")
Back to Journals » International Journal of Nanomedicine » Volume 14
3,5,4′-trimethoxy-trans-stilbene loaded PEG-PE micelles for the treatment of colon cancer
Authors Wu JY, Li YJ , Liu XY, Cai JX, Hu XB , Wang JM , Tang TT, Xiang DX
Received 2 July 2019
Accepted for publication 30 August 2019
Published 12 September 2019 Volume 2019:14 Pages 7489—7502
DOI https://doi.org/10.2147/IJN.S221625
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Jun-Yong Wu,1–3* Yong-Jiang Li,1–3* Xin-Yi Liu,1–3 Jia-Xin Cai,1–3 Xiong-Bin Hu,1–3 Jie-Min Wang,1–3 Tian-Tian Tang,1–3 Da-Xiong Xiang1–3
1Department of Pharmacy, The Second Xiangya Hospital, Central South University, Changsha 410011, Hunan, People’s Republic of China; 2Institute of Clinical Pharmacy, Central South University, Changsha 410011, Hunan, People’s Republic of China; 3Hunan Provincial Engineering Research Center of Translational Medicine and Innovative Drug, Changsha, Hunan, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Da-Xiong Xiang
Department of Pharmacy, The Second Xiangya Hospital, Central South University, Changsha 410011, Hunan, People’s Republic of China
Email [email protected]
Background: 3,5,4′-trimethoxy-trans-stilbene (BTM) is a methylated derivative of resveratrol. To improve the pharmaceutical properties of BTM, BTM loaded PEG-PE micelles (BTM@PEG-PE) were fabricated and its anti-cancer efficacy against colon cancer was evaluated.
Methods: BTM@PEG-PE micelles were prepared by the solvent evaporation method and were characterized by nuclear magnetic resonance (NMR), size, zeta potential, polymer disperse index (PDI) and transmission electron microscopy (TEM). Cellular uptake, cell viability assay, caspase-3 activity assay and flow cytometry were performed to evaluate the cell internalization and anti-cancer efficacy of BTM@PEG-PE micelles in vitro. Pharmacokinetic profiles of BTM and BTM@PEG-PE micelles were compared and in vivo anti-cancer therapeutic efficacy and safety of BTM@PEG-PE micelles on CT26 xenograft mice were evaluated.
Results: BTM was successfully embedded in the core of PEG-PE micelles, with a drug loading capacity of 5.62±0.80%. PEG-PE micelles facilitated BTM entering to the CT26 cells and BTM@PEG-PE micelles exerted enhanced anti-cancer efficacy against CT26 cells. BTM@PEG-PE micelles showed prolonged half-life and increased bioavailability. More importantly, BTM@PEG-PE micelles treatment suppressed tumor growth in tumor-bearing mice and prolonged survival with minimal damage to normal tissues.
Conclusion: Altogether, the BTM@PEG-PE micelles might be a promising strategy to enhance the pharmacokinetic and pharmacodynamic potentials of BTM for colon cancer therapy.
Keywords: 3,5,4′-trimethoxy-trans-stilbene, bioavailability, colon cancer, drug delivery, micelles
Background
Colorectal cancer is the third most common cancer and the third leading cause of cancer death in the United States.1 Despite improvements in cancer prevention and diagnostic techniques, new cases were not decreased and current treatment options for colon cancer is limited.2 Colon cancer is an advanced carcinoma difficult to be fully eradicated by surgery and chemotherapy still plays an important role in the management of colon cancer.3,4 However, drug toxicity and undesirable side effects limit desirable anti-cancer effects and there is still a need for potent chemotherapeutics and strategies.
Resveratrol, a naturally occurring antioxidant, has been extensively studied for possible health benefits,5,6 and its role in colon cancer management have recently been explored in clinical7,8 and preclinical studies.9–11 However, poor absorption and low bioavailability limit its potential benefits in cancer treatment.12,13 It has been reported that the methylated derivatives of flavonoids may exhibit higher anti-proliferative potency than the corresponding hydroxylated molecules.14 Herein, 3,5,4′-Trimethoxy-trans-stilbene (BTM), the fully methylated derivative of resveratrol, has been developed and has shown potent anti-cancer efficacy.15,16 However, poor water solubility, short half-life, and low bioavailability also hindered the application of BTM.17
Micelles are widely used nano vectors for drug delivery, they are formed through the self-assembly of amphiphilic polymers in aqueous media.18 The hydrophilic PEG2000 groups that form the shell of the micelles may help the vector escaping the reticuloendothelial system (RES) to prolong the circulation,19 while the core of highly hydrophobic PE residues can solubilize hydrophobic drugs.20 Moreover, the dense PEG shell provides a shelter for the entrapped molecules and the long fatty acyl chains of PEG-PE micelles can protect the embedded molecules from degradation in circulation and release the drug slowly by controlling the mobility of the entrapped drug.21,22
Therefore, the PEG-PE micelles could be a promising delivery system for prolonging circulation and increasing the bioavailability of BTM to enhance its anti-tumor effect. In this study, we developed BTM@PEG-PE micelles by forming PE as the core and PEG as the shell. The BTM@PEG-PE micelles exhibited increased therapeutic efficacy against colon cancer with prolonged survival but minimal damage to normal tissues.
Materials and methods
Cell culture
The mouse colon carcinoma cell line CT26 and human fetal hepatocyte line L02 were obtained from the Cell Bank of the Chinese Academy of Sciences (SIBS, Shanghai, China). CT26 cells were maintained in RPMI-1640 (Thermo Fisher Scientific, USA) and L02 cells were maintained in DMEM (Thermo Fisher Scientific, USA), supplemented with 10% FBS (fetal bovine serum, Thermo Fisher Scientific, USA) and 1% penicillin−streptomycin (Thermo Fisher Scientific, USA). Cells were incubated at 37 °C with 5% CO2.
Preparation of BTM-loaded micelles
BTM@PEG-PE micelles were prepared by the solvent evaporation method.23 Briefly, 100 mg of 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000] (PEG2000-PE, Lipoid GmbH, Ludwigshafen, Germany) and BTM (8 mg) were co-dissolved in 20 mL of methanol. The solvent was dried under reduced pressure in a round-bottom flask at 37 °C to form a thin film. Immediately, the obtained film was kept in vacuum for over 6 h to remove remaining solvent. The dry thin film was hydrated in 5 mL of distilled water at 37 °C and then cooled to 10 °C and filtrated through a 0.22 nm Millipore filter to remove undissolved drug and copolymer and to formulate BTM@PEG-PE in clear solution.
