")
Back to Journals » Drug Design, Development and Therapy » Volume 12
A novel multifunctional anti-CEA-IL15 molecule displays potent antitumor activities
Authors Liu Y, Wang Y, Xing J, Li Y, Liu J , Wang Z
Received 23 February 2018
Accepted for publication 9 May 2018
Published 29 August 2018 Volume 2018:12 Pages 2645—2654
DOI https://doi.org/10.2147/DDDT.S166373
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Tuo Deng
Yue Liu,1,2 Yanlan Wang,1,2 Jieyu Xing,1,2 Yumei Li,1,2 Jiayu Liu,1,2 Zhong Wang1,2
1School of Pharmaceutical Sciences, 2Center for Cellular and Structural Biology, Sun Yat-Sen University, Guangzhou, People’s Republic of China
Introduction: Interleukin-15 (IL-15) is an immunomodulatory cytokine. It can activate and expand cytotoxic CD8 T lymphocytes and natural killer cells, leading to potent antitumor effects. Various forms of IL-15 are now in different stages of development for cancer immunotherapy. One of the major issues with IL-15 or IL15–IL15Rα fusion is high toxicity due to systemic activation of immune cells.
Materials and methods: In this study, we engineered a nanobody–cytokine fusion molecule, anti-CEA-IL15, in which an anti-CEA nanobody was linked to an IL15Rα–IL15 fusion. The nanobody–cytokine fusion exhibited multiple mechanisms to kill tumor cells, including promoting immune cell proliferation and directing antibody-dependent cytotoxicity against CEA-positive tumor cells.
Results: In xenograft models, anti-CEA-IL15 was localized in the tumor microenvironment and exhibited more potent antitumor activities than non-targeting IL-15, supporting potential application of this multifunctional fusion molecule in tumor immunotherapy.
Conclusion: We generated and validated a tumortargeting fusion protein, anti-CEA-IL15, which has potent cytokine activity to activate and mobilize the immune system to fight cancer cells. Such strategies may also be applied to other cytokines and tumor-targeting molecules to increase antitumor efficacy.
Keywords: immunotherapy, IL-15, nanobody, CEA, antibody–cytokine fusion
Introduction
Cytokines are key regulators of the immune system, and a number of them can activate and drive immune cells to kill tumor cells.1 Thus, much effort has been focused on the application of a variety of cytokines in cancer therapy. Among them, interleukin-2 (IL-2) has already been approved by the US Food and Drug Administration for use in metastatic melanoma and renal cell carcinoma.2,3 However, broad application of IL-2 was hindered by significant toxicity, stimulation of regulatory T cells, and activation of cell death activity.4–8
Recently, interleukin-15 (IL-15) has been reported as a potential antitumor cytokine.9 IL-15 belongs to the same cytokine family as IL-2;10,11 however, IL-15 may have more potent antitumor activities as it does not share the immune-suppressive feature with IL-2.11 Recombinant IL-15 has been clinically studied for cancer therapy,12 but has shown limited efficacy due to its short half-life. In addition, high doses are needed to achieve biological responses, but they can lead to increased toxicity.12–14
Many novel attempts have been undertaken to enhance and prolong therapeutic activity of IL-15, including gene therapy15 and engineering cells to secrete IL-15.16 Complexes of IL-15 and its soluble receptor IL-15Rα have also been extensively studied,17,18 as binding with IL-15Rα can increase IL-15 activity ~50-fold. Fusion of IL-15 to another larger protein fragment has been proposed,19 such as the Fc domain of IgG, which has been widely used to increase plasma half-life of many proteins in vivo.20–22
Given the diverse functions of IL-15, which include increasing the number of activated natural killer (NK) cells, monocytes, and granulocytes, systemic increases in IL-15 activity may conceivably lead to high toxicity.12–14 A more attractive strategy would be to target IL-15 to the tumor microenvironment, engaging immune cells specifically in the tumor microenvironment to enhance the antitumor functions of IL-15.23–28
Carcinoembryonic antigen (CEA), also called CEACAM5 or CD66e, is a heavily glycosylated protein involved in cell adhesion. CEA facilitates bacterial colonization of the intestine and protects the colon from microbial infection by binding and trapping infectious microorganisms.29–31 While exhibiting little or no expression in normal tissues,29 CEA overexpression has been observed in most lung and breast carcinomas, ~95% of gastrointestinal and pancreatic cancers,31 and the majority of colorectal cancers.32 Moreover, in normal colon tissue, CEA is only expressed on the luminal surface of the epithelium, which is inaccessible to IgG antibody. Throughout tumorigenesis, CEA expression pattern changes, and it becomes expressed on the basal and lateral membranes of tumor cells,31 making it accessible to antibody. Thus, CEA-expressing cells are an ideal target for antibody-based cancer therapy because this strategy allows avoidance of inappropriate cytotoxicity against normal tissue.33
In this study, we constructed an anti-CEA-IL15 structure by fusing an anti-CEA nanobody-Fc with an IL15Rα–IL15. This fusion protein recognized CEA-positive tumor cells and promoted proliferations of immune cells in vitro. In xenograft models, anti-CEA-IL15 was targeted to the tumor microenvironment, where it exerted potent antitumor activity. These data support further development of anti-CEA-IL15 for use in cancer immunotherapy.
Materials and methods
Antibody design and purification
To generate the recombinant protein anti-CEA-IL15, the anti-CEA nanobody34 was fused with the IgG1 Fc domain and then linked to the IL15Rα–IL15 fusion protein.35 Next, the fusion gene was cloned into the pcDNA3.1(+) vector with an IL-2 signal peptide. The plasmid was then transiently transfected into 293F cells. Anti-CEA-IL15 fusion protein was purified using a Protein-A-agarose affinity purification system. Control molecules, anti-CEA-Fc and Fc-IL15, were also expressed in 293F cells and purified using a Protein-A-agarose affinity purification system.
