")
Back to Journals » Drug Design, Development and Therapy » Volume 13
6,7-Dihydroxy-2-(4′-hydroxyphenyl)naphthalene induces HCT116 cell apoptosis through activation of endoplasmic reticulum stress and the extrinsic apoptotic pathway
Authors Chiu CF, Lai GY, Chen CH , Chiu CC, Hung SW, Chang CF
Received 8 November 2018
Accepted for publication 21 February 2019
Published 9 May 2019 Volume 2019:13 Pages 1609—1621
DOI https://doi.org/10.2147/DDDT.S193914
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Qiongyu Guo
Ching-Feng Chiu,1–3 Guan-Ying Lai,4 Chung-Hwan Chen,5–7 Chien-Chao Chiu,8 Shao-Wen Hung,8,9 Chi-Fen Chang10
1Graduate Institute of Metabolism and Obesity Sciences, College of Nutrition, Taipei Medical University, Taipei 11031, Taiwan; 2TMU Research Center of Cancer Translational Medicine, Taipei Medical University, Taipei 11031, Taiwan; 3TMU Research Center of Cancer Translational Medicine, Taipei Medical University, Taipei, Taiwan; 4Master Program for Pharmaceutical Manufacture, China Medical University, Taichung 40402, Taiwan; 5Department of Orthopedics, Kaohsiung Municipal Ta-Tung Hospital, Kaohsiung Medical University, Kaohsiung 80145, Taiwan; 6Department of Orthopedics, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung 80708, Taiwan; 7Orthopaedic Research Center, Kaohsiung Medical University, Kaohsiung 80708, Taiwan; 8Division of Animal Industry, Animal Technology Laboratories, Agricultural Technology Research Institute, Xiangshan, Hsinchu 300, Taiwan; 9Nursing Department, Yuanpei University, Xiangshan, Hsinchu 300, Taiwan; 10Department of Anatomy, School of Medicine, China Medical University, Taichung 40402, Taiwan
Background: Colorectal cancer is the third leading cause of cancer-related deaths worldwide, and therefore, the development of novel drugs for its prevention and therapy are urgently required. This study aimed to determine the molecular mechanism of 6,7-dihydroxy-2-(4'-hydroxyphenyl)naphthalene (PNAP-6)-induced cytotoxicity in human colorectal cancer (HCT116) cells.
Methods: The effects of 2-phenylnaphthalene derivatives on HCT116 cell growth and viability were assessed by MTT assays. The mechanisms involved in the regulation of the extrinsic apoptosis and endoplasmic reticulum (ER) stress pathways by PNAP-6 were analyzed by annexin-V/propidium iodide flow cytometric analysis, Hoechst 33342 fluorescent staining, and Western blotting.
Results: PNAP-6 was shown to have an IC50 value 15.20 µM. It induced G2/M phase arrest in HCT116 cells, associated with a marked decrease in cyclin B and CDK1 protein expression and increased caspase activation, PARP cleavage, chromatin condensation, and sub-G1 apoptosis. Moreover, we found that the apoptotic effects of PNAP-6 proceeded through extrinsic apoptosis and ER stress pathways, by increasing the expression of Fas protein and ER stress markers, including PERK, ATF4, CHOP, p-IRE1α, and XBP-1s.
Conclusion: These results suggest that 2-phenylnaphthalene derivatives, such as PNAP-6, have potential as new treatments for colorectal cancer.
Keywords: colorectal cancer, endoplasmic reticulum stress, extrinsic apoptosis pathway, 2-phenylnaphthalenes
Introduction
Colorectal cancer is the third most common cancer worldwide and the third leading cause of cancer-related deaths worldwide.1 Therefore, it is imperative to develop drugs that are more effective than those currently in use. The spatial and conformational requirements of 2-phenylnaphthalenes (PNAPs) and genistein are pharmacologically similar.2 Genistein is well known as an anticancer agent. Genistein effectively suppresses cancer progression via several mechanisms, including increasing apoptosis, controlling invasion and metastasis, adjusting intracellular signaling pathways, and arresting the cell cycle of cancer cells.3 In the past few years, a series of PNAP derivatives have been synthesized in our laboratory and we have assessed their effects on breast cancer and macrophage cells. Our research has shown that 6,7-dihydroxy-2-(4′-hydroxyphenyl)naphthalene (PNAP-6) exhibits potent anti-breast cancer activity, inducing cell cycle arrest and death of breast cancer cells.2 Moreover, PNAP-6 reduces the production of lipopolysaccharide-induced pro-inflammatory mediators, such as IL-6, tumor necrosis factor-alpha, inducible nitric oxide synthase, and COX-II.4 However, it is unclear whether PNAPs also possess anticancer effects on human colorectal carcinoma cells.
Cell proliferation is associated with cell cycle regulation. Cell cycle progression is regulated by a series of cyclin-dependent kinases (CDKs), which rely on cyclin subunits.5 Passage through G1 into S phase is regulated by activation of the cyclin D1-CDK4/6 and cyclin E-CDK2 complexes.6,7 Passage through G2 into M phase is regulated by activation of the cyclin B1-CDK1 complex.8,9 Deregulation of the cell cycle causes aberrant cell proliferation and tumor growth. Therefore, any compound that regulates cell cycle processes is also potentially capable of inhibiting tumor progression.10
Apoptosis is considered a normal physiological process, whereas impaired apoptosis and interruptions in cell cycle regulation are indications of cancer growth and aggressiveness.11 Caspase activation is known to be important during apoptosis, and therefore has been targeted by many anticancer agents.12 An extrinsic pathway that regulates caspase-dependent apoptosis has been identified.13 Fas, a death receptor, plays an initial role in the extrinsic apoptotic pathway.14 Fas-associated protein with death domain (FADD) is an adaptor protein for death receptor-mediated apoptosis. It bridges the Fas receptor with the downstream effector, caspase-8, to form the death-inducing signaling complex.15,16 Activated caspase-8 then activates the downstream effectors, caspase-3 and caspase-7, resulting in apoptosis. In addition, an increasing number of studies have indicated that disruption of endoplasmic reticulum (ER) function by various anticancer agents, such as farnesol, paclitaxel, bortezomib, and dipyridamole, significantly alters the expression of ER stress-related proteins, such as PERK, eif2a, ATF4, CHOP, XBP-1, and IRE, which leads to apoptosis.17–19 However, it is unclear whether the ER stress and extrinsic pathways are activated in PNAP-mediated apoptosis. Therefore, this study aimed to elucidate the molecular mechanism underlying PNAP-6-induced cytotoxicity in human colorectal cancer cells.
Materials and methods
Chemicals
Eight PNAP derivatives (PNAP-1–PNAP-8) were provided by Dr Ta-Hsien Chuang (School of Pharmacy, China Medical University, Taiwan). The molecular structures of 2-phenylnaphthalene (PNAP-1), 6-hydroxy-2-phenylnaphthalene (PNAP-2), 6,7-dihydroxy-2-phenylnaphthalene (PNAP-3), 2-(4′-hydroxyphenyl)naphthalene (PNAP-4), 6-hydroxy-2-(4′-hydroxyphenyl)naphthalene (PNAP-5), 6,7-dihydroxy-2-(4′-hydroxyphenyl)naphthalene (PNAP-6), 6,7-dihydroxy-2-(3′,4′-dihydroxyphenyl)naphthalene (PNAP-7), and 2-(4′-aminophenyl)-6,7-dimethoxynaphthalene (PNAP-8) are shown in Figure 1A.