Characterization of BTM-loaded micelles
Nuclear magnetic resonance spectroscopic studies (NMR)
For confirmation of the core-shell structure of BTM@PEG-PE micelles, the 1H NMR spectra of free BTM, blank PEG-PE and lyophilized BTM@PEG-PE micelles in DMSO were recorded by an Eclipse 400 MHz NMR spectrometer (JEOL Ltd., Japan).
Micelles size, zeta potential, polymey disperse index (PDI) and transmission electron microscopy (TEM)
To obtain the size distribution, zeta potential and PDI of BTM@PEG-PE micelles, the Zetasizer Nano ZS90 (Malvern, UK) was used. To observe the morphology of BTM@PEG-PE micelles, 10 μL of BTM@PEG-PE micelles was dropped onto an ultrathin carbon film-coated 200 mesh copper grid. After drying excess liquid, the BTM@PEG-PE micelles-coated grid was imaged by a multipurpose field emission transmission electron microscope (2100F, JEOL Ltd., Japan).
Entrapment efficiency (EE) and drug loading (DL)
The amount of BTM loaded in PEG-PE micelles was measured by a high performance liquid chromatography (HPLC) system (H20-AT, SHIMADZU, Japan) equipped with a UV detector. The detection wavelength was set at 318 nm. The mobile phase consisted of methanol and water (75:25, v/v) at a flow rate of 1.0 mL/min. The column temperature was maintained at 30 °C. Drug loading efficiency (DL%) and entrapment efficiency (EE%) were calculated as previously described.23
Drug release
To measure BTM release, 5.5 mg of BTM@PEG-PE was placed in a 3,000 MWCO centrifugal filter (Millipore, USA) with 0.8 mL of PBS. The filter was then immersed in a tube with 7.2 mL of PBS, at pH 7.4 or pH 5.5, under the sink conditions at 37 °C with stirring (100 rpm). Samples (500 μL) were taken at 1, 2, 4, 6, 8, 10, 12, 24, 36, 48, 60, 72 h time points from inside the tube and replaced with an equal amount of fresh medium (500 μL). Samples were analyzed by HPLC as described.17 The amount of BTM released from BTM@PEG-PE was expressed as a percentage of total BTM loaded.
Cellular uptake
For cellular uptake studies, Coumarin-6 (C6) (J&K Chemical Ltd, Shanghai, China) was loaded into PEG-PE using the method for preparation of BTM-loaded micelles as described above to formulate C6@PEG-PE micelles. CT26 cells were seeded into 12-well culture plates at 1.0×105 cells per well and cultured for 24 h. Cells were then incubated with C6@PEG-PE micelles or C6 solution at an equivalent C6 concentration (0.1 mg/L). After 1 h, 2 h or 4 h of incubation, cells were washed with PBS (pH 7.4) three times to eliminate residual C6, followed by fixing with paraformaldehyde (PFA) and staining with 4ʹ6-diamidino-2-phenylindole (DAPI, Beyotime, China). C6 and DAPI showed green and blue colorations, respectively. Cellular internalization of C6@PEG-PE micelles in CT26 cells was observed using Axio Vert A1 fluorescence microscope and then analyzed by using ZEN software (Carl Zeiss, Germany). Also, to investigate the efficiency of cellular uptake, the fluorescence intensity of CT26 cells was analyzed 1 h, 2 h or 4 h post-incubation using flow cytometry (BD, USA).
Cell viability assay
CT26 cells were seeded into 96-well plates at a density of 5×104 cells per well. We first investigated the IC50 of BTM on CT26 cells by treating with various doses of BTM for 24 h, 48 h or 72 h. Then we compared the in vitro anti-tumor efficacy of BTM and BTM@PEG-PE at the same dose for 24 h or 48 h. The cellular viability was determined using MTS Cell Proliferation Colorimetric Assay (Promega, USA) by measuring the absorbance at 490 nm using a Multiskan FC (Thermo Fisher Scientific, USA). Experiments were repeated three times, and the data are representative of three experiments. The same cell viability assay was performed on L02 cells as well.
Caspase-3 activity assay
The activity of caspase 3 was determined by using a caspase-3 activity kit (Beyotime Institute of Biotechnology, China). The caspase-3 activity based on the ability of caspase-3 to change Ac-DEVDpNA into the yellow formazan product, p-nitroaniline (pNA), was detected for CT26 cells treated with different formulations for 24 h. Briefly, CT26 cells after treatments were resuspended in lysis buffer and left on ice for 10 min and the lysate was centrifuged at 14,000 g for 10 min at 4 °C. The supernatants were extracted into a microcentrifuge tube and 10 μL of caspase 3 substrate (Ac-DEVDpNA, 2 mM) was added for incubation in the dark at 37 °C for 1 h. The release of pNA was measured at 405 nm by a Multiskan Spectrum (Thermo, Waltham, MA). The activity of caspase-3 was presented as the fold of enzyme activity compared to that of synchronized control cells.
Apoptosis and cell cycle assay
To assess the induction of apoptosis, CT26 cells were treated with free BTM or BTM@PEG-PE and then stained by Annexin V-FITC/PI (BD Pharmingen) or Cell-cycle assay kit (BD Pharmingen) according to the manufacturer’s instructions. The percentage of the apoptotic cells and cell cycles were analyzed using flow cytometry (BD Biosciences).
Pharmacokinetic study
The animal experiment was approved by the Animal Ethics Committee at the Institute of Clinical Pharmacy, Central South University (No. 2018sydw0208). All animal studies were carried out using the Institutional Animal Care and Use Committee (IACUC)-approved procedures. For the convenience of continuous blood collection, we used rats for the pharmacokinetic study. Female Sprague–Dawley rats (200–220 g) were obtained from Hunan Slack Scene of Laboratory Animal Co., Ltd. Five rats were treated with free BTM or BTM@PEG-PE micelles at an equivalent dose of 20 mg/kg by tail vein injection. After administration, blood samples (0.4 mL) were collected into microcentrifuge tubes containing heparin as anti-coagulant at 30, 60, 90, 120, 180, 240, 360 and 480 min. Blood samples were centrifuged at 3,000 rpm for 10 min, the supernatant plasma samples were pretreated as previously described17 before analysis.