Cell lines and animals
SKOV3, LS174T, HT29, CHO, Mo7e, and CTLL-2 cell lines were obtained from Shanghai Cell Bank (Shanghai, People’s Republic of China). MC38 cells with stable CEA expression (MC38-CEA) were purchased from Kerafast (Boston, MA, USA). Mo7e cells were cultured in RPMI 1640 supplemented with 10% fetal bovine serum (FBS), 10 ng/mL granulocyte-macrophage colony-stimulating factor, and 1% non-essential amino acids (NEAA). CTLL-2 cells were cultured in RPMI 1640 supplemented with 20% FBS, 30 ng/mL IL-2, and 1% NEAA. SKOV3, LS174T, HT29, CHO, and MC38-CEA cells were cultured in DMEM or RPMI 1640 (Thermo Fisher Scientific, Waltham, MA, USA) with 10% HI FBS (Thermo Fisher Scientific) and 1% penicillin/streptomycin (HyClone) at 37°C with 5% CO2. Peripheral blood mononuclear cells (PBMCs) were isolated using Ficoll-Paque plus (GE health care, Chicago, IL, USA) and cultured in RPMI 1640 supplemented with 10% FBS.
Nonobese diabetic-severe combined immunodeficiency disease (NOD/SCID) mice (female, 18–22 g) were purchased from the Vital River Laboratory Animal Technology Co. Ltd. (Beijing, People’s Republic of China). C57bl/6 mice were purchased from the Animal Experiment Facility of Sun Yat-Sen University. Animals were housed in the Laboratory Animal Center, Sun Yat-Sen University under sterile and standardized environmental conditions (20°C–26°C room temperature, 40%–70% relative humidity, and 12 h light–dark rhythm).
The animal care and animal experiments were approved by the Institutional Animal Ethics Committee of Sun Yat-Sen University and were performed according to the committee’s guidelines. The use of human blood was approved by the Health and Family Planning Commission of Guangdong Province and the Institutional Animal Ethics Committee of Sun Yat-Sen University. The human blood samples were obtained from the Health and Family Planning Commission of Guangdong Province, and all donors had provided written informed consent for this study.
Flow cytometry analysis
CEA binding by anti-CEA-IL15 was analyzed using the following flow cytometry method. Exactly 1×106 cells per sample were collected by centrifugation at 1,000 rpm for 5 min and then washed with 1×PBS containing 0.2% bovine serum albumin (BSA). Cell pellets were resuspended in 100 μL of ice-cold PBS+0.1% BSA and incubated with 10 μg anti-CEA-IL15 on ice for 1 h followed by washing twice with ice-cold PBS+0.1% BSA. After washing, cells were incubated with Alexa 488-conjugated anti-human IgG1 (A11013; Thermo Fisher Scientific) for a further 1 h on ice. Cells were then washed and resuspended in 500 μL 1×PBS buffer. Flow cytometry analysis was performed on an FC500 (Beckman Coulter, Brea, CA, USA).
Mouse lymphocytes and tumor tissues were analyzed using the following flow cytometry procedure. Peripheral blood was collected from orbital vein, and tumor tissues were collected with forceps, following which they were subjected to enzymatic digestion using 0.2 mg/mL collagenase IV with 0.1 mg/mL DNase I in RPMI 1640 at 37°C for 15 min. Single cell suspensions were filtered with 70 μM nylon mesh. Cell samples from blood and tumor tissue were then incubated with corresponding antibodies at 1 h on ice. Cells were then washed and resuspended in 500 μL 1×PBS buffer. Flow cytometry analysis was performed on an FC500 (Beckman Coulter).
Cytokine-dependent cell proliferation assay
To measure cytokine-dependent cell proliferation, Mo7e and CTLL-2 cells were harvested during their logarithmic growth phase, washed twice with PBS, and incubated for 4 h with cytokine starvation in assay medium (RPMI 1640 supplemented with 10% FBS and 1% NEAA) at 37°C and 5% CO2. After incubation, the cells were collected, and a cell suspension (2×104 cells/well) was seeded immediately into wells with anti-CEA-IL15 or Fc-IL15 at different concentrations. Cells were then incubated at 37°C and 5% CO2 for 48 h, followed by application of the Cell Counting Kit-8 (Dojindo, Kumamoto, Japan) assay to quantify live cells.
Isolation of PBMCs and NK cells
Human PBMCs were prepared from healthy donors’ blood using Ficoll density centrifugation as previously described.36 NK cells were then isolated from PBMCs using an Easy Sep™ Human NK Cell Enrichment Kit according to the manufacturer’s instructions (Stem Cell Co. Ltd [Carlsbad, CA, USA], Cat#19055). Isolated NK cells were cultured in complete RPMI 1640 with 10% FBS and 1% penicillin/streptomycin at 37°C in a 5% CO2 humidified incubator prior to assays.
Immunofluorescence assay
SKOV3 and LS174T cells were cultured in 30 mm glass bottom dishes (Cellvis, Mountain View, CA, USA) to 70% confluence. Cells were then incubated with 5 μg of purified anti-CEA-IL15 at 0°C for 1 h. The cells were then washed with cold PBS and fixed with 4% paraformaldehyde. The fixed cells were incubated with goat anti-human IgG-Alexa Fluor 488 for 1 h at room temperature. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole. Cells were then washed with PBS and imaged with a Zeiss LSM710 confocal microscope.
In vitro cytotoxicity assay
In vitro cytotoxicity assays were performed as previously described.15 Briefly, tumor cells were seeded in 96-well microplates at 2,500 cells per well and incubated overnight at 37°C in the presence of 5% CO2. PBMCs or NK cells (25,000 per well) with or without anti-CEA-IL15, Fc-IL15, or anti-CEA-Fc were then mixed in growth medium and added to each well. After 72 h of culture, the supernatant was removed, and cells were washed twice with PBS. The Cell Counting Kit-8 reagent (Dojindo) was then applied. After 1–4 h of incubation, the OD450 nm was measured using a TECAN microplate reader and Fc-IL15 was used as control. Cell survival rate was calculated as [(OD450 nm treated−OD450 nm medium)/(OD450 nm untreated−OD450 nm medium)]×100%.
In vivo antitumor experiments
To study the antitumor efficacy of anti-CEA-IL15 in vivo, the human tumor cell LS174T xenograft model was constructed.34 In brief, fresh cultured LS174T cells (1×106) were mixed with freshly isolated human PBMCs (5×106) in 200 μL and subcutaneously co-implanted into the right flank of 5-week-old male NOD/SCID mice. Mice were then treated with anti-CEA-IL15 or PBS every 3 days by intraperitoneal injection for a total of four doses. Mice were weighed, and tumor growth was measured twice a week using an electro caliper.
