")
Back to Journals » Drug Design, Development and Therapy » Volume 9
A bioinformatic and mechanistic study elicits the antifibrotic effect of ursolic acid through the attenuation of oxidative stress with the involvement of ERK, PI3K/Akt, and p38 MAPK signaling pathways in human hepatic stellate cells and rat liver
Authors Li W, Shi F, Zhou Z, Li B, Zhang K, Zhang X, Ouyang C, Zhou S, Zhu X
Received 25 March 2015
Accepted for publication 29 April 2015
Published 31 July 2015 Volume 2015:9 Pages 3989—4104
DOI https://doi.org/10.2147/DDDT.S85426
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Wenhua He,1,* Feng Shi,1,* Zhi-Wei Zhou,2,* Bimin Li,1 Kunhe Zhang,1 Xinhua Zhang,1 Canhui Ouyang,1 Shu-Feng Zhou,2 Xuan Zhu1
1Department of Gastroenterology, the First Affiliated Hospital of Nanchang University, Nanchang, Jiangxi, People’s Republic of China; 2Department of Pharmaceutical Sciences, College of Pharmacy, University of South Florida, Tampa, FL, USA
*These authors contributed equally to this work
Abstract: NADPH oxidases (NOXs) are a predominant mediator of redox homeostasis in hepatic stellate cells (HSCs), and oxidative stress plays an important role in the pathogenesis of liver fibrosis. Ursolic acid (UA) is a pentacyclic triterpenoid with various pharmacological activities, but the molecular targets and underlying mechanisms for its antifibrotic effect in the liver remain elusive. This study aimed to computationally predict the molecular interactome and mechanistically investigate the antifibrotic effect of UA on oxidative stress, with a focus on NOX4 activity and cross-linked signaling pathways in human HSCs and rat liver. Drug–drug interaction via chemical–protein interactome tool, a server that can predict drug–drug interaction via chemical–protein interactome, was used to predict the molecular targets of UA, and Database for Annotation, Visualization, and Integrated Discovery was employed to analyze the signaling pathways of the predicted targets of UA. The bioinformatic data showed that there were 611 molecular proteins possibly interacting with UA and that there were over 49 functional clusters responding to UA. The subsequential benchmarking data showed that UA significantly reduced the accumulation of type I collagen in HSCs in rat liver, increased the expression level of MMP-1, but decreased the expression level of TIMP-1 in HSC-T6 cells. UA also remarkably reduced the gene expression level of type I collagen in HSC-T6 cells. Furthermore, UA remarkably attenuated oxidative stress via negative regulation of NOX4 activity and expression in HSC-T6 cells. The employment of specific chemical inhibitors, SB203580, LY294002, PD98059, and AG490, demonstrated the involvement of ERK, PI3K/Akt, and p38 MAPK signaling pathways in the regulatory effect of UA on NOX4 activity and expression. Collectively, the antifibrotic effect of UA is partially due to the oxidative stress attenuating effect through manipulating NOX4 activity and expression. The results suggest that UA may act as a promising antifibrotic agent. More studies are warranted to evaluate the safety and efficacy of UA in the treatment of liver fibrosis.
Keywords: ursolic acid, liver fibrosis, NADPH oxidase, ROS, DDI-CPI, DAVID
Introduction
Liver fibrosis remains the major cause of morbidity and mortality worldwide. It causes several notorious complications, including ascites, portal hypertension, encephalopathy, and liver failure, and it accelerates the risk of hepatocellular carcinoma, placing a substantial burden to individual, society, and health care system.1,2 The liver fibrosis-driven chronic liver disease and cirrhosis cause 36,427 deaths and with a ratio of 11.5/100,000 in 2013 in USA.1 This is a complicated etiology of hepatic fibrosis. Viral infection is the most common contributing factor to liver fibrosis affecting 1%–2% of the US population,3 and liver fibrosis resultant cirrhosis is projected to reach 45% of those infected with hepatitis C virus patients in 2030.4 Hepatitis B virus is the main type of hepatitis virus in the People’s Republic of China with a substantial contribution to the incidence of liver fibrosis.5,6 Moreover, nonalcoholic fatty liver disease and nonalcoholic steatohepatitis are also important causes of liver fibrosis, and over 20% of patients with nonalcoholic steatohepatitis progress to cirrhosis worldwide.7 In addition, other etiologies of liver fibrosis and its late-stage liver injury include alcohol-induced disease, drug-induced toxicity, other liver infections (eg, schistosomiasis), immune-mediated liver diseases (eg, autoimmune hepatitis), metabolic disorders (eg, lipid, glycogen, or metal storage disorders), and cholestasis (eg, secondary biliary cirrhosis).
To date, the molecular mechanisms that underlie the development of liver fibrosis have been extensively investigated, including epithelial to mesenchymal transition and inflammatory response.8–10 Notably, increasing evidence suggests that oxidative stress has been implicated in the pathogenesis of liver fibrosis, with the intracellular accumulation of excessive reactive oxygen species (ROS), and the compelling evidence shows the involvement of ROS in the development of liver fibrosis with a regulatory role in a variety of cellular processes.7,10–13 However, the specific targets and signaling pathways have not been fully mapped yet, and the sources of ROS have not yet been conclusively determined in the pathogenesis of liver fibrosis. There are several enzymatic sources of ROS, including NADPH oxidase (NOX) family of oxidoreductases, nitric oxide synthases, xanthine oxidase, and cytochrome P450, of which NOX is the most important ROS-generating enzyme in both plants and animals, especially in mammals. In the liver, NOX has been involved in fibrogenesis. In particular, NOX4 is one of seven NOX isoforms and is the most widely distributed isoform.11 It has been reported that a functionally active form of NOX is expressed in hepatic stellate cells (HSCs) and that NOX-generated ROS serve as a second messenger for profibrogenic factor signal transduction in HSCs. However, regulation of NOX in HSCs remains largely elusive.
Recently, it has been proposed that antioxidants hold promise as potential antifibrotic therapies due to the ROS-attenuating effect.12,13 Ursolic acid (UA), a natural pentacyclic triterpenoid carboxylic acid, is the major component of certain traditional medicinal herbs and possesses a wide range of biological functions, such as antioxidative, anti-inflammatory, and anticancer activities.14 UA exerts a protective effect against ethanol-induced toxicity in isolated rat hepatocytes and ethanol-mediated experimental liver damage in rats.15,16 Our previous studies showed that UA significantly inhibited the proliferation of HSCs and induced apoptosis in vitro.17 Moreover, Steinkamp-Fenske et al18 showed that UA downregulated the expression level of NOX4 and suppressed ROS generation in human endothelial cells. However, there is a lack of study on the antifibrotic effect of UA, and the underlying mechanisms of the antifibrotic effect of UA are largely unknown.
Therefore, the present study aimed to evaluate the molecular targets of UA and analyze the molecular interactome of UA using computational and bioinformatic approaches and, subsequently, examine the antifibrotic effect of UA and delineate the underlying mechanisms in HSCs and Sprague Dawley rats.
Materials and methods
Prediction of the interactome of UA and pathway analysis by molecular docking and bioinformatic approach
The prediction of interactome of UA was performed using the drug–drug interaction via chemical–protein interactome (DDI-CPI) tool (http://cpi.bio-x.cn/ddi/) as previously described.19 DDI-CPI is a web-based server that can be used to predict DDI via CPI.20,21 In brief, protein targets were obtained from a third-party protein structure database named PDBbind (http://sw16.im.med.umich.edu/databases/pdbbind/index.jsp), which was based on the contents of Protein Data Bank (PDB).22 There are a total of 1,780 PDB entries of human proteins available in PDBbind, and a total of 301 nonredundant PDBs corresponding to 353 ligand-binding pockets were identified from it, with 86% of which have resolutions <2.5 Å. The docking boxes for each of the pockets were defined by expanding the circumscribed cube of the pocket with a margin of 8 Å at six directions (up, down, front, back, left, and right). For the preparation of the UA, the 2D structure of the UA was downloaded from PubChem. The hydrogen and Gasteiger charge were added, and the PDB file was converted into mol2 format using VEGA ZZ. The docking program AutoDock 4.2 was used to dock the prepared UA molecule into all 353 pockets, generating a score vector of 353 dimensions. Z′-scores were then calculated using the methodologies we applied previously.20,23,24 Here, an empirical threshold of −0.6 of the Z′-score was set to indicate that the binding of UA toward this target was likely to be true.
Pathway analysis by Database for Annotation, Visualization, and Integrated Discovery
Following the computational target prediction, the Database for Annotation, Visualization, and Integrated Discovery version 6.7 (DAVID, http://david.abcc.ncifcrf.gov/) was employed to interpret the biological function of the potential targets of UA derived from DDI-CPI.19–21 The protein IDs of these targets from UniProtKB, NCBI, and other sources were converted into gene lists by using the Gene ID Conversion Tool in DAVID. The DAVID database adds biological function annotation including gene ontology, pathway, protein–protein interactions, functional groups of genes (ie, clustering), and disease association derived from main public data sources. The genes of interest were visualized using BioCarta and Kyoto encyclopedia of Genes and Genomes (KEGG) pathway maps. The high classification stringency was selected for functional annotation clustering. Enrichment scores and Fisher’s exact test P-values (and corresponding false discovery rate) were then calculated to identify which functional-related gene groups were significantly enriched in the target list. These significant enriched gene groups could provide clues on how UA interacts with molecular targets in a systematic way.
Chemicals and reagents
Recombinant rat leptin was purchased from Peprotech Inc. (Rocky Hill, NJ, USA). UA, diphenyleneiodonium (DPI), dimethyl sulfoxide (DMSO), Thiazolyl blue tetrazolium bromide (MTT) and protease inhibitor and phosphatase inhibitor cocktails were purchased from Sigma-Aldrich Co., (St Louis, MO, USA). Dulbecco’s Modified Eagle’s Medium (DMEM), fetal bovine serum, and AG490 were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Antioxidant N-acetyl-L-cysteine (NAC) was purchased from Solarbio Inc. (Beijing, People’s Republic of China). PD98059, an extracellular signal-regulated kinase (ERK) inhibitor, was purchased from Promega Corporation (Fitchburg, WI, USA). SB203580, a p38 mitogen-activated protein kinase (p38 MAPK) inhibitor, LY294002, a phosphoinositide 3-kinase (PI3K) inhibitor, and the ROS assay kit (which contains 2′,7′-dichlorofluorescin diacetate [DCF-DA]) were purchased from Beyotime Inc. (Jiangsu, People’s Republic of China). p47phox antibody was purchased from Bioworld Inc. (St Louis Park, MN, USA); gp91phox, p22phox, and Rac1 antibodies were purchased from Abcam Inc. (Cambridge, MA, USA). p67phox, PI3K (p110α), protein kinase B (Akt), phosphorylated (p)-Akt, p-ERK1/2, p-p38 MAPK, and p38 MAPK antibodies were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). TIMP metallopeptidase inhibitor 1 (TIMP-1) and matrix metalloproteinase-1 (MMP-1) antibodies were purchased from PL Laboratories Inc. (Port Moody, British Columbia, Canada). The α-smooth muscle actin (α-SMA) antibody was purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA), and the β-Actin antibody and goat anti-rabbit secondary antibodies were purchased from Zhongshan Golden Bridge Biotechnology Co., Ltd. (Beijing, People’s Republic of China).
Cell culture and treatment
The immortalized rat liver stellate cell line (HSC-T6) was kindly provided by Dr L-M Xu, Shanghai University of Chinese Traditional Medicine (Shanghai, People’s Republic of China). These cells exhibit the phenotype that most closely resembles primary rat stellate cells.25 HSC-T6 cells were cultured in DMEM supplemented with 10% fetal bovine serum, penicillin (120 μg/mL), and streptomycin (100 μg/mL) at a humidified atmosphere of 5% CO2 at 37°C. The culture medium was replaced with serum-free DMEM (serum starvation) for 24 hours before the start of the experiments.
Animals and experimental design
A total of 32 male Sprague Dawley rats (weighing between 180 g and 200 g) were experimented in the present study. All animals were housed in plastic cages containing wood shaving and cotton bedding and maintained in a room at 22°C–25°C with a 12-hour day/night cycle and had free access to standard laboratory diet and water. The animal study protocol was approved by the Institutional Animal Care and Use Committee of the First Affiliated Hospital of Nanchang University. Dimethylnitrosamine (DMN) and UA were freshly dissolved in saline at predetermined concentrations.
A liver fibrosis model was established in rats by administering 1% DMN at a dosage of 1 mL/kg body weight by intraperitoneal injection for 3 consecutive days per week for 4 weeks. At week 5, the DMN-treated rats with liver fibrosis were randomly divided into three groups with or without UA treatment for another 4-week treatment. The UA groups were administered by intraperitoneal injections at dosages of 20 mg/kg (UA-2) and 40 mg/kg (UA-3) per day. The DMN group received DMN alone for 8 weeks. The vehicle group was treated with a volume of saline that was equivalent to the volume used per day in UA groups. The rats were euthanized by saline perfusion and exsanguinated via the inferior vena cava 1 day after the last treatment. The liver and blood samples were collected and stored at −80°C for further analysis. During the period of experiment, if the weight loses 10%, mice will be euthanized. Also, the mice will be immediately euthanatized if other symptoms such as pale mucous membranes, shivering, failure to respond to stimuli, piloerection, matted hair coat, soiled anogenital/vent area, and vocalization are observed.
MTT assay
To examine the effect of UA on the proliferation of HSC-T6 cells, the MTT assay was performed. Briefly, HSC-T6 cells were seeded in to 96-well plates with medium containing leptin at a confluence of ~60% following a 24-hour starvation. The cells were treated with UA (50 μM), AG490 (50 μM), or DPI (20 μM) for 12 hours, 24 hours, or 48 hours, respectively. After the treatment, a volume of 10 μL of thiazolyl blue tetrazolium bromide (MTT) (5 mg/mL) was added and incubated for an additional 4 hours. Then, the medium was carefully aspirated, and the 100 μL of DMSO was added to dissolve the formazan particle. The absorption intensity was measured using a microplate reader (Bio-Rad 550; Bio-Rad Laboratories Inc., Hercules, CA, USA) at 490 nm.
Histological evaluation
To examine the effect of UA on the general characterization, inflammatory cell infiltration, and disposition of connective tissue in rat liver, the hematoxylin and eosin and van Gieson staining were performed.
Determination of serum maleic dialdehyde and superoxide dismutase levels
In order to assess the antioxidative effect of UA in rats, the blood level of maleic dialdehyde (MDA) and superoxide dismutase (SOD) activity were examined using the thiobarbituric acid method.
Measurement of intracellular ROS level
To further examine the antioxidative effect of UA in HSC-T6 cells, the intracellular ROS level was determined using flow cytometry by the oxidation-sensitive probe DCF-DA as previously described.26,27 DCF-DA, a ROS probe that undergoes intracellular deacetylation followed by ROS-mediated oxidation to a fluorescent DCF, was used to measure ROS generation in the cytoplasm and cellular organelles.28,29 HSC-T6 cells were treated with UA (50 μM), NAC (10 mM), DPI (20 μM), or AG490 (50 μM) in the absence or presence of leptin. After the treatment, cells were incubated with DCF-DA (10 μM) for 20 minutes. Then, cellular fluorescence intensity was measured using a FACScan flow cytometer (Becton Dickinson, San Jose, CA, USA) at excitation and emission wavelengths of 488 nm and 525 nm, respectively. The intracellular ROS level was calculated as a percentage of DCF fluorescence intensity relative to the vehicle controls (untreated HSC-T6 cells).
Determination of NOX activity
Following the examination of ROS generation in HSC-T6 cells, the NOX activity was determined to address the regulating effect of UA on the enzymatic source of ROS in HSC-T6 cells as previously described with some modifications.27,30,31 Briefly, HSC-T6 cells were incubated in the culture medium in the absence or presence of leptin and treated with UA (50 μM), NAC (10 mM), DPI (20 μM), or AG490 (50 μM). After the treatment, the cells were harvested by trypsinization, pelleted by centrifugation at 2,500× g for 5 minutes at 4°C and resuspended in PBS. Subsequently, the cells were incubated with 250 μM of NADPH. NADPH consumption was monitored by the decrease in absorbance at λ=340 nm for 10 minutes. For the specific analysis of NOX activity, the rate of NADPH consumption specifically inhibited by DPI was measured by pretreated with 10 μM of DPI for 30 minutes. An aliquot of cells was lysed by adding sodium dodecyl sulfate, and the protein concentration of the cell lysate was determined. The absorption extinction coefficient used to calculate the amount of NADPH consumed was 6.22 mM−1 cm−1. The data of NOX activity were expressed as picomol per liter of substrate per minute per milligram of protein.
Western blotting analysis
Whole-cell extracts were obtained using Triton lysis buffer that contained protease inhibitor and phosphatase inhibitor cocktails. Liver extracts were obtained in modified radioimmunoprecipitation buffer. The proteins were loaded and separated by sodium dodecyl sulfate/polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane. Then, the membrane was blocked at room temperature for 1 hour with 5% nonfat milk in Tris-buffered saline with Tween (TBST), followed by the incubation with indicated primary antibodies overnight at 4°C. Next, the membranes were washed and incubated with the corresponding secondary antibody conjugated to horseradish peroxidase (HRP) at room temperature for 1 hour. Visualization was performed using Bio-Rad ChemiDoc™ XRS system (Hercules, CA, USA) with enhanced chemiluminescence substrate, and the blots were analyzed using Image Lab 3.0 (Bio-Rad Laboratories Inc., Hercules, CA, USA). Protein level was normalized to the matching densitometric value of internal control.
Reverse transcription polymerase chain reaction
Total RNA was extracted using TRNzol A+ total reagent (Tiangen Biotech, Beijing, People’s Republic of China) and subject to reverse transcription with dT15-oligonucleotide and Moloney Murine Leukemia Virus (M-MLV) reverse transcriptase (Promega Corporation, Fitchburg, WI, USA). The primers for type I collagen and β-Actin used in the reverse transcription polymerase chain reaction are shown in Table 1. The mRNA levels of the type I collagen gene were normalized to the β-Actin mRNA level. The number of amplification cycles was 30, and the specific amplicons were analyzed by 1% agarose gel electrophoresis and visualized with ethidium bromide.
 | Table 1 Primer sequences for type I collagen and β-Actin |
Statistical analysis
The results are expressed as the mean ± standard deviation. Differences between groups were compared using one-way analysis of variance followed by the Tukey’s test. Data with a skewed distribution or heterogeneity of variance were analyzed by the Kruskal–Wallis nonparametric test followed by a Nemenyi test. A P-value <0.05 was considered to be statistically significant.
Results
UA likely interacts with a number of important functional proteins
First, we computationally predicted the molecular targets of UA using our web-based DDI-CPI tool. There were 611 proteins that possibly interacted with UA (Table 2), including those involved in MAPK signaling pathway (FGFR2, TRAF2, FGFR1, HRAS, GRB2, MAPKAPK3, MAPKAPK2, AKT1, CDC42, TNFRSF1A, CASP3, RAC1, PRKACA, PAK1, TRAF6, AKT2, EGFR, PRKCA, MAP2K1, BRAF, TGFBR1, TP53, RAF1, MAPK10, PRKCB, MAPK1, DUSP3, RPS6KA1, MAPK14, MAPK3, PLA2G2A, MAPK9, and MAPK8), apoptosis (PIK3CG, TRAF2, XIAP, TP53, BCL2L1, AKT1, IRAK4, TNFRSF1A, CASP3, CASP7, BCL2, CASP8, PRKACA, and AKT2), energy metabolism (PPARA, PDPK1, PPARD, CHKB, RXRA, PPARG, FABP3, FABP4, FABP7, MMP-1, PCK1, NR1H3, NAMPT, CD38, NT5M, PNP, and NNMT), and cell proliferation (YWHAZ, TP53, TTK, CDK6, CHEK1, RB1, CHEK2, SFN, WEE1, CDK2, HDAC2, PLK1, GSK3B, MDM2, and ABL1). Furthermore, as shown in Table 3, our DAVID analysis showed that there were 49 functional clusters that were identified to be significantly enriched (enrichment score >3) in the target list derived from molecular docking calculations, including energy metabolism, signal transduction, vascular regulation, and carbohydrate metabolism. Furthermore, there were 76 KEGG pathways significantly enriched in the target list (Table 4), such as MAPK, p53, and mTOR signaling pathways.