In vivo anti-cancer efficacy study
For the generation of CT26 xenografts, 5×106 cells mixed with 50% Matrigel solution (Corning, USA) were injected subcutaneously into right underarm of female BALB/C nude mice (6 weeks old), obtained from Hunan Slack Scene of Laboratory Animal Co., Ltd. and housed according to the regulations of the IACUC. After the tumor volume reached approximately 100 mm3, mice were randomly divided into four groups: PBS, control PEG-PE, free BTM and BTM@PEG-PE at equivalent dose of BTM (5 mg/kg), all formulations were administrated through tail vein injection using a microsyringe for five times with an interval of two days (day 5 to day 13). Tumor volume and mice weight were measured every two days for 15 days from the initial tumor cells injection. Then, five mice in each group were randomly selected, and tumors and major organs and blood samples were collected from the selected mice after sacrifice. Major organs and tumors were subjected to histopathological assessment, plasma levels of ALT, AST, Cr, and BUN were measured using assay kits (Huili Biotech, China), levels of TNF-α and IL-6 in plasma were measured using enzyme-linked immunosorbent assay (ELISA) kits (Boster, China) according to the manufacturer’s instructions. The remaining mice in each group were housed without further intervention for survival observation.
Statistical analysis
Data were presented as mean values ± SD. Pharmacokinetics parameters were obtained using the Drug and Statistics (DAS) Version 3.3 software. One-way analysis of variance (ANOVA) and Tukey’s HSD post-hoc test were performed at the significance level α=0.05.
Results
Characterization of BTM@PEG-PE micelles
The morphology of BTM@PEG-PE micelles was presented in Figure 1A, and a spheroidal shape with small size was observed. The size distribution of blank PEG-PE micelles (15.53±0.08 nm) and the BTM@PEG-PE micelles (17.73±1.08 nm) tested by DLS was presented in Figure 1B, indicated that the loading of BTM increased the size of micelles very slightly. The DL and EE of BTM@PEG-PE micelles were 5.62±0.80% and 79.34±5.61% respectively (Figure 1C), showing a relatively high drug loading efficiency. BTM@PEG-PE micelles showed increased PDI and decreased absolute value of zeta potential of the micelles (Figure 1C), indicating that the loading of BTM may alter the dispersity of the micelles and this may attribute to the high drug loading capacity.
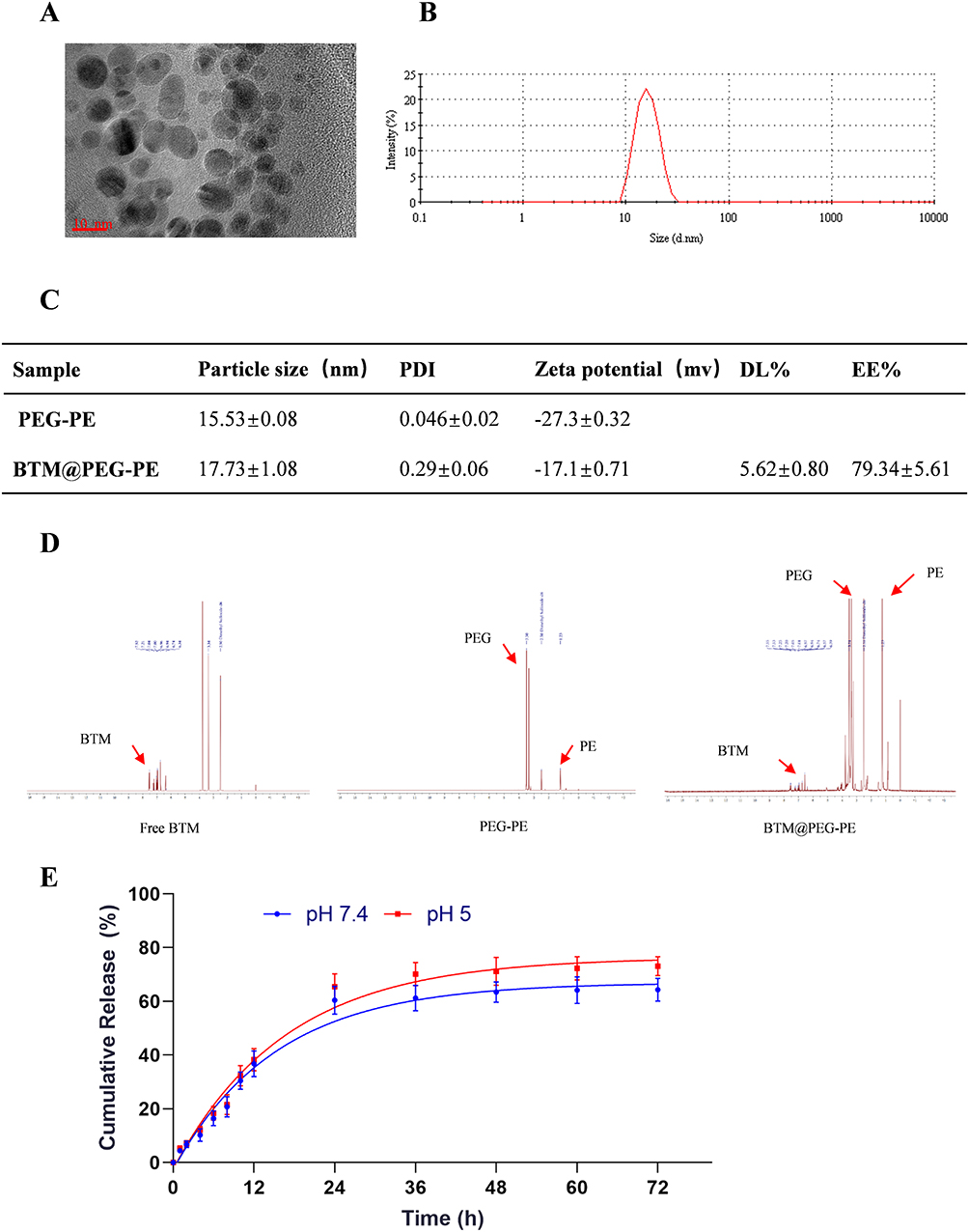 |
Figure 1 Characterization and loading and releasing properties of BTM@PEG-PE micelles. (A) The TEM image of BTM@PEG-PE micelles. (B) The size distribution of BTM@PEG-PE micelles. (C) Drug loading properties of BTM@PEG-PE micelles. (D) The core-shell structure of BTM@PEG-PE micelles. (E) Drug releasing properties of BTM@PEG-PE micelles at pH 7.4 and pH 5.0. |
Core-shell structure of BTM@PEG-PE micelles and drug release
The NMR results were presented in Figure 1D. Free BTM showed a cluster of peaks with aromatic protons around 7.00 ppm while blank PEG-PE micelles showed strong PEG peaks at 3.50 ppm and relatively weak peaks of PE at 1.23 in DMSO, suggesting a PEG shell and PE core structure of the blank micelles. The 1H NMR spectra of BTM@PEG-PE micelles showed both peaks at about 7.00 ppm (aromatic protons of BTM) and peaks at 3.51 ppm (PEG) and 1.23 ppm (PE), suggesting that the BTM was successfully encapsulated into the core-shell of PEG-PE.