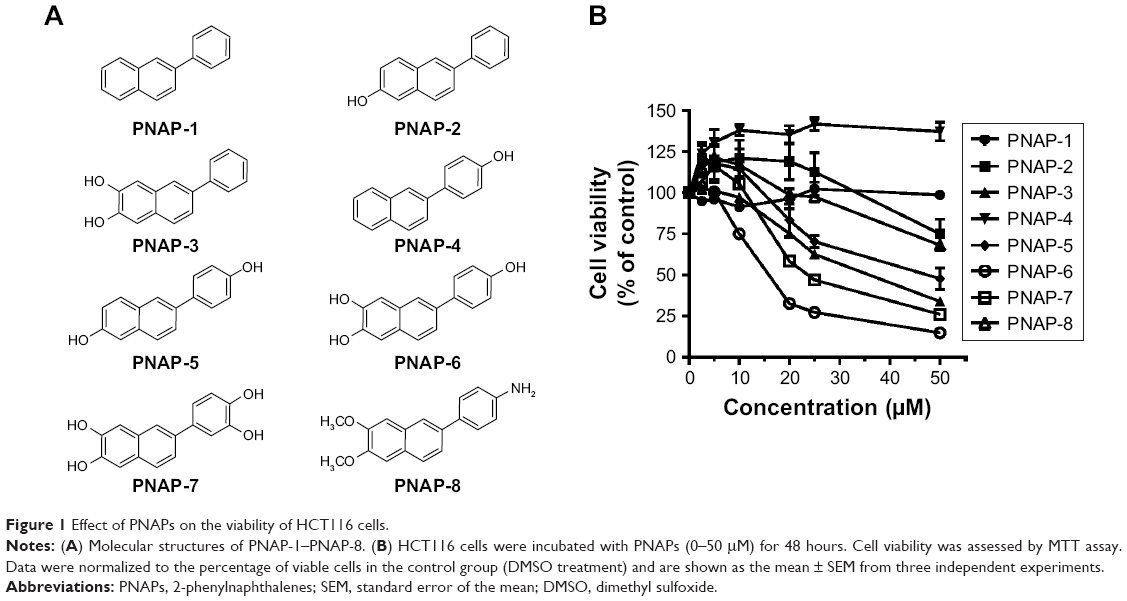 | Figure 1 Effect of PNAPs on the viability of HCT116 cells. |
Cell culture
HCT116 cells were obtained from the laboratory of Dr Yang-Chang Wu (School of Pharmacy, China Medical University, Taiwan), confirmed by short tandem repeat profiling and free of mycoplasma contamination. HCT116 cells were cultured and grown in DMEM/F12 medium containing 10% FBS, 1% penicillin–streptomycin, and maintained in a humidified 5% CO2 incubator at 37°C.
Cell viability assay
HCT116 cells were plated at a density of 5×103 cells per well in 96-well plates, incubated overnight, and then treated with different concentrations (0, 2.5, 5, 10, 20, 25, and 50 μM) of eight PNAPs (PNAP-1−PNAP-8) which were dissolved in dimethyl sulfoxide (DMSO) to obtain a final concentration of 50 mM, and DMSO was used for untreated control. After 48 hours of treatment, MTT (0.5 mg/mL) was added to each well, and the cells were incubated for 4 hours at 37°C. Supernatants were then removed, and 50 μL of DMSO was added to each well to dissolve the formazan product. The absorbance at a wavelength of 550 nm was then measured using a MQX200R microplate reader (BioTek, Winooski, VT, USA).
Cell cycle analysis
HCT116 cells were seeded at a density of 3×105 cells per well in six-well plates, incubated overnight, and then treated with PNAP-6 at three different concentrations (10, 15, and 20 μM) or with 20 μM PNAP-6 at different time intervals (12, 24, and 48 hours). Both floating and adherent cells were collected and washed with PBS. The cells were centrifuged, and the pellets were suspended with 70% ethanol for 1 hour at −20°C. The fixed cells were washed with ice-cold PBS, suspended in 0.5 mL of PBS containing 0.2 mg/mL RNase, and 0.1% Triton X-100 for 30 minutes at room temperature, and stained with 20 μg/mL propidium iodide (PI). The stained cells were analyzed using a FACScan flow cytometer (Becton Dickinson, San Jose, CA, USA). The fluorescence emitted from the PI-DNA complex was estimated from a minimum of 10,000 cells per sample and analyzed using the Cell Quest Alias software (BD Biosciences, San Jose, CA, USA).
Annexin V/PI cell death assay
HCT116 cells were seeded at a density of 3×105 cells per well in six-well plates, incubated overnight, and then treated with PNAP-6 at three different concentrations (10, 15, and 20 μM). After 48 hours of treatment, the cells were trypsinized, washed once with PBS, and then suspended in 1X binding buffer. Annexin V-fluorescein isothiocyanate (5 μL) and PI (5 μL) were then added to each sample, followed by an incubation for 15 minutes at room temperature in the dark. Cells were analyzed using flow cytometry (BD Biosciences).
Cell morphology studies
The cells were seeded in 12-well plates at a density of 1×105 cells/well. Different treatment groups and the control group were cultured for 48 hours. The cells were washed once with PBS and stained with Hoechst 33342 (10 μg/mL for 15 minutes). The morphological changes were then observed by light microscopy at 100× magnification and fluorescence microscopy at 200× magnification.
Western blot analysis
HCT116 cells were seeded at a density of 3×105 cells per well in six-well plates, incubated overnight, and then treated with PNAP-6 at different concentrations for 24 or 48 hours or with 20 μM PNAP-6 at different time intervals. The cells were washed with PBS and lysed in ice-cold RIPA buffer for 30 minutes. The supernatant was collected and centrifuged at 15,000 rpm and 4°C for 30 minutes. Proteins were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA, USA). Membranes were incubated overnight at 4°C with the following antibodies: cyclin D1 (1:2,000; Cell Signaling Technology, Beverly, MA, USA); CDK4 (1:2,000; Cell Signaling Technology); cyclin E (1:2,000; Cell Signaling Technology); CDK2 (1:2,000; Cell Signaling Technology); p21 (1:2,000; Cell Signaling Technology); p27 (1:2,000; Cell Signaling Technology); cyclin B1 (1:2,000; Cell Signaling Technology); CDK1 (1:2,000; Cell Signaling Technology); cleaved caspase-12 (1:1,000; Cell Signaling Technology); cleaved caspase-7 (1:1,000; Cell Signaling Technology); cleaved caspase-8 (1:1,000; Cell Signaling Technology); cleaved caspase-3 (1:1,000; Cell Signaling Technology); cleaved PARP (1:1,000; Cell Signaling Technology); Fas (1:1,000; Cell Signaling Technology); FADD (1:2,000; Cell Signaling Technology); ATF4 (1:2,000; Cell Signaling Technology); CHOP (1:2,000; Cell Signaling Technology); XBP-1s (1:2,000; Cell Signaling Technology); PERK (1:2,000; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA); p53 (1:3,000; Santa Cruz Biotechnology Inc.); p-IREα (1:1,000; Thermo Fisher Scientific, Inc., Waltham, MA, USA); actin (1:5,000, Sigma-Aldrich Co., Inc., St Louis, MO, USA). The secondary antibodies: horseradish peroxidase (HRP)-linked goat anti-rabbit IgG (1:2,000; Cell Signaling Technology); HRP-linked goat anti-mouse IgG (1:2,000; Cell Signaling Technology). The membranes were then washed three times with TBST and probed with HRP-conjugated secondary antibody for 1 hour at room temperature. After three washes in TBST, the bound antibody was visualized using the ECL Western Blotting Reagent (PerkinElmer Inc., Waltham, MA, USA), and the chemiluminescence was detected using Fuji Medical X-ray film (Tokyo, Japan). Expression levels of target proteins were quantified using a Gel-Pro Analyzer image analysis system and normalized with cellular actin protein level.
Statistical analysis
The data are presented as the mean ± standard error of the mean from three replicate experiments. The differences among the groups were compared by ANOVA using GraphPad Prism 5 software (GraphPad Software, Inc., La Jolla, CA, USA). P-values <0.05 were considered to be significant.
Results
Effect of PNAP-6 on survival of HCT116 cells
To investigate the effects of the eight PNAPs (PNAP-1–PNAP-8, Figure 1A) on cell viability, HCT116 cells were treated with different doses (0, 2.5, 5, 10, 20, 25, and 50 μM) for 48 hours. Cell viability was then determined by MTT assay and the results are shown in Figure 1B. First, PNAP-1 and -4 showed no cytotoxic effects on HCT116 cells. PNAP-2 and -8 showed a low level of cytotoxicity against HCT116 cells, whereas PNAP-3, -5, -6, and -7 showed potent cytotoxic activity against HCT116 cells, with IC50 values of 33.83, 46.95, 15.20, and 23.61 μM, respectively. These results indicated that PNAP-6 exhibited the greatest activity against HCT116 cells. Thus, we further investigated the underlying mechanism for the decrease in cell viability induced by PNAP-6.