To further test the antitumor effects of anti-CEA-IL15 in vivo, the mouse colon cancer cell line MC38 stably transfected with human CEA gene was used. Briefly, 4–6-week-old female C57bl/6 mice were injected with 1×106 MC38-CEA cells in the right flank. Mice were then treated with different dosages of anti-CEA-IL15 by intraperitoneal injection for five doses in total. Mice were weighed, and tumor growth was measured twice a week using an electro caliper.
To compare the antitumor efficacy of anti-CEA-IL15 with that of Fc-IL15, MC38-CEA, the mouse colon cancer cell line MC38 stably transfected with human CEA gene was used. After transplantation with MC38-CEA cells, the mice were treated with anti-CEA-IL15 or Fc-IL15 in 200 μL PBS every 3 days until the tumor volume reached 5–8 mm in diameter (~5–7 days). Tumor diameters were measured every 3 days with an electro caliper. Tumor volume was calculated as 1/2 (length × width2). At the end of the experiment, tumor tissues were harvested for immunohistochemical (IHC) analysis.
IHC analysis
IHC staining was carried out according to the manufacturer’s protocol. Briefly, processed sections were incubated in a moistened box with goat anti-human IgG Fc (HRP; 1:1,000; Abcam, Cambridge, UK) in PBS for 1 h at room temperature after washing with PBS. Then, diaminobenzidine was used for colorimetric development, and sections were counterstained with hematoxylin, followed by mounting with distyrene plasticizer xylene.
Results
Anti-CEA-IL15 stimulates immune cell proliferation in vitro
To target IL-15 in the tumor microenvironment and generate immune activation, anti-CEA-IL15 was constructed to contain three functional modules (Figure 1A): the IL15–IL15Rα complex to activate and expand immune cells, the human IgG1 Fc domain to prolong protein half-life and facilitate purification, and an anti-CEA single domain to specifically target tumor cells with CEA overexpression (Figure 1A). The fusion gene (anti-CEA-IL15) was cloned into the pcDNA3.1(+) vector and then transiently transfected into 293F cells. The protein was purified from the cell culture medium using Protein-A affinity chromatography. A single band of ~70 kDa was observed under reducing conditions (Figure 1B), which was in accordance with the predicted molecular weight of anti-CEA-IL15, ~68.3 kDa.
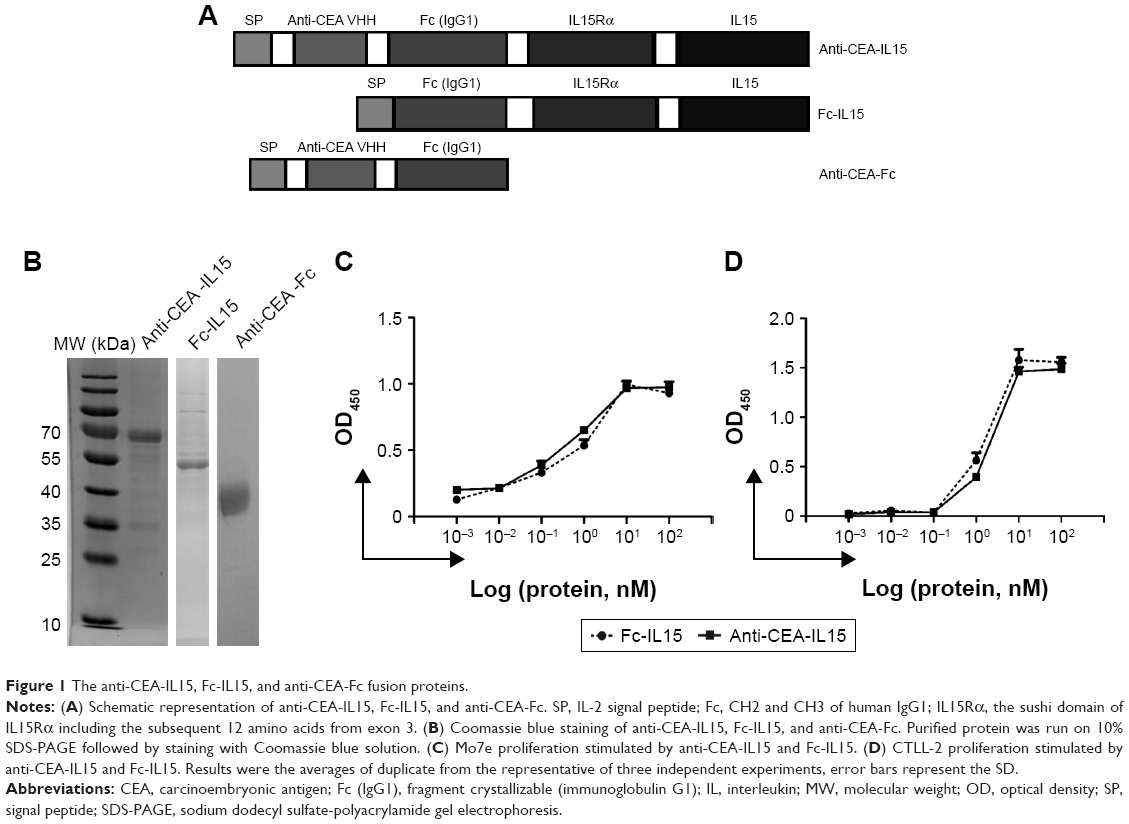 | Figure 1 The anti-CEA-IL15, Fc-IL15, and anti-CEA-Fc fusion proteins. |
Cytokine activity of anti-CEA-IL15 was then analyzed using a cytokine-dependent cell proliferation assay in Mo7e and CTLL-2 cell lines, which have previously been used to assess IL-15 activity.37 The results showed that anti-CEA-IL15 significantly stimulated the proliferation of Mo7e and CTLL-2 cell lines at a similar level to Fc-IL15,35 demonstrating that the IL15–IL15Rα complex of anti-CEA-IL15 maintains IL-15 cytokine activity (Figure 1C and D).