For drug release, a burst release was observed of about 60% of BTM in BTM@PEG-PE micelles in the first 24 h (Figure 1E). At pH 7.4, the release of BTM was slightly slower than at pH 5.0, suggesting that the PEG-PE micelles were stable under circulation condition (37 °C, pH 7.4). After 24 h, the encapsulated BTM was released in a controlled manner (Figure 1E).
Cellular uptake
To test whether the micelles could be effectively uptake by the target cells, Coumarin-6 (C6) loaded PEG-PE micelles (green) was formulated. CT26 cells uptake of C6 and C6@PEG-PE micelles were increased with incubation time, from 1 h to 4 h (Figure 2); however, the fluorescent intensity for C6@PEG-PE micelles was significantly higher than free C6 (Figure 2). As for the results of flow cytometry (FACS), a time-dependent uptake was confirmed (Figure 2B). At 4 h, nearly all CT26 cells have uptake C6 or C6@PEG-PE (Figure 2C). The mean fluorescence intensity for CT26 cells incubated with C6@PEG-PE was significantly higher than free C6 (Figure 2B and C).
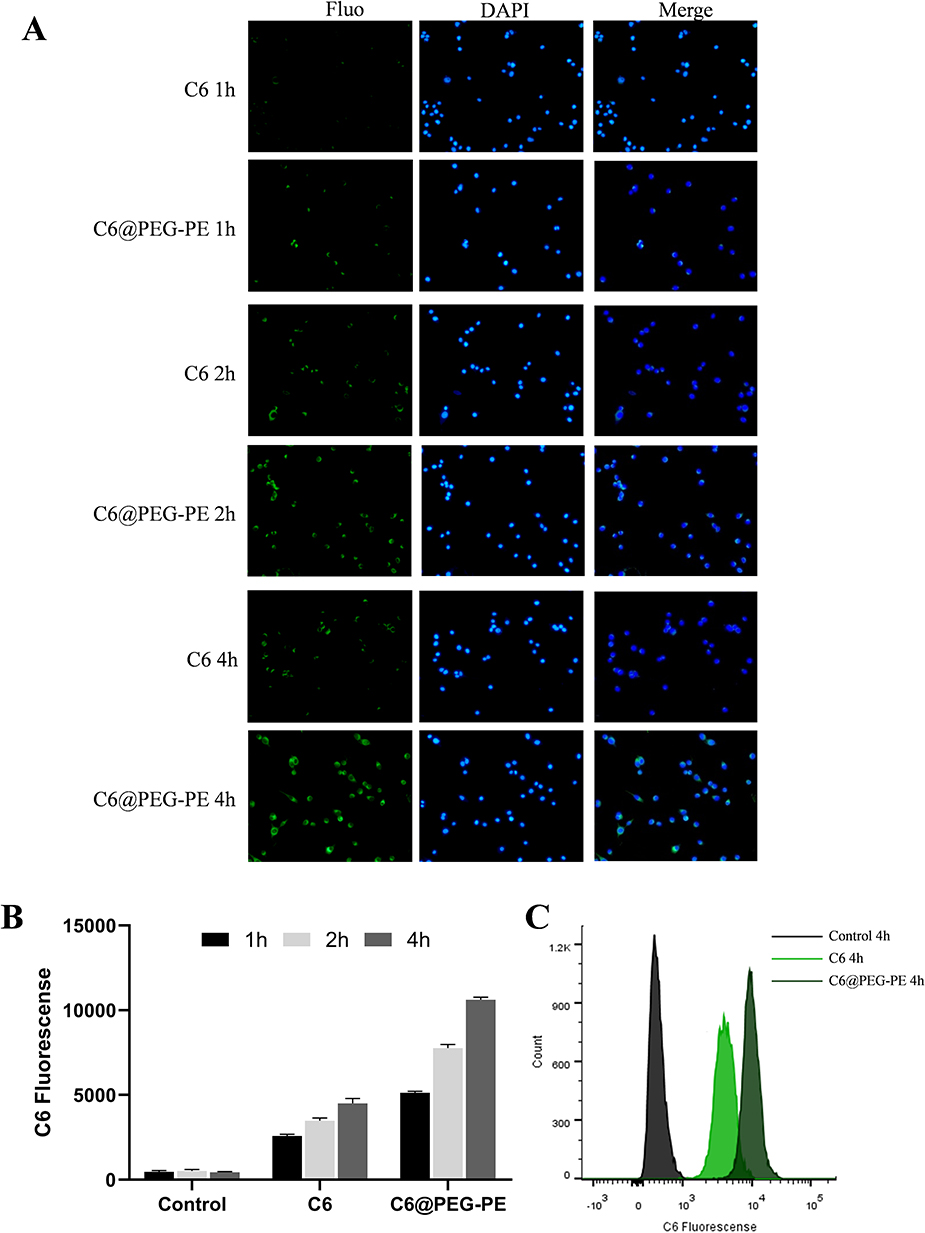 |
Figure 2 In vitro cellular uptake of C6@PEG-PE micelles. Cells were incubated with C6@PEG-PE micelles for 1 h, 2 h or 4 h before imaging and flow cytometry analysis. (A) uptake of C6 and C6@PEG-PE micelles incubation in CT26 cells, green indicates C6 or C6@PEG-PE micelles while blue indicates the nucleus. (B) Mean C6 fluorescence of CT26 cells incubated with C6 or C6@PEG-PE for 1, 2, 4 h. (C) Fluorescence intensity when CT26 cells were treated with C6 or C6@PEG-PE for 4 h. Data were presented as mean ± SD of three independent experiments. |
Cell viability assay and caspase-3 activity
As the solubility of BTM is very low in water, we first investigated the cytotoxic effect of BTM@PEG-PE on CT26 cells. When CT26 cells were treated with blank PEG-PE without the drug, the cell viability was not decreased even at high concentration (Figure 3A), suggesting a minimum effect of the vehicle on cell viability. A dose-response cytotoxic effect of BTM@PEG-PE micelles on CT26 cells was observed (Figure 3A); however, BTM@PEG-PE showed reduced cytotoxic effects on L02 cells, even at relatively high dose (Figure 3B). The calculated IC50 of BTM@PEG–PE micelles on CT26 cells were 39.24 μM for 24 h and 11.53 μM 48 h (Figure 3C). Then, we investigated the cytotoxic effects of free BTM using the maximum concentration of BTM in DMSO (0.3%) and compared it with BTM@PEG-PE micelles at equivalent BTM dose. At the relatively low dose (4.97 μM BTM), free BTM and BTM@PEG-PE exhibited similar cytotoxic effects on CT26 cells (Figure 3D).