Effect of PNAP-6 on cell cycle progression
To further explore the inhibitory effects of PNAP-6 on colon cancer cells, the viability of HCT116 (p53 wild type) and HT-29 (p53 mutant) cells were examined by MTT assay after PNAP-6 treatment. Expression of the apoptosis-related protein (p53) and the p53-induced proteins (p21 and p27) was also assessed by Western blotting. PNAP-6 treatment significantly decreased cell viability (Figure 2A) and increased p53, p21, and p27 protein expression in HCT116 cells compared to HT-29 cells (Figure 2B). This suggested that the PNAP-6-induced growth suppression may occur via the regulation of p53-dependent signaling in HCT116 cells. We further examined the cell cycle modulating properties of PNAP-6 in HCT116 cells. As shown in Figure 3, treatment of cells with PNAP-6 resulted in a dose-dependent inhibition of cell viability, which was accompanied by an accumulation of cells at the G2/M and sub-G1 phases, as determined by flow cytometric analysis. Approximately, 34.28% untreated control cells were at the G2/M phase, whereas >50% of HCT116 cells were at the G2/M phase following 48 hours of PNAP-6 treatment, which suggested the existence of a block at this phase of the cell cycle (Figure 3A). The percentage of cells at the sub-G1 phase (apoptotic cells) significantly increased from 1.4% in controls to >20% after treatment with PNAP-6 (Figure 3A). Thus, PNAP-6 may induce apoptosis in colon cancer cells. The increase in the percentage of HCT116 cells at the G2/M and sub-G1 phases following PNAP-6 treatment was found to be time-dependent (Figure 4A). In order to confirm the results of flow cytometry experiments, we analyzed cell cycle-coordinating proteins, such as cyclin D1, CDK4, cyclin E, CDK2, CDK1, cyclin B1, p21, and p27, by immunoblotting. PNAP-6 treatment resulted in a significant dose- and time-dependent decrease in the expression of cyclin D1, CDK4, cyclin E, CDK2, CDK1, and cyclin B1 (Figure 3B and C; Figure 4B and C). Moreover, PNAP-6 caused a significant time-dependent induction of p21 and p27 protein activity in HCT116 cells (Figure 4B). Taken together, these results indicated that the growth inhibition of HCT116 cells in response to PNAP-6 is due to growth arrest and cell death.
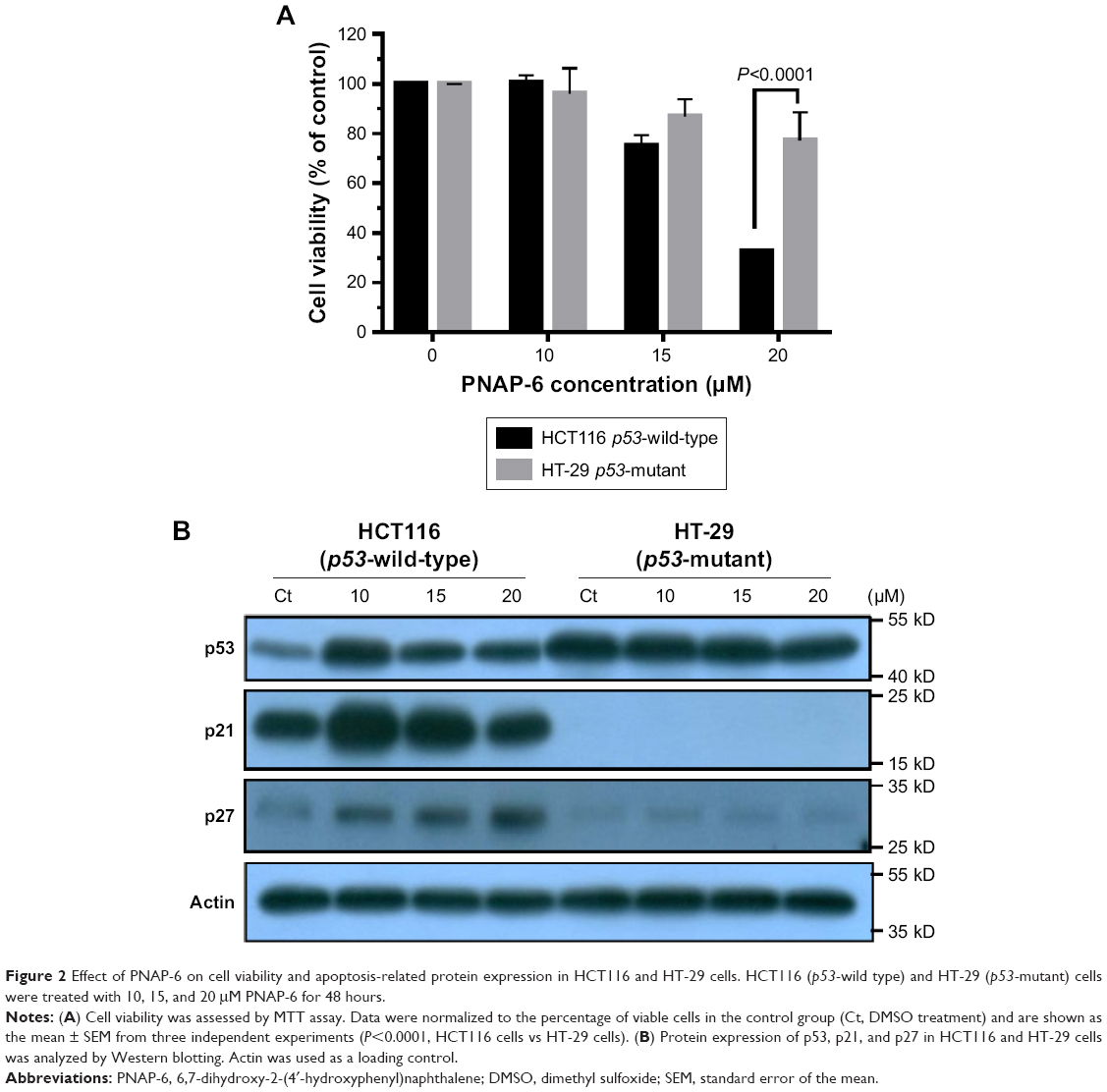 | Figure 2 Effect of PNA P-6 on cell viability and apoptosis-related protein expression in HCT116 and HT-29 cells. HCT116 (p53-wild type) and HT-29 (p53-mutant) cells were treated with 10, 15, and 20 μM PNAP-6 for 48 hours. |