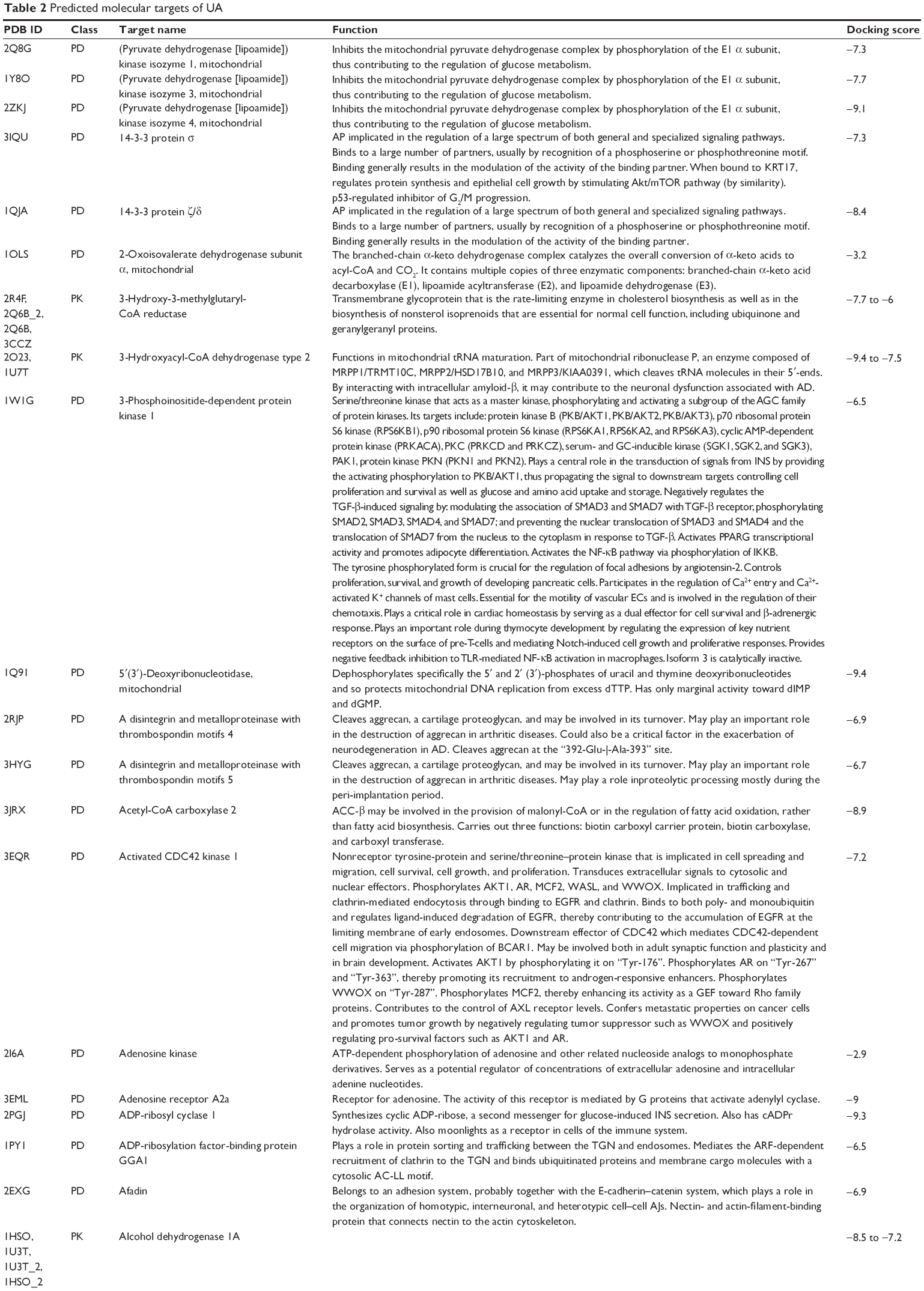 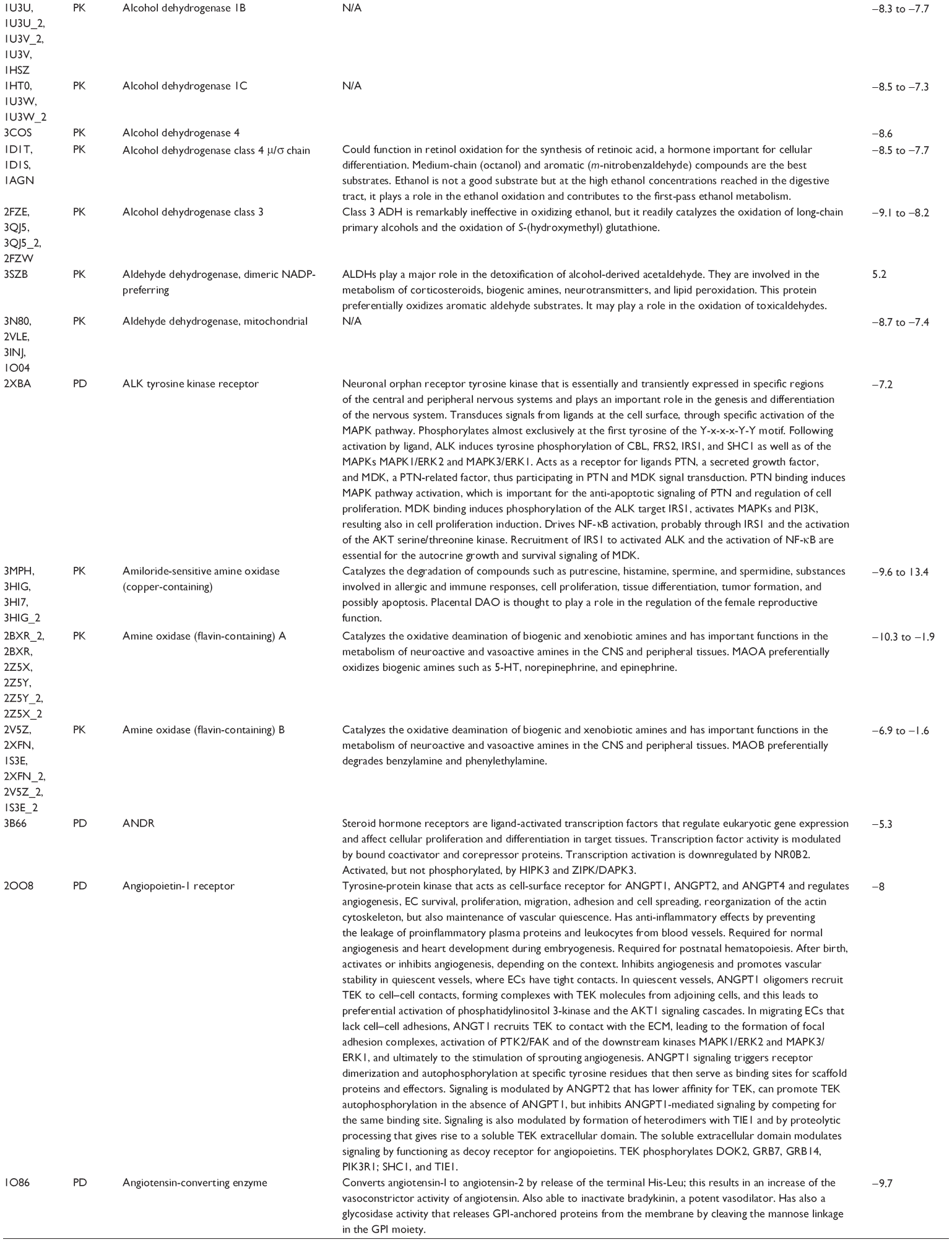 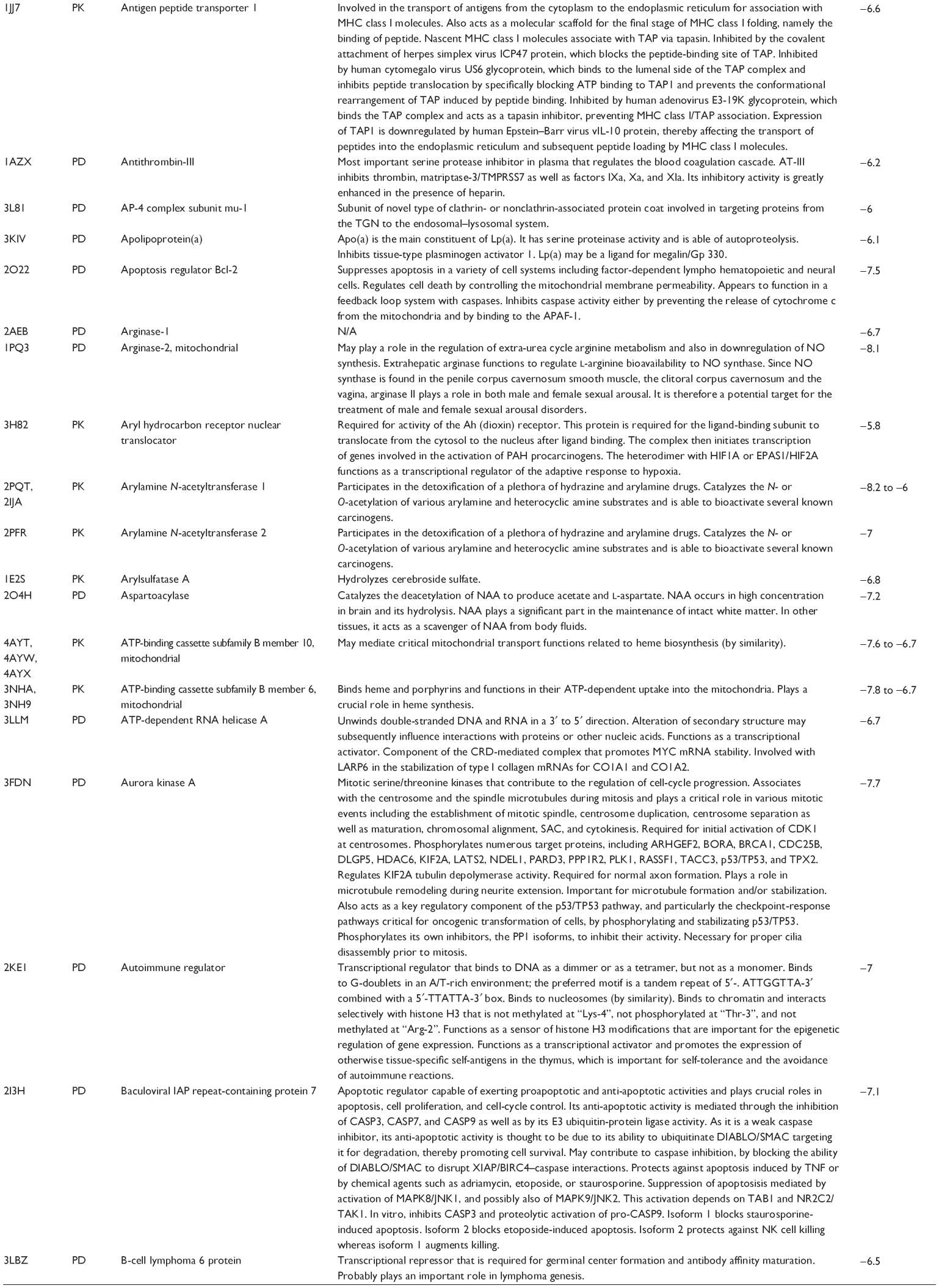 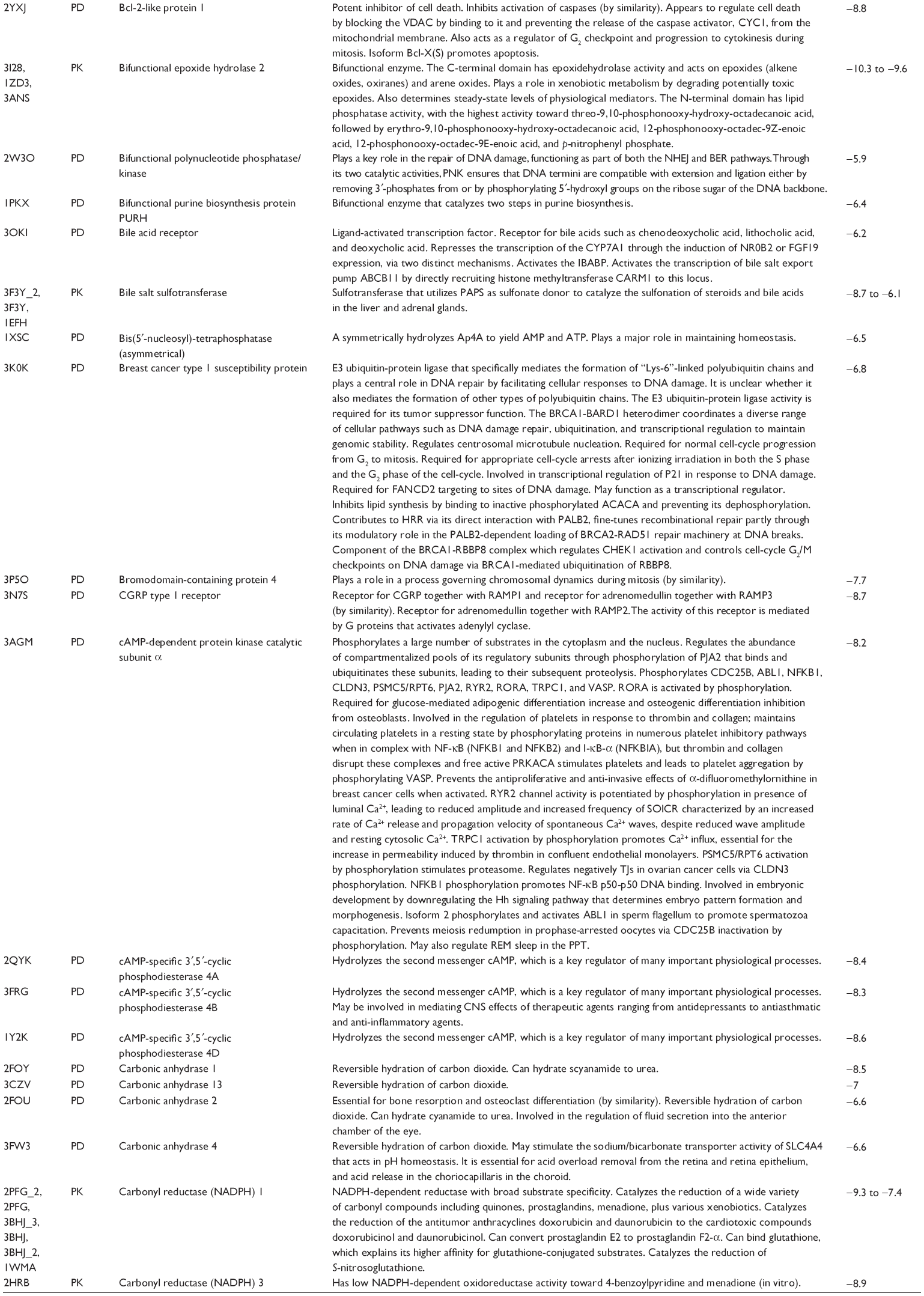 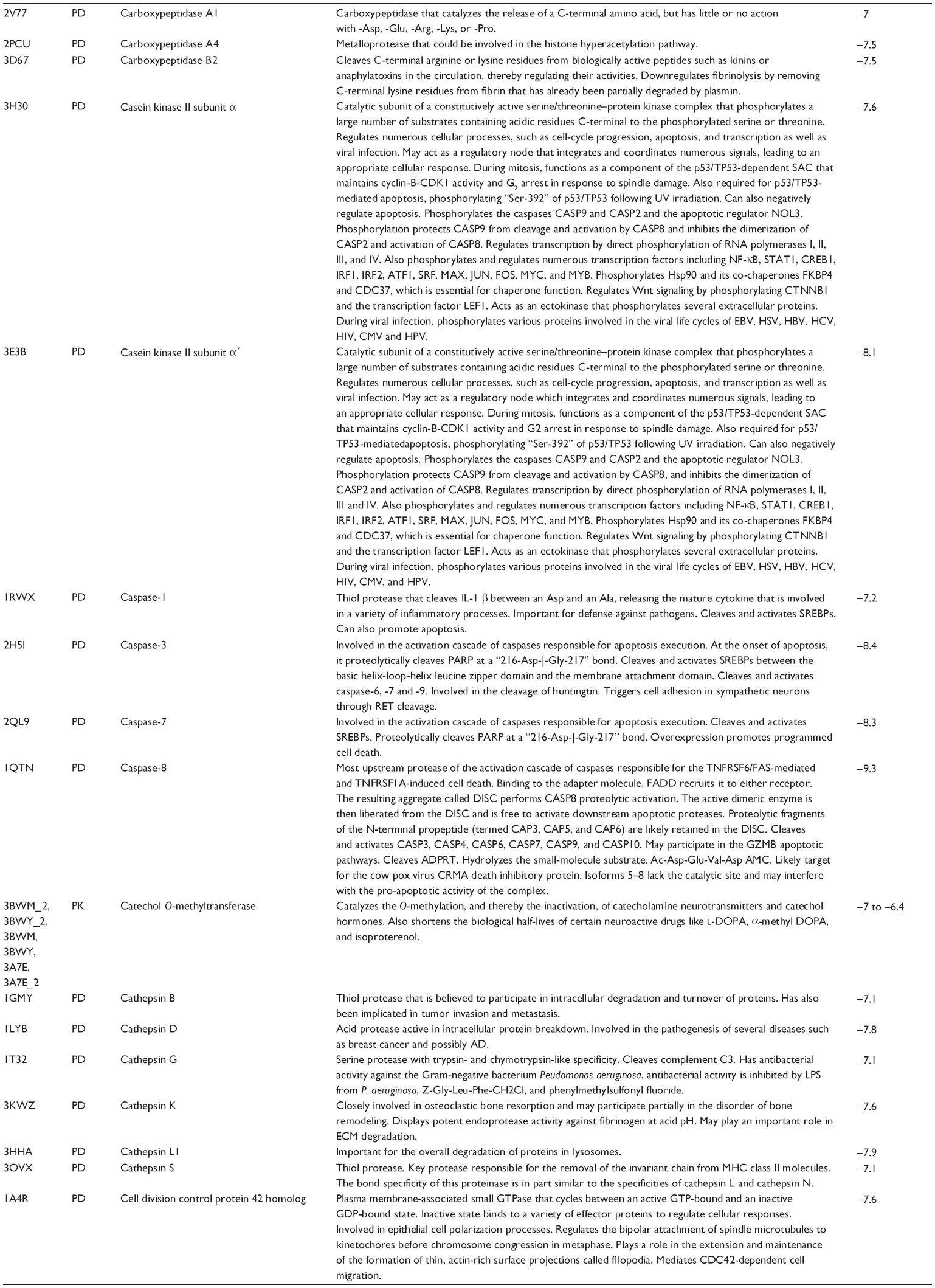 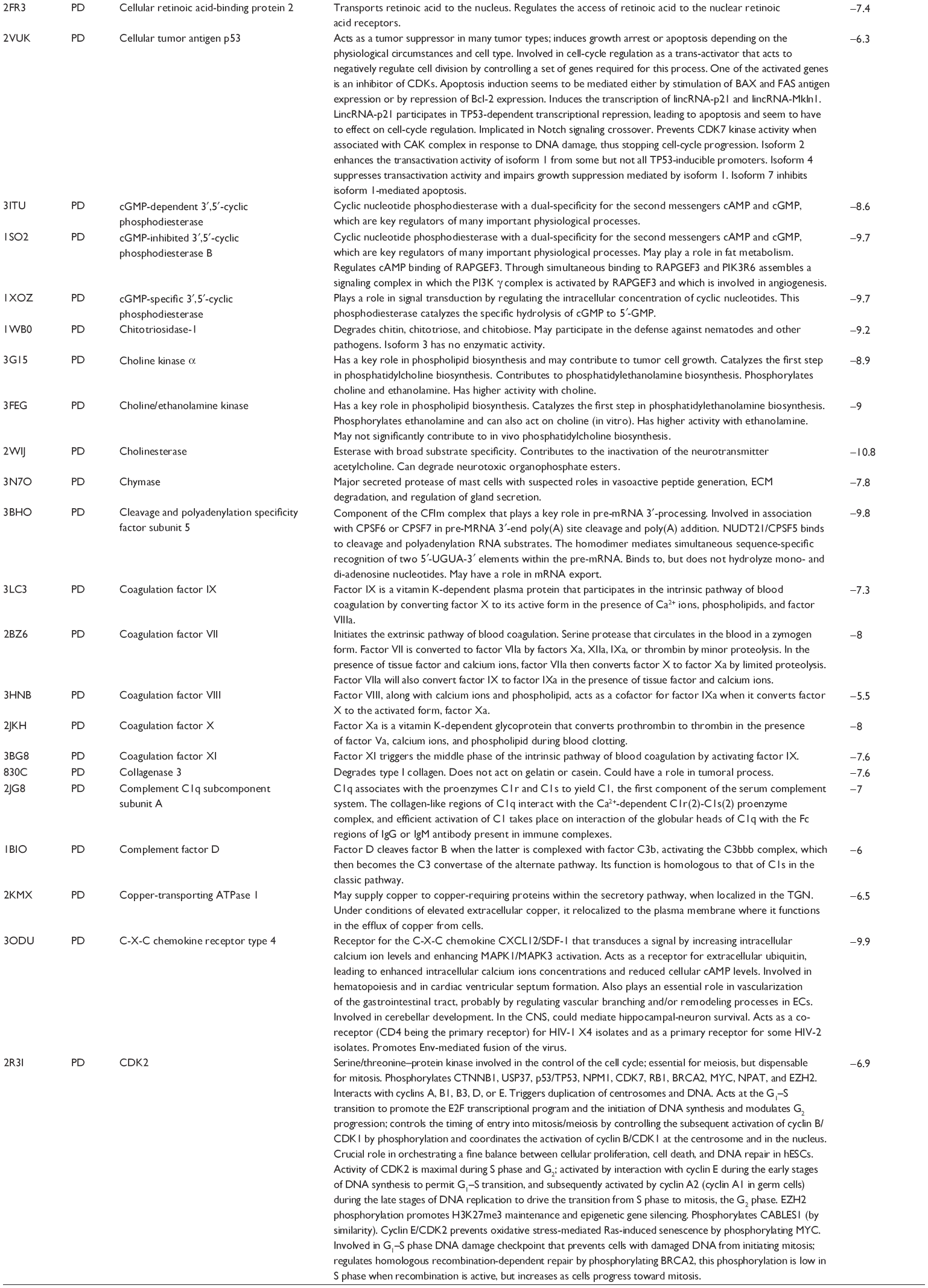  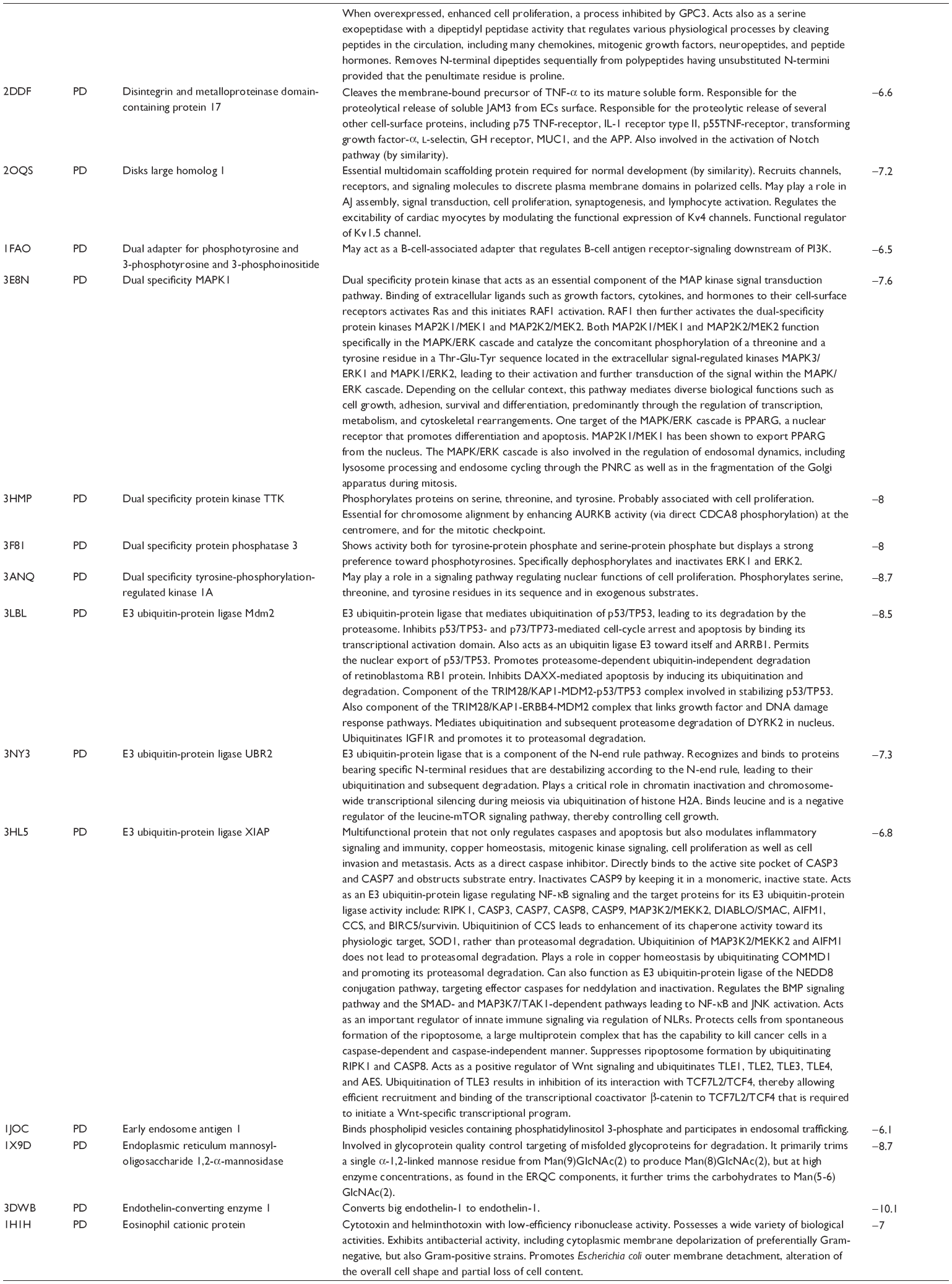  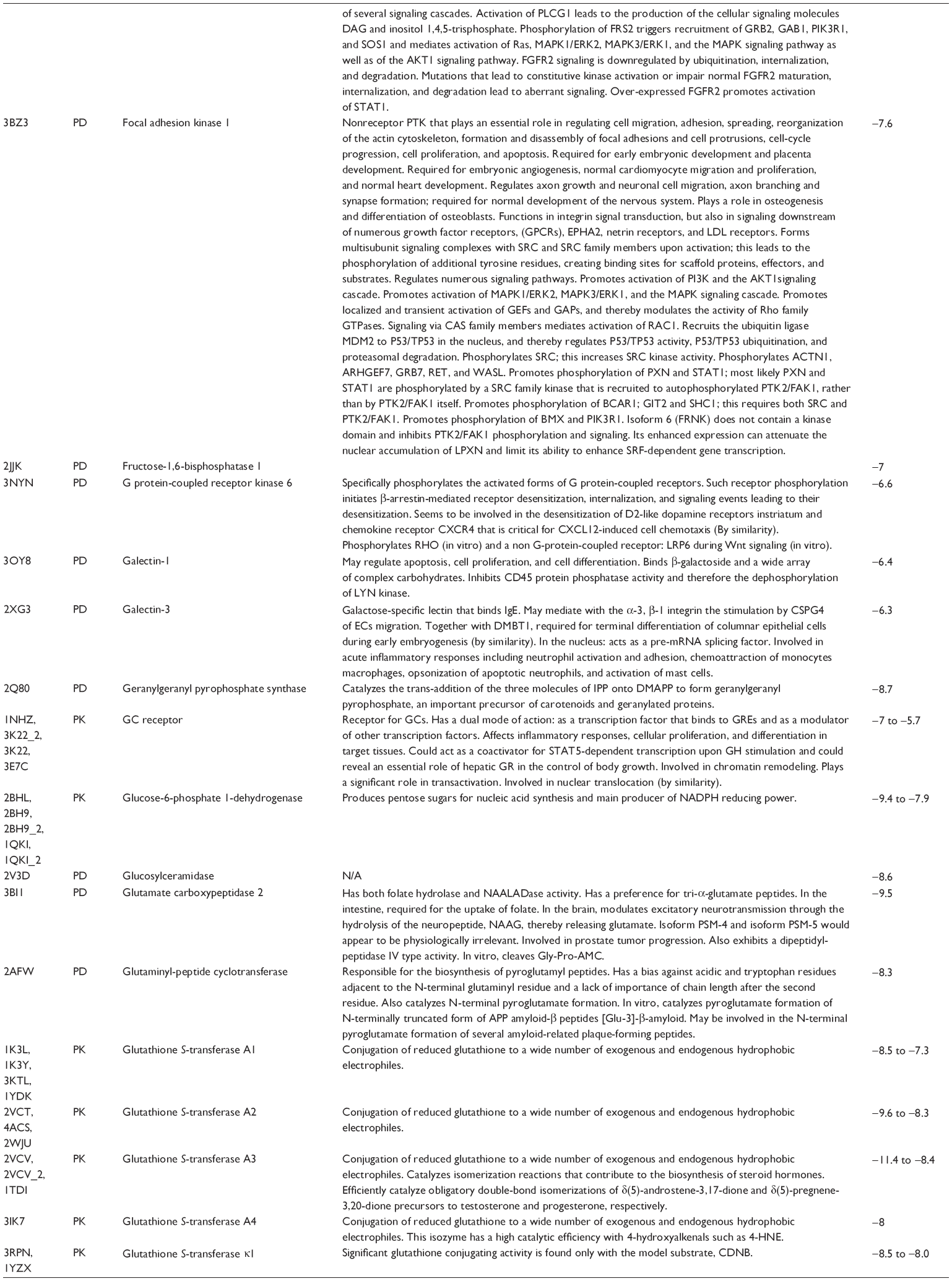      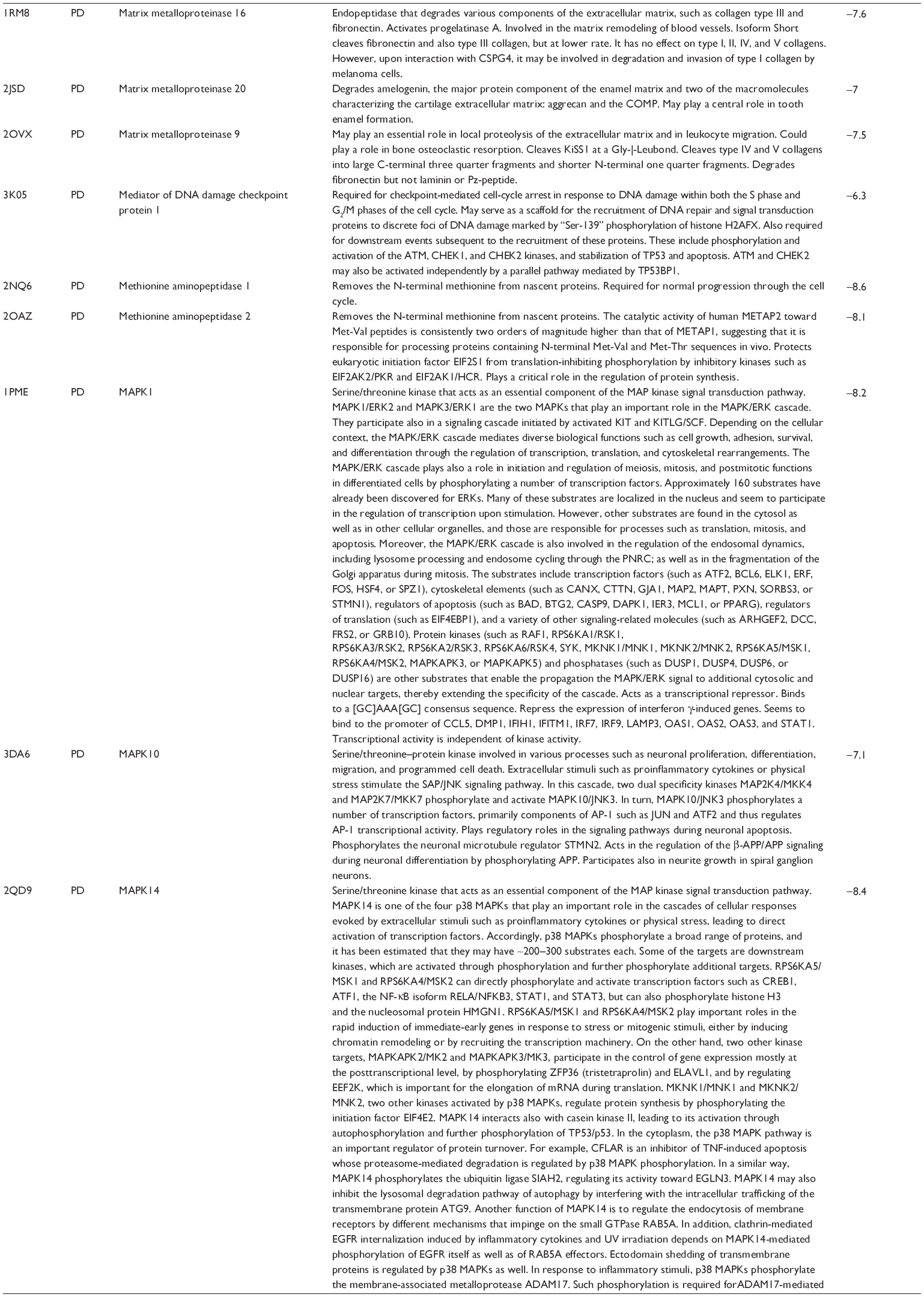  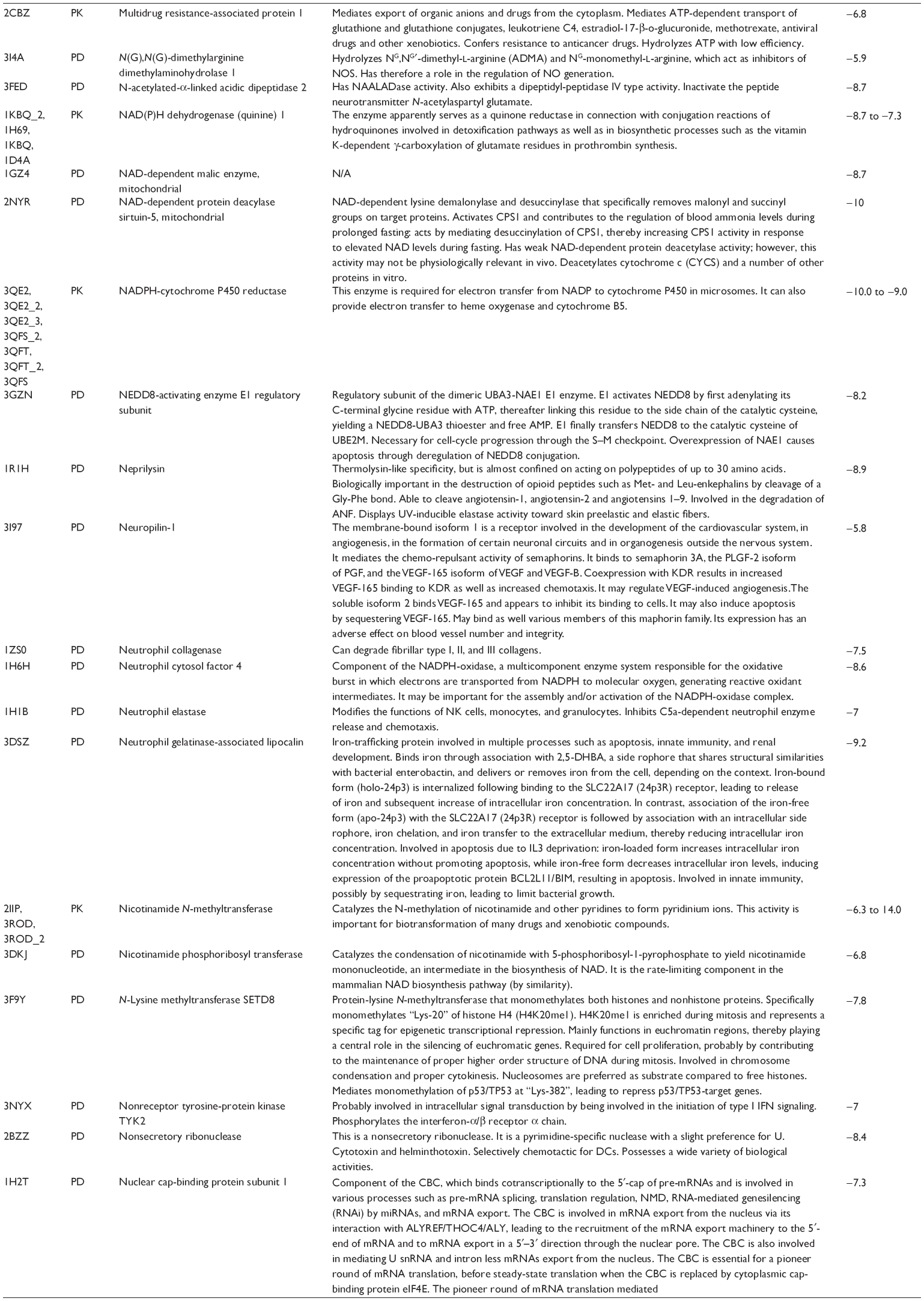 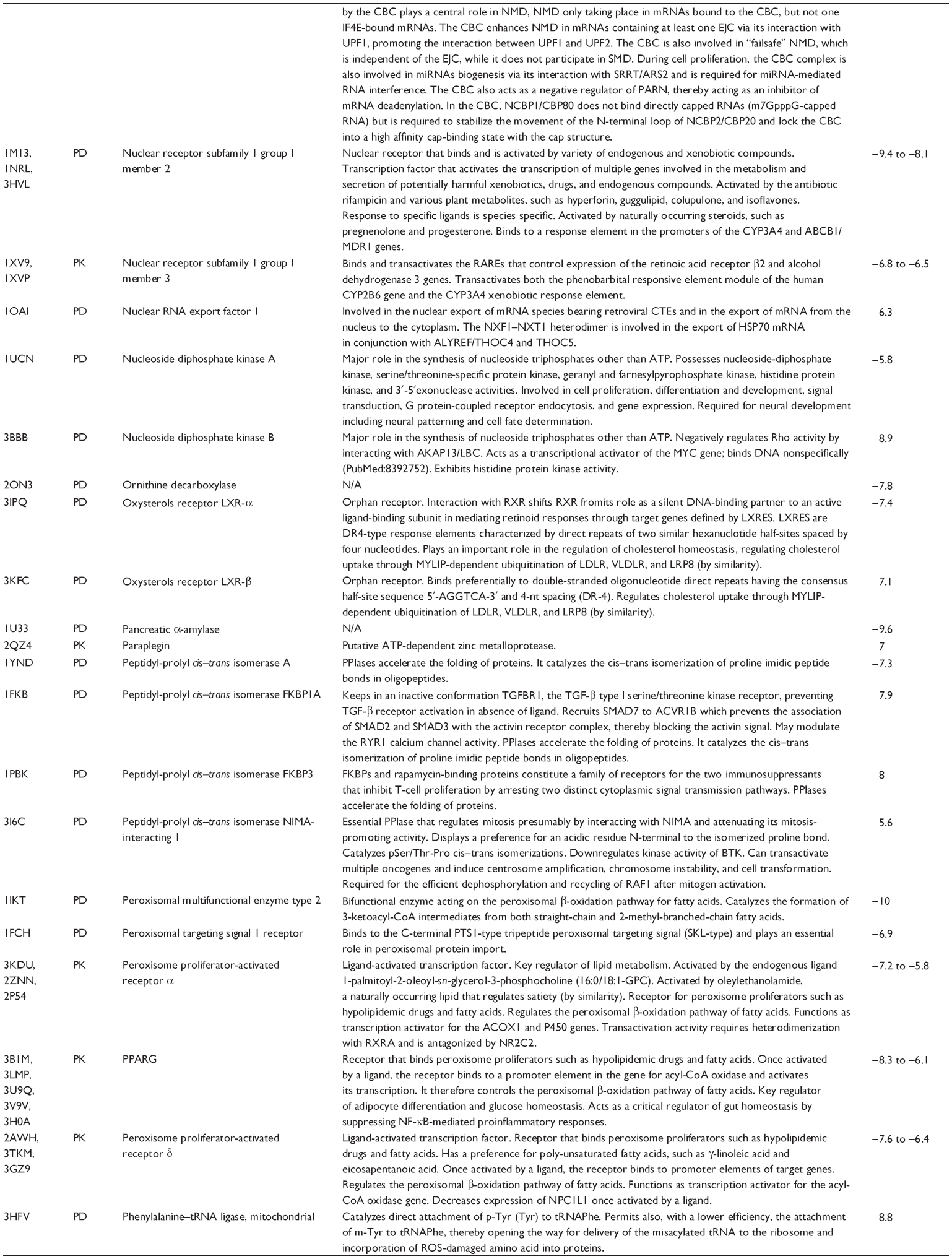 