The key role of caspase-3 in apoptosis has been reported.24 We measured the activity of caspase-3 in CT26 cells treated with various formulations. As a result, the PEG-PE without the drug did not increase the caspase-3 activity, free BTM and BTM@PEG-PE micelles at the maximum equivalent concentration induced a 2-fold caspase-3 activity, while the higher concentration of BTM@PEG-PE micelles induced about 3.5-fold caspase-3 activity (Figure 3E). These results demonstrated the PEG-PE micelles increased the solubility of BTM and exhibited superior cytotoxic effects against CT26 cells.
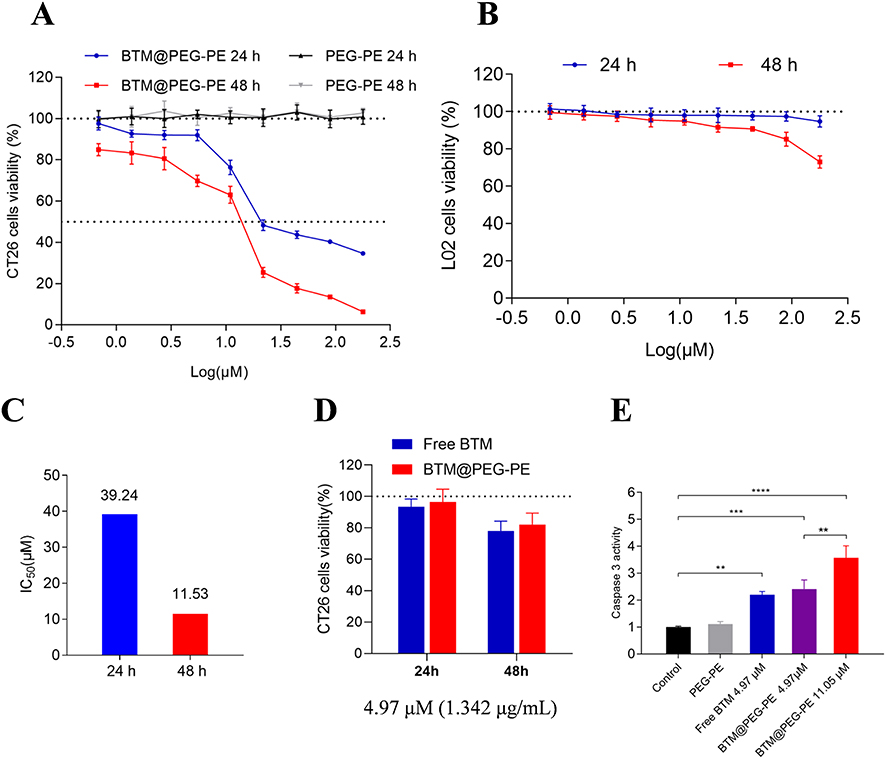 |
Figure 3 In vitro antitumor activity of BTM and BTM@PEG-PE micelles. CT26 cells were treated over 24 or 48 h with PEG-PE or free BTM or BTM@PEG-PE micelles. Relative viability was measured by MTS assay and each point represents the mean ± SD of six independent experiments. (A) Effects of increasing concentrations of BTM@PEG-PE micelles on CT26 cells viability. (B) Effects of increasing concentrations of BTM@PEG-PE micelles on L02 cells viability. (C) Calculated IC50 of BTM@PEG-PE micelles on CT26 cells viability. (D) Comparison of effects of free BTM BTM@PEG-PE micelles on CT26 cells viability at the equivalent highest free BTM concentration (4.97 μM). (E) Caspase 3 activity of CT26 cells after treatment of 24 h. Each point represents the mean ± SD, **P<0.01, ***P<0.001, ****P<0.0001. |
Apoptosis and cell cycle assay
To further confirm the anti-cancer effects of BTM@PEG-PE micelles, we examined the cell death induced by free BTM or BTM@PEG-PE micelles (Figure 4A). FITC-Annexin V/propidium iodide (PI) was used to label apoptotic cells and necrotic cells. Cells incubated with a high dose of BTM@PEG-PE micelles (11.05 μM) showed the highest apoptotic ratio (20.83%). At the equivalent BTM dose (4.97 μM), BTM@PEG-PE micelles showed higher apoptotic ratio (14.16%) than free BTM (8.04%), while PEG-PE without BTM showed the lowest apoptotic ratio (4.36%). Cell cycle assay also confirmed the significant cytotoxic effects of BTM@PEG-PE micelles against CT26 cells (Figure 4B) as the proportion of cells in S-phase was significantly increased. These flow cytometry results were consistent with the results of the MTS cell viability assay. Taken together, the PEG-PE micelles mediated delivery enhanced the solubility of BTM and contributed to a dramatically increased tumor cell killing effect.