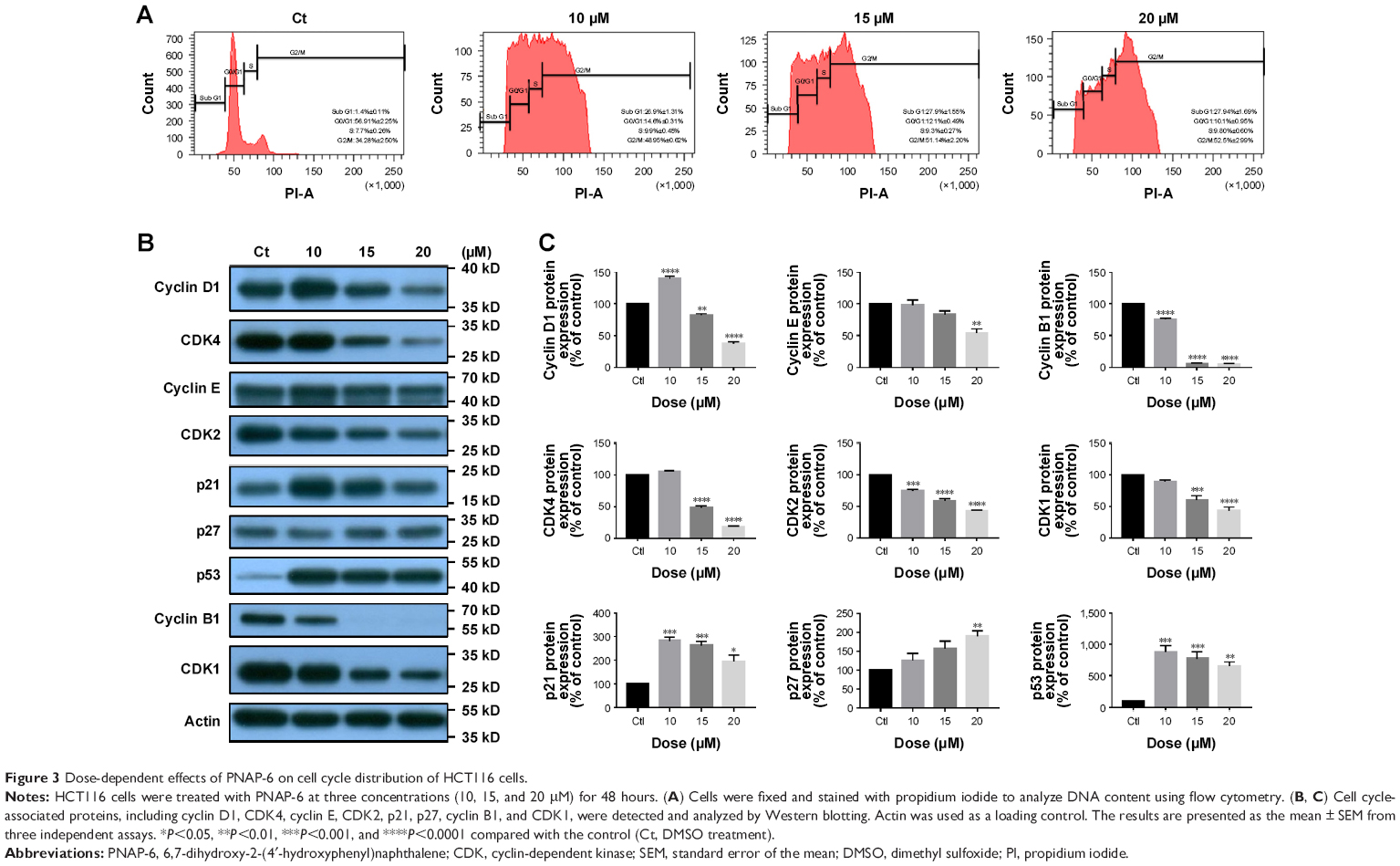 | Figure 3 Dose-dependent effects of PNAP-6 on cell cycle distribution of HCT116 cells. |
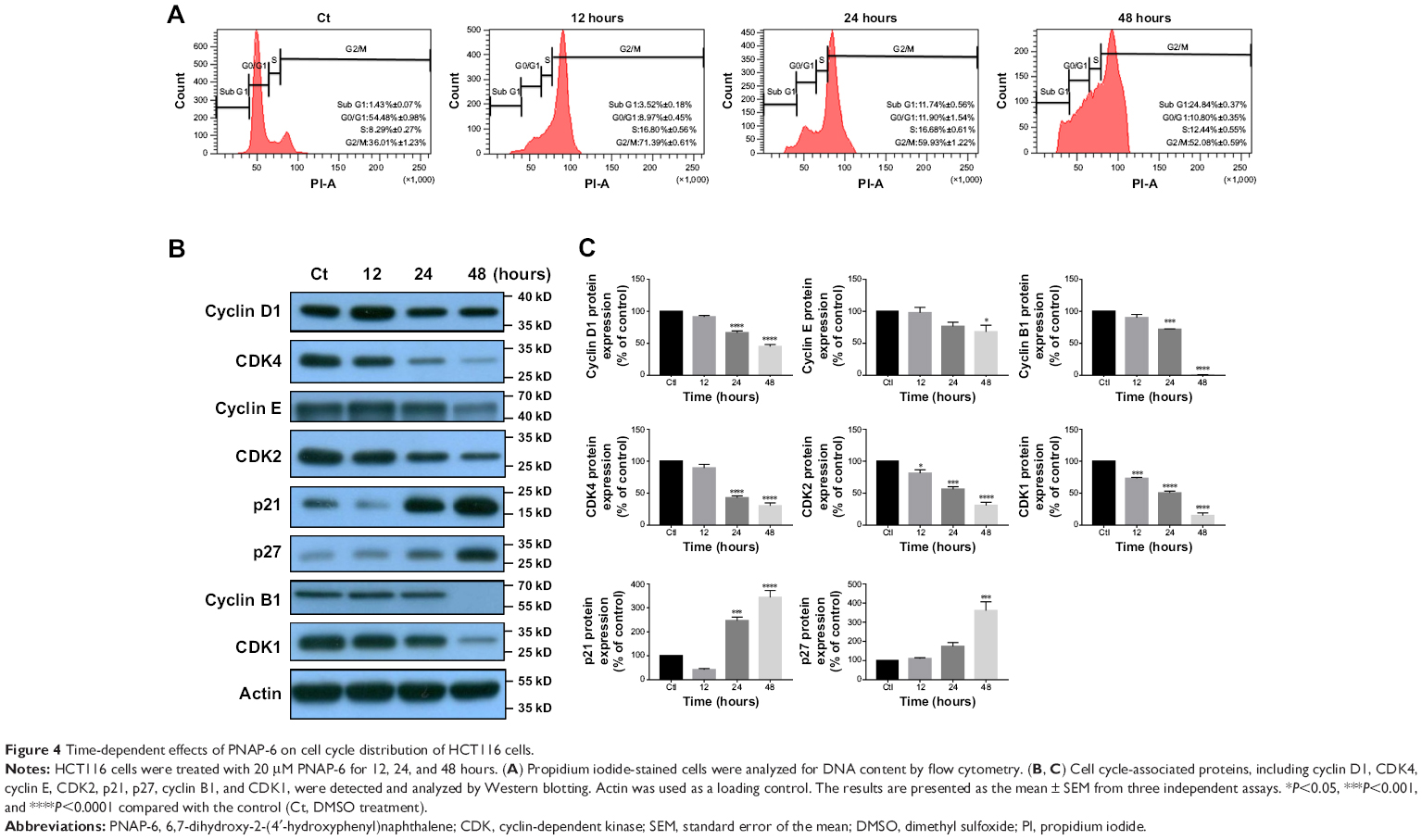 | Figure 4 Time-dependent effects of PNAP-6 on cell cycle distribution of HCT116 cells. |
Induction of apoptosis by PNAP-6
Flow cytometric analysis showed a dose- and time-dependent increase in the number of cells with sub-G1 amounts of DNA (apoptotic cells) following PNAP-6 treatment. To further verify that apoptosis was induced by PNAP-6, HCT116 cells were treated with 10, 15, and 20 μM PNAP-6, and morphological changes were observed by phase-contrast microscopy (Figure 5A–D). Prior to treatment, HCT116 cells were regular in shape and size, with large nuclei and a relatively small amount of cytoplasm. The density and structure of cells after 48 hours of treatment with 10–20 μM PNAP-6 showed obvious changes. Cells became shrunken and lost contact with adjacent cells. Moreover, the apoptotic cells no longer attached to the substrate, but detached from the culture plates and floated in the medium. Moreover, we stained HCT116 cells with Hoechst 33342 fluorescent dye to detect apoptotic morphology after 48 hours of treatment with different concentrations of PNAP-6. As seen in Figure 5E–H, cells treated with PNAP-6 showed typical apoptotic features, such as nuclear shrinkage, nuclear fragmentation, and nuclear hypercondensation. We further performed annexin-V and PI staining to quantitate the effects of PNAP-6. During the apoptotic process, annexin V binds to phosphatidylserine on apoptotic cells, whereas PI can stain the nucleus of apoptotic cells due to the loss of membrane integrity. In Figure 5I–L, cells with annexin V(+)/PI(−) staining were classified as early apoptotic, cells with annexin V(+)/PI(+) staining were classified as late apoptotic, and cells with annexin V(−)/PI(+) staining were classified as necrotic. After 48 hours of treatment with 0, 10, 15, or 20 μM PNAP-6, early apoptotic HCT116 cells were 2.23%±0.37%, 8.93%±0.28%, 9.93%±0.23%, and 11.93%±0.55% of the total cell population, respectively (Q4 region). With the same PNAP-6 treatment concentrations, late apoptotic cells made up 2.3%±0.29%, 21.6%±0.78%, 22.1%±0.68%, and 22.6%±0.47% of the total cell population, respectively (Q2 region). There were no significant differences in the percentage of necrotic cells (Q1 region) between PNAP-6-treated cells and control cells. These findings indicated that PNAP-6 induced early/late stages of apoptosis, but does not induce necrosis in HCT116 cells.
