects and implications for future treatment research. Curr Opin HIV AIDS. 2013;8:572–578. | ||
Haqqani AA, Tilton JC. Entry inhibitors and their use in the treatment of HIV-1 infection. Antiviral Res. 2013;98:158–170. | ||
Moulard M, Lortat-Jacob H, Mondor I, et al. Selective interactions of polyanions with basic surfaces on human immunodeficiency virus type 1 gp120. J Virol. 2000;74:1948–1960. | ||
Arthos J, Cicala C, Martinelli E, et al. HIV-1 envelope protein binds to and signals through integrin alpha4beta7, the gut mucosal homing receptor for peripheral T cells. Nat Immunol. 2008;9:301–309. | ||
Wu L, KewalRamani VN. Dendritic-cell interactions with HIV: infection and viral dissemination. Nat Rev Immunol. 2006;6:859–868. | ||
Wilen CB, Tilton JC, Doms RW. Molecular mechanisms of HIV entry. Adv Exp Med Biol. 2012;726:223–242. | ||
Klasse PJ. The molecular basis of HIV entry. Cell Microbiol. 2012;14:1183–1192. | ||
Pollakis G, Paxton WA. Use of (alternative) coreceptors for HIV entry. Curr Opin HIV AIDS. 2012;7:440–449. | ||
Blumenthal R, Durell S, Viard M. HIV entry and envelope glycoprotein-mediated fusion. J Biol Chem. 2012;287:40841–40849. | ||
Este JA, Telenti A. HIV entry inhibitors. Lancet. 2007;370:81–88. | ||
Melby T, Westby M. Inhibitors of viral entry. Handb Exp Pharmacol. 2009;189:177–202. | ||
Matthews T, Salgo M, Greenberg M, Chung J, DeMasi R, Bolognesi D. Enfuvirtide: the first therapy to inhibit the entry of HIV-1 into host CD4 lymphocytes. Nat Rev Drug Discov. 2004;3:215–225. | ||
Eggink D, Berkhout B, Sanders RW. Inhibition of HIV-1 by fusion inhibitors. Curr Pharm Des. 2010;16:3716–3728. | ||
Forssmann WG, The YH, Stoll M, et al. Short-term monotherapy in HIV-infected patients with a virus entry inhibitor against the gp41 fusion peptide. Sci Transl Med. 2010;2:63re63. | ||
Dorr P, Westby M, Dobbs S, et al. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob Agents Chemother. 2005;49:4721–4732. | ||
Nichols WG, Steel HM, Bonny T, et al. Hepatotoxicity observed in clinical trials of aplaviroc (GW873140). Antimicrob Agents Chemother. 2008;52:858–865. | ||
Caseiro MM, Nelson M, Diaz RS, et al. Vicriviroc plus optimized background therapy for treatment-experienced subjects with CCR5 HIV-1 infection: final results of two randomized phase III trials. J Infect. 2012;65:326–335. | ||
Lalezari J, Gathe J, Brinson C, et al. Safety, efficacy, and pharmacokinetics of TBR-652, a CCR5/CCR2 antagonist, in HIV-1-infected, treatment-experienced, CCR5 antagonist-naive subjects. J Acquir Immune Defic Syndr. 2011;57:118–125. | ||