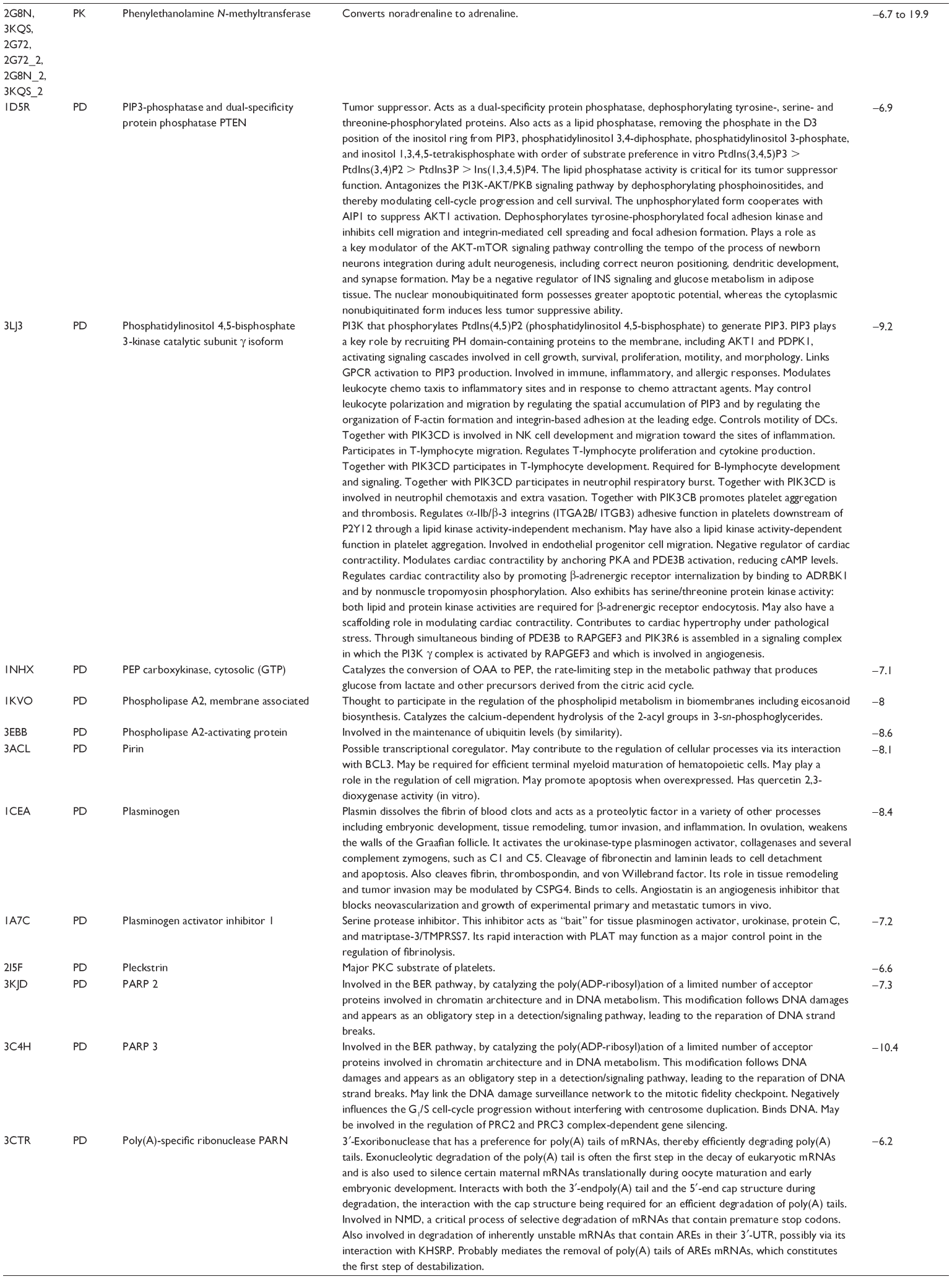 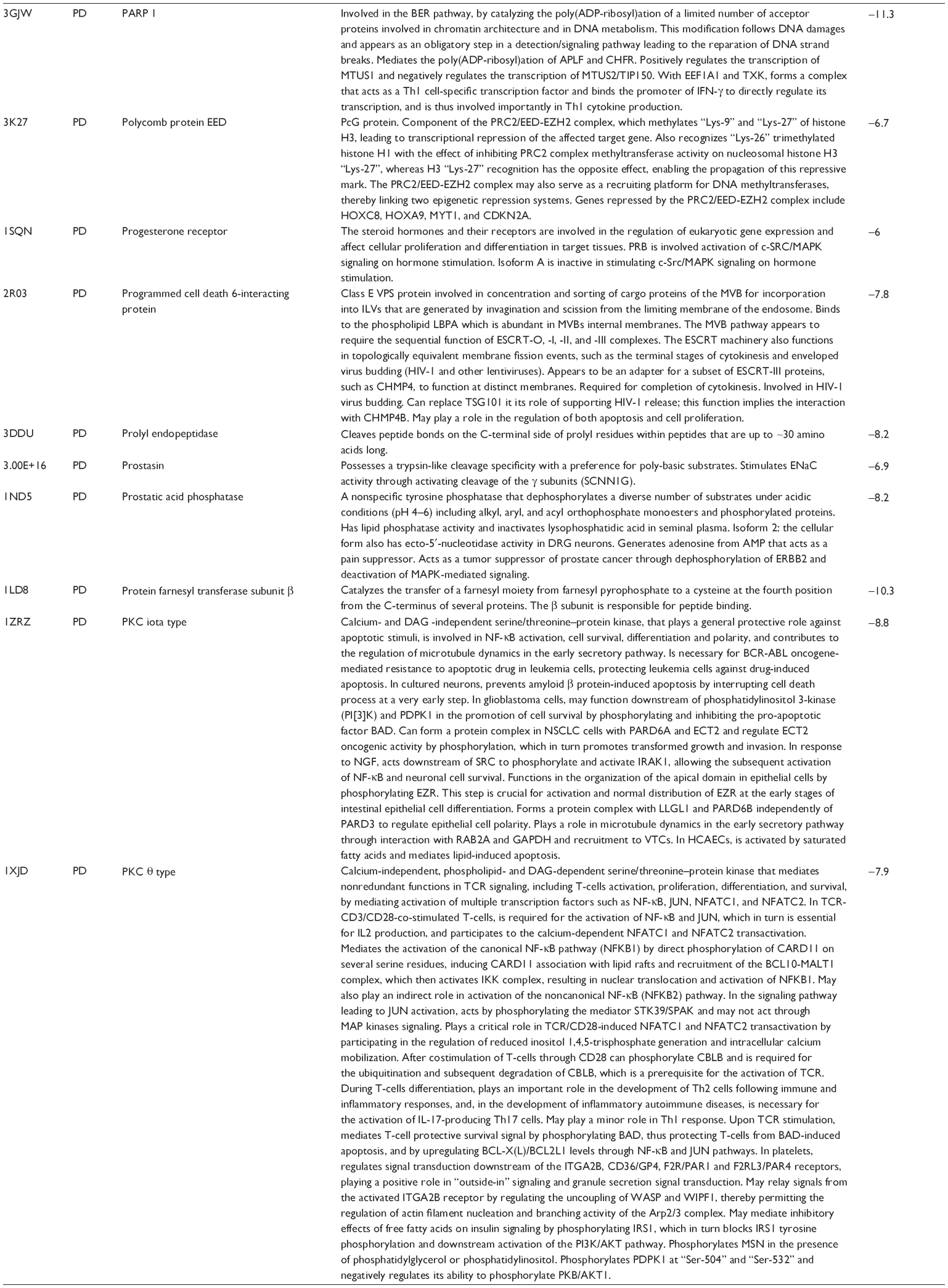  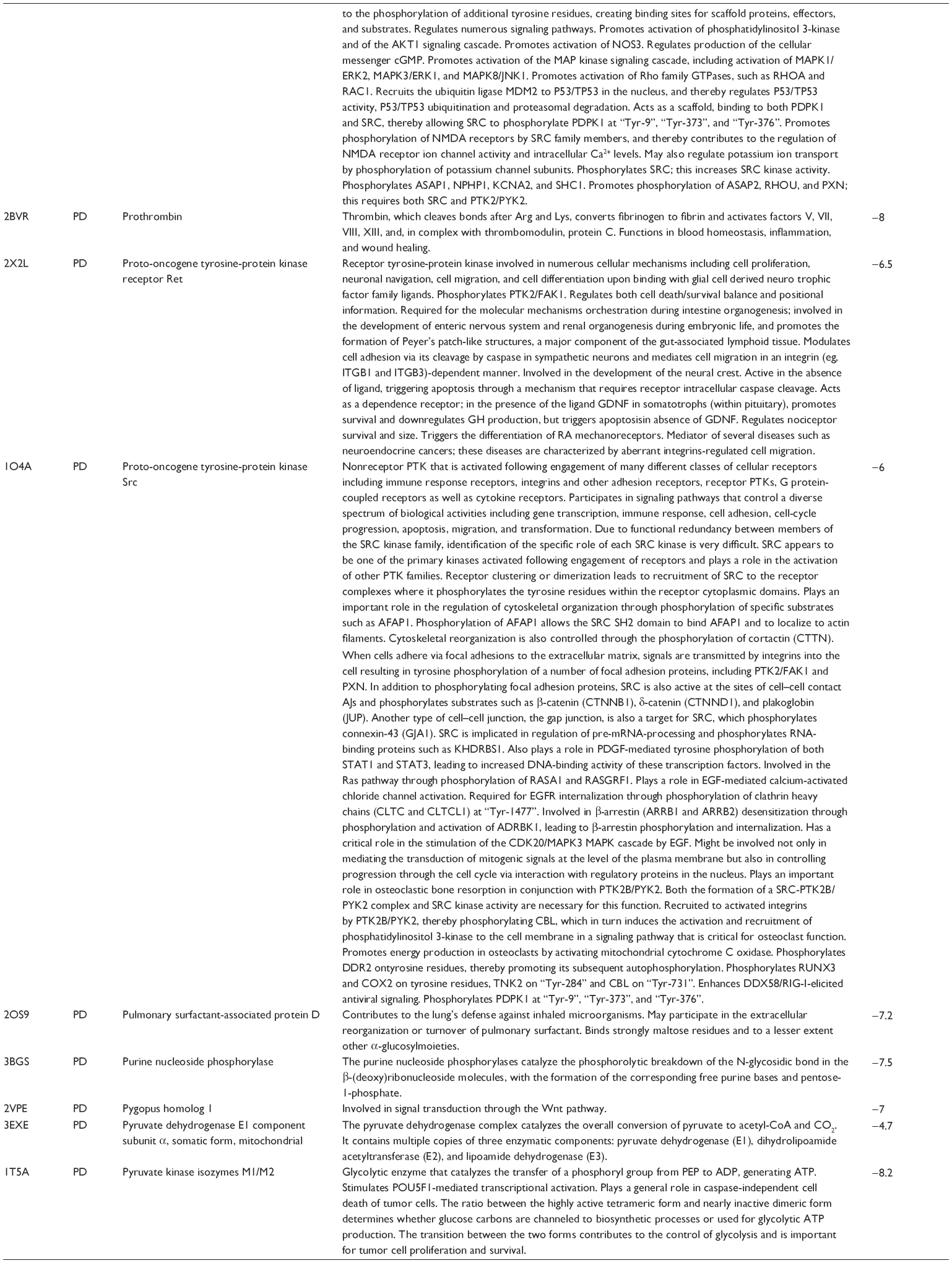  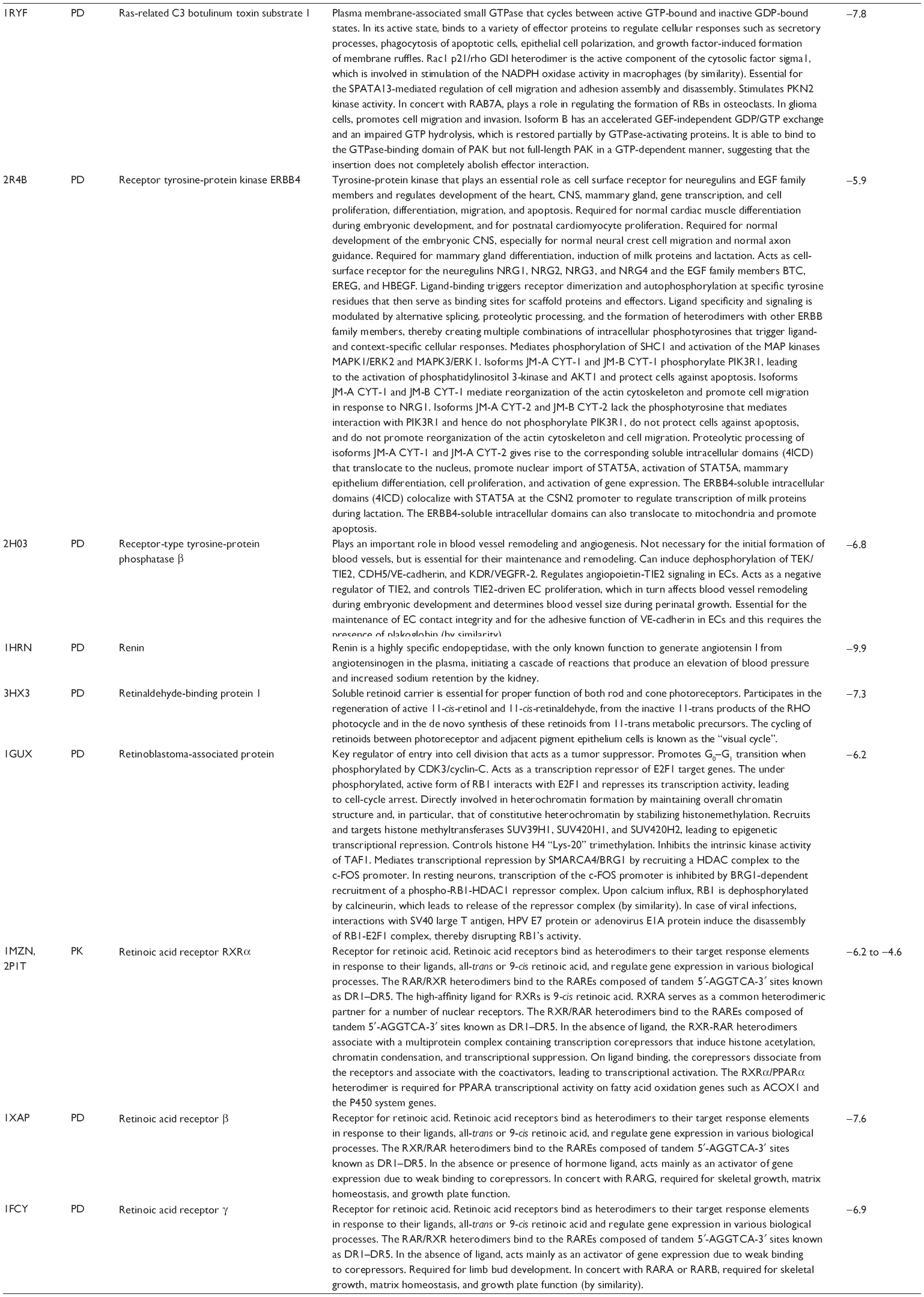 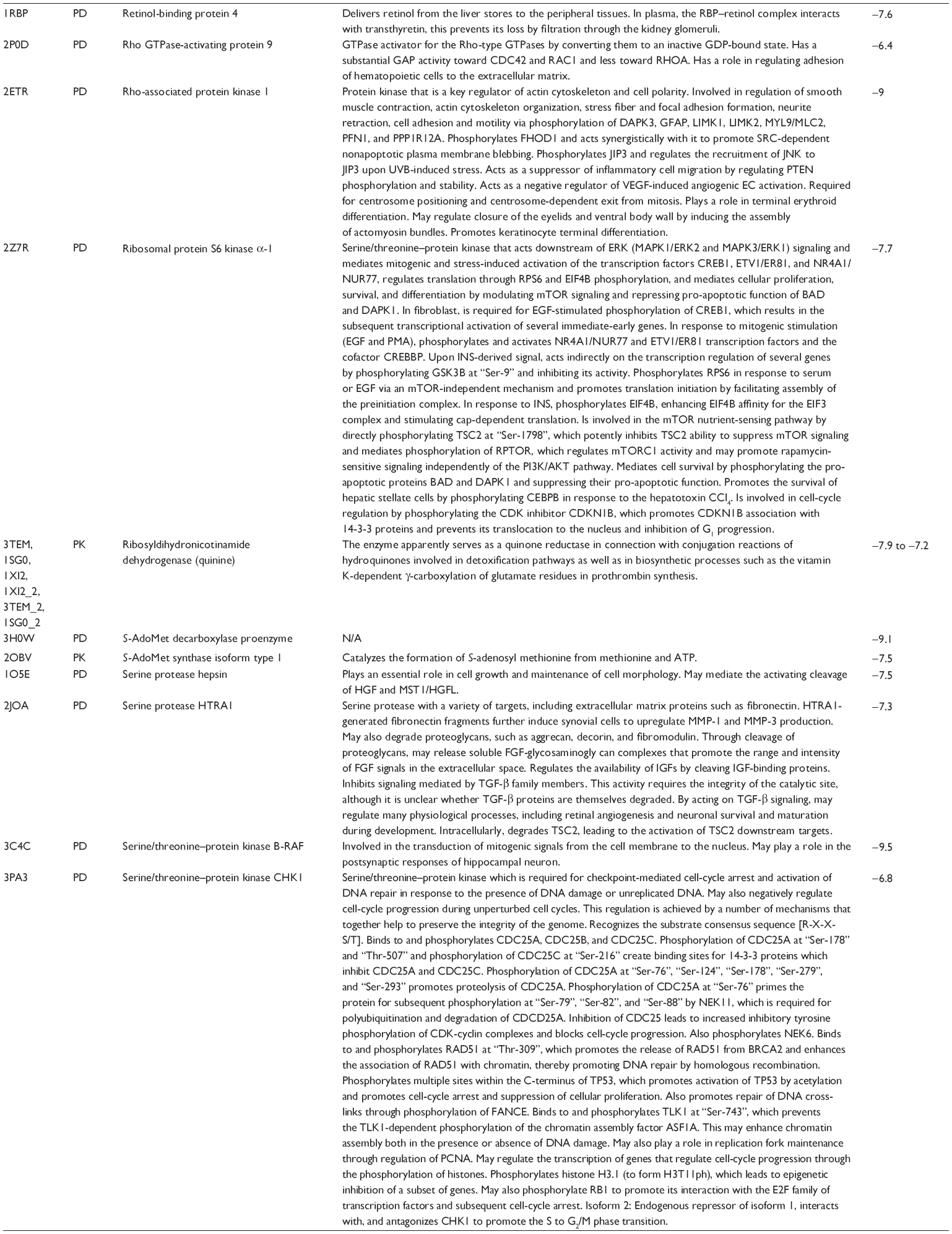   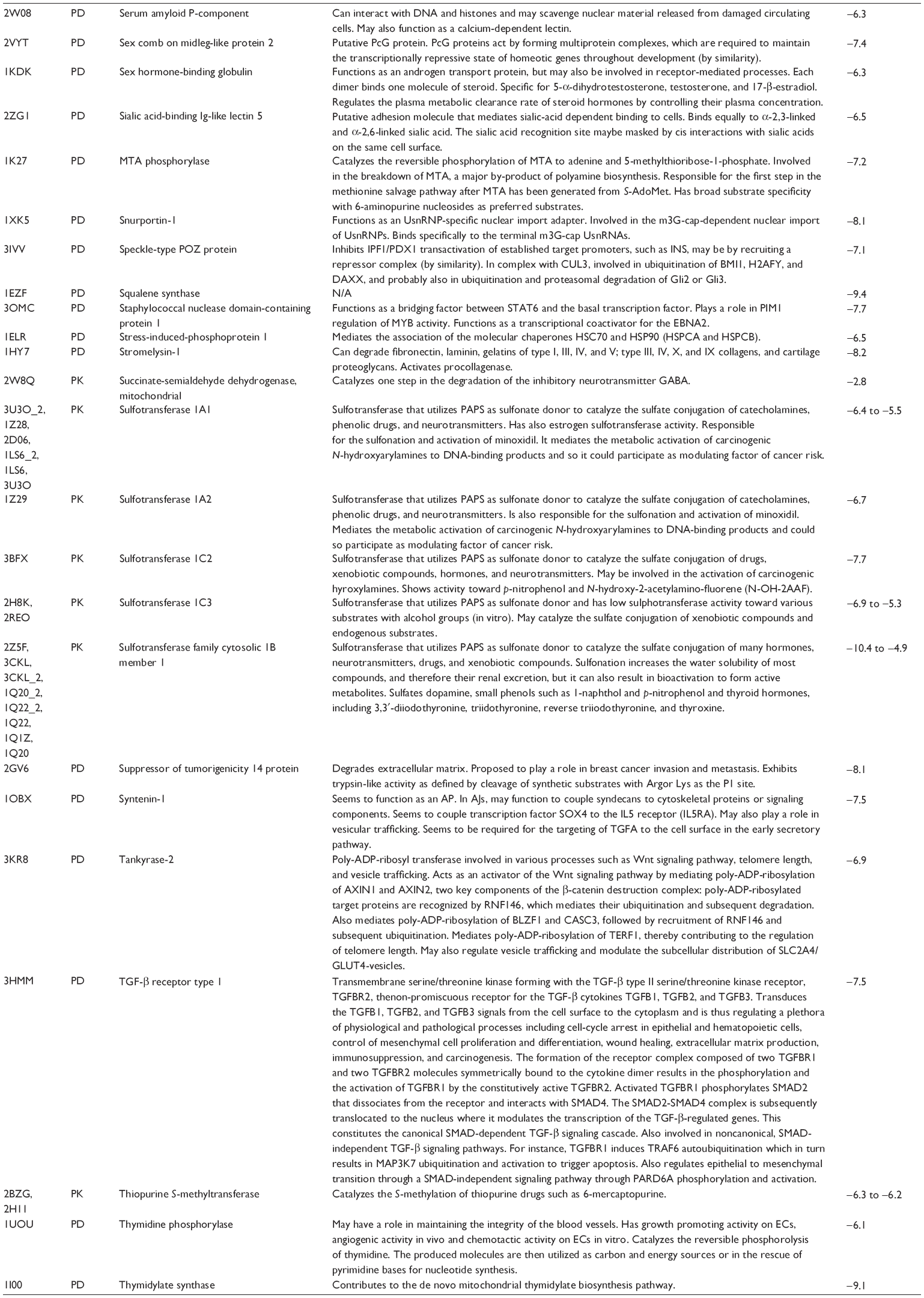 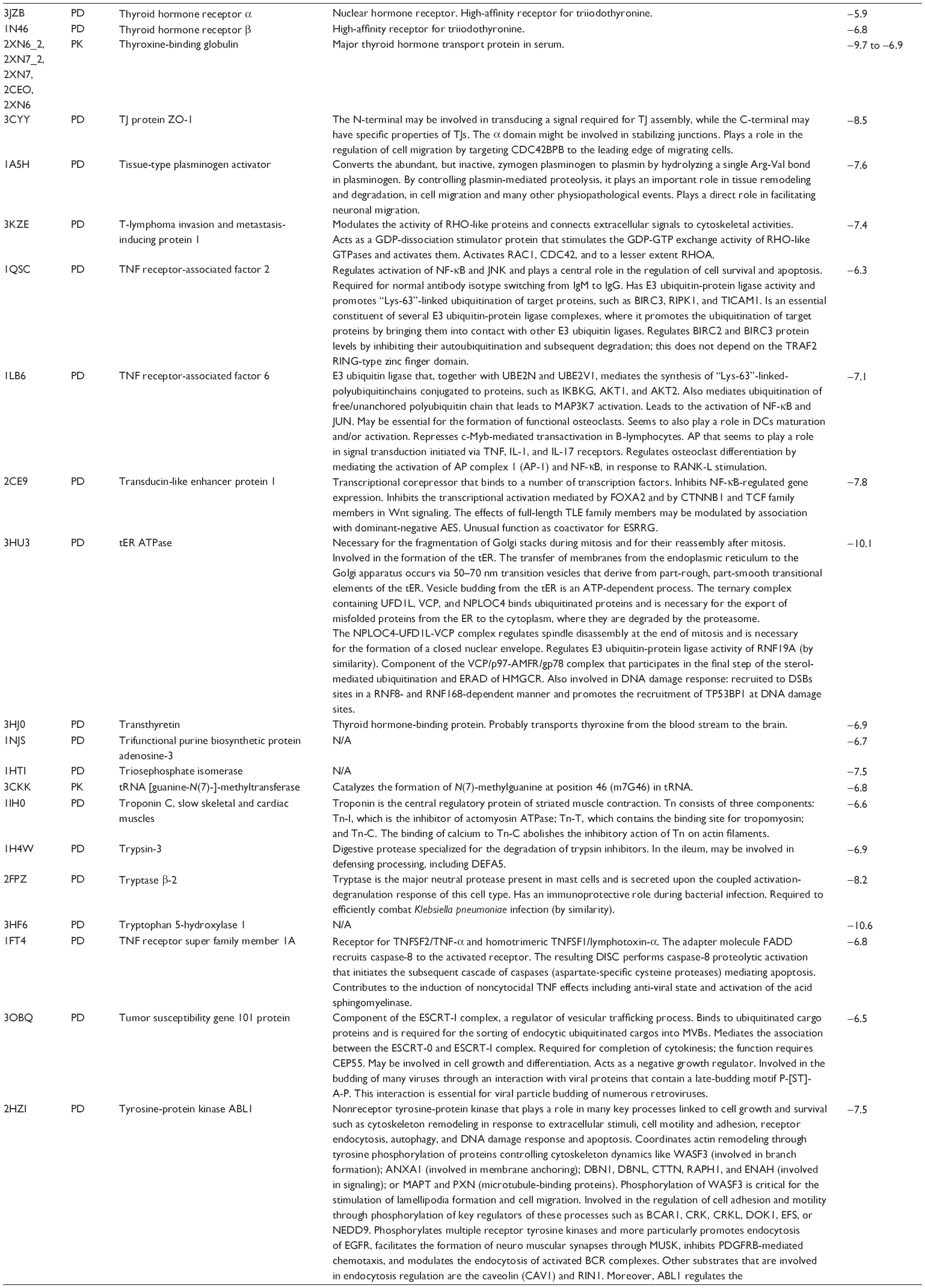   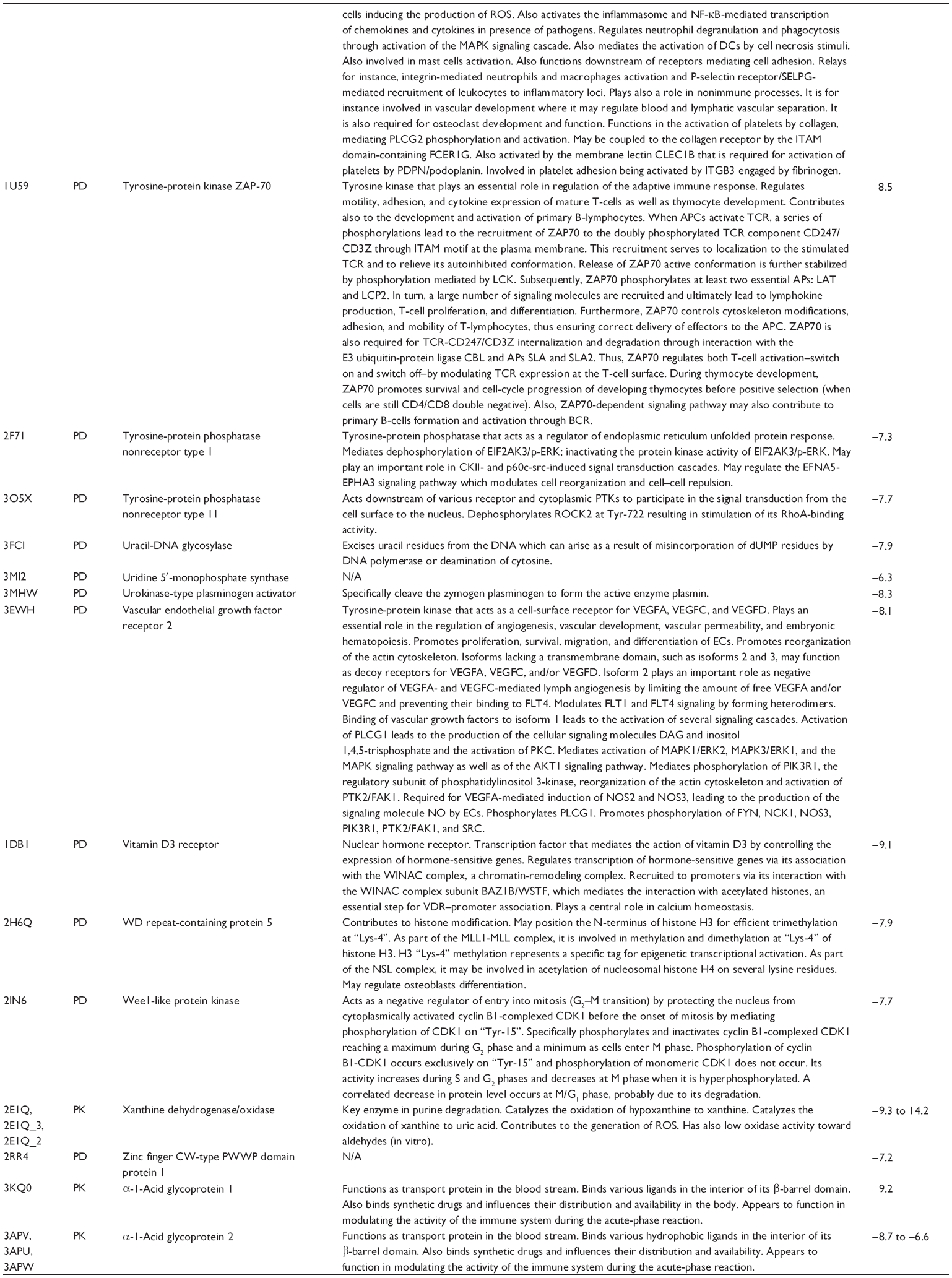  | Table 2 Predicted molecular targets of UA |