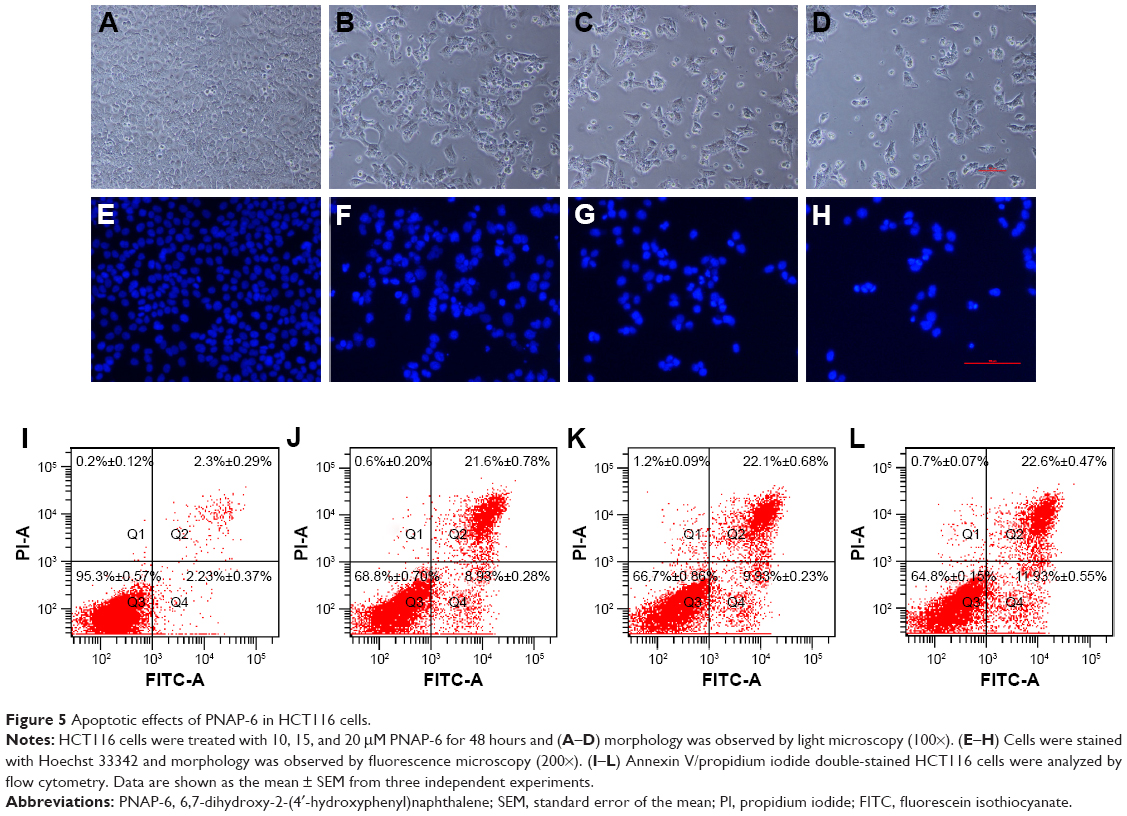 | Figure 5 Apoptotic effects of PNAP-6 in HCT116 cells. |
PNAP-6 triggers caspase-dependent apoptosis in HCT116 cells
Generally, caspases play essential roles in mediating different apoptotic responses.20 One of the cellular substrates of caspases is PARP-1. Its activation causes apoptosis of tumor cells.21 To determine whether PNAP-induced apoptosis in HCT116 cells occurs via caspase activation, we examined the activation of caspase-3, -7, -8, and -12 and PARP by Western blotting after incubation with 10, 15, and 20 μM PNAP-6 for 48 hours or 20 μM PNAP-6 for 12, 24, and 48 hours. As depicted in Figure 6A and B, the activity of caspase-8 in HCT116 cells treated with PNAP-6 at 10, 15, and 20 μM increased by 2.8-fold, 3.2-fold, and 3.6-fold, respectively, compared to control cells. Intracellular caspase-3, -7, -12, and PARP activities were also enhanced by PNAP-6 treatment in a dose-dependent manner. Moreover, the levels of cleaved caspase-3, -7, -8, -12, and PARP increased significantly after 24 and 48 hours of PNAP-6 treatment, in a time-dependent manner (Figure 6C and D). These results suggested that activation of the caspase cascade and PARP cleavage are involved in PNAP-6-mediated apoptosis.
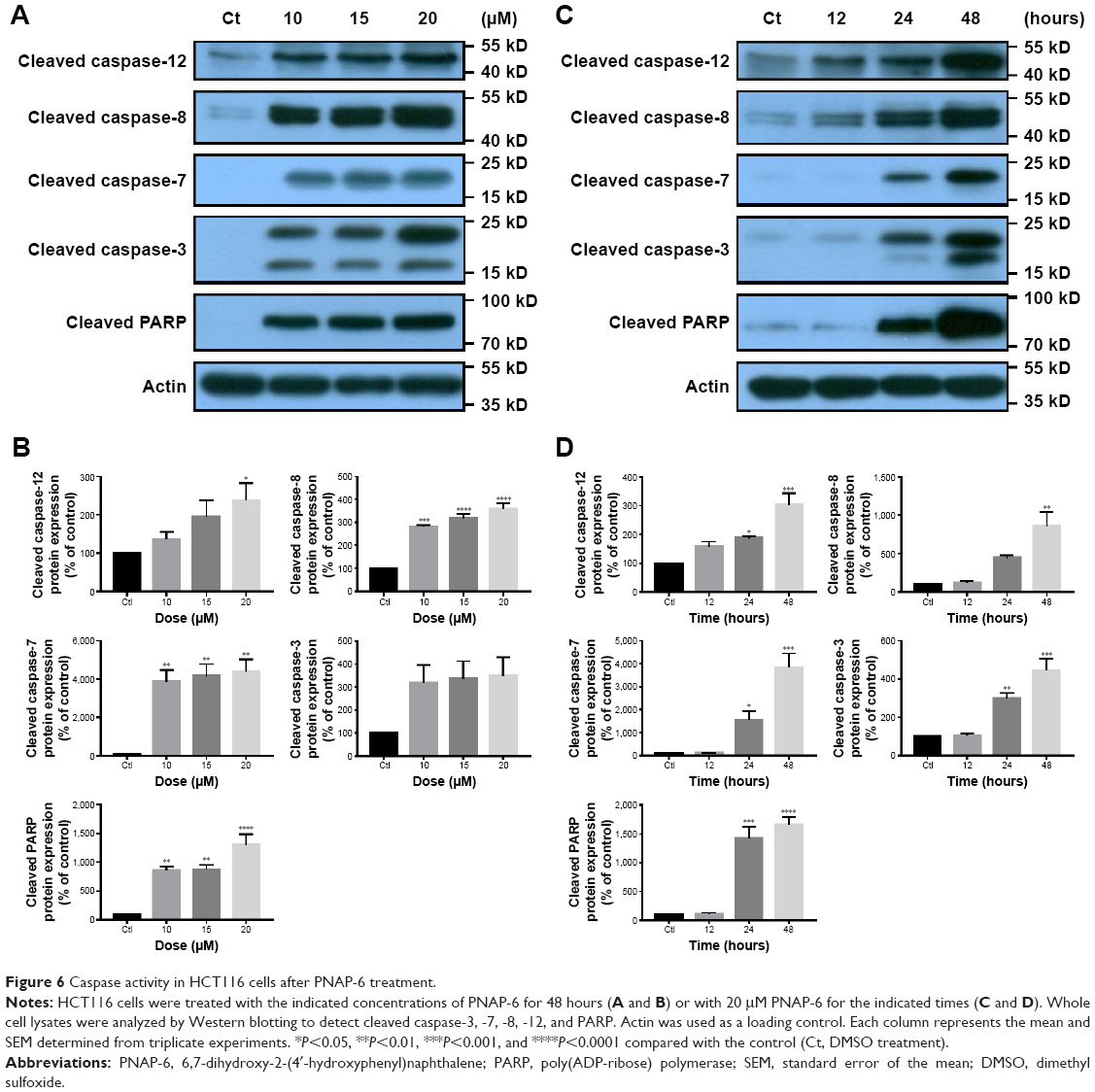 | Figure 6 Caspase activity in HCT116 cells after PNAP-6 treatment. |
PNAP-6 induces ER stress and the extrinsic apoptosis pathway in HCT116 cells
Caspase-8 plays an essential part in the extrinsic apoptotic signaling pathway,16 which is initiated by the activation of death receptors, such as Fas.22 This process forms a death-inducing signaling complex, in which caspase-8 is triggered by its adaptor, FADD. Subsequently, caspase-8 directly cleaves caspase-3 to induce apoptosis.23,24 Western blotting experiments were performed to determine whether caspase-8 activation was associated with the formation of complexes involving Fas and FADD. HCT116 cells were treated with PNAP-6 for 1, 3, 6, 12, and 24 hours, and Fas and FADD proteins were detected using specific antibodies. The results showed that PNAP-6 treatment caused a significant increase in Fas protein expression and a decrease in FADD protein expression (Figure 7A and B), but did not affect Bcl-xL or Bax protein levels (Figure 7A), suggesting that PNAP-6 induced HCT116 apoptosis through the extrinsic pathway via Fas/FADD signaling. ER stress-mediated cell death involves caspase-12 activation.25 To investigate whether ER stress is involved in PNAP-6-induced apoptosis, we initially examined the expression of ER stress markers, including PERK, ATF4, CHOP, p-IRE1α, and XBP-1s. The ER membrane receptor, PERK, was found to be upregulated in a time-dependent manner following PNAP-6 treatment. ATF-4 and CHOP expression were upregulated after 6 hours of PNAP-6 treatment, and this effect increased thereafter in a time-dependent manner (Figure 7A and B). Moreover, 1 hour of PNAP-6 treatment induced p-IRE1α and XBP-1s expression. Taken together, these data indicated that PNAP-6 promoted apoptosis by activating both the extrinsic and ER stress pathways.