Veazey RS, Ling B, Green LC, et al. Topically applied recombinant chemokine analogues fully protect macaques from vaginal simian-human immunodeficiency virus challenge. J Infect Dis. 2009;199:1525–1527. | ||
Cerini F, Landay A, Gichinga C, et al. Chemokine analogues show suitable stability for development as microbicides. J Acquir Immune Defic Syndr. 2008;49:472–476. | ||
Hendrix CW, Collier AC, Lederman MM, et al; AMD3100 HIV Study Group. Safety, pharmacokinetics, and antiviral activity of AMD3100, a selective CXCR4 receptor inhibitor, in HIV-1 infection. J Acquir Immune Defic Syndr. 2004;37:1253–1262. | ||
Daar ES, Li XL, Moudgil T, Ho DD. High concentrations of recombinant soluble CD4 are required to neutralize primary human immunodeficiency virus type 1 isolates. Proc Natl Acad Sci U S A. 1990;87:6574–6578. | ||
Lin PF, Blair W, Wang T, et al. A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc Natl Acad Sci U S A. 2003;100:11013–11018. | ||
Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393:648–659. | ||
Nowicka-Sans B, Gong YF, McAuliffe B, et al. In vitro antiviral characteristics of HIV-1 attachment inhibitor BMS-626529, the active component of the prodrug BMS-663068. Antimicrob Agents Chemother. 2012;56:3498–3507. | ||
Nettles RE, Schürmann D, Zhu L, et al. Pharmacodynamics, safety, and pharmacokinetics of BMS-663068, an oral HIV-1 attachment inhibitor in HIV-1-infected subjects. J Infect Dis. 2012;206:1002–1011. | ||
Klotman ME, Chang TL. Defensins in innate antiviral immunity. Nat Rev Immunol. 2006;6:447–456. | ||
Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415:389–395. | ||
Latinovic O, Reitz M, Le NM, et al. CCR5 antibodies HGS004 and HGS101 preferentially inhibit drug-bound CCR5 infection and restore drug sensitivity of Maraviroc-resistant HIV-1 in primary cells. Virology. 2011;411:32–40. | ||
Heredia A, Latinovic O, Gallo RC, et al. Reduction of CCR5 with low-dose rapamycin enhances the antiviral activity of vicriviroc against both sensitive and drug-resistant HIV-1. Proc Natl Acad Sci U S A. 2008;105:20476–20481. | ||
LaLonde JM, Kwon YD, Jones DM, et al. Structure-based design, synthesis, and characterization of dual hotspot small-molecule HIV-1 entry inhibitors. J Med Chem. 2012;55:4382–4396. | ||
Wickstrom L, Okur A, Simmerling C. Evaluating the performance of the ff99SB force field based on NMR scalar coupling data. Biophys J. 2009;97:853–856. | ||
Schrödinger Release 2013-2: Maestro, version 9.5, Schrödinger, LLC, New York, NY; 2013. | ||
Jakalian A, Bush BL, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J Comput Chem. 2000;21:132–146. | ||
Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J Comput Chem. 2002;23:1623–1641. | ||
Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004; 25:1157–1174. | ||
Raccoon|AutoDock VS: an automated tool for preparing AutoDock virtual screenings; 2010. Available from: http://autodock.scripps.edu/resources/raccoon | ||
Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–2791. | ||
Morris GM, Green LG, Radic Z, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19:1639–1662. | ||
Teixeira C, Serradji N, Maurel F, Barbault F. Docking and 3D-QSAR studies of BMS-806 analogs as HIV-1 gp120 entry inhibitors. Eur J Med Chem. 2009;44:3524–3532. | ||
Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(33–38):27–38. | ||
Barbault F, Landon C, Guenneugues M, et al. Solution structure of Alo-3: a new knottin-type antifungal peptide from the insect Acrocinus longimanus. Biochemistry. 2003;42:14434–14442. | ||
Lehrer RI, Lu W. Alpha-Defensins in human innate immunity. Immunol Rev. 2012;245:84–112. | ||
Wang W, Owen SM, Rudolph DL, et al. Activity of alpha- and theta-defensins against primary isolates of HIV-1. J Immunol. 2004;173:515–520. | ||
Wu Z, Cocchi F, Gentles D, et al. Human neutrophil alpha-defensin 4 inhibits HIV-1 infection in vitro. FEBS Lett. 2005;579:162–166. | ||
Furci L, Sironi F, Tolazzi M, Vassena L, Lusso P. Alpha-defensins block the early steps of HIV-1 infection: interference with the binding of gp120 to CD4. Blood. 2007;109:2928–2935. | ||
Demirkhanyan LH, Marin M, Padilla-Parra S, et al. Multifaceted mechanisms of HIV-1 entry inhibition by human alpha-defensin. J Biol Chem. 2012;287:28821–28838. | ||
Varney KM, Bonvin AM, Pazgier M, et al. Turning defense into offense: defensin mimetics as novel antibiotics targeting lipid II. PLoS Pathog. 2013;9:e1003732. | ||
Chang TL, Klotman ME. Defensins: natural anti-HIV peptides. AIDS Rev. 2004;6:161–168. | ||
Cavrois M, De Noronha C, Greene WC. A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat Biotechnol. 2002;20:1151–1154. | ||
Chen L, Kwon YD, Zhou T, et al. Structural basis of immune evasion at the site of CD4 attachment on HIV-1 gp120. Science. 2009;326:1123–1127. |
Supplementary materials
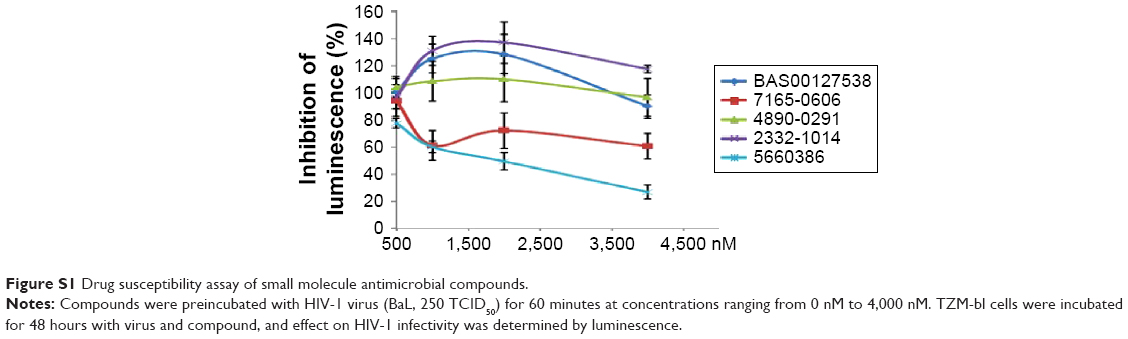 | Figure S1 Drug susceptibility assay of small molecule antimicrobial compounds. |
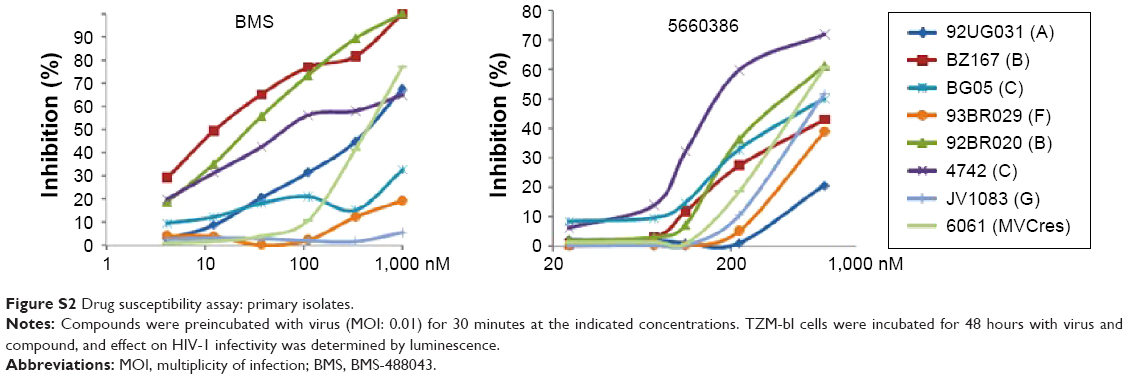 | Figure S2 Drug susceptibility assay: primary isolates. |
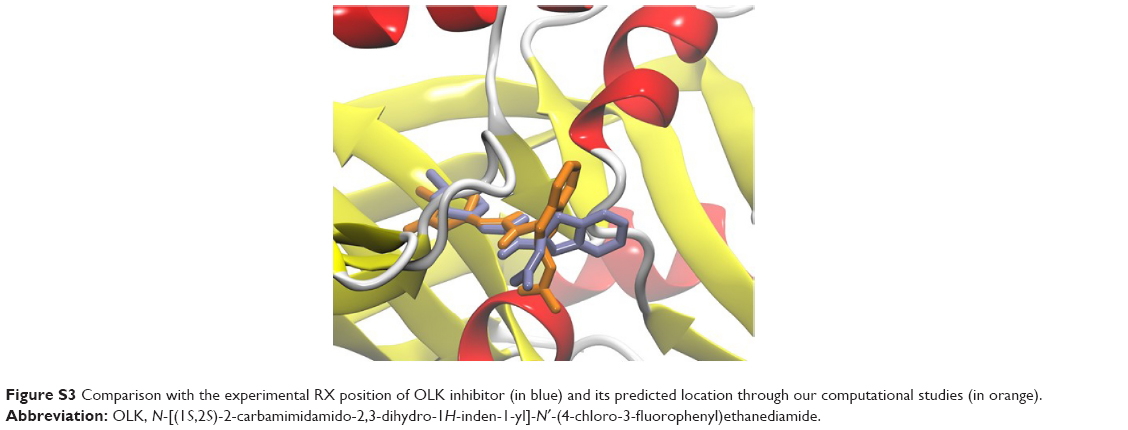 | Figure S3 Comparison with the experimental RX position of OLK inhibitor (in blue) and its predicted location through our computational studies (in orange). |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.