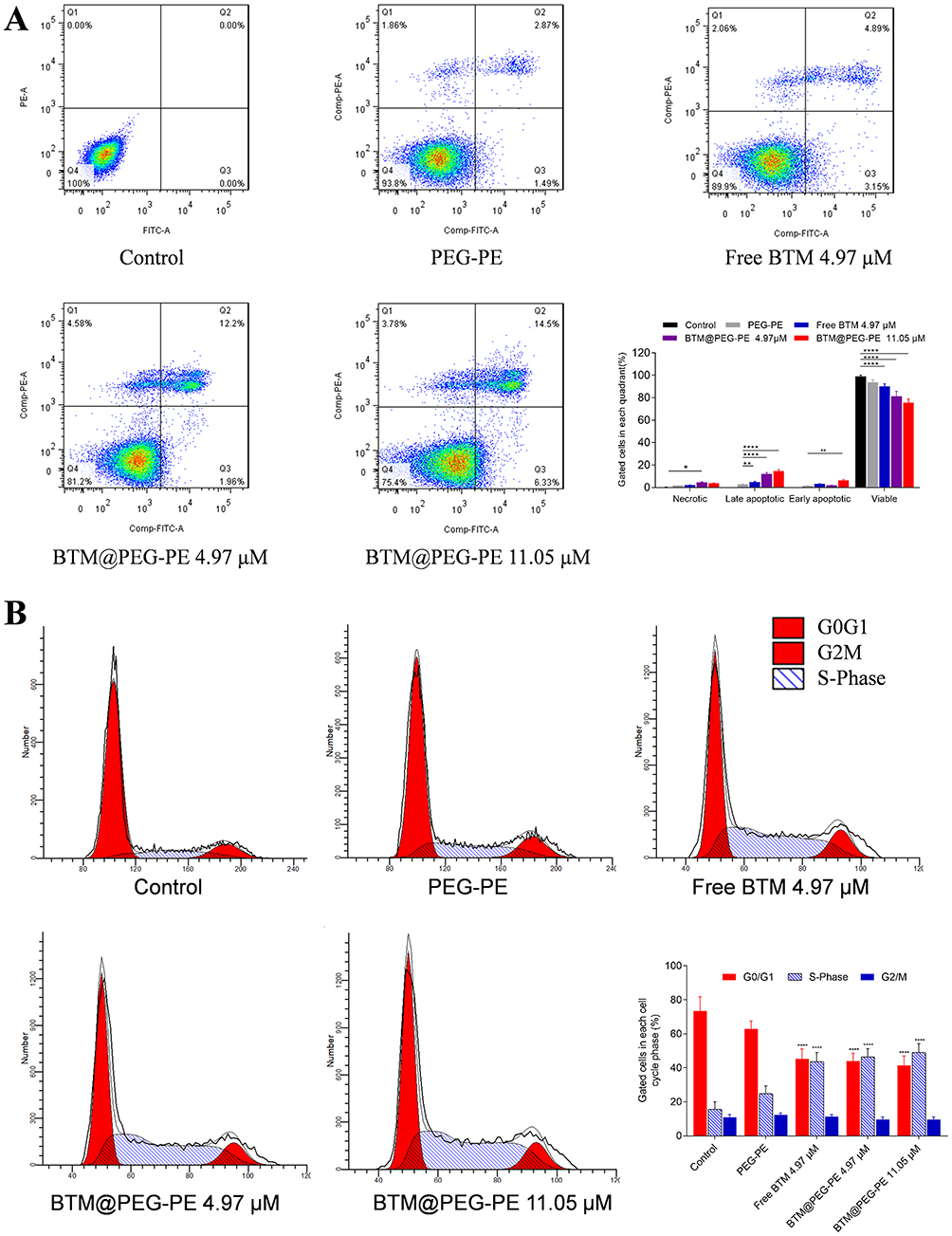 |
Figure 4 Apoptosis and cell cycle assay. (A) Apoptosis was analyzed using an Annexin V/PI staining kit. (B) Cell cycle was analyzed using a BrdU FITC assay. |
Pharmacokinetics
The plasma concentration-time curve of BTM@PEG-PE micelles and free BTM were presented in Figure 5A and detailed pharmacokinetic parameters were summarized in Figure 5B. The clearance of BTM in circulation was not very quick, but BTM was almost undetectable 480 min after free BTM administration, whereas it remained a relatively high concentration in plasma of rats after BTM@PEG-PE micelles administration. For free BTM, the AUC0–∞ was 14,323.8±3,314 µg/L·h and the half-life (t1/2) was 1.6±0.25 h. However, BTM@PEG-PE micelles exhibited a controlled drug release of BTM. the AUC0–∞ was 26,583.1±4,962 µg/L·h and the half-life (t1/2) was 3.1±0.42 h, which was 1.8 times higher and 1.9 times longer than free BTM, with significantly decreased plasma clearance.