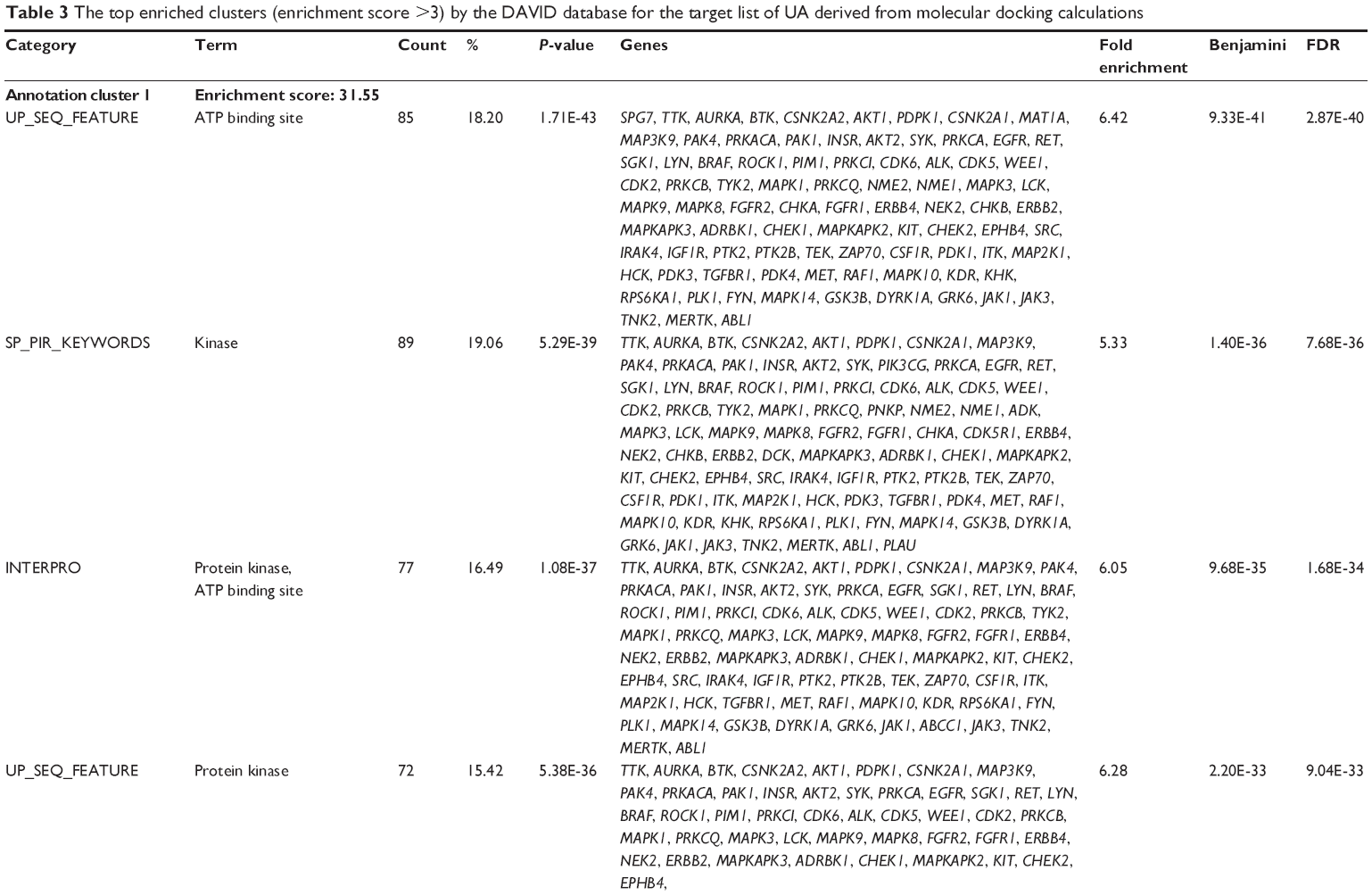  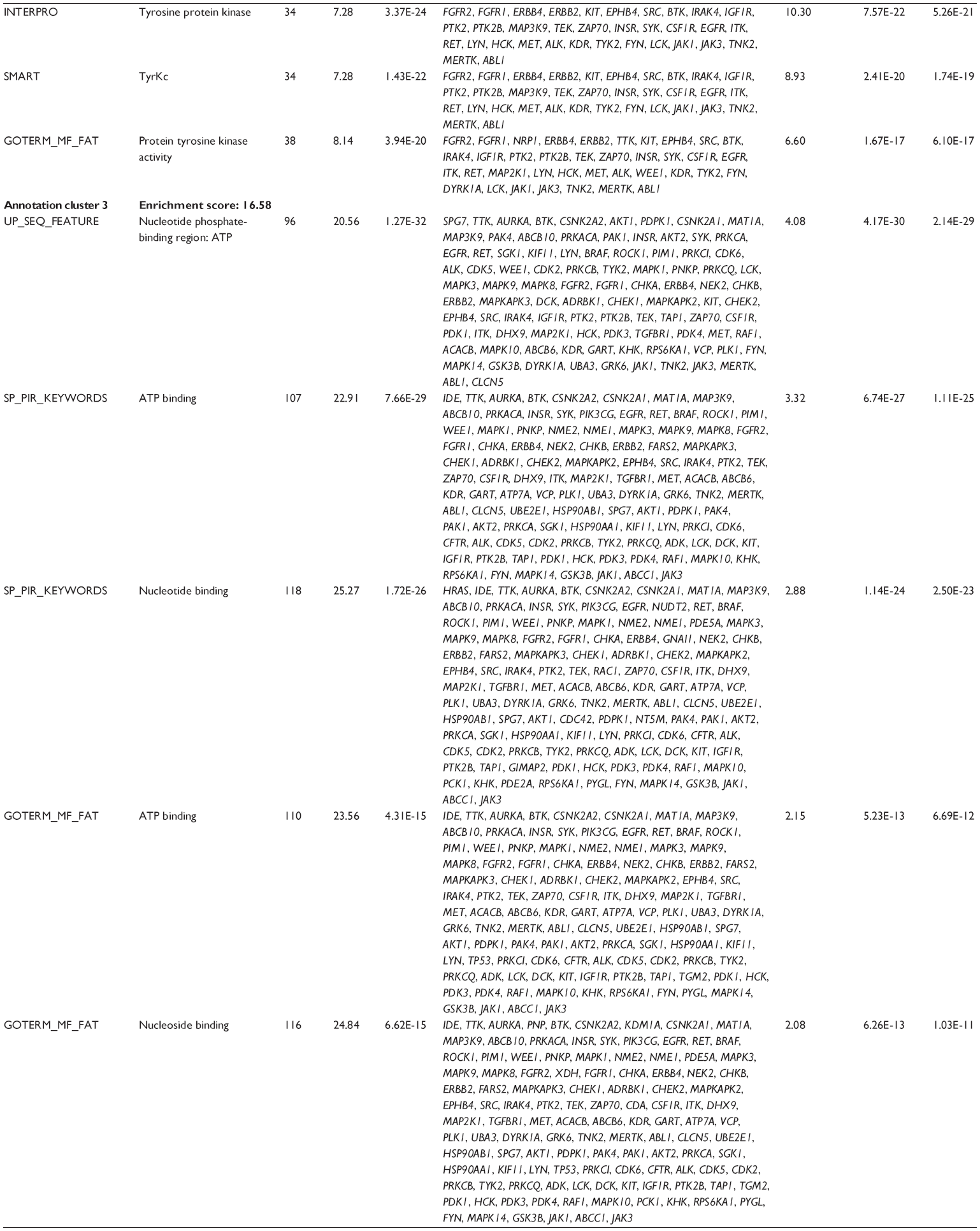  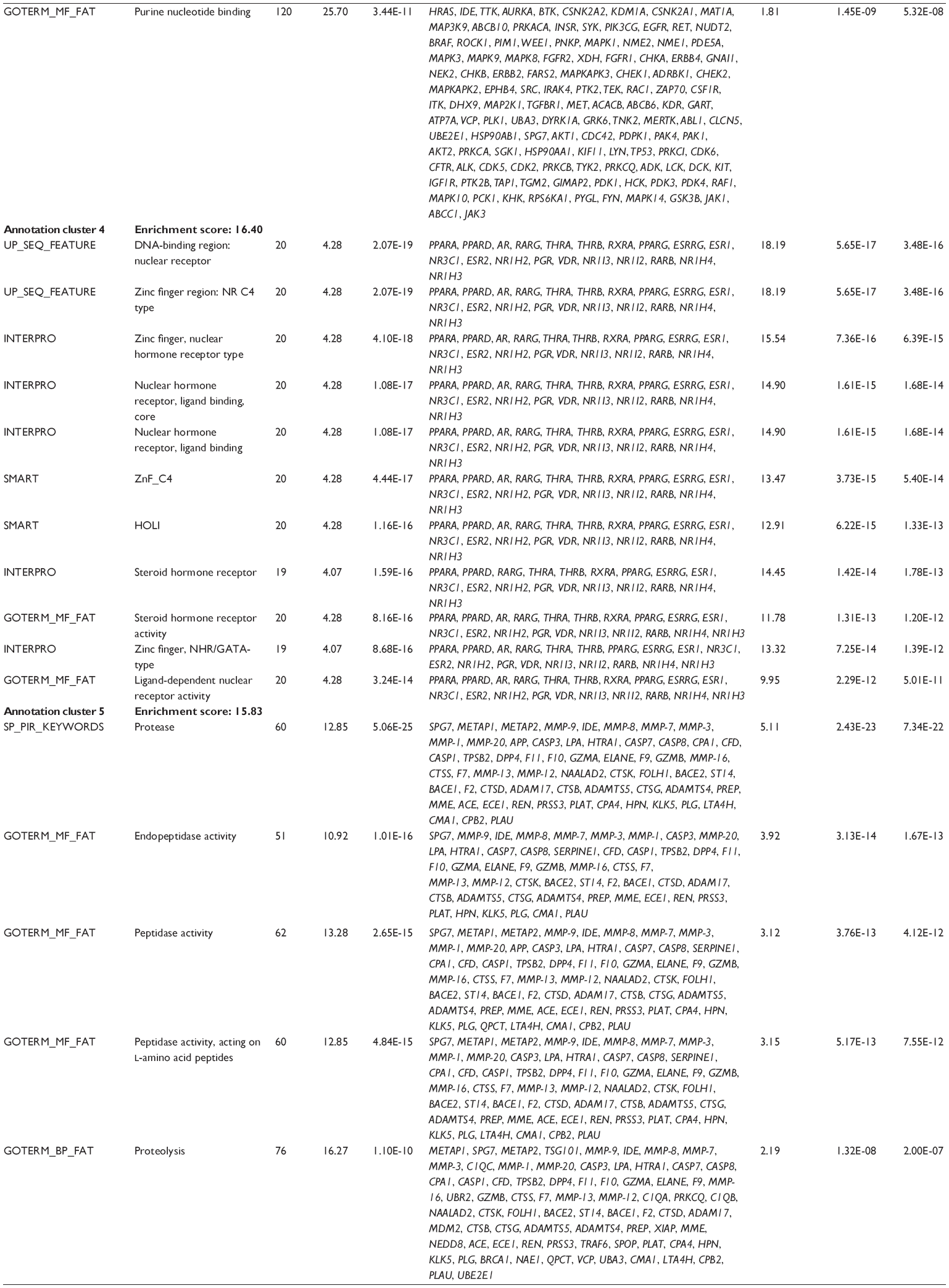 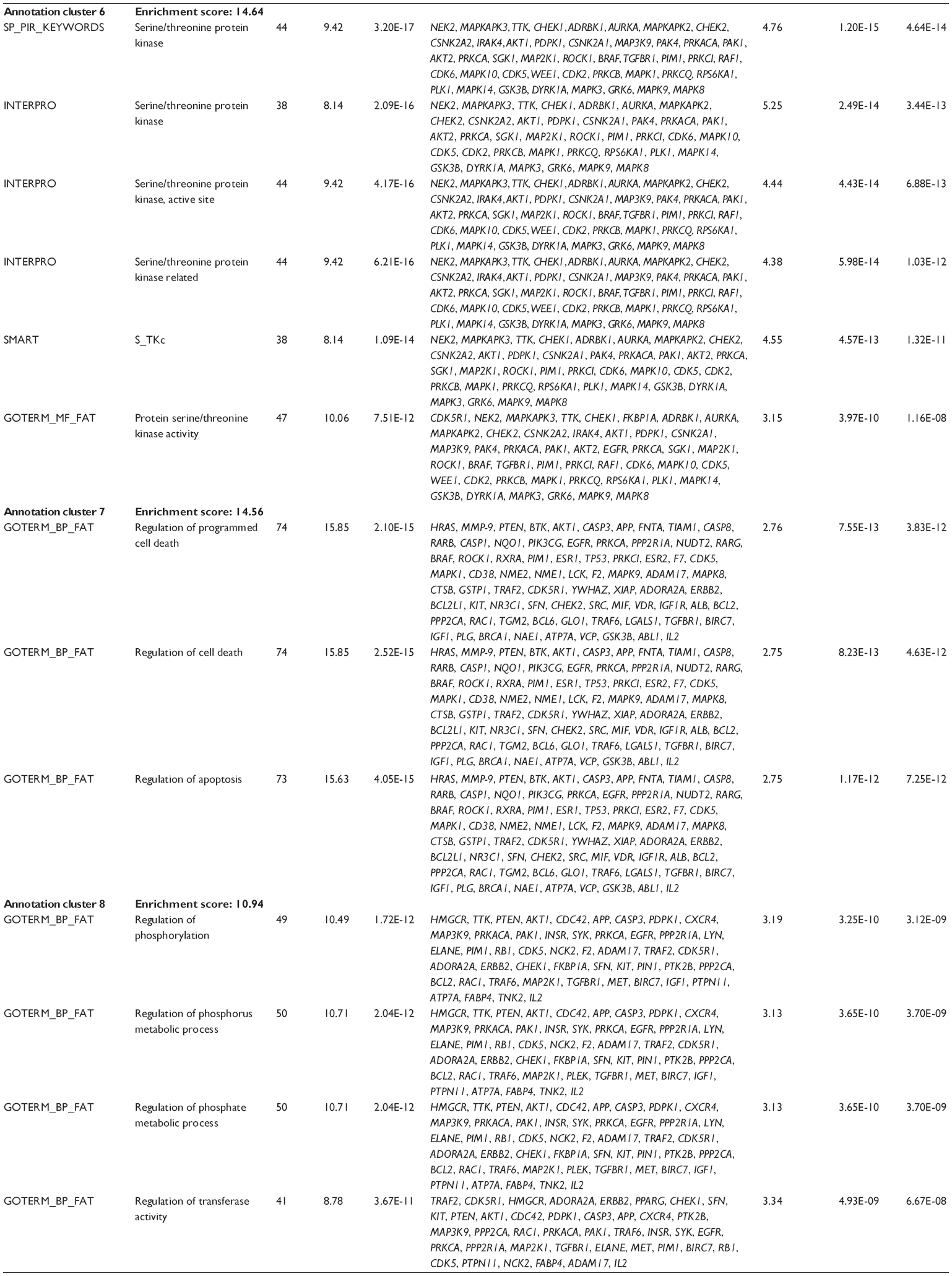 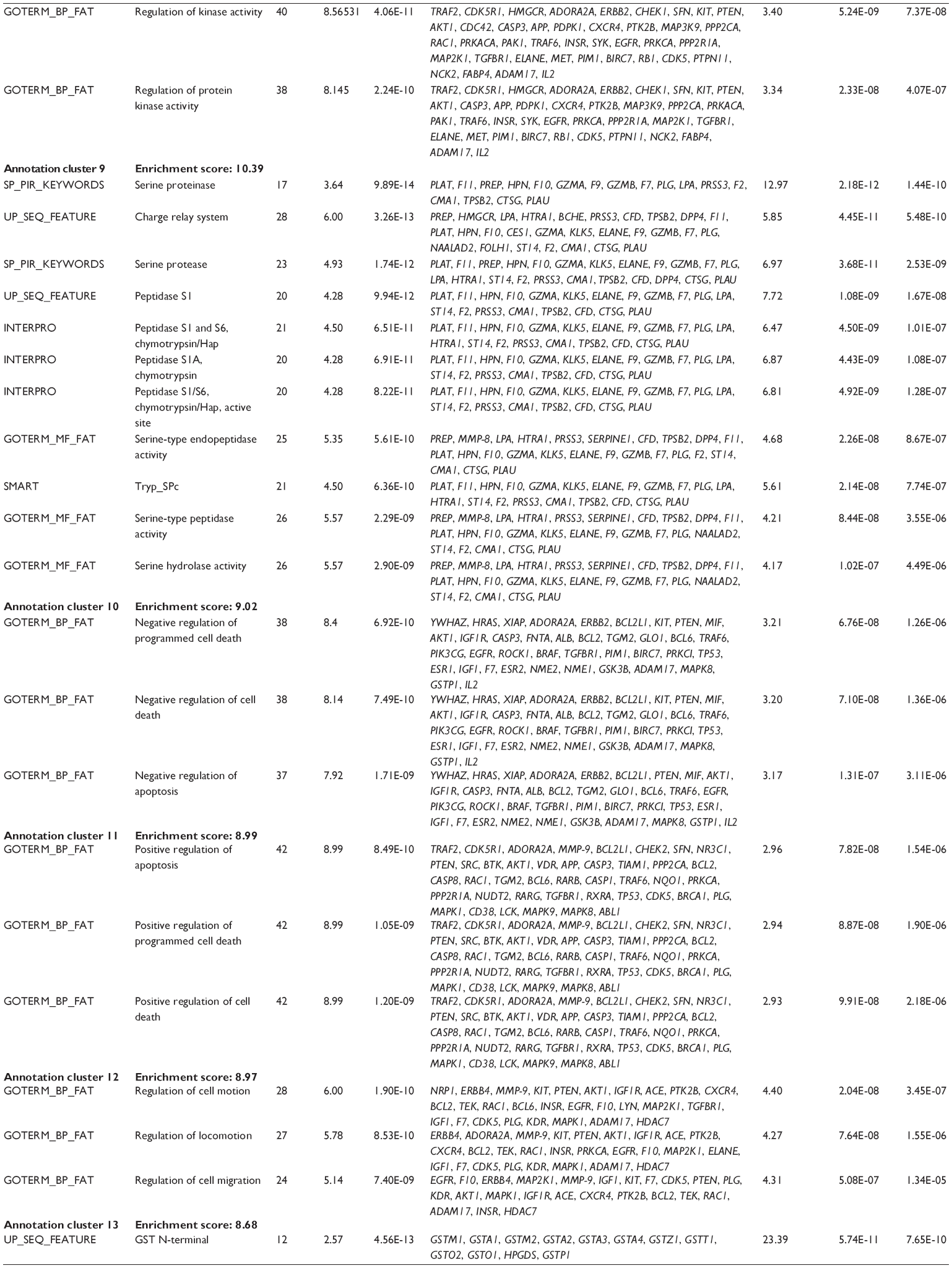     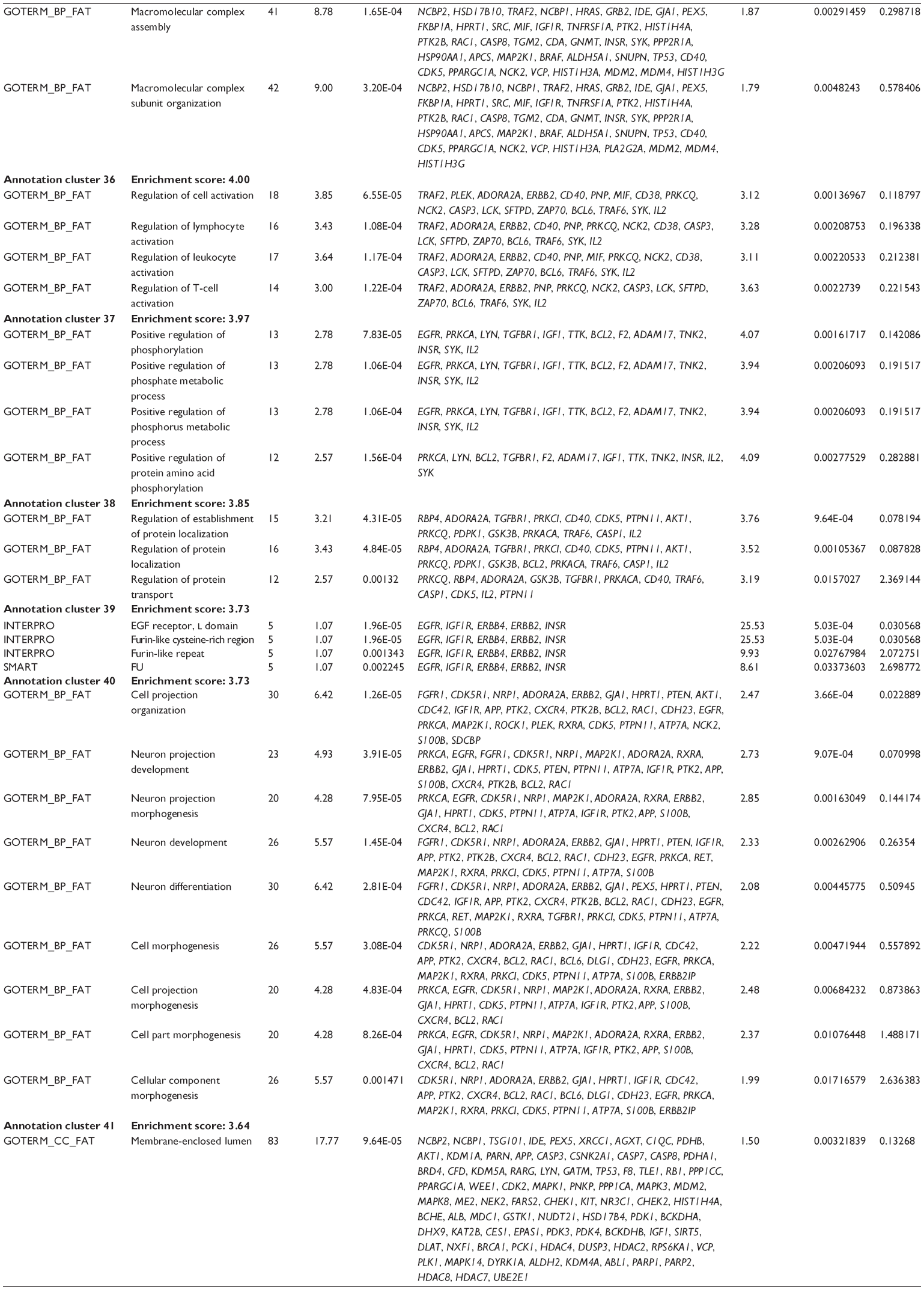 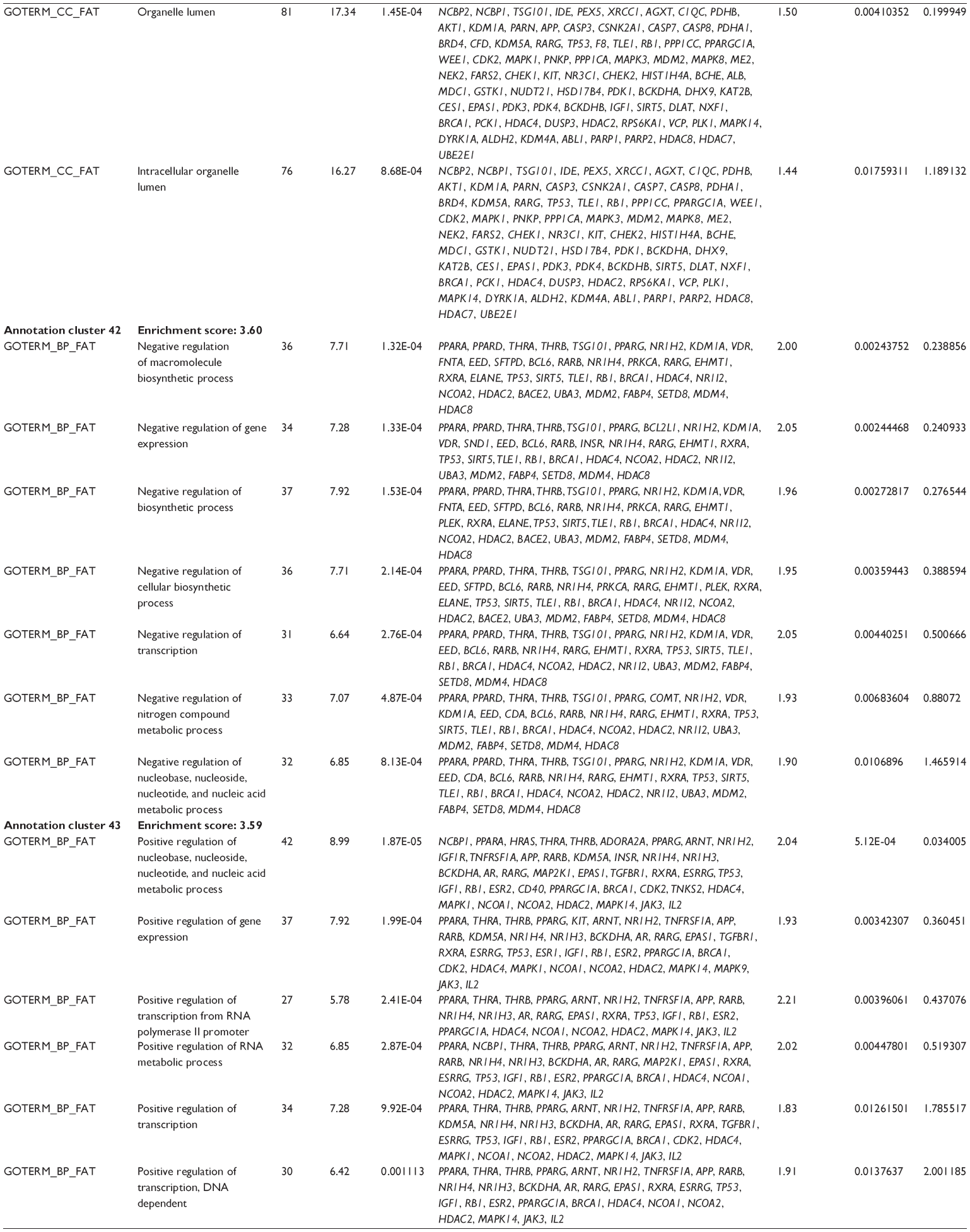 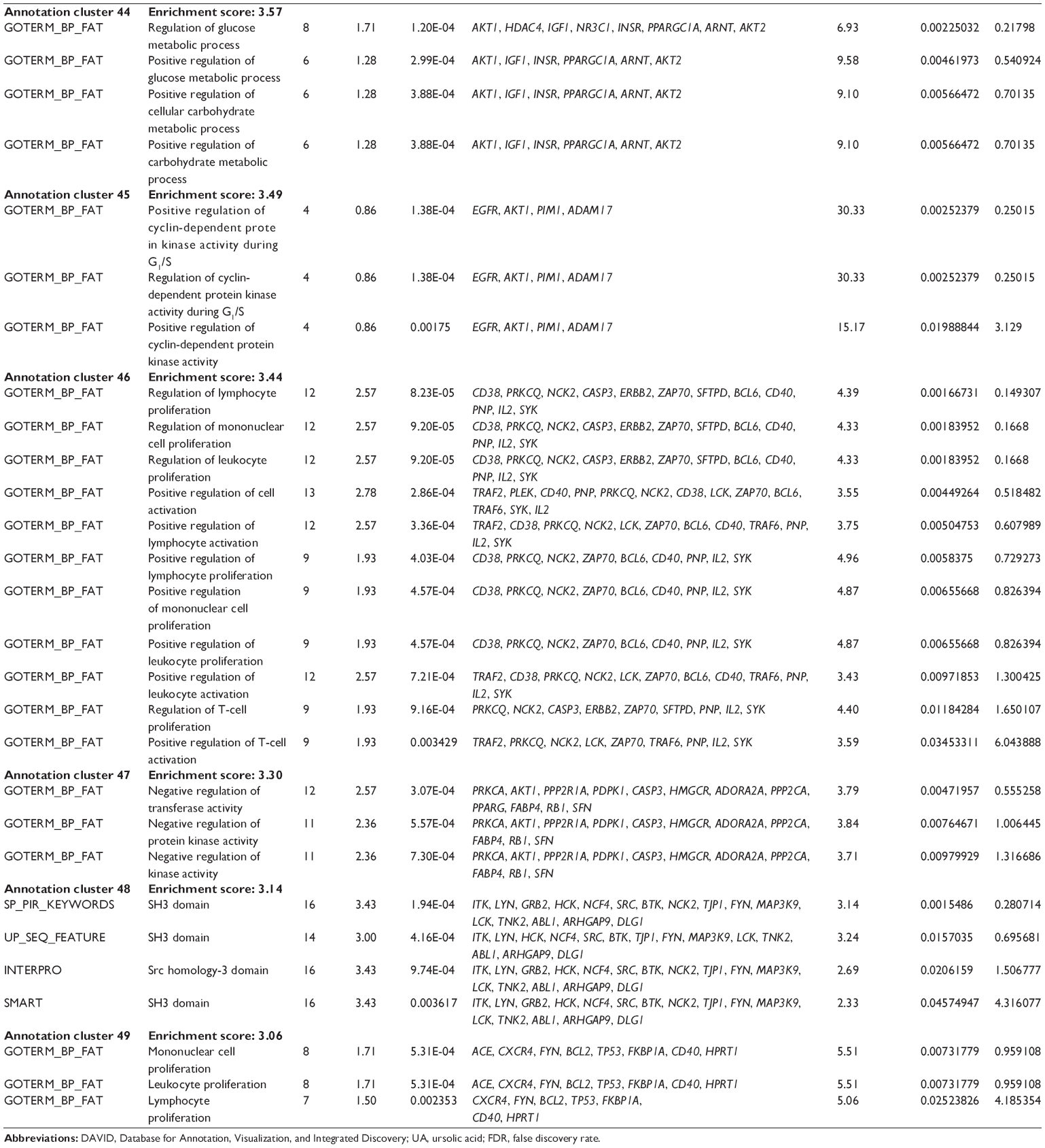 | Table 3 The top enriched clusters (enrichment score >3) by the DAVID database for the target list of UA derived from molecular docking calculations |