Anti-CEA-IL15 binds to CEA-positive tumor cells
To evaluate whether anti-CEA-IL15 can bind to CEA tumor cells, flow cytometry analysis was performed using CEA-positive LS174T and HT29 cells and CEA-negative SKOV3 and CHO cells. Based on flow cytometry analysis, anti-CEA-IL15 binds to CEA-positive cells, LS174T and HT29, but not CEA-negative cells, SKOV3 and CHO (Figure 2A), suggesting specific recognition of CEA tumor cells by anti-CEA-IL15.
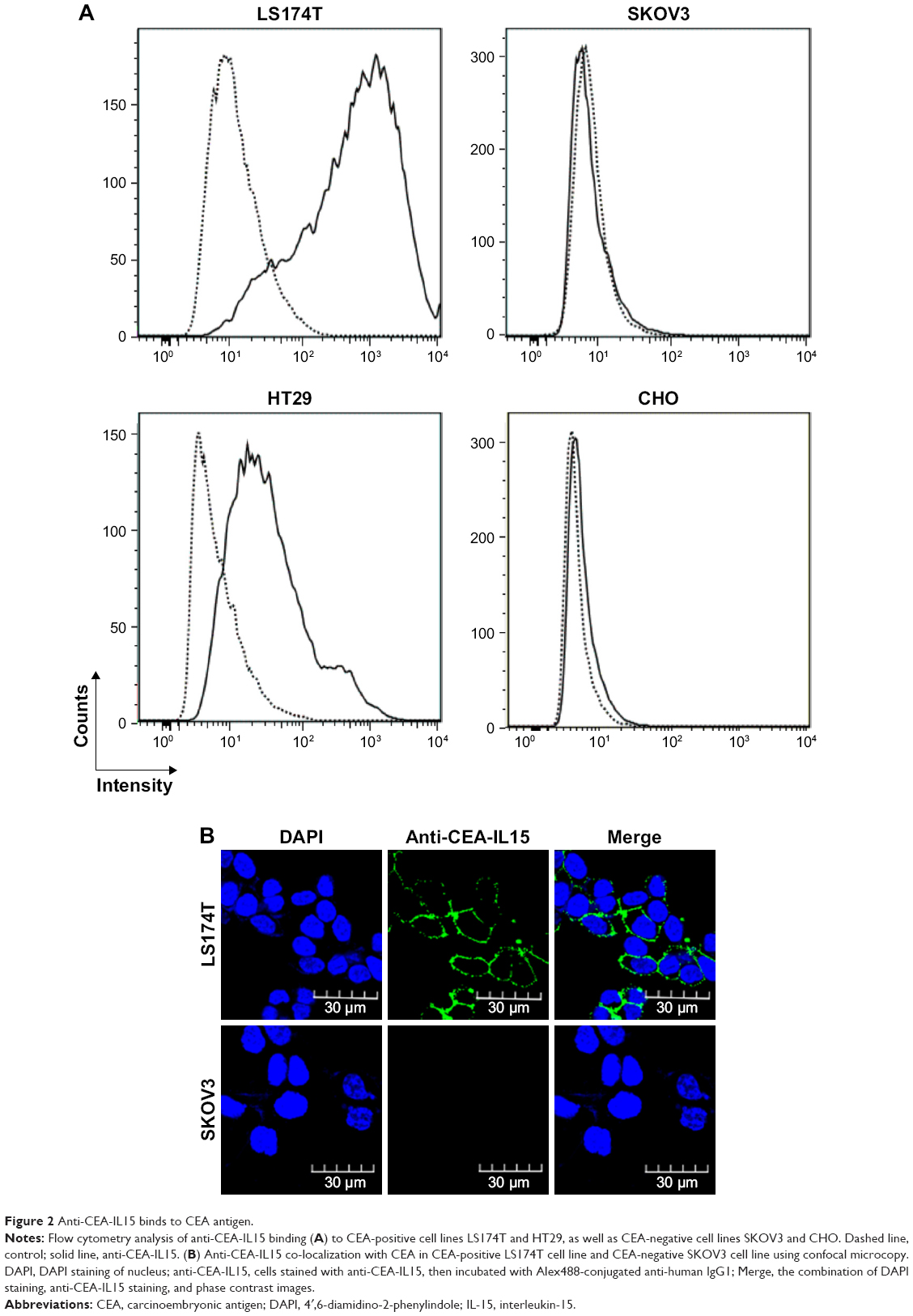 | Figure 2 Anti-CEA-IL15 binds to CEA antigen. |
Confocal microscopy analysis was also performed to further confirm that anti-CEA-IL15 binds specifically on the surface of CEA-positive cells, but not CEA-negative cells (Figure 2B). These data support that anti-CEA-IL15 can specifically recognize CEA-positive cells.
Anti-CEA-IL15 mediates potent cytotoxicity against tumor cells in vitro
One feature of many antitumor antibodies is antibody-dependent cell cytotoxicity. As anti-CEA-IL15 can bind tumor cells with full-length human IgG1, we tested whether anti-CEA-IL15 can directly mediate cell killing by immune cells. Cytotoxic assays were performed using freshly isolated non-stimulated human PBMCs (Figure 3A) or NK cells (Figure 3B) as effector cells. The control, FC-IL15, which has no anti-CEA fragment, had no cytotoxic activity against LAS174T or SKOV3 cells. In the presence of effector cells, potent cytotoxicity against CEA-positive cells, LS174T, was observed, but not against CEA-negative SKOV3 cells (Figure 3A and B), while anti-CEA-IL15 alone had no cytotoxicity against either LS174T or SKOV3 cells (Figure 3A and B).