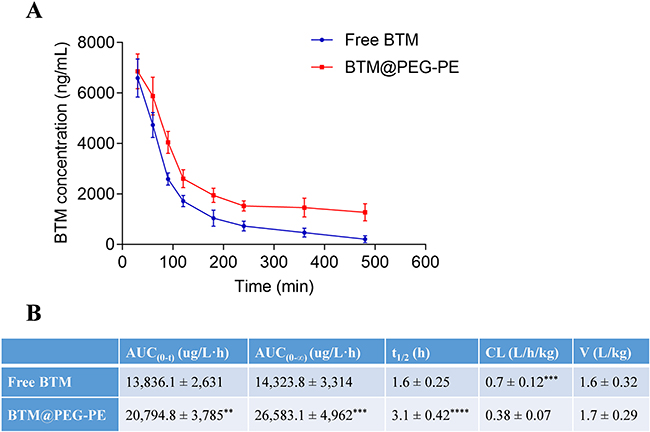 |
Figure 5 Pharmacokinetic profiles of free BTM and BTM@PEG-PE micelles after intravenous injection (N=5). (A) Plasma concentration-time curve of free BTM and BTM@PEG-PE micelles. (B) Pharmacokinetic parameters of free BTM and BTM@PEG-PE micelles. Each point represents the mean ± SD, **P<0.01, ***P<0.001, ****P<0.0001. |
In vivo anti-tumor effects of BTM@PEG-PE
For the investigation of the in vivo anti-cancer therapeutic efficacy of BTM@PEG-PE micelles on CT26 xenograft mice, mice were treated with various formulations intravenously for six times with an interval of two days. Very fast tumor growth was observed in mice receiving PBS as controls, while mice receiving PEG-PE without BTM showed similar tumor growth to that of the control group. BTM treatment was effective, BTM@PEG-PE micelles showed improved therapeutic efficacy at suppressing tumor growth than free BTM (Figures 6A and S1). As for the bodyweight of mice during the treatment, a steady increasing trend was observed for all groups, without significant differences between groups, suggesting potential safety of low dose BTM and PEG-PE mediated drug delivery (Figure 6B). Excised tumors (Figure 6C) and tumor weight (Figure 6D) also confirmed the enhanced anti-cancer efficacy of BTM@PEG-PE as compared to free BTM. As for the survival after treatment, while both mice receiving free BTM and BTM@PEG-PE micelles showed prolonged survival, BTM@PEG-PE micelles further improved the survival as compared to free BTM, and the median survival for mice receiving PBS, PEG-PE, free BTM, and BTM@PEG-PE micelles were 25, 26, 35 and 40 days respectively (Figure 6E). Also, the H&E staining and TUNEL immunohistochemistry showed clear cell death in the tumor of BTM@PEG-PE treated mice (Figure 7).
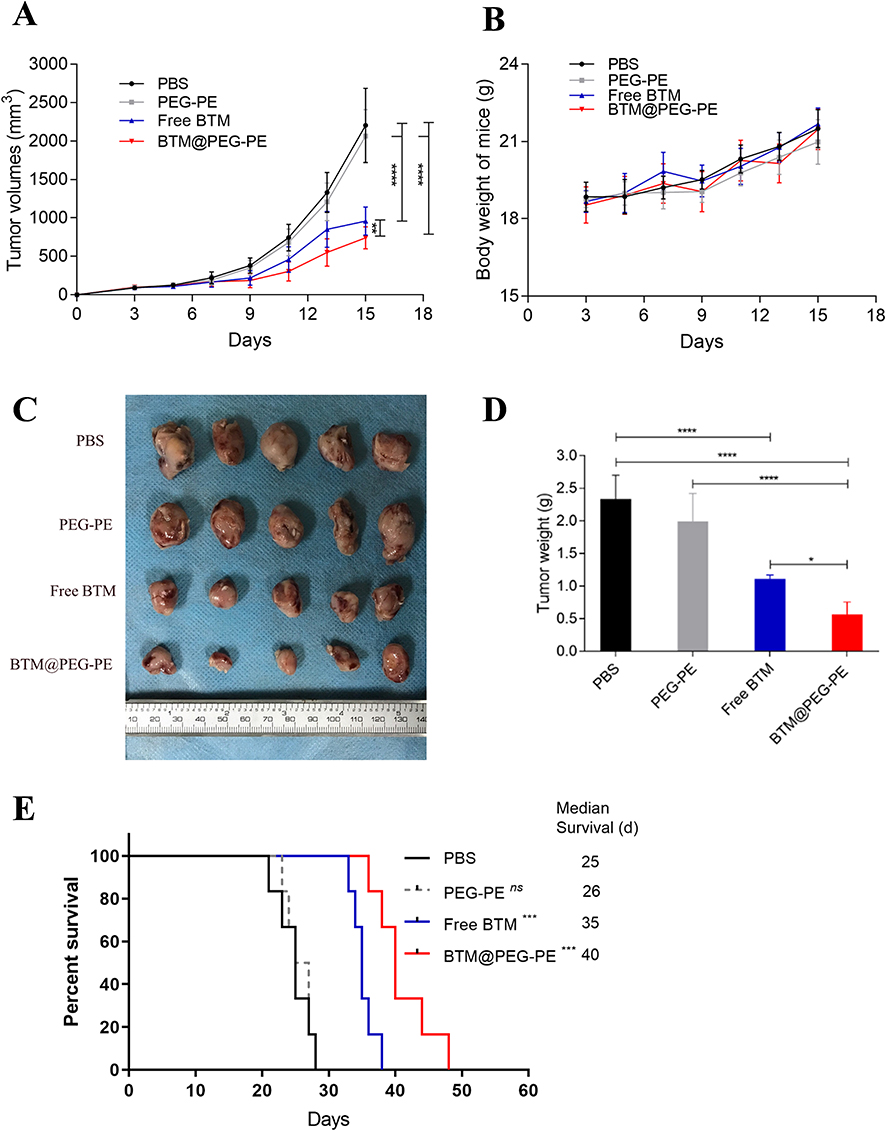 |
Figure 6 Therapeutic efficacy of BTM@PEG-PE micelles against CT26 tumors (N=10). (A) Tumor growth in mice during the experimental period. (B) Change of bodyweight of tumor-bearing mice (N=10). (C) Excised tumors at the end of experiments (N=5). (D) Tumor weight at the end of experiments (N=5). (E) Survival analysis of tumor-bearing mice after treatment of different formulations (N=6). Each point represents the mean ± SD, *P<0.05, ***P<0.001, ****P<0.0001; ns, not significant. |
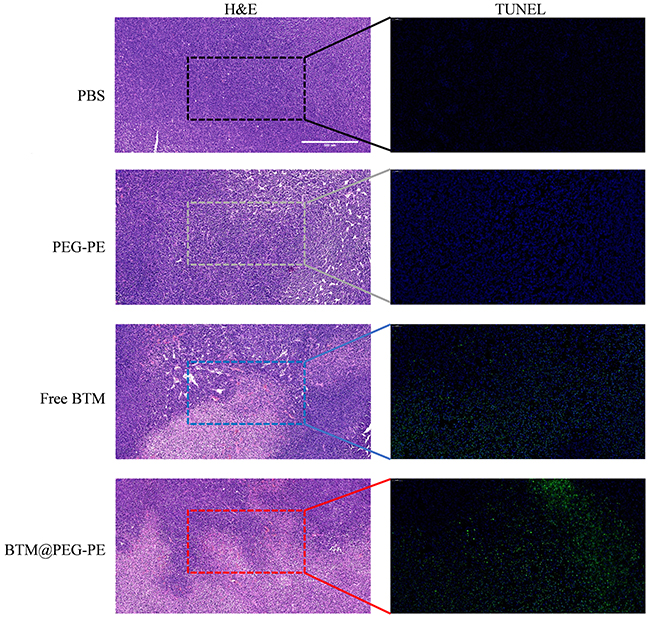 |
Figure 7 Histopathological confirmation of efficient therapeutic efficacy of BTM@PEG-PE micelles against CT26 tumor. H&E and TUNEL-stained tumors of mice after treatment (scale bar =500 μm). |
Safety evaluation
We analyzed the relative levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) to reflect liver functions, and blood urea nitrogen (BUN), creatinine (Cr) and uric acid (UA) to reflect renal functions. Tumor-bearing mice showed elevated levels of all measures. Mice receiving PBS and blank PEG-PE exhibited similar liver and kidney functions as detected. However, all measures were not significantly different for normal mice and BTM or BTM@PEG-PE micelles treated mice (Figure 8A–E), demonstrating that suppressing tumor growth may also be of benefit to other organs, such as liver and kidney.