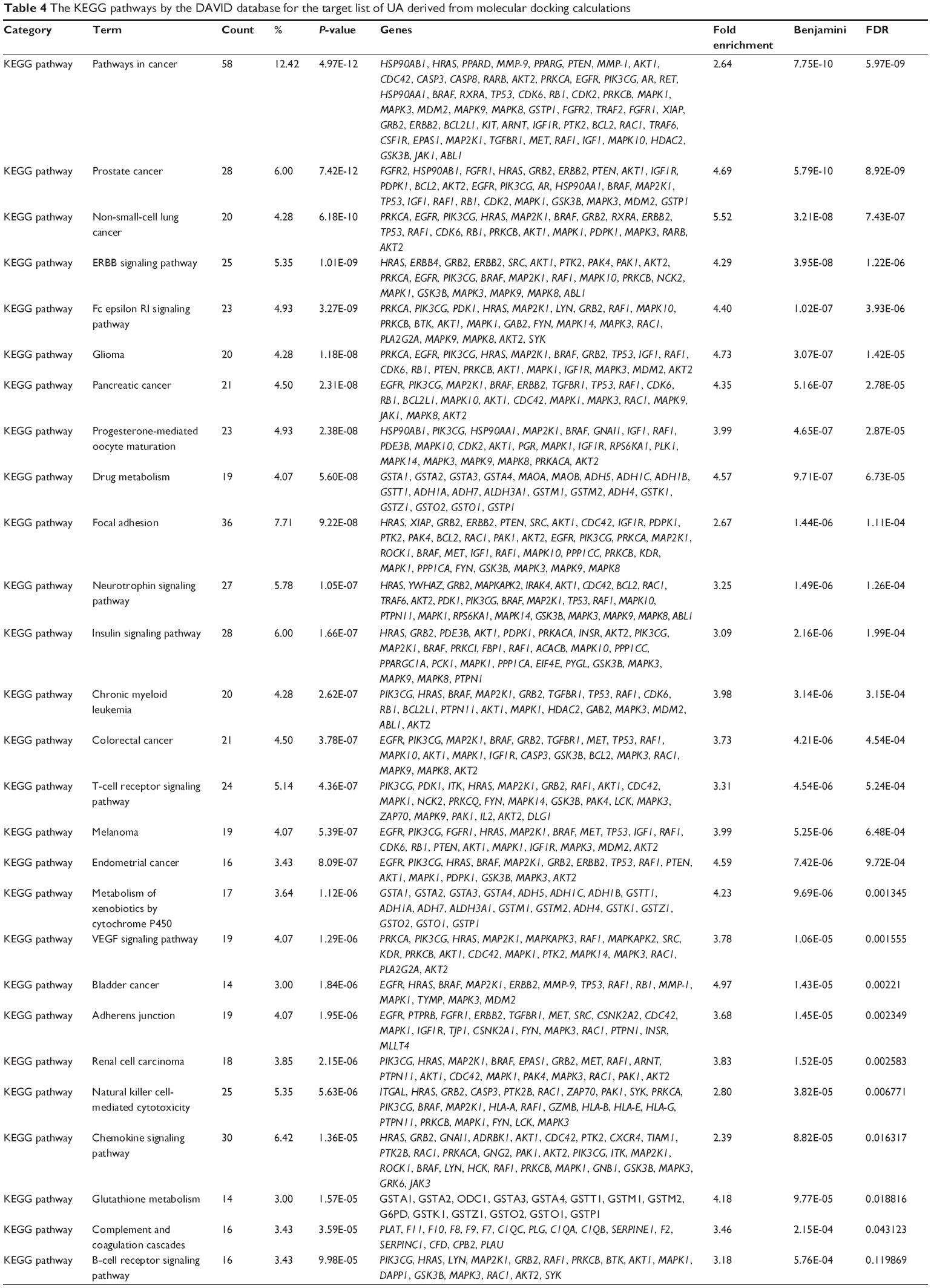 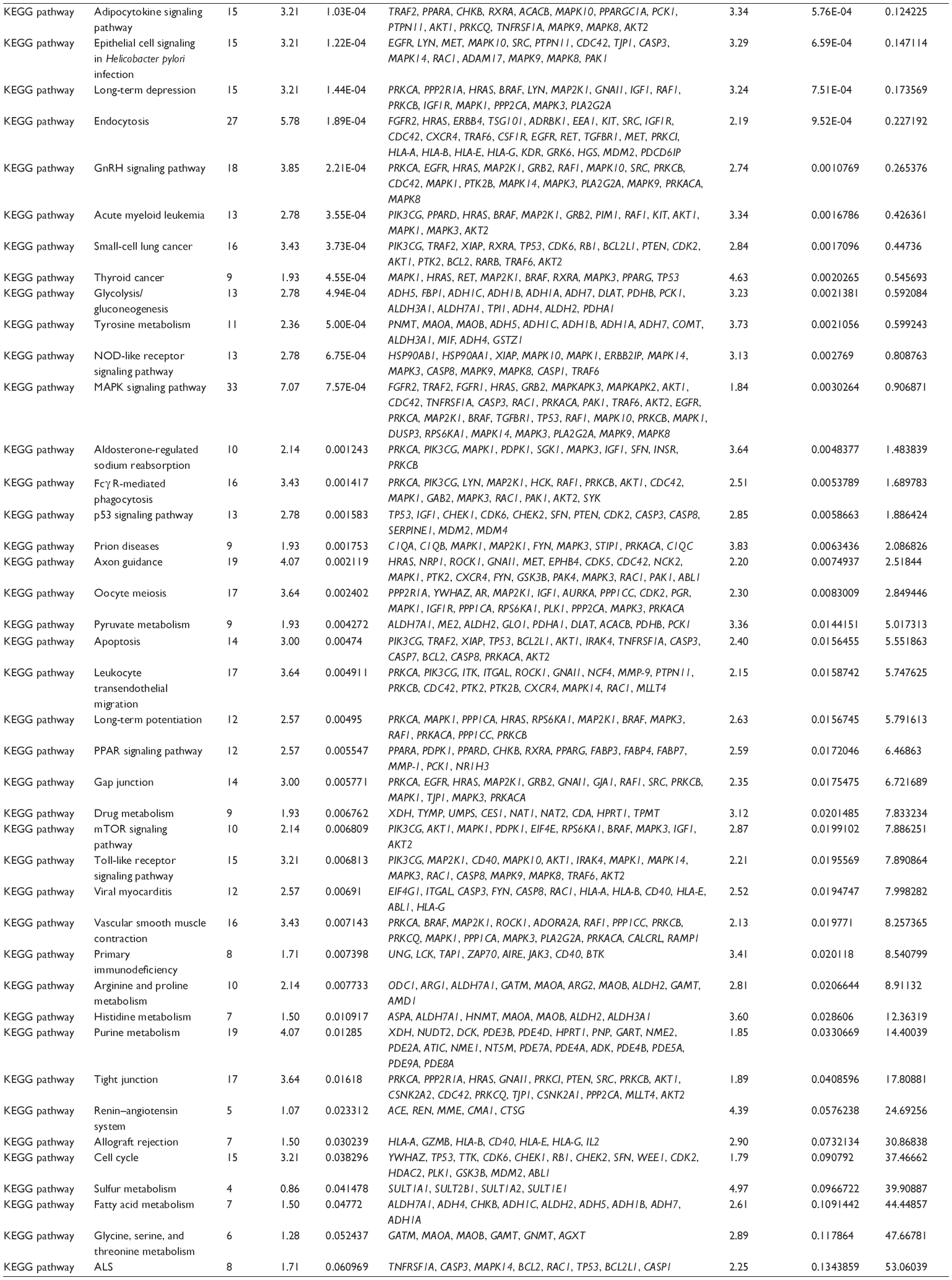  | Table 4 The KEGG pathways by the DAVID database for the target list of UA derived from molecular docking calculations |
Following the bioinformatical prediction of the molecular interactome of UA, we functionally and mechanistically examined the effect of UA on the liver fibrosis through the assessment of collagen accumulation and ROS generation in vitro and in vivo.