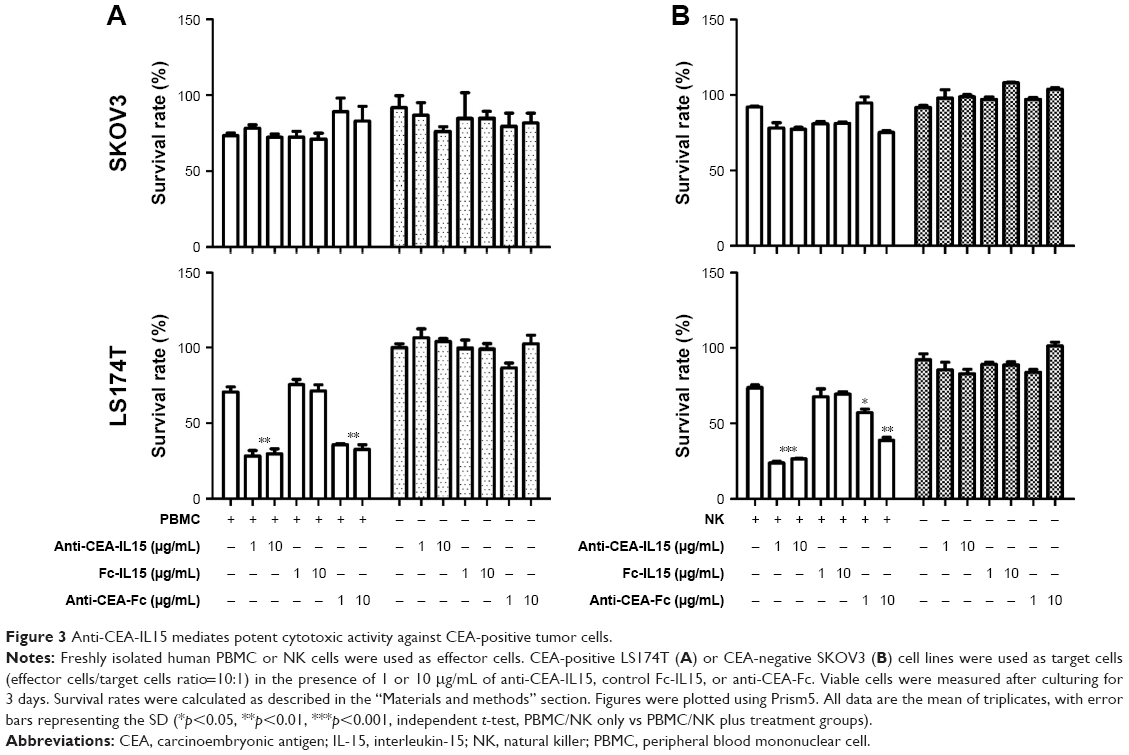 | Figure 3 Anti-CEA-IL15 mediates potent cytotoxic activity against CEA-positive tumor cells. |
To assess whether cytotoxicity against tumor cells in vitro was due to the presence of IL15–IL15Rα, anti-CEA-Fc without IL15–IL15Rα was constructed and tested for cytotoxicity (Figure 3A and B). Anti-CEA-Fc exhibited potent and specific cytotoxicity against LS174T cells, suggesting that the in vitro tumor cell cytotoxicity of anti-CEA-IL15 was conferred by anti-CEA-Fc. No difference in tumor cell killing between anti-CEA-IL15 and anti-CEA-Fc was observed under current conditions (Figure 3A and B), even though anti-CEA-IL15 can expand immune cells, suggesting that the number of immune cells was not a limiting factor under current culture conditions. These data demonstrate that in addition to activating immune cells, anti-CEA-IL15 also mediates potent direct PBMC or NK cell-dependent cytotoxicity against CEA-positive tumor cells.
Anti-CEA-IL15 targets tumor tissues and has potent antitumor activities in vivo
To evaluate the in vivo antitumor activity of anti-CEA-IL15, LS174T human cancer cells were transplanted with freshly prepared human PBMCs into NOD/SCID mice. When mice were treated only with PBS, rapid tumor growth was observed (Figure 4A); however, tumor growth was significantly inhibited by treatment with anti-CEA-IL15 in a dose-dependent manner (Figure 4A). No weight loss or apparent toxicity was observed in any mouse (data not shown). These results demonstrate that anti-CEA-IL15 inhibits tumor growth in the human tumor transplantation model.
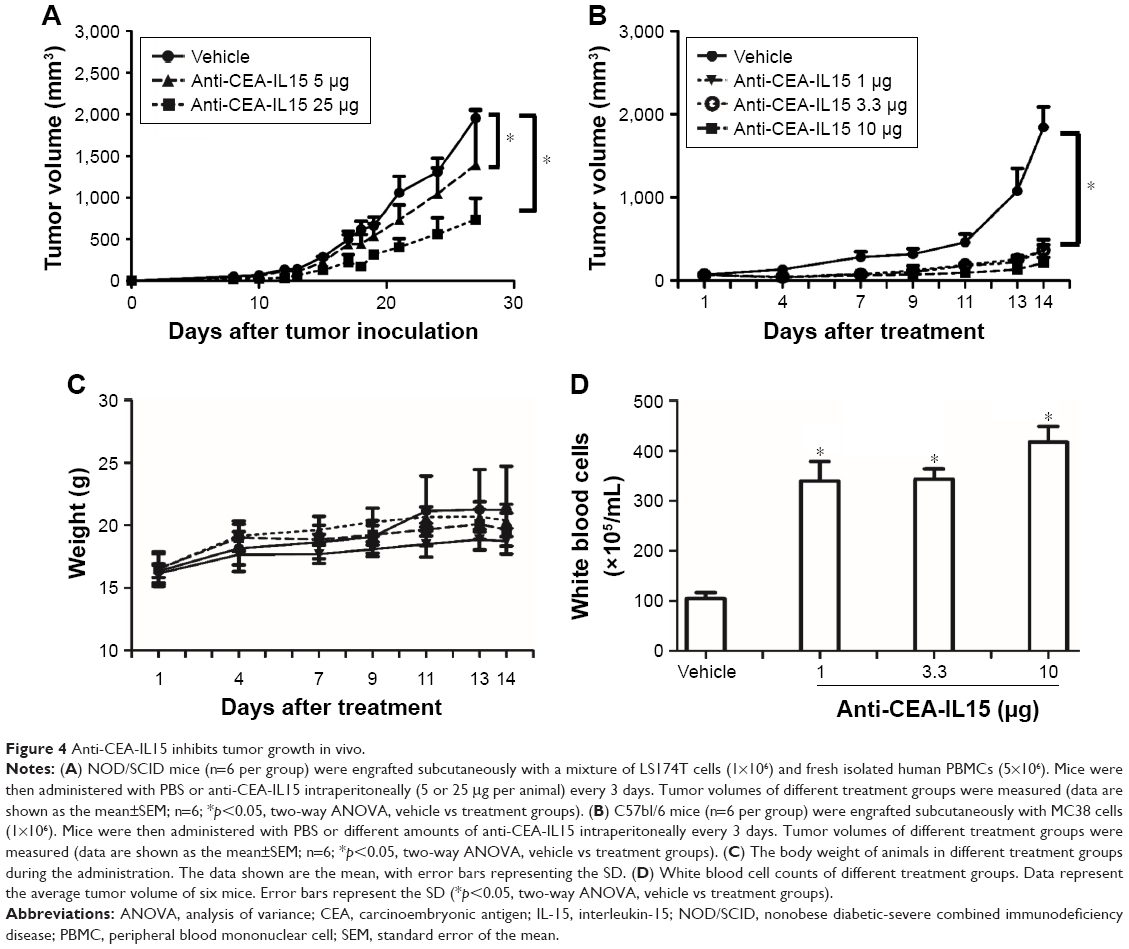 | Figure 4 Anti-CEA-IL15 inhibits tumor growth in vivo. |
To further evaluate anti-CEA-IL15 antitumor activity, a synergic model using the mouse colon cancer cell line MC38 was also evaluated. After transplanting MC38 cells in C57bl/6 mice, rapid tumor growth was observed. In contrast, when mice were treated with anti-CEA-IL15 at 1, 3.3, or 10 μg per animal, potent tumor inhibition was observed for all treatment groups, with 1 μg anti-CEA-IL15 treatments inhibiting tumor growth by over 80% (Figure 4B), indicating strong antitumor effects of anti-CEA-IL15 in vivo. No weight loss or apparent toxicity was observed in any mouse (Figure 4C). Moreover, white blood cell numbers increased ~4-fold in mice treated with anti-CEA-IL15 (Figure 4D), confirming the biological activity of IL-15. These results demonstrate that anti-CEA-IL15 activates immune cells and has potent antitumor activities in vivo, similar to previous tumor-targeting IL-15 molecule.35