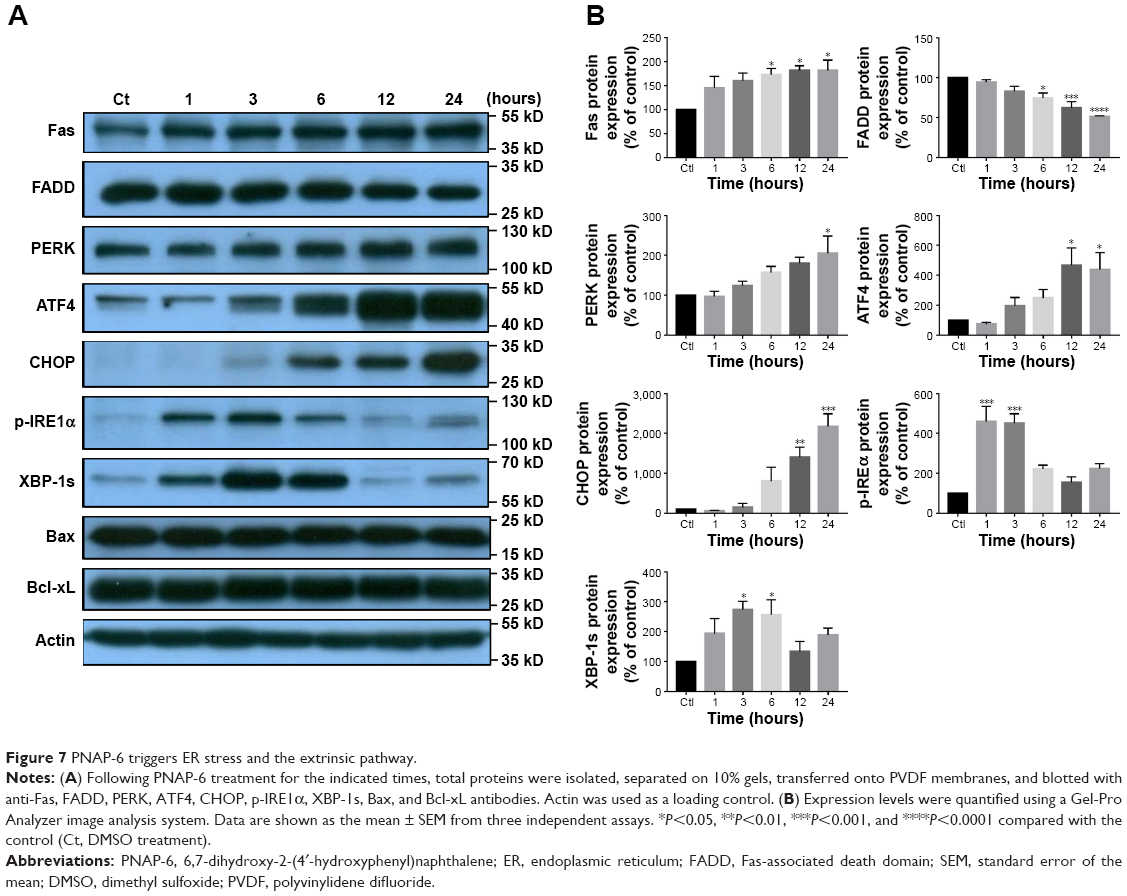 | Figure 7 PNAP-6 triggers ER stress and the extrinsic pathway. |
Discussion
In this study, we evaluated the anticancer effects of PNAPs using the human colorectal carcinoma cell line, HCT116. MTT assays were first performed to measure HCT116 cell viability after treatment with PNAP-1–PNAP-8. Among the PNAPs tested, PNAP-6 exhibited the greatest cytotoxicity, with an IC50 value of 15.20 μM (Figure 1B). In addition, PNAP-6 suppressed cell proliferation, caused G2/M phase arrest, and induced apoptosis via extrinsic and ER stress pathways in HCT116 cells. Previous studies have shown that naphthylchalcones R7, R13, and R15 increase CHOP expression and subsequently, induce caspase-12 activity and apoptosis, but decrease p53 and p21 protein levels in leukemic cells.26 Additionally, the flavonoid derivative, LZ-205, was also shown to induce ER stress and the exogenous apoptotic pathway in H460 cells, which are known to harbor the wild-type p53 allele.27,28 These data indicate that the influence of these compounds on protein folding and subsequently ER stress and apoptosis may proceed through a p53-independent pathway. Our data showed that PNAP-6 increased p53 expression and led to the induction of extrinsic apoptosis and ER stress pathways in p53-wild-type HCT116 cells, but not in p53-mutant HT-29 cells, suggesting that PNAP-6 specifically increased p53, p21, and p27 expression and then induced cell cycle arrest in p53-wild-type cancer cells.
The G2/M checkpoint serves to prevent cells from entering mitosis when DNA is damaged.29 According to flow cytometric analysis, PNAP-6 treatment of HCT116 cells resulted in arrest at the G2/M phase of the cell cycle. We examined the effect of PNAP-6 on cell cycle regulatory molecules operative at the G2/M phase (Figures 3A and 4A). PNAP-6 treatment resulted in a significant time- and dose-dependent upregulation of CDK1 and cyclin B1 (Figures 3B and 4B). The CDK inhibitors, p21 and p27, have an important role in blocking the activation of CDK1/cyclin B.29 Our data also demonstrated a significant upregulation of p21 and p27 by PNAP-6 treatment (Figures 3B and 4B). These results were consistent with a previous study, which reported that PNAP-6 decreases CDK1 and cyclin B1 expression, leading to G2/M arrest in MCF-7 cells.2
Apoptosis plays an essential role in cancer suppression and drug treatment response.30 Apoptotic cells have specific characteristics, such as cell shrinkage, membrane blebbing, chromatin condensation, and an increased population of sub-G1 phase cells.31 PNAP-6 induced nuclear morphological alterations, such as nuclear shrinkage and nuclear hypercondensation in HCT116 cells (Figure 5E–H). When HCT116 cells were treated with PNAP-6 (20 μM for 48 hours), the sub-G1 hypodiploid cell population increased by 20% (Figure 4A). PARP cleavage is another major marker of apoptosis.32 Through Western blotting analysis, PNAP-6 treatment was also shown to increase the levels of cleaved PARP (Figure 6A and B). Collectively, these results suggested that PNAP-6 induced apoptosis.