UA ameliorates DMN-induced hepatic fibrosis in rats
First, we established the model of liver fibrosis and examined the effect of UA on liver fibrosis in rats through examining the accumulation of collagen in rat HSCs. As shown in Figure 1A and B and Table 5, there is marked difference in the collagen disposition in rat HSCs. Compared to the vehicle group, rats that received 1% DMN treatment remarkably increased the accumulation of collagen in HSCs (Figure 1B). Administration of rats with UA at 20 mg/kg and 40 mg/kg decreased the collagen accumulation in rat HSCs (Figure 1C and D). Notably, high dose (40 mg/kg) of UA normalized the level of collagen in rat HSCs (Figure 1D).
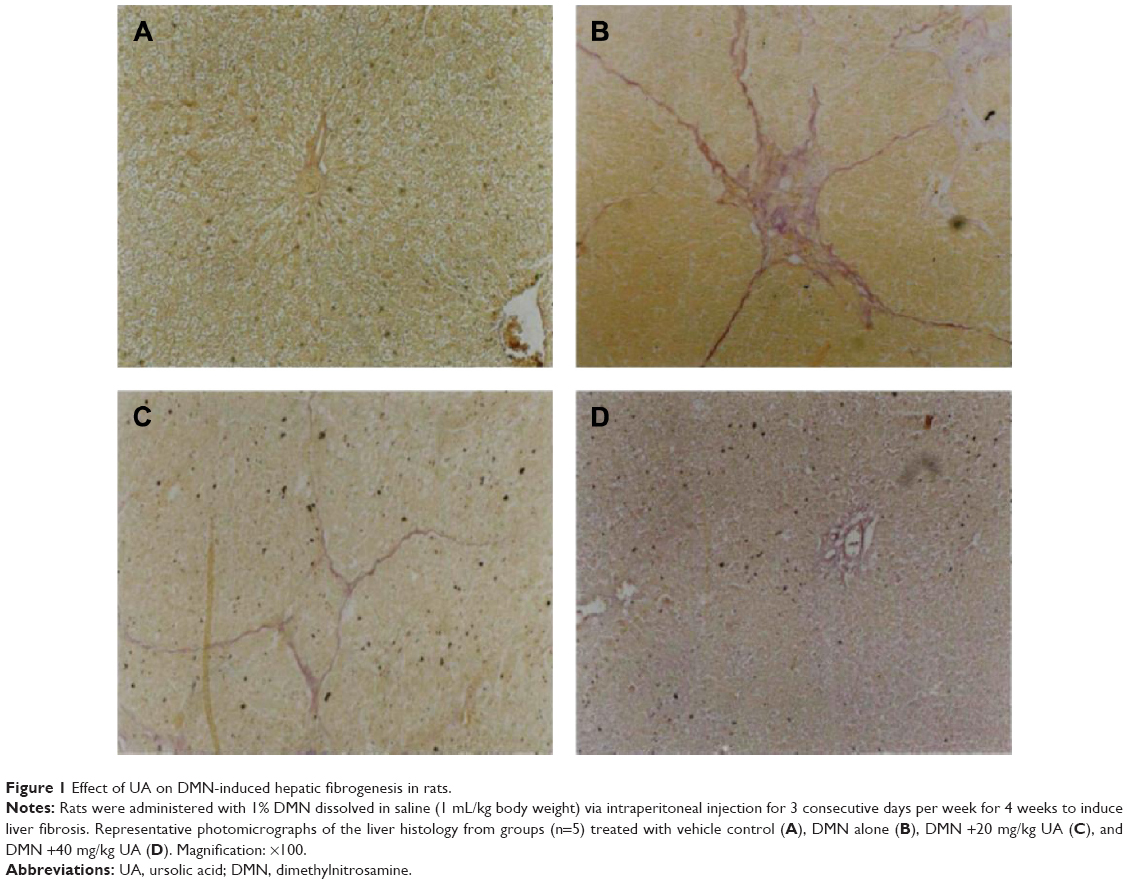 | Figure 1 Effect of UA on DMN-induced hepatic fibrogenesis in rats. |
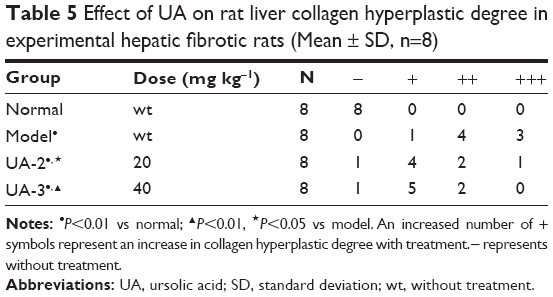 | Table 5 Effect of UA on rat liver collagen hyperplastic degree in experimental hepatic fibrotic rats (Mean ± SD, n=8) |
Furthermore, we examined the expression level of α-SMA, a marker of HSCs activation and fibrogenesis in rat liver. Treatment of rats with 1% DMN gave a remarkable increase (5.7-fold) in the expression lever of α-SMA in rat liver, compared to the vehicle control (Figure 2A and B, P<0.001). However, administration of rats with UA at 20 mg/kg and 40 mg/kg markedly decreased UA-induced expression level of α-SMA by 54.9% and 70.6% in rat liver, respectively (Figure 2A and B, P<0.001). Higher dose (40 mg/kg) of UA exhibited a more potent inhibitory effect on the expression level of α-SMA than that of low dose (20 mg/kg) of UA in rat liver (Figure 2A and B). Taken together, it shows that UA exerts an ameliorating effect on DMN-induced liver fibrosis in rats through reducing the accumulation of collagen and suppressing the expression of α-SMA in rat HSCs.
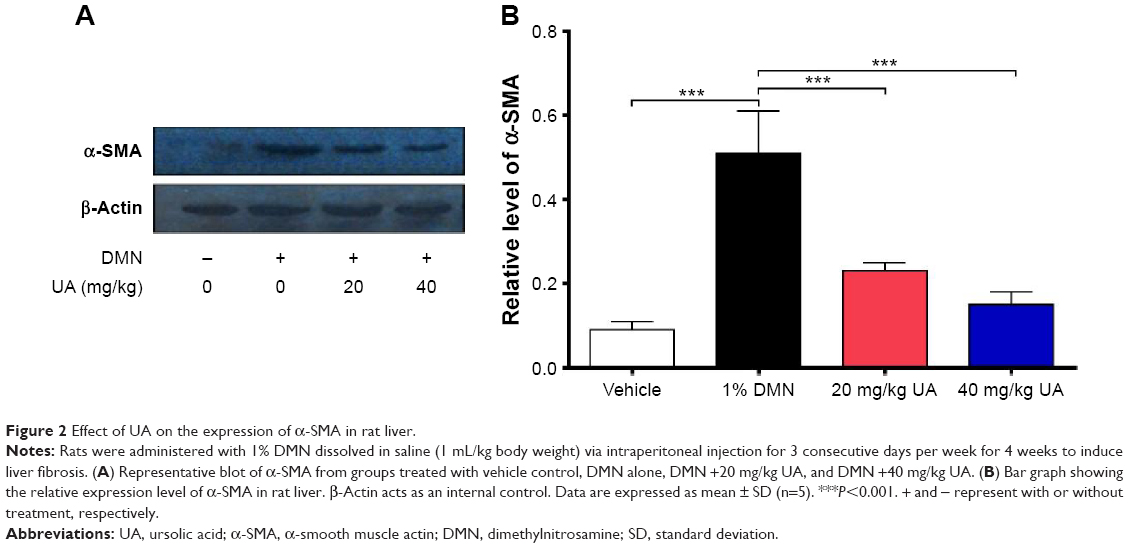 | Figure 2 Effect of UA on the expression of α-SMA in rat liver. |
UA restores serum SOD level and suppresses serum MDA in DMN-induced liver fibrotic rats
Growing evidence shows that oxidative stress has been implicated in the pathogenesis of liver fibrosis.32 The antioxidative effect of UA was examined in rats treated with 1% DMN by determining the serum levels of SOD and MDA. SOD has a capability of detoxifying superoxide and protecting cells against oxidative stress, whereas MDA is a principal toxic product of lipid peroxidation under oxidative stress.33 As shown in Figure 3A, DMN-alone treated group exhibited low serum level of SOD, and there was a 29.4% decrease (P<0.001), compared to the vehicle group; whereas UA treatment remarkably increased the serum level of SOD. In comparison to the DMN group, there was a 1.6- and 2.0-fold rise in the serum level of SOD (P<0.001). On the contrary, there was an 8.4-fold increase in the serum level of MDA in DMN-alone treated group, compared to the vehicle group (Figure 3B, P<0.001). UA treatment suppressed DMN-induced elevation in the serum level of MDA. There was a 78.4% and 84.2% reduction in the serum level of MDA, compared to DMN group (Figure 3B, P<0.001). Taken together, the data show that UA treatment can attenuate oxidative stress through the regulation of serum level of SOD and MDA in DMN-induced liver fibrotic rats.
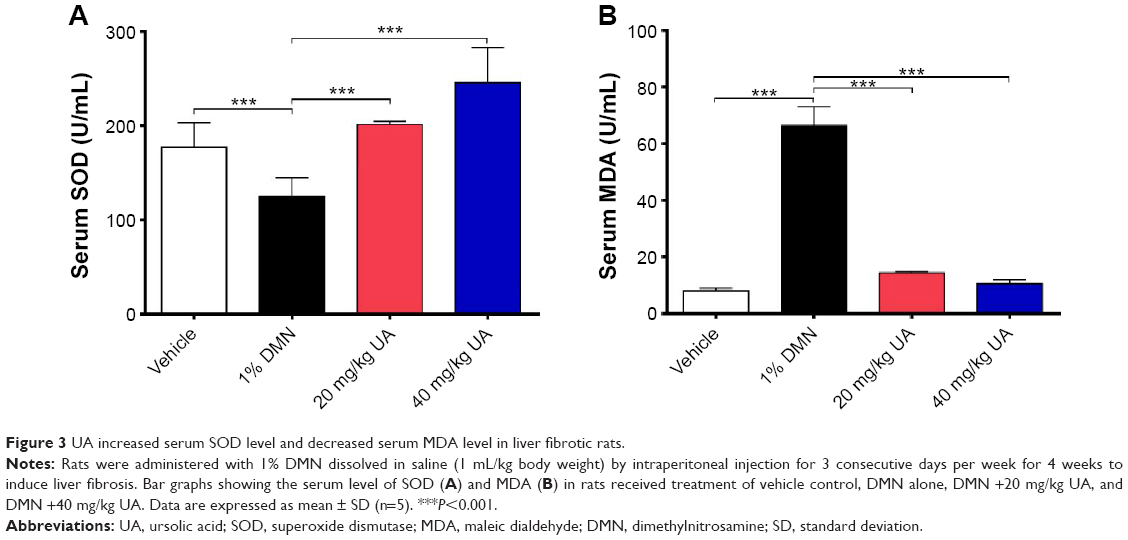 | Figure 3 UA increased serum SOD level and decreased serum MDA level in liver fibrotic rats. |
UA inhibits proliferation in HSC-T6 cells
Since we have observed the beneficial effects of UA in DMN-induced liver fibrotic rats, we further investigated the molecular mechanisms that underlie the antifibrotic effect of UA in vitro. We first examined the effect of UA (50 μM), DPI (20 μM, an NOX inhibitor), AG490 (50 μM, a specific and potent inhibitor of JAK2), and PD98059 (30 μM, a potent and selective inhibitor of MAP kinase) on the proliferation of HSC-T6 cells using MTT assay (Figure 4). Treatment of HSC-T6 cells with leptin (100 ng/mL) markedly induced cell proliferation over 48 hours (P<0.001). Treatment of HSC-T6 cells with UA, AG490, DPI, or PD98059 prevented leptin-induced proliferation over 48 hours (Figure 4). DPI exhibited the most potent preventive effect, and the data showed that UA exerted a comparable inhibitory effect to DPI to the cell proliferation. Taken together, the initial data suggest that ROS, JAK2, and MAPK/ERK signaling pathways are involved in the leptin-promoted proliferation in HSC-T6 cells.
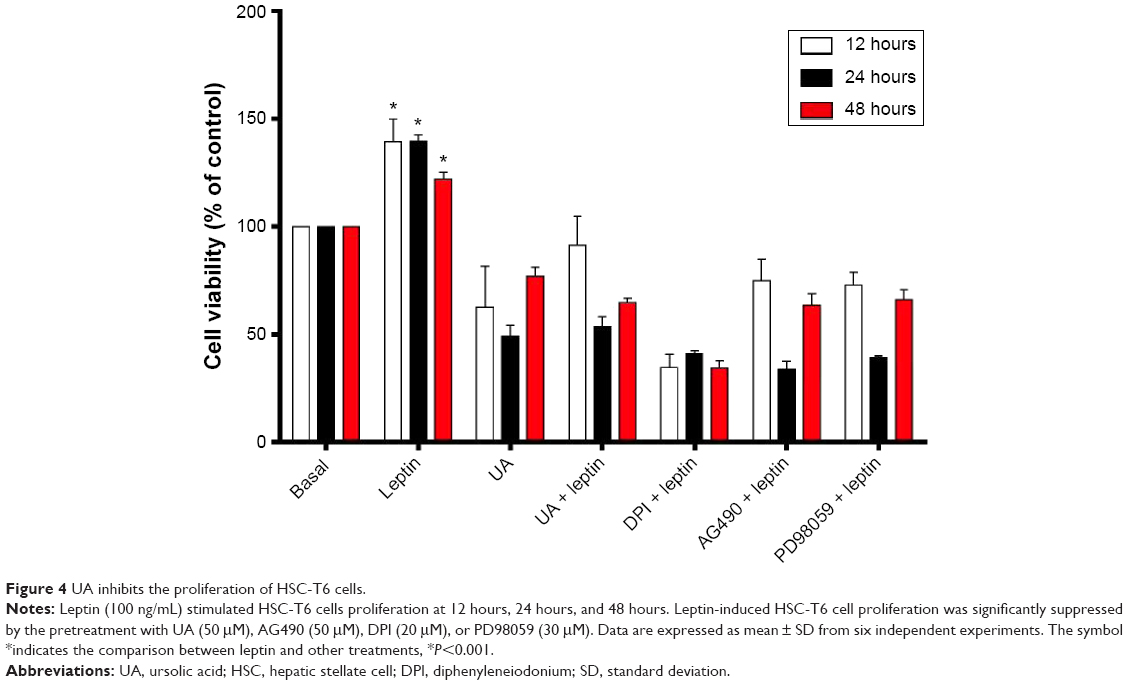 | Figure 4 UA inhibits the proliferation of HSC-T6 cells. |
UA suppresses intracellular ROS generation in HSC-T6 cells
We also examined the effect of UA on the ROS generation in the HSC-T6 cells using DCF-DA fluorescence (Figure 5A and B). Within 1 hour, leptin-stimulated HSC-T6 cells showed a significant increase in ROS production compared to the vehicle control group. There was a 3.4-fold elevation in the intracellular ROS level in leptin (100 ng/mL)-treated cells, compared to basal level (P<0.001; Figure 5A). The leptin-induced effect on ROS generation was abolished by pretreatment with UA (50 μM), NAC (10 mM, a ROS scavenger), DPI (20 μM), or AG490 (50 μM) in HSC-T6 cells (P<0.001; Figure 5A). Of note, UA suppressed leptin-induced ROS generation in a time-dependent manner over 24 hours, which was similar to DPI (Figure 5B). NAC potently scavenged intracellular ROS level within 1 hour; however, the ROS scavenging effect of NAC was gradually attenuated over 24 hours (Figure 5B). Taken together, the data suggest that UA reduces intracellular ROS level mainly through the inhibition of enzymatic source of ROS, rather than the scavenging of intracellular ROS.
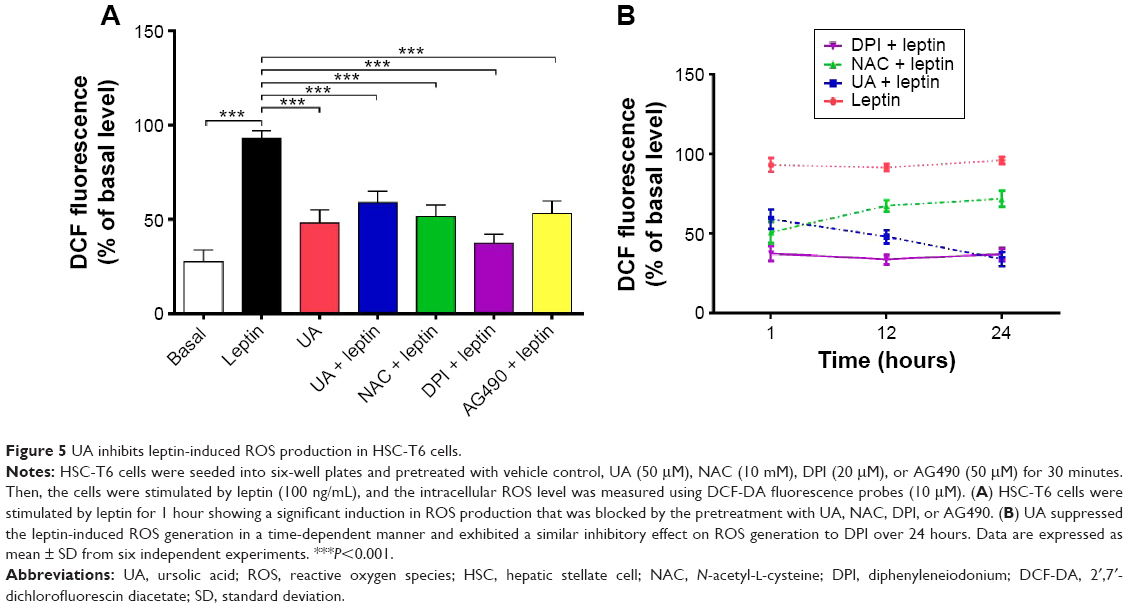 | Figure 5 UA inhibits leptin-induced ROS production in HSC-T6 cells. |
UA inhibits leptin-induced NOX activation in HSC-T6 cells
Since we observed the inhibitory effect of UA on intracellular ROS generation via the regulation of enzymatic source, we further examined the effect of UA on the activity of NOX in HSC-T6 cells. As shown in Figure 6, the NOX activity remarkably increased in the HSC-T6 cells when treated with leptin (100 ng/mL) for 1 hour, 12 hours, or 24 hours, compared to the basal level (P<0.001). UA (50 μM) markedly inhibited leptin-induced NOX activation after 1 hour, 12 hours, or 24 hours. UA’s inhibitory effect on NOX activity was similar to that of DPI (20 μM) and AG490 (50 μM). However, NAC showed no effect on leptin-induced NOX activation. Taken together, the data further showed that UA suppresses intracellular ROS production via the modulation of enzymatic source, NOX, in HSC-T6 cells.