In the next step, whether anti-CEA-IL15 has better antitumor efficacy than non-targeting Fc-IL15 was evaluated using MC38-CEA, in which the mouse colon cancer cell line MC38 was stably transfected with human CEA gene. Flow cytometry analysis was performed to confirm that anti-CEA-IL15 binds to MC38-CEA cells, but not non-targeting Fc-IL15 (Figure 5A). After MC38-CEA cells were transplanted into C57bl/6 mice, the mice were treated with either vehicle, anti-CEA-IL15, or non-targeting Fc-IL15. Both anti-CEA-IL15 and non-targeting Fc-IL15 exhibited significant antitumor activity. However, tumor-targeting anti-CEA-IL15 displayed more significant tumor inhibition than did non-targeting Fc-IL15, indicating the anti-CEA module of anti-CEA-IL15 leads to superior antitumor efficacy compared to Fc-IL15 in vivo (Figure 5B). No weight loss or apparent toxicity was observed in any mouse (data not shown). To analyze whether anti-CEA-IL15 was located in the tumor microenvironment, tumor tissues were fixed and processed. IHC analysis using a specific anti-human IL-15 antibody was performed. The IHC analysis confirmed that anti-CEA-IL15, not Fc-IL15, was localized to the tumor tissue (Figure 5C). These results demonstrated that anti-CEA-IL15 was targeted to the tumor microenvironment and enhanced the antitumor functions of IL-15.
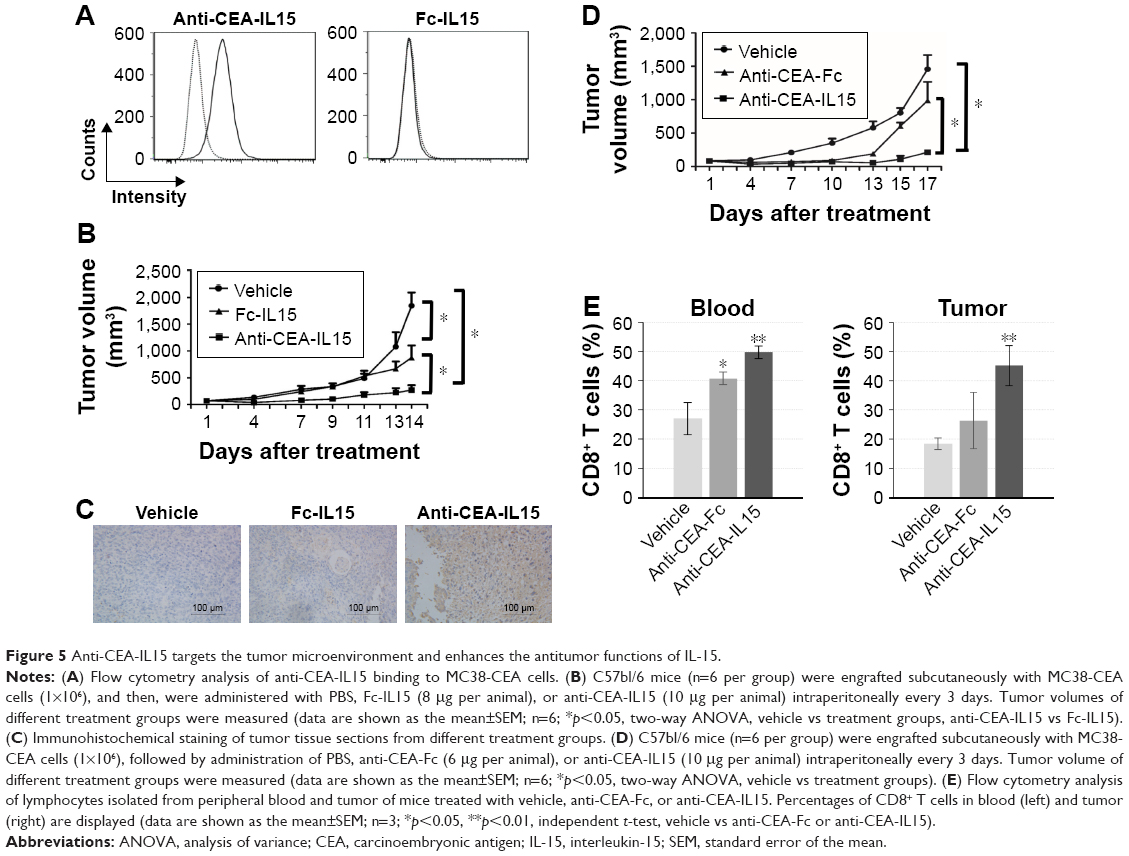 | Figure 5 Anti-CEA-IL15 targets the tumor microenvironment and enhances the antitumor functions of IL-15. |
To further study whether the immune cell proliferation and activation conferred by IL-15 is important for anti-CEA-IL15 function, the same molar concentration of anti-CEA-Fc (6 μg per animal) and anti-CEA-IL15 (10 μg per animal) was used to treat mice transplanted with MC38-CEA (Figure 5D). Potent antitumor proliferation was observed for anti-CEA-IL15 (Figure 5D). Only modest tumor inhibition was observed with the same dosage of anti-CEA-Fc (Figure 5D), suggesting that IL-15 is important for the antitumor function of anti-CEA-IL15. An increase of CD8+ T cells in peripheral blood and tumor was observed in mice treated with anti-CEA-IL15 (Figure 5E). All the data suggested anti-CEA-IL15 can stimulate immune cells and have potent antitumor activities.