Extracellular stress stimulation, sensed and generated through Fas activation, can activate the extrinsic pathway,33 and it positively correlates with drug sensitivity in lung cancer.34 The death receptor-initiated pathway results in caspase-8 activation, which can directly induce the activation of caspase-3 and -7, leading to apoptosis.35 In this study, we showed that PNAP-6 treatment upregulated the cell surface expression of Fas and the expression of caspase-3, -8, and -7. In addition, apoptosis can be induced by the ER, as a major organelle for the synthesis and folding of proteins.36 ER stress, the accumulation of unfolded or misfolded proteins inside the ER, can be induced by a variety of perturbations.37 ER stress activates a series of signaling pathways and transcriptional events, known as the unfolded protein response (UPR).38 The UPR is triggered by three ER transmembrane protein sensors, PERK, IRE, and ATF6.39 Activated PERK increases eIF-2a phosphorylation, which attenuates the translation of proteins, such as ATF4, to relieve the ER workload during stress.40 Through the activation of CHOP, a downstream transcriptional factor, ATF-4 can activate pro-apoptotic signals.41 Actions of the pro-apoptotic factor, CHOP, include suppression of the anti-apoptotic Bcl-2/Bcl-xL family and upregulated expression of the pro-apoptotic proteins, Bax, Bak, and Bok.42 Our data showed that PNAP-6 significantly increased CHOP expression from 6 to 24 hours of treatment, but did not affect Bcl-xL or Bax expression in HCT116 cells (Figure 7A), suggesting that PNAP-6 may increase CHOP expression to induce the ER stress pathway, but does not affect the CHOP-mediated regulation of Bcl-xL and Bax protein expression. In addition, to cope with ER stress, the endonuclease activity of IRE-1 increases XBP-1 splicing. Spliced XBP-1 can then upregulate genes that are crucial for cell survival during ER stress.43 Using Western blotting analysis, we found a time-dependent activation of ER stress-related pro-apoptotic signaling proteins, including PERK, ATF4, and CHOP, in PNAP-6 treated HCT116 cells. Similarly, PNAP-6 treatment increased the expression of p-IRE1α and XBP-1s (Figure 7). Thus, PNAP-6 induces the apoptosis of HCT116 cells through the extrinsic and ER stress pathways.
In summary, PNAP-6 exhibited the strongest cytotoxicity among a series of hydrophilic PNAP derivatives tested. PNAP-6 induced the apoptosis of HCT116 cells through the activation of extrinsic and ER stress pathways. PNAP-6 also inhibited HCT116 cell proliferation by inducing G2/M arrest and modulating the cyclin B1 and CDK1 network (Figure 8).
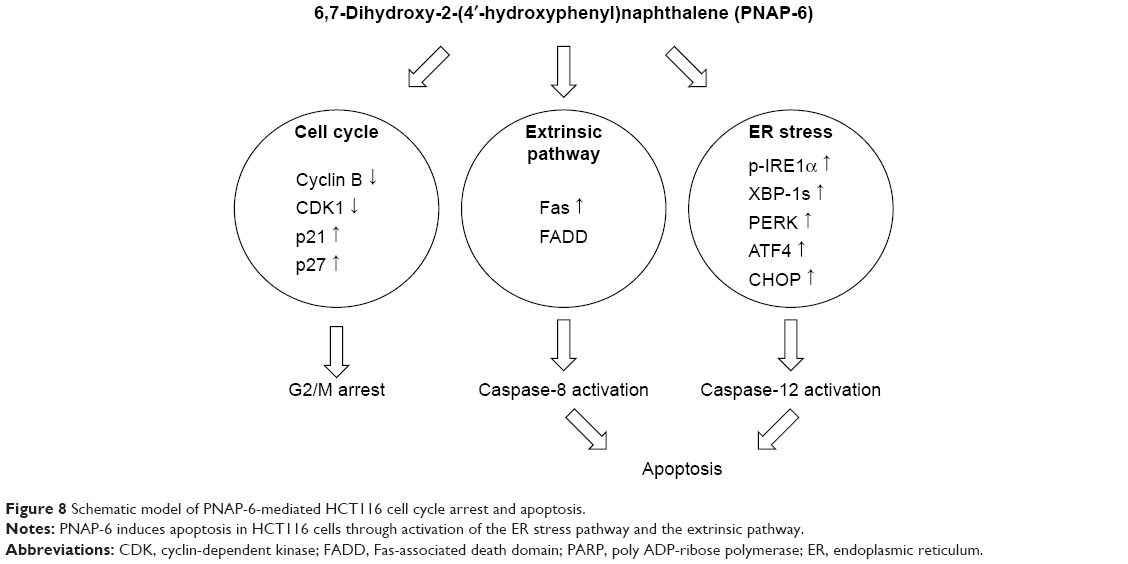 | Figure 8 Schematic model of PNAP-6-mediated HCT116 cell cycle arrest and apoptosis. |
Conclusion
This is the first study to show the novel mechanism of PNAP derivatives inducing p53-mediated cell cycle arrest and apoptotic signaling in colorectal cancer cells. These results indicate that the activation of the extrinsic and ER stress pathways is an attractive strategy for the treatment of colorectal carcinoma. We propose that PNAP-6 may have therapeutic potential for colon cancer treatment.
Ethical approval
This article does not include any studies with human participants or animals.
Acknowledgments
We would like to acknowledge Dr Ta-Hsien Chuang for providing the compounds (PNAP-1—PNAP-8). We also thank Dr Yang-Chang Wu for kindly providing HCT116 cell lines. This work was financially supported by Ministry of Science and Technology (MOST 104-2321-B-038-012-MY3 and 107-2320-B-038-065), China Medical University (CMU107-SR-104), and the TMU Research Center of Cancer Translational Medicine from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan.
Author contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Ye Q, Feng B, Peng YF, et al. Expression of gamma-synuclein in colorectal cancer tissues and its role on colorectal cancer cell line HCT116. World J Gastroenterol. 2009;15(40):5035–5043. | ||
Chang CF, Ke CY, Wu YC, Chuang TH. Structure-activity relationship of synthetic 2-phenylnaphthalenes with hydroxyl groups that inhibit proliferation and induce apoptosis of MCF-7 cancer cells. PLoS One. 2015;10(10):e0141184. doi:10.1371/journal.pone.0141184 | ||
Li QS, Li CY, Li ZL, Zhu HL. Genistein and its synthetic analogs as anticancer agents. Anticancer Agents Med Chem. 2012;12(3):271–281. | ||
Chang CF, Liao KC, Chen CH. 2-phenylnaphthalene derivatives inhibit lipopolysaccharide-induced pro-inflammatory mediators by downregulating of MAPK/NF-kappaB pathways in RAW 264.7 macrophage cells. PLoS One. 2017;12(1):e0168945. doi:10.1371/journal.pone.0168945 | ||
Barnum KJ, O’Connell MJ. Cell cycle regulation by checkpoints. Methods Mol Biol. 2014;1170:29–40. doi:10.1007/978-1-4939-0888-2_2 | ||
Resnitzky D, Reed SI. Different roles for cyclins D1 and E in regulation of the G1-to-S transition. Mol Cell Biol. 1995;15(7):3463–3469. | ||
Korzelius J, The I, Ruijtenberg S, et al. Caenorhabditis elegans cyclin D/CDK4 and cyclin E/CDK2 induce distinct cell cycle re-entry programs in differentiated muscle cells. PLoS Genet. 2011;7(11):e1002362. doi:10.1371/journal.pgen.1002138 | ||
Jang SH, Kim AR, Park NH, Park JW, Han IS. DRG2 regulates G2/M progression via the cyclin B1-Cdk1 complex. Mol Cells. 2016;39(9):699–704. doi:10.14348/molcells.2016.0149 | ||
Huang Y, Sramkoski RM, Jacobberger JW. The kinetics of G2 and M transitions regulated by B cyclins. PLoS One. 2013;8(12):e80861. doi:10.1371/journal.pone.0080861 | ||
Zhang Z, Wang CZ, Du GJ, et al. Genistein induces G2/M cell cycle arrest and apoptosis via ATM/p53-dependent pathway in human colon cancer cells. Int J Oncol. 2013;43(1):289–296. doi:10.3892/ijo.2013.1946 | ||
Huang WS, Kuo YH, Kuo HC, et al. CIL-102-induced cell cycle arrest and apoptosis in colorectal cancer cells via upregulation of p21 and GADD45. PLoS One. 2017;12(1):e0168989. doi:10.1371/journal.pone.0168989 | ||
Schmitt E, Sane AT, Bertrand R. Activation and role of caspases in chemotherapy-induced apoptosis. Drug Resist Updat. 1999;2(1):21–29. doi:10.1054/drup.1999.0065 | ||
Wang L, Liu L, Shi Y, et al. Berberine induces caspase-independent cell death in colon tumor cells through activation of apoptosis-inducing factor. PLoS One. 2012;7(5):e36418. doi:10.1371/journal.pone.0036418 | ||