We also analyzed the TNF-α and IL-6 cytokines to reflect the immunological response of the formulates. As a result, Both TNF-α and IL-6 were higher in PBS group than normal mice, suggesting that the tumor growth was associated with elevated TNF-α and IL-6 levels and PEG-PE caused minor induction of TNF-α and IL-6. Free BTM significantly increased the production of TNF-α and IL-6 while BTM@PEG-PE micelles group showed reduced TNF-α and IL-6 levels, suggesting that the vector, PEG-PE micelles, reduced BTM-mediated induction of cytokines (Figure 8F and G). The H&E staining results showed no significant difference of major organs (heart, spleen, lung, and kidney) between treatment groups (Figure S2), most likely due to the low BTM dose (5 mg/kg).
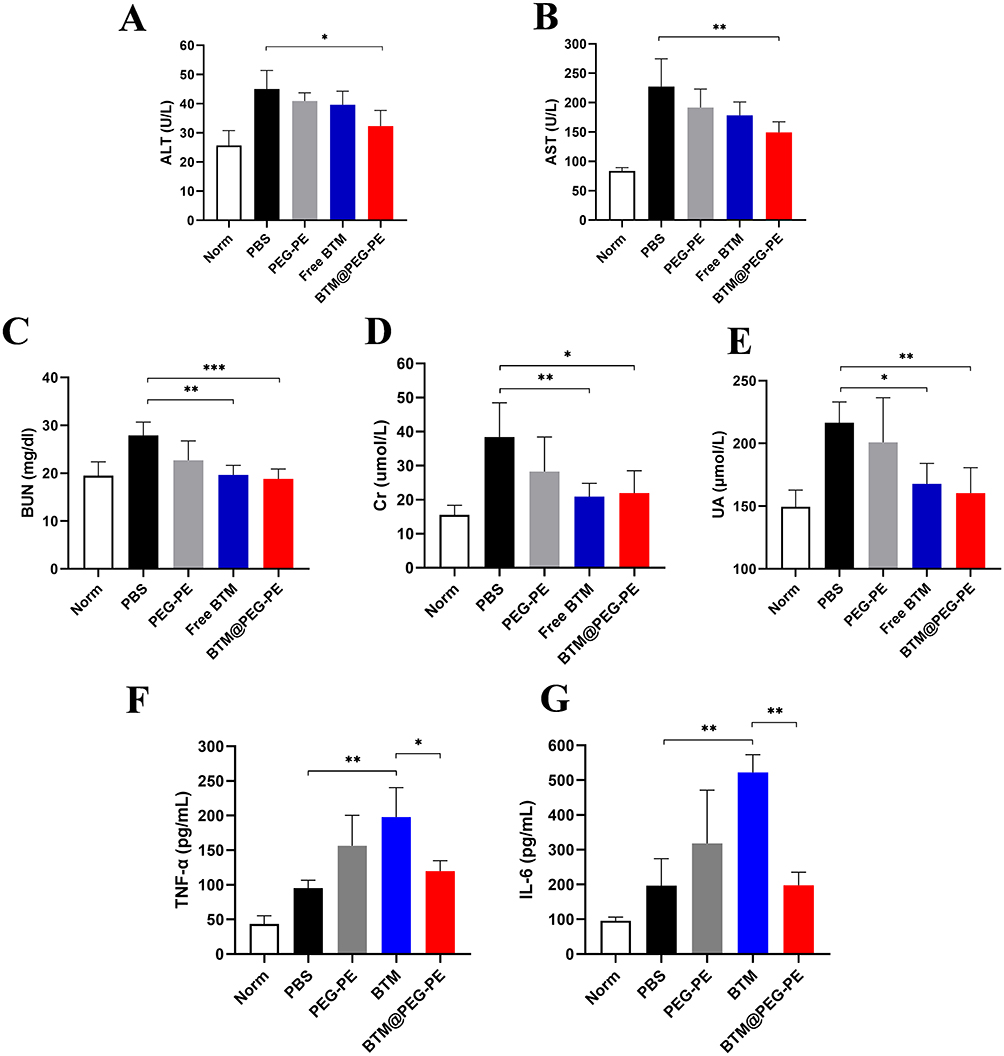 |
Figure 8 Safety and biocompatibility evaluation of free BTM and BTM@PEG-PE micelles. Plasma levels of (A) ALT, (B) AST, (C) BUN, (D) Cr, (E) UA, (F) TNF-α and (G) IL-6 in tumor-bearing mice two days after treatment, n=5. Each point represents the mean ± SD, *P<0.05, **P<0.01, ***P<0.001. |
Discussion
PEG