Discussion
In this study, we utilized a tumor-targeting cytokine fusion, anti-CEA-IL15, as a potential immunotherapeutic agent for CEA-positive tumors. The potent antitumor activity of anti-CEA-IL15 supported anti-CEA-IL15 as a promising anticancer therapeutic.
Cytokines play important roles in regulating the immune system, including mediating antitumor immune responses.1 Among cytokines, IL-15 has been extensively studied as a promising antitumor candidate;1,2 however, IL-15 has a short plasma half-life.13 In addition, high dosages of IL-15 are required to achieve biological antitumor responses in vivo, which are likely to induce toxic side effects.13 To reduce the toxicity of IL-15, localizing IL-15 selectively within tumors is desired to reduce toxicity. Thus, a tumor microenvironment targeting IL-15 using RGD peptide was constructed and previously shown to exhibit strong antitumor activity.35 In this study, we constructed anti-CEA-IL15, a targeted cytokine fusion that comprises 1) IL15Rα–IL15 linked with a GGGGS peptide to mimic the physiological trans-presentation of IL-15, 2) an Fc domain to enhance protein half-life, and 3) an anti-CEA single domain antibody to lead the fusion protein to tumor cells with overexpression of CEA.
In addition to IL-15, many tumor-targeting cytokine fusions have been studied to minimize dose-limiting systemic toxicities and enhance the efficacy of these cytokines.38,39 Most of those cytokine fusions employed IL-2, tumor necrosis factor, or IL-12 to activate immune cells. Some of them have shown promising results in vitro and now in clinical trials.38 Compared to these cytokines, the function of IL-15 is more restricted to NK and CD8 cytotoxic T cells, potentially reducing toxicity. We also employed the anti-CEA-VHH single-domain antibody or nanobody,40 which enhances tumor targeting and reduces toxicity. Furthermore, using single-domain antibody or nanobody (25 kDa) rather than conventional IgG, the molecular weight of anti-CEA-IL15 was reduced from ~200 to ~70 kDa, which may improve tumor tissue penetration.
Anti-CEA-IL15 exhibited strong immune cell proliferative activation in vitro (Figure 1). Proliferation activity was also observed in vivo with the expansion of white blood cells (Figure 4), suggesting that the IL15–IL15α portion of anti-CEA-IL15 causes activation and expansion of immune cells. Interestingly, in contrast to the previous tumor-targeting PFC1, which only binds to integrins on tumor cells or within the tumor microenvironment, anti-CEA-IL15 exerted direct cytotoxic activity against tumor cells (Figure 3), similar to the anti-CEA bispecific antibodies.34,40 These data suggest that anti-CEA-IL15 possesses multiple functions to inhibit tumor growth, including expansion and activation of immune cells, localization to the tumor microenvironment, and direct induction of tumor cytotoxicity.
Conclusion
In conclusion, we generated and validated a tumor-targeting fusion protein, anti-CEA-IL15, which has potent cytokine activity to activate and mobilize the immune system to fight cancer cells. Such strategies may also be applied to other cytokines and tumor-targeting molecules to increase antitumor efficacy.
Acknowledgment
This project was financially supported by the Department of Science and Technology of Guangdong Province (People’s Republic of China) (2016A050503028).
Disclosure
The authors report no conflicts of interest in this work.
References
Smyth MJ, Cretney E, Kershaw MH, Hayakawa Y. Cytokines in cancer immunity and immunotherapy. Immunol Rev. 2004;202(1):275–293. | ||
Bhatia S, Tykodi SS, Thompson JA. Treatment of metastatic melanoma: an overview. Oncology (Williston Park). 2009;23(6):488–496. | ||
Noble S, Goa KL. Aldesleukin (recombinant interleukin-2). BioDrugs. 1997;7(5):394–422. | ||
Antony PA, Paulos CM, Ahmadzadeh M, et al. Interleukin-2-dependent mechanisms of tolerance and immunity in vivo. J Immunol. 2006;176(9):5255–5266. | ||
Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005;6(11):1142–1151. | ||
Oh S, Berzofsky JA, Burke DS, Waldmann TA, Perera LP. Coadministration of HIV vaccine vectors with vaccinia viruses expressing IL-15 but not IL-2 induces long-lasting cellular immunity. Proc Natl Acad Sci U S A. 2003;100(6):3392–3397. | ||
Waldmann TA. The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat Rev Immunol. 2006;6(8):595–601. | ||
Waldmann TA, Dubois S, Tagaya Y. Contrasting roles of IL-2 and IL-15 in the life and death of lymphocytes: implications for immunotherapy. Immunity. 2001;14(2):105–110. | ||
Comes A, Di Carlo E, Musiani P, et al. IFN-gamma-independent synergistic effects of IL-12 and IL-15 induce anti-tumor immune responses in syngeneic mice. Eur J Immunol. 2002;32(7):1914–1923. | ||
Fehniger TA, Cooper MA, Caligiuri MA. Interleukin-2 and interleukin-15: immunotherapy for cancer. Cytokine Growth Factor Rev. 2002;13(2):169–183. | ||
Ma A, Koka R, Burkett P. Diverse functions of IL-2, IL-15, and IL-7 in lymphoid homeostasis. Annu Rev Immunol. 2006;24:657–679. | ||
Conlon KC, Lugli E, Welles HC, et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J Clin Oncol. 2015;33(1):74–82. | ||