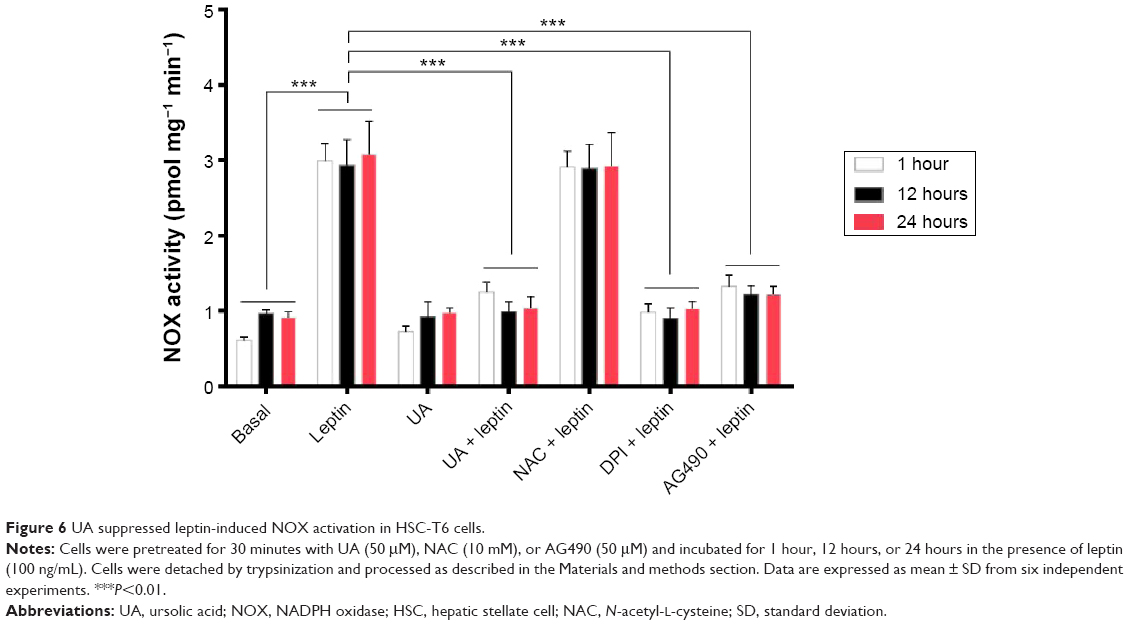 | Figure 6 UA suppressed leptin-induced NOX activation in HSC-T6 cells. |
UA inhibits leptin-induced p47phox membrane translocation
p47phox translocation from cytoplasm to cell membrane occurs during NOX activation; thus, the translocation was examined through testing the content of the membrane-bound p47phox and the total cellular p47phox in HSC-T6 cells. There was a marked alteration in the leptin (100 ng/mL)-induced translocation of p47phox, whereas the total level of p47phox did not dramatically change in the presence of UA (50 μM), DPI (20 μM), or AG490 (50 μM) (Figure 7A and B). Stimulation of HSC-T6 cells with leptin for 30 minutes remarkably induced the translocation of p47phox from cytoplasm to cell membrane. There was a 1.5-fold increase in the membrane- bound p47phox in leptin-stimulated cells, compared to the control cells (P<0.01; Figure 7A and B). Pretreatment of cells with UA blocked the leptin-induced translocation of p47phox and normalized the membrane-bound p47phox. These effects were observed in DPI- and AG490-treated cells as well (P<0.05; Figure 7A and B). Collectively, UA blocks the translocation of p47phox from cytoplasm to cell membrane, contributing to the inhibitory effect of NOX activity and ROS generation in HSC-T6 cells.
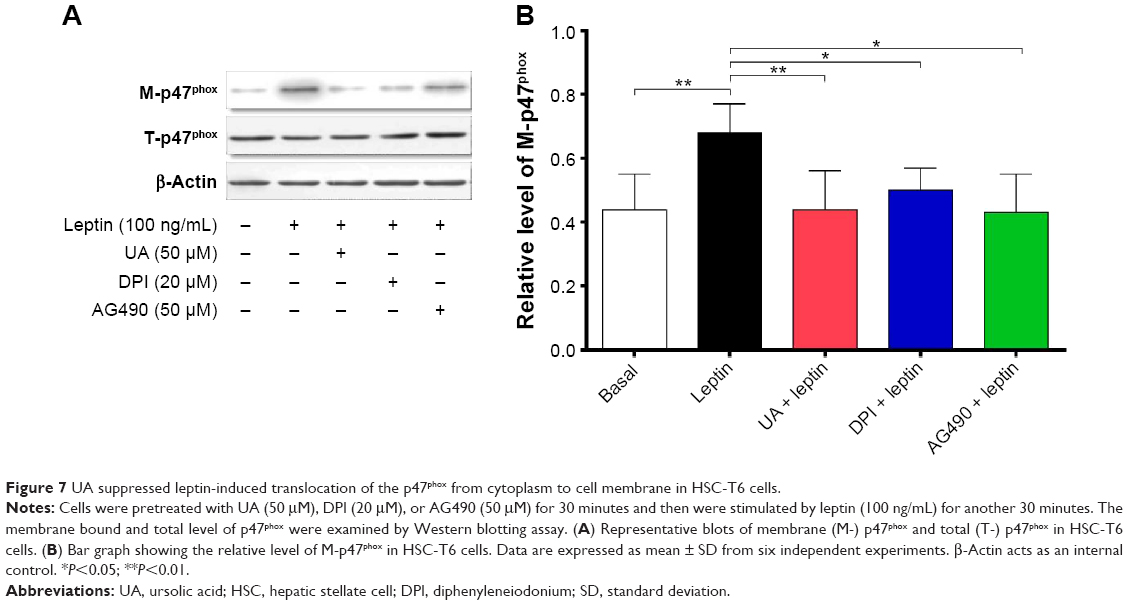 | Figure 7 UA suppressed leptin-induced translocation of the p47phox from cytoplasm to cell membrane in HSC-T6 cells. |
UA inhibits the leptin-induced expression of gp91phox, p22phox, p67phox, and Rac1
To further investigate the effect of UA on the activity of NOX in HSC-T6 cells, we examined the effect of UA on the expression level of NOX subunits, including gp91phox, p22phox, p67phox, and Rac1, which are important to NOX activity. As shown in Figure 8A and B, after HSC-T6 cells were stimulated with leptin (100 ng/mL) for 12 hours, the expression level of gp91phox, p22phox, p67phox, and Rac1 was remarkably increased 1.9-, 1.7-, 2.1-, and 1.9-fold compared to the control group, respectively (P<0.01 or <0.001). Pretreatment of cells with UA (50 μM) markedly decreased the leptin-induced protein expression of gp91phox, p22phox, p67phox, and Rac1 by 71.9%, 62.5%, 61.0%, and 71.9%, compared to the leptin-treated cells, respectively (P<0.001). These results suggest that UA potently negatively regulate the expression level of NOX4 subunits, contributing to the reducing effect of ROS generation.
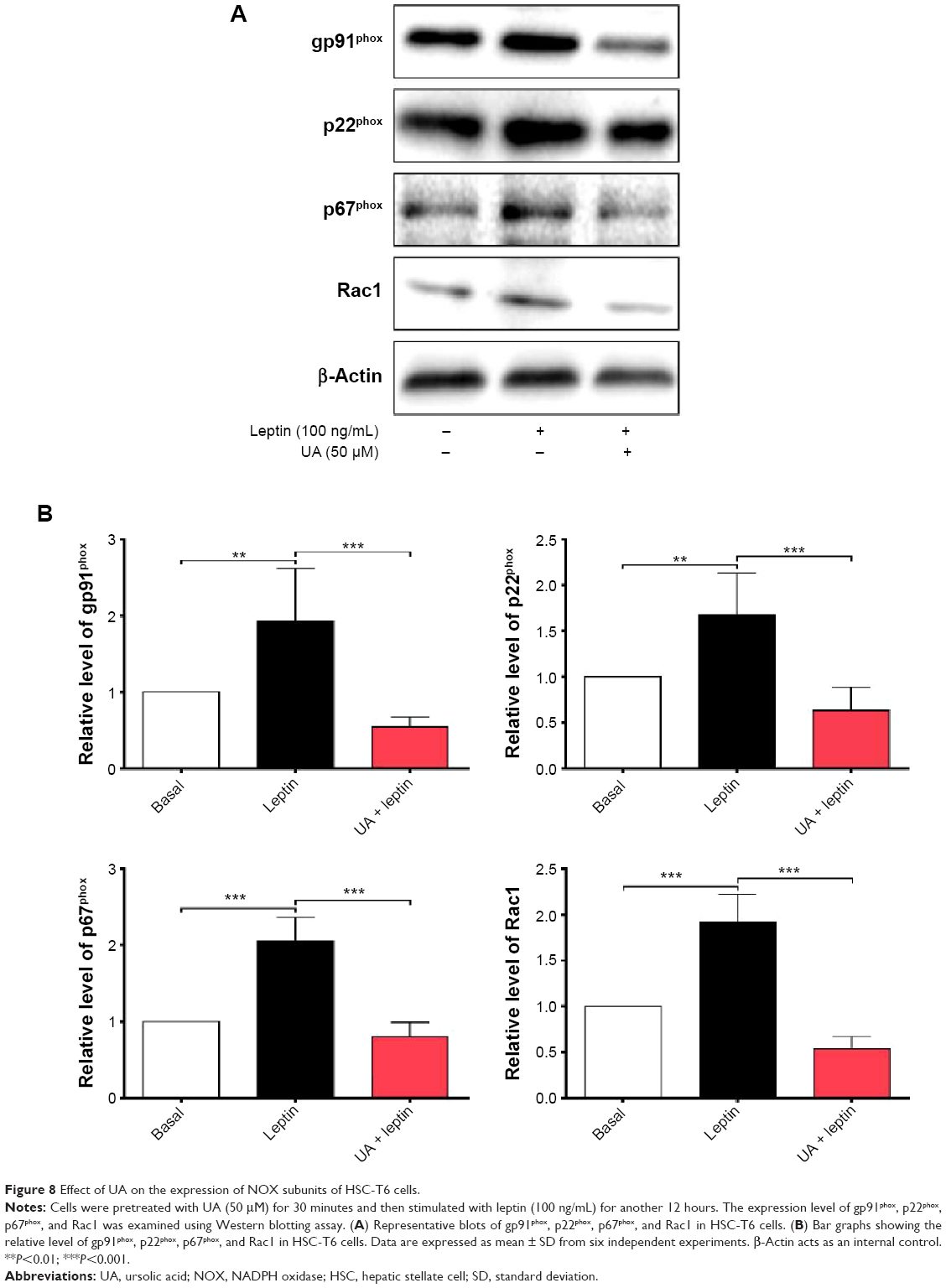 | Figure 8 Effect of UA on the expression of NOX subunits of HSC-T6 cells. |
UA inhibits ERK, PI3K/Akt, and p38 MAPK signaling pathways in HSC-T6 cells
NOX-derived ROS regulate signal transduction involved in liver fibrosis in HSCs. We evaluated the effect of UA on the modulation of PI3K/Akt, p38 MAPK, and ERK1/2 signaling pathways in HSC-T6 cells using Western blotting analysis. As shown in Figure 9A and B, 30-minute stimulation of HSC-T6 cells with 100 ng/mL leptin remarkably increased the level of p-ERK1 and p-ERK2 by 1.6- and 1.4-fold, compared to the control group, respectively (P<0.05 or 0.01). Pretreatment of cells with UA (50 μM) completely blocked the leptin-induced phosphorylation of ERK1/2 in HSC-T6 cells (P<0.01; Figure 9A and B). Incubation of HSC-T6 cells with DPI (20 μM), AG490 (50 μM), or PD98059 (30 μM) markedly inhibited leptin-induced phosphorylation of ERK1 (P<0.05 or 0.01; Figure 9A and B). There was a decrease in the level of p-ERK2 but without statistical significance when treated with DPI, AG490, or PD98059 (P>0.05; Figure 9A and B). Furthermore, the effect of UA on PI3K/Akt and p38 MAPK signaling pathways was examined in HSC-T6 cells. As shown in Figure 10A and B, stimulation of HSC-T6 cells with leptin markedly increased the level of p-p38 MAPK, PI3K, and p-Akt (P<0.05 or 0.001). However, pretreatment of cells with UA dramatically suppressed leptin-induced increase in the level of p-p38 MAPK, PI3K, and p-Akt (P<0.05 or 0.001; Figure 10A and B). Notably, DPI, SB203580, and LY294002 showed a similar effect to UA on leptin-induced elevation in the level of p-p38 MAPK, PI3K, and p-Akt (P<0.001; Figure 10A and B). Taken together, UA negatively regulates ERK, PI3K/Akt, and p38 MAPK signaling pathways in HSC-T6 cells, which contributes, at least in part, to the underlying mechanism of the antifibrotic effect of UA.
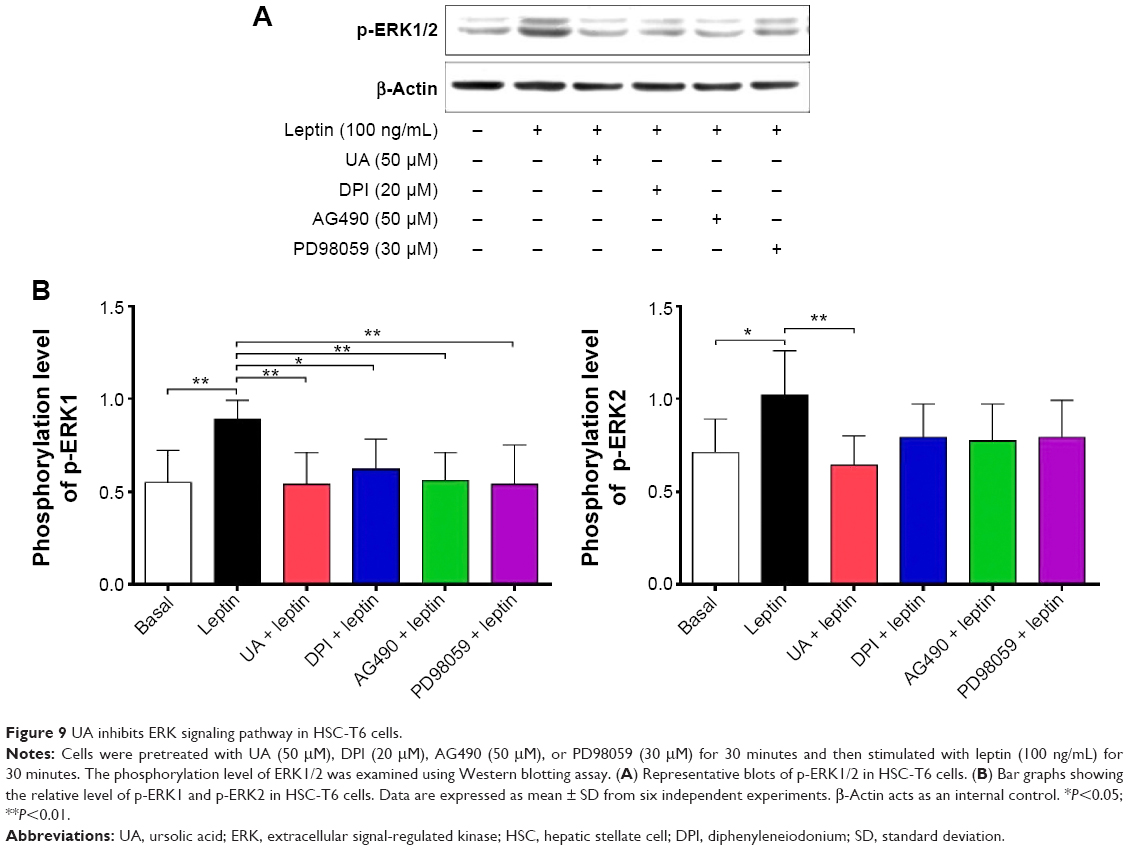 | Figure 9 UA inhibits ERK signaling pathway in HSC-T6 cells. |
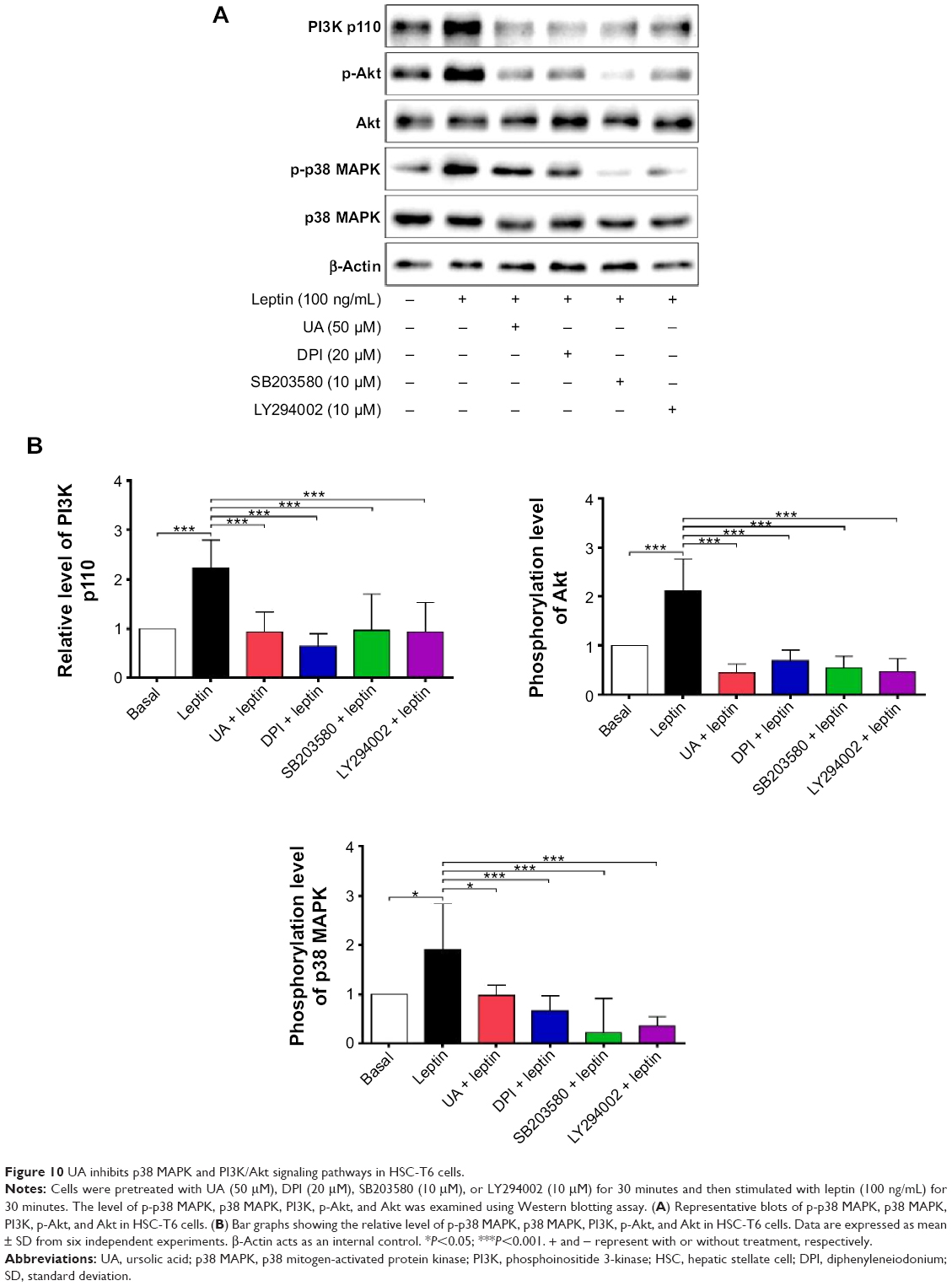 | Figure 10 UA inhibits p38 MAPK and PI3K/Akt signaling pathways in HSC-T6 cells. |
UA promotes the expression of MMP-1 but inhibits the expression of TIMP-1 and type I collagen in HSC-T6 cells
Finally, we examined the effect of UA on the protein expression of TIMP-1 and MMP-1 and mRNA expression of type I collagen in HSC-T6 cells. HSC-T6 cells were treated with leptin at 100 ng/mL for 24 hours, the TIMP-1 protein expression level and the type I collagen mRNA expression level markedly increased, but the MMP-1 protein expression level significantly decreased (Figures 11A and B and 12A and B). Pretreatment of cells with UA (50 μM), DPI (20 μM), SB203580 (10 μM), or LY294002 (10 μM) inhibited leptin-promoted expression of TIMP-1 (P<0.001; Figure 11A and B), while UA and DPI remarkably increased leptin-suppressed expression level of MMP-1 (P<0.001; Figure 11A and B). SB203580 and LY294002 did not show significant regulatory effect on the expression of MMP-1 in HSC-T6 cells (P>0.05; Figure 11A and B). Moreover, pretreatment of HSC-T6 cells with UA, DPI, AG490, or PD98059 markedly blocked the leptin-induced mNRA expression of type I collagen (P<0.05 or <0.01; Figure 12A and B). Collectively, UA inhibits liver fibrosis with the involvement of ERK, PI3K/Akt, and p38 MAPK signaling pathways.