Pirzad G, Jafari M, Tavana S, et al. The role of Fas-FasL signaling pathway in induction of apoptosis in patients with sulfur mustard-induced chronic bronchiolitis. J Toxicol. 2010;2010:7. doi:10.1155/2010/373612 | ||
Lee EW, Kim JH, Ahn YH, et al. Ubiquitination and degradation of the FADD adaptor protein regulate death receptor-mediated apoptosis and necroptosis. Nat Commun. 2012;3:978. doi:10.1038/ncomms1981 | ||
Kominami K, Nakabayashi J, Nagai T, et al. The molecular mechanism of apoptosis upon caspase-8 activation: quantitative experimental validation of a mathematical model. Biochim Biophys Acta. 2012;1823(10):1825–1840. doi:10.1016/j.bbamcr.2012.07.003 | ||
Liao PC, Tan SK, Lieu CH, Jung HK. Involvement of endoplasmic reticulum in paclitaxel-induced apoptosis. J Cell Biochem. 2008;104(4):1509–1523. doi:10.1002/jcb.21730 | ||
Joo JH, Liao G, Collins JB, Grissom SF, Jetten AM. Farnesol-induced apoptosis in human lung carcinoma cells is coupled to the endoplasmic reticulum stress response. Cancer Res. 2007;67(16):7929–7936. doi:10.1158/0008-5472.CAN-07-0931 | ||
Goda AE, Erikson RL, Sakai T, Ahn JS, Kim BY. Preclinical evaluation of bortezomib/dipyridamole novel combination as a potential therapeutic modality for hematologic malignancies. Mol Oncol. 2015;9(1):309–322. doi:10.1016/j.molonc.2014.08.010 | ||
Parrish AB, Freel CD, Kornbluth S. Cellular mechanisms controlling caspase activation and function. Cold Spring Harb Perspect Biol. 2013;5(6):a008672. doi:10.1101/cshperspect.a008672 | ||
Chaitanya GV, Steven AJ, Babu PP. PARP-1 cleavage fragments: signatures of cell-death proteases in neurodegeneration. Cell Commun Signal. 2010;8:31. doi:10.1186/1478-811X-8-5 | ||
Castro L, Gao X, Moore AB, et al. A high concentration of genistein induces cell death in human uterine leiomyoma cells by autophagy. Expert Opin Environ Biol. 2016;5(Suppl1). | ||
Seo HS, Choi HS, Choi YK, et al. Phytoestrogens induce apoptosis via extrinsic pathway, inhibiting nuclear factor-kappaB signaling in HER2-overexpressing breast cancer cells. Anticancer Res. 2011;31(10):3301–3313. | ||
Hentze H, Schmitz I, Latta M, Krueger A, Krammer PH, Wendel A. Glutathione dependence of caspase-8 activation at the death-inducing signaling complex. J Biol Chem. 2002;277(7):5588–5595. doi:10.1074/jbc.M110766200 | ||
Momoi T. Caspases involved in ER stress-mediated cell death. J Chem Neuroanat. 2004;28(1–2):101–105. doi:10.1016/j.jchemneu.2004.05.008 | ||
Winter E, Chiaradia LD, Silva AH, Nunes RJ, Yunes RA, Creczynski-Pasa TB. Involvement of extrinsic and intrinsic apoptotic pathways together with endoplasmic reticulum stress in cell death induced by naphthylchalcones in a leukemic cell line: advantages of multi-target action. Toxicol In Vitro. 2014;28(5):769–777. doi:10.1016/j.tiv.2014.02.002 | ||
Tsui KC, Chiang TH, Wang JS, et al. Flavonoids from Gynostemma pentaphyllum exhibit differential induction of cell cycle arrest in H460 and A549 cancer cells. Molecules. 2014;19(11):17663–17681. doi:10.3390/molecules191117663 | ||
Zhang Y, Xu X, Li W, et al. Activation of endoplasmic reticulum stress and the extrinsic apoptotic pathway in human lung cancer cells by the new synthetic flavonoid, LZ-205. Oncotarget. 2016;7(52):87257–87270. doi:10.18632/oncotarget.13535 | ||
Wang H, Zhang T, Sun W, et al. Erianin induces G2/M-phase arrest, apoptosis, and autophagy via the ROS/JNK signaling pathway in human osteosarcoma cells in vitro and in vivo. Cell Death Dis. 2016;7(6):e2247. doi:10.1038/cddis.2016.138 | ||
Wong RS. Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res. 2011;30(1):87. doi:10.1186/1756-9966-30-24 | ||
Boo HJ, Hyun JH, Kim SC, et al. Fucoidan from Undaria pinnatifida induces apoptosis in A549 human lung carcinoma cells. Phytother Res. 2011;25(7):1082–1086. doi:10.1002/ptr.3489 | ||
Casao A, Mata-Campuzano M, Ordas L, Cebrian-Perez JA, Muino-Blanco T, Martinez-Pastor F. Cleaved PARP-1, an apoptotic marker, can be detected in ram spermatozoa. Reprod Domest Anim. 2015;50(4):688–691. doi:10.1111/rda.12549 | ||
Zhou X, Jiang W, Liu Z, Liu S, Liang X. Virus infection and death receptor-mediated apoptosis. Viruses. 2017;9(11):316. doi:10.3390/v9110316 | ||
Chiu CF, Chang YW, Kuo KT, et al. NF-kappaB-driven suppression of FOXO3a contributes to EGFR mutation-independent gefitinib resistance. Proc Natl Acad Sci U S A. 2016;113(18):E2526–E2535. doi:10.1073/pnas.1522612113 | ||
Huang X, Lu Z, Lv Z, et al. The Fas/Fas ligand death receptor pathway contributes to phenylalanine-induced apoptosis in cortical neurons. PLoS One. 2013;8(8):e71553. doi:10.1371/journal.pone.0071553 | ||
Zhang L, Cheng X, Xu S, Bao J, Yu H. Curcumin induces endoplasmic reticulum stress-associated apoptosis in human papillary thyroid carcinoma BCPAP cells via disruption of intracellular calcium homeostasis. Medicine (Baltimore). 2018;97(24):e11095. doi:10.1097/MD.0000000000011095 | ||
Manie SN, Lebeau J, Chevet E. Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. 3. Orchestrating the unfolded protein response in oncogenesis: an update. Am J Physiol Cell Physiol. 2014;307(10):C901–C907. doi:10.1152/ajpcell.00292.2014 | ||
McGuckin MA, Eri RD, Das I, Lourie R, Florin TH. ER stress and the unfolded protein response in intestinal inflammation. Am J Physiol Gastrointest Liver Physiol. 2010;298(6):G820–G832. doi:10.1152/ajpgi.00063.2010 | ||
Sanderson TH, Gallaway M, Kumar R. Unfolding the unfolded protein response: unique insights into brain ischemia. Int J Mol Sci. 2015;16(4):7133–7142. doi:10.3390/ijms16047133 | ||
Kang KA, Piao MJ, Madduma Hewage SR, et al. Fisetin induces apoptosis and endoplasmic reticulum stress in human non-small cell lung cancer through inhibition of the MAPK signaling pathway. Tumour Biol. 2016;37(7):9615–9624. doi:10.1007/s13277-016-4864-x | ||
Zhang J, Feng Z, Wang C, et al. Curcumin derivative WZ35 efficiently suppresses colon cancer progression through inducing ROS production and ER stress-dependent apoptosis. Am J Cancer Res. 2017;7(2):275–288. | ||
Rozpedek W, Pytel D, Mucha B, Leszczynska H, Diehl JA, Majsterek I. The role of the PERK/eIF2alpha/ATF4/CHOP signaling pathway in tumor progression during endoplasmic reticulum stress. Curr Mol Med. 2016;16(6):533–544. | ||
Chen S, Zhao Y, Zhang Y, Zhang D. Fucoidan induces cancer cell apoptosis by modulating the endoplasmic reticulum stress cascades. PLoS One. 2014;9(9):e108157. doi:10.1371/journal.pone.0108157 |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.