Kobayashi H, Carrasquillo JA, Paik CH, Waldmann TA, Tagaya Y. Differences of biodistribution, pharmacokinetics, and tumor targeting between interleukins 2 and 15. Cancer Res. 2000;60(13):3577–3583. | ||
Zamai L, Ponti C, Mirandola P, et al. NK cells and cancer. Adv Cancer Res. 2007;178(7):4011–4016. | ||
Yu YL, Wei CW, Chen YL, Chen MH, Yiang GT. Immunotherapy of breast cancer by single delivery with rAAV2-mediated interleukin-15 expression. Int J Oncol. 2010;36(2):365–370. | ||
Vera M, Razquin N, Prieto J, Melero I, Fortes P, Gonzalez-Aseguinolaza G. Intratumoral injection of dendritic cells transduced by an SV40-based vector expressing interleukin-15 induces curative immunity mediated by CD8+ T lymphocytes and NK cells. Mol Ther. 2005;12(5):950–959. | ||
Rubinstein MP, Kovar M, Purton JF, et al. Converting IL-15 to a superagonist by binding to soluble IL-15R{alpha}. Proc Natl Acad Sci U S A. 2006;103(24):9166–9171. | ||
Stoklasek TA, Schluns KS, Lefrancois L. Combined IL-15/IL-15Ralpha immunotherapy maximizes IL-15 activity in vivo. J Immunol. 2006;177(9):6072–6080. | ||
Kaspar M, Trachsel E, Neri D. The antibody-mediated targeted delivery of interleukin-15 and GM-CSF to the tumor neovasculature inhibits tumor growth and metastasis. Cancer Res. 2007;67(10):4940–4948. | ||
Czajkowsky DM, Hu J, Shao Z, Pleass RJ. Fc-fusion proteins: new developments and future perspectives. EMBO Mol Med. 2012;4(10):1015–1028. | ||
Kontermann RE. Antibody-cytokine fusion proteins. Expert Opin Biol Ther. 2012;526(2):194–205. | ||
Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7(9):715–725. | ||
Keilholz U, Szelenyi H, Siehl J, Foss HD, Knauf W, Thiel E. Rapid regression of chemotherapy refractory lymphocyte predominant Hodgkin’s disease after administration of rituximab (anti CD 20 mono-clonal antibody) and interleukin-2. Leuk Lymphoma. 1999;35(5–6):641–642. | ||
Moga E, Alvarez E, Canto E, et al. NK cells stimulated with IL-15 or CpG ODN enhance rituximab-dependent cellular cytotoxicity against B-cell lymphoma. Exp Hematol. 2008;36(1):69–77. | ||
Roberti MP, Barrio MM, Bravo AI, et al. IL-15 and IL-2 increase cetuximab-mediated cellular cytotoxicity against triple negative breast cancer cell lines expressing EGFR. Breast Cancer Res Treat. 2011;130(2):465–475. | ||
Shak S. Overview of the trastuzumab (Herceptin) anti-HER2 monoclonal antibody clinical program in HER2-overexpressing metastatic breast cancer. Herceptin Multinational Investigator Study Group. Semin Oncol. 1999;26(4 Suppl 12):71–77. | ||
Soiffer RJ, Chapman PB, Murray C, et al. Administration of R24 monoclonal antibody and low-dose interleukin 2 for malignant melanoma. Clin Cancer Res. 1997;3(1):17–24. | ||
Sondel PM, Hank JA. Combination therapy with interleukin-2 and antitumor monoclonal antibodies. Cancer J Sci Am. 1997;3(Suppl 1):S121–S127. | ||
Beauchemin N, Benchimol S, Cournoyer D, Fuks A, Stanners CP. Isolation and characterization of full-length functional cDNA clones for human carcinoembryonic antigen. Mol Cell Biol. 1987;7(9):3221–3230. | ||
Oikawa S, Nakazato H, Kosaki G. Primary structure of human carcinoembryonic antigen (CEA) deduced from cDNA sequence. Biochem Biophys Res Commun. 1987;142(2):511–518. | ||
Hammarstrom S. The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and malignant tissues. Semin Cancer Biol. 1999;9(2):67–81. | ||
Wagener C, Petzold P, Kohler W, Totovic V. Binding of five monoclonal anti-CEA antibodies with different epitope specificities to various carcinoma tissues. Int J Cancer. 1984;33(4):469–475. | ||
Khare PD, Shao-Xi L, Kuroki M, et al. Specifically targeted killing of carcinoembryonic antigen (CEA)-expressing cells by a retroviral vector displaying single-chain variable fragmented antibody to CEA and carrying the gene for inducible nitric oxide synthase. Cancer Res. 2001;61(1):370–375. | ||
Li L, He P, Zhou C, et al. A novel bispecific antibody, S-Fab, induces potent cancer cell killing. J Immunother. 2015;38(9):350–356. | ||
Chen S, Huang Q, Liu J, et al. A targeted IL-15 fusion protein with potent anti-tumor activity. Cancer Biol Ther. 2015;16(9):1415–1421. | ||
Behar G, Siberil S, Groulet A, et al. Isolation and characterization of anti-FcgammaRIII (CD16) llama single-domain antibodies that activate natural killer cells. Protein Eng Des Sel. PEDS. 2008;21(1):1–10. | ||
Kermer V, Baum V, Hornig N, Kontermann RE, Muller D. An antibody fusion protein for cancer immunotherapy mimicking IL-15 trans-presentation at the tumor site. Mol Cancer Ther. 2012;11(6):1279–1288. | ||
Neri D, Sondel PM. Immunocytokines for cancer treatment: past, present and future. Curr Opin Immunol. 2016;40:96–102. | ||
Young PA, Morrison SL, Timmerman JM. Antibody-cytokine fusion proteins for treatment of cancer: engineering cytokines for improved efficacy and safety. Semin Oncol. 2014;41(5):623–636. | ||
Dong B, Zhou C, He P, et al. A novel bispecific antibody, BiSS, with potent anti-cancer activities. Cancer Biol Ther. 2016;17(4):364–370. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.