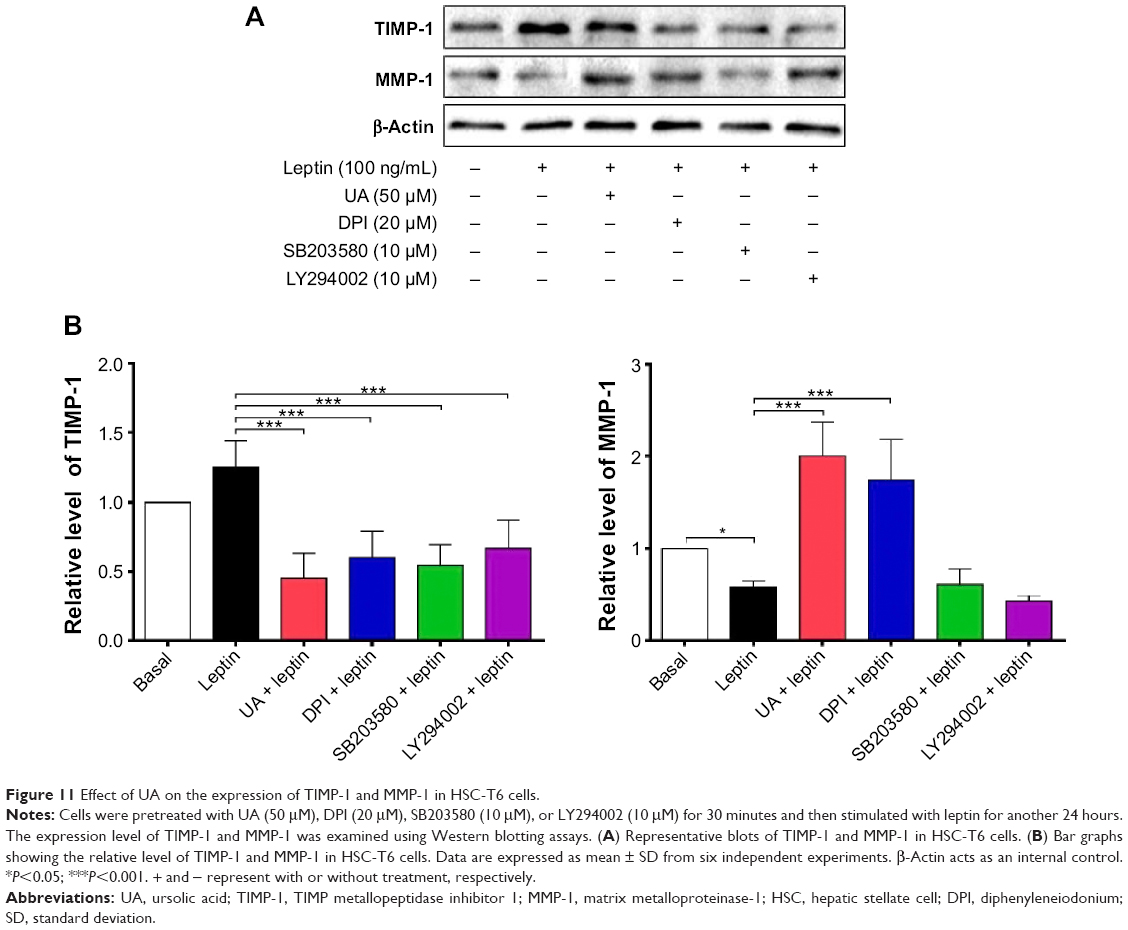 | Figure 11 Effect of UA on the expression of TIMP-1 and MMP-1 in HSC-T6 cells. |
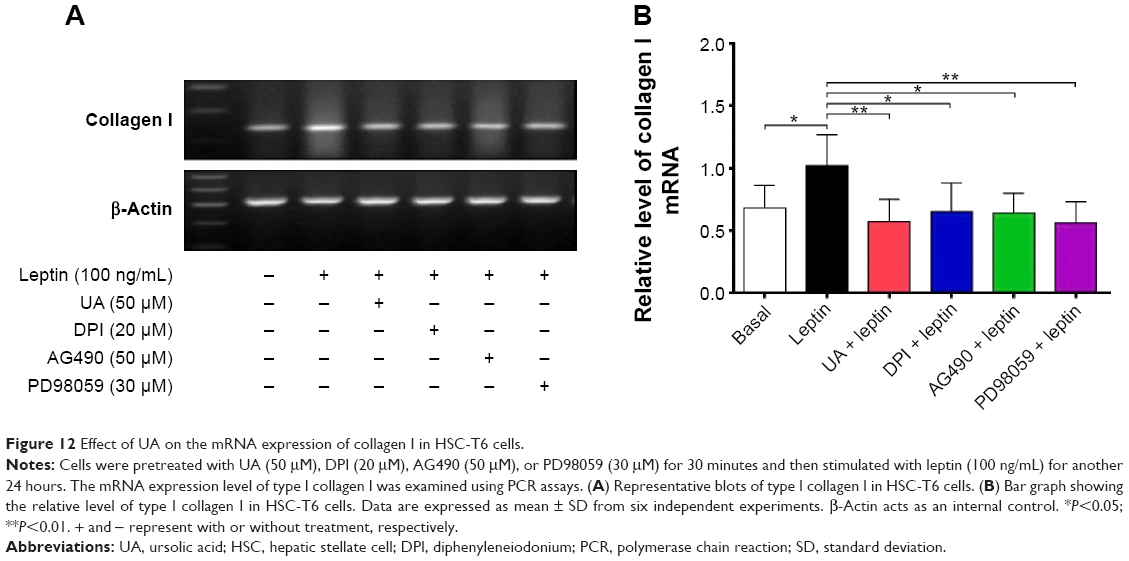 | Figure 12 Effect of UA on the mRNA expression of collagen I in HSC-T6 cells. |
Discussion
Liver fibrosis represents a major challenge with considerable morbidity and mortality worldwide, due to the complexity of etiology.34 Compelling evidence indicates that oxidative stress plays a causal role in the pathogenesis of liver fibrosis11,35,36 and that antioxidant may be a promising agent to treat liver fibrosis through the attenuation of oxidative stress.13,37 In the present study, we have predicted the molecular interactome of UA, and there are 611 molecular proteins possibly interacting with UA. The subsequential functional and mechanistic experiments show that UA reverses DMN-induced liver fibrosis through the attenuation of oxidative stress with the involvement of ERK, PI3K/Akt, and p38 MAPK signaling pathways in vitro and in vivo.
In recent years, there is a growing interest in the investigation of the beneficial effects and molecular mechanisms of UA that is a natural pentacyclic triterpenoid carboxylic acid and a major component of some traditional medicinal herbs.38 Increasing evidence shows that UA possesses a variety of bioactivities, including antioxidative, anti-inflammation, and anticancer activities, although the full spectrum of molecular targets has not yet been revealed.38,39 In the present study, the computational experiment provided an interactome of UA, with 611 potential molecular targets been predicted. The bioinformatic analysis showed various signaling pathways that may be able to explain the beneficial effects of UA. These results can provide a clue for us to investigate the beneficial effects and underlying mechanisms of UA.
Employment of computational and bioinformatic approaches have become a practical and valuable way to efficiently predict the molecular interactome of a chemical molecule. The DDI-CPI tool has been used to predict the potential targets, and DAVID has been employed to analyze the molecular targets and related signaling pathways that are regulated by UA. An approach using DDI-CPI server offers a fast, efficient, and inexpensive strategy to predict the potential targets, identify drug repositioning potential, and evaluate and determine adverse drug reactions of a chemical/drug via molecular docking of small compound across human proteome,19–21,24,40,41 although this web-based program has limitations that may affect the accuracy of the outcome.21 Our findings showed that UA may modulate a number of functional proteins and related signaling pathways. These proteins and signaling pathways have important roles in the regulation of redox homeostasis, cell proliferation, apoptosis, energy metabolism, xenobiotics metabolism, lipid and carbohydrate metabolism, and inflammatory response.
Oxidative stress plays a causal role in the development of liver fibrosis.11,35,36,42 It is caused by an increase in the generation of pro-oxidant molecules and/or a decrease in the antioxidants in a given cellular compartment, in which the pro-oxidants overweigh the capability of antioxidant systems, resulting in an imbalance in the amounts of oxidants and antioxidants. NOXs are multicomponent enzymes mediating the transfer of electrons from cytosolic NADPH to O2 to produce O2−,43–45 and they are the primary enzymatic source of ROS.46 NOXs are differentially expressed and distributed among the tissues and subject to regulation by various factors. Moreover, NOXs derived from ROS are also important signaling molecules under pathophysiological conditions. It is known that ROS is involved in a wide range of cellular processes,46–49 including host defence, inflammation, cellular signaling, gene expression, cellular death, cellular senescence, regulation of cell death, O2 sensing, biosynthesis, protein cross-linking, regulation of cellular redox potential, reduction of metal ions, regulation of matrix metalloproteinases, angiogenesis, and cross-link with the nitric oxide system. This role suggests that NOXs should be considered a potential pharmacological target for antifibrotic therapies.26,50
To date, seven members of the NOX family have been distinguished on the basis of the membrane spanning catalytic subunits of NOX or DUOX that they utilize to transfer electrons from NADPH to O2 and produce ROS. They are NOX1, 2, 3, 4, 5 as well as DUOX1- and 2-containing oxidases.46,51–54 The core structure of all NOX isoforms consists of six conserved transmembrane domains and a cytosolic C terminal. Increased activity and expression of NOX isoforms has been demonstrated in a wide variety of diseases and/or disorders, which has been marked by the excessive NOX-generated ROS resulted from upregulation of activity and/or expression of various NOX family members.55–57 In the last decade, extensive attention has been focused on the redox control and the underlying molecular mechanisms of NOX-dependent ROS generation. In addition, the regulation on NOX isoforms has also been studied.55–57 It has been shown that the regulation of NOX isoforms varies from transcriptional level to posttranslational level, although the underlying mechanisms of NOX regulation have not been fully revealed.55–57
NOX4, like the other NOX isoforms, its structure consists mainly of a six-transmembrane domain known as the gp9phox domain; NOX4 predominantly generates hydrogen peroxide rather than superoxide, although it is able to generate superoxide under specific conditions.35,42,58 Despite their extensive similarity in structure and enzymatic function, members of the NOX family differ in their mechanism of activation. In particular, NOX4 requires interaction with a second membrane-bound subunit, p22phox.54,59 NOX4 is a multiprotein complex that generates ROS in both phagocytic and nonphagocytic cells, regulating intracellular signaling.11,36 In the liver, NOX4 plays a central role in fibrogenesis.11 A functionally active form of NOX4 is expressed in HSCs, and the ROS derived from NOX4 act as a second messenger for profibrogenic factor signal transduction in HSCs. HSCs activation is a critical event in the occurrence and development of fibrosis, because activated HSCs are the primary source of extracellular matrix in liver upon injury.2 This role suggests that NOX4 may act as a potential therapeutic target for antifibrotic therapies.26,50
The NOX subunits and regulatory protein are essential for the superoxide generating function and activity of NOX isoforms. The subunits and regulatory proteins required for NOX4 activation include membrane-bound and cytosolic proteins – p22phox, which is a membrane-bound protein helping to stabilize the NOX proteins and dock cytosolic factors, and p47phox, p67phox, the small GTPase Rac, and the modulatory p40phox, which are cytosolic proteins working together to activate the NOX enzymes.46,47,60 Rac1, a member of the Rho family of small GTPase proteins, regulates the activation of NOX4 and the production of ROS and plays an important role in regulating NOX activity.59 Over-expression of Rac1 in HSCs promotes liver injury and fibrosis in mice,61 and induces the phagocytosis of apoptotic bodies by HSCs.62 The level of ROS in cells and tissues is controlled by the tissue- and stimulus-specific expressions of NOX protein and their regulatory subunits and by the acute regulation via calcium, protein phosphorylation, guanine nucleotide exchange on Rac, and the assembly of regulatory subunits.54 Our results demonstrated that leptin induced an increase in the expression level of the NOX4 regulatory subunits, including gp91phox, p22phox, p67phox, and Rac1 in HSC-T6 cells and that pretreatment with UA blocked the leptin-induced increase in the expression level of gp91phox, p22phox, p67phox, and Rac1. Furthermore, pretreatment of UA inhibited the translocation of p47phox from cytoplasm to cell membrane. The negative regulatory effect of UA on the expression of NOX4 subunits explains the reduction of ROS generation in HSC-T6 cells and rats. Consequently, the UA-attenuated oxidative stress leads to a reversal of liver fibrosis and protection of HSC-T6 cells, evident from the decrease in the accumulation of type I collagen in HSCs and the expression of TIMP-1 and type I collagen at transcriptional or translational level, but increase the level of MMP-1.
Furthermore, increasing evidence shows that posttranslational modification of NOX represents an important mechanism for NOX regulation. The phosphorylation of NOX subunits and their assembly into active complexes are mediated by various mechanisms, of which many of them are redox sensitive. It has been demonstrated that a number of stimuli and pathways are involved in the activation of NOX by phosphorylation. They comprise phospholipases (PLC/γ, PLD), arachidonic acid metabolites, GTP-binding proteins (Ras, Rac1/2), PKC, PI3K, MAPK, and nonreceptor protein tyrosine kinases.63,64 Previous studies showed that NOX-mediated ROS generation through the activation of Akt and MAPK phosphorylation, leading to an increase in AP-1-DNA-binding activity, promotion in the expression of type I collagen, TGF-β1, and other inflammatory cytokines26 and that NOX-mediated ROS production interplays with p38 MAPK50 and JAK1/2-STAT3/ERK signaling pathways in HSCs.65 In agreement with previous studies, the data clearly show that UA modulates the NOX activity and expression with the involvement of ERK, PI3K/Akt, and p38 MAPK signaling pathways in HSCs via the employment of specific chemical inhibitors. In aggregate, it suggests that targeting NOX4 represents a promising strategy for liver fibrosis treatment. Notably, recent studies have indicated that the NOX1/4 dual inhibitor GKT137831 can significantly inhibit the activation of HSCs and the development of liver fibrosis.12,66–68 This inhibitor is currently being investigated in Phase II clinical trials. Therefore, NOX-targeting drugs with low toxicity that are effective against fibrosis are expected to lead to a breakthrough in the treatment of liver fibrosis. However, there are many factors that may affect the therapeutic outcome of antifibrotic agents in the treatment of liver fibrosis.69 Thus, more well-designed studies are needed to investigate the therapeutic effect and underlying mechanism of the antifibrotic effect.
In summary, the present study has depicted a full spectrum of molecular targets and related signaling pathways that possibly respond to UA treatment. The benchmarking results have clearly shown that UA exhibits a potent antifibrotic effect in rats evident from the reduction in the accumulation of type I collagen in rat HSCs. The underlying molecular mechanism and beneficial effect of UA can be ascribed to the oxidative stress attenuating effect through negative regulation of NOX4 activity and expression with the involvement of ERK, PI3K/Akt, and p38 MAPK signaling pathways in HSCs. These results render UA as a promising antifibrotic agent via targeting NOX4, and further studies are warranted to evaluate the safety and efficacy of UA in the treatment of liver fibrosis.
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Murphy SL, Xu J, Kochanek KD. Deaths: final data for 2010. Natl Vital Stat Rep. 2013;61(4):1–117. | ||
Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115(2):209–218. | ||
Mitchell AE, Colvin HM, Palmer Beasley R. Institute of Medicine recommendations for the prevention and control of hepatitis B and C. Hepatology. 2010;51(3):729–733. | ||
Davis GL, Alter MJ, El-Serag H, Poynard T, Jennings LW. Aging of hepatitis C virus (HCV)-infected persons in the United States: a multiple cohort model of HCV prevalence and disease progression. Gastroenterology. 2010;138(2):e1–e6. | ||
Liu J, Fan D. Hepatitis B in China. Lancet. 2007;369(9573):1582–1583. | ||
Zhang F, Zhu H, Wu Y, et al. HIV, hepatitis B virus, and hepatitis C virus co-infection in patients in the China national free antiretroviral treatment program, 2010–2012: a retrospective observational cohort study. Lancet Infect Dis. 2014;14(11):1065–1072. | ||
Edmison J, McCullough AJ. Pathogenesis of non-alcoholic steatohepatitis: human data. Clin Liver Dis. 2007;11(1):75–104,ix. | ||
Brenner DA. Molecular pathogenesis of liver fibrosis. Trans Am Clin Climatol Assoc. 2009;120:361–368. | ||
Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134(6):1655–1669. | ||
Novo E, Cannito S, Paternostro C, Bocca C, Miglietta A, Parola M. Cellular and molecular mechanisms in liver fibrogenesis. Arch Biochem Biophys. 2014;548:20–37. | ||
De Minicis S, Brenner DA. NOX in liver fibrosis. Arch Biochem Biophys. 2007;462(2):266–272. | ||
Cohen-Naftaly M, Friedman SL. Current status of novel antifibrotic therapies in patients with chronic liver disease. Therap Adv Gastroenterol. 2011;4(6):391–417. | ||
Rosenbloom J, Mendoza FA, Jimenez SA. Strategies for anti-fibrotic therapies. Biochim Biophys Acta. 2013;1832(7):1088–1103. | ||
Dufour D, Pichette A, Mshvildadze V, et al. Antioxidant, anti-inflammatory and anticancer activities of methanolic extracts from Ledum groenlandicum Retzius. J Ethnopharmacol. 2007;111(1):22–28. | ||
Saravanan R, Viswanathan P, Pugalendi KV. Protective effect of ursolic acid on ethanol-mediated experimental liver damage in rats. Life Sci. 2006;78(7):713–718. | ||
Saraswat B, Visen PK, Agarwal DP. Ursolic acid isolated from Eucalyptus tereticornis protects against ethanol toxicity in isolated rat hepatocytes. Phytother Res. 2000;14(3):163–166. | ||
Shen YM, Zhu X, Zhang KH, et al. [Effect of ursolic acid on proliferation and apoptosis of hepatic stellate cells in vitro]. Zhonghua Gan Zang Bing Za Zhi. 2008;16(4):298–301. | ||
Steinkamp-Fenske K, Bollinger L, Völler N, et al. Ursolic acid from the Chinese herb danshen (Salvia miltiorrhiza L.) upregulates eNOS and downregulates Nox4 expression in human endothelial cells. Atherosclerosis. 2007;195(1):e104–e111. | ||
Huang da W, Shermann BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. | ||
Luo H, Chen J, Shi L, et al. DRAR-CPI: a server for identifying drug repositioning potential and adverse drug reactions via the chemical-protein interactome. Nucleic Acids Res. 2011;39(Web Server issue): W492–W498. | ||
Luo H, Zhang P, Huang H, et al. DDI-CPI, a server that predicts drug-drug interactions through implementing the chemical-protein interactome. Nucleic Acids Res. 2014;42(Web Server issue): W46–W52. | ||
Wang R, Fang X, Lu Y, Wang S. The PDBbind database: collection of binding affinities for protein-ligand complexes with known three-dimensional structures. J Med Chem. 2004;47(12):2977–2980. | ||
Yang L, Luo H, Chen J, Xing Q, He L. SePreSA: a server for the prediction of populations susceptible to serious adverse drug reactions implementing the methodology of a chemical-protein interactome. Nucleic Acids Res. 2009;37(Web Server issue):W406–W412. | ||
Yang L, Chen J, Shi L, Hudock MP, Wang K, He L. Identifying unexpected therapeutic targets via chemical-protein interactome. PLoS One. 2010;5(3):e9568. | ||
Vogel S, Piantedosi R, Frank J, et al. An immortalized rat liver stellate cell line (HSC-T6): a new cell model for the study of retinoid metabolism in vitro. J Lipid Res. 2000;41(6):882–893. | ||
Bataller R, Schwabe RF, Choi YH, et al. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J Clin Invest. 2003;112(9):1383–1394. | ||
Herrera B, Murillo MM, Alvarez-Barrientos A, Beltrán J, Fernández M, Fabregat I. Source of early reactive oxygen species in the apoptosis induced by transforming growth factor-β in fetal rat hepatocytes. Free Radic Biol Med. 2004;36(1):16–26. | ||
Wang H, Joseph JA. Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radic Biol Med. 1999;27(5–6):612–616. | ||
LeBel CP, Ischiropoulos H, Bondy SC. Evaluation of the probe 2′,7′-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem Res Toxicol. 1992;5(2):227–231. | ||
Proell V, Carmona-Cuenca I, Murillo MM, Huber H, Fabregat I, Mikulits W. TGF-β dependent regulation of oxygen radicals during transdifferentiation of activated hepatic stellate cells to myofibroblastoid cells. Comp Hepatol. 2007;6:1. | ||
Zhou ZW, Xie XL, Zhou SF, Li CG. Mechanism of reversal of high glucose-induced endothelial nitric oxide synthase uncoupling by tanshinone IIA in human endothelial cell line EA.hy926. Eur J Pharmacol. 2012;697(1–3):97–105. | ||
Sanchez-Valle V, Chavez-Tapia NC, Uribe M, Mendez-Sanchez N. Role of oxidative stress and molecular changes in liver fibrosis: a review. Curr Med Chem. 2012;19(28):4850–4860. | ||
Van Raamsdonk JM, Hekimi S. Superoxide dismutase is dispensable for normal animal lifespan. Proc Natl Acad Sci U S A. 2012;109(15):5785–5790. | ||
Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annu Rev Pathol. 2011;6:425–456. | ||
Galli F, Battistoni A, Gambari R, et al; Working Group on Inflammation in Cystic Fibrosis. Oxidative stress and antioxidant therapy in cystic fibrosis. Biochim Biophys Acta. 2012;1822(5):690–713. | ||
Paik YH, Kim J, Aoyama T, De Minicis S, Bataller R, Brenner DA. Role of NADPH oxidases in liver fibrosis. Antioxid Redox Signal. 2014;20(17):2854–2872. | ||
Maraldi T. Natural compounds as modulators of NADPH oxidases. Oxid Med Cell Longev. 2013;2013:271602. | ||
Ikeda Y, Murakami A, Ohigashi H. Ursolic acid: an anti- and pro-inflammatory triterpenoid. Mol Nutr Food Res. 2008;52(1):26–42. | ||
Mazumder K, Tanaka K, Fukase K. Cytotoxic activity of ursolic acid derivatives obtained by isolation and oxidative derivatization. Molecules. 2013;18(8):8929–8944. | ||
Yang L, Wang KJ, Wang LS, et al. Chemical-protein interactome and its application in off-target identification. Interdiscip Sci. 2011;3(1):22–30. | ||
Yang L, Wang K, Chen J, et al. Exploring off-targets and off-systems for adverse drug reactions via chemical-protein interactome–clozapine-induced agranulocytosis as a case study. PLoS Comput Biol. 2011;7(3):e1002016. | ||
Jiang F, Liu GS, Dusting GJ, Chan EC. NADPH oxidase-dependent redox signaling in TGF-beta-mediated fibrotic responses. Redox Biol. 2014;2:267–272. | ||
Babior BM, Lambeth JD, Nauseef W. The neutrophil NADPH oxidase. Arch Biochem Biophys. 2002;397(2):342–344. | ||
Babior BM, Kipnes RS, Curnutte JT. Biological defense mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent. J Clin Invest. 1973;52(3):741–744. | ||
Babior BM. The NADPH oxidase of endothelial cells. IUBMB Life. 2000;50(4–5):267–269. | ||
Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87(1):245–313. | ||
Katsuyama M, Matsuno K, Yabe-Nishimura C. Physiological roles of NOX/NADPH oxidase, the superoxide-generating enzyme. J Clin Biochem Nutr. 2012;50(1):9–22. | ||
Lassegue B, Griendling KK. NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol. 2010;30(4):653–661. | ||
Nauseef WM. Biological roles for the NOX family NADPH oxidases. J Biol Chem. 2008;283(25):16961–16965. | ||
Adachi T, Togashi H, Suzuki A, et al. NAD(P)H oxidase plays a crucial role in PDGF-induced proliferation of hepatic stellate cells. Hepatology. 2005;41(6):1272–1281. | ||
Ris-Stalpers C. Physiology and pathophysiology of the DUOXes. Antioxid Redox Signal. 2006;8(9–10):1563–1572. | ||
Kawahara T, Quinn MT, Lambeth JD. Molecular evolution of the reactive oxygen-generating NADPH oxidase (Nox/Duox) family of enzymes. BMC Evol Biol. 2007;7:109. | ||
Kawahara T, Lambeth JD. Molecular evolution of Phox-related regulatory subunits for NADPH oxidase enzymes. BMC Evol Biol. 2007;7:178. | ||
Lambeth JD, Kawahara T, Diebold B. Regulation of Nox and Duox enzymatic activity and expression. Free Radic Biol Med. 2007;43(3):319–331. | ||
Cave AC, Brewer AC, Narayanapanicker A, et al. NADPH oxidases in cardiovascular health and disease. Antioxid Redox Signal. 2006;8(5–6):691–728. | ||
Streeter J, Thiel W, Brieger K, Miller FJ Jr. Opportunity Nox: the future of NADPH oxidases as therapeutic targets in cardiovascular disease. Cardiovasc Ther. 2012;31(3):125–137. | ||
Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov. 2011;10(6):453–471. | ||
Sancho P, Mainez J, Crosas-Molist E, et al. NADPH oxidase NOX4 mediates stellate cell activation and hepatocyte cell death during liver fibrosis development. PLoS One. 2012;7(9):e45285. | ||
Hordijk PL. Regulation of NADPH oxidases: the role of Rac proteins. Circ Res. 2006;98(4):453–462. | ||
Katsuyama M. NOX/NADPH oxidase, the superoxide-generating enzyme: its transcriptional regulation and physiological roles. J Pharmacol Sci. 2010;114(2):134–146. | ||
Choi SS, Sicklick JK, Ma Q, et al. Sustained activation of Rac1 in hepatic stellate cells promotes liver injury and fibrosis in mice. Hepatology. 2006;44(5):1267–1277. | ||
Mehal W, Imaeda A. Cell death and fibrogenesis. Semin Liver Dis. 2010;30(3):226–231. | ||
Yamamori T, Inanami O, Nagahata H, Kuwabara M. Phosphoinositide 3-kinase regulates the phosphorylation of NADPH oxidase component p47(phox) by controlling cPKC/PKCdelta but not Akt. Biochem Biophys Res Commun. 2004;316(3):720–730. | ||
Kilpatrick LE, Sun S, Li H, Vary TC, Korchak HM. Regulation of TNF-induced oxygen radical production in human neutrophils: role of delta-PKC. J Leukoc Biol. 2010;87(1):153–164. | ||
De Minicis S, Seki E, Oesterreicher C, Schnabl B, Schwabe RF, Brenner DA. Reduced nicotinamide adenine dinucleotide phosphate oxidase mediates fibrotic and inflammatory effects of leptin on hepatic stellate cells. Hepatology. 2008;48(6):2016–2026. | ||
Schuppan D, Kim YO. Evolving therapies for liver fibrosis. J Clin Invest. 2013;123(5):1887–1901. | ||
Jiang JX, Chen X, Serizawa N, et al. Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free Radic Biol Med. 2012;53(2):289–296. | ||
Babalola O, Mamalis A, Lev-Tov H, Jagdeo J. NADPH oxidase enzymes in skin fibrosis: molecular targets and therapeutic agents. Arch Dermatol Res. 2014;306(4):313–330. | ||
Guy J, Peters MG. Liver disease in women: the influence of gender on epidemiology, natural history, and patient outcomes. Gastroenterol Hepatol (N Y). 2013;9(10):633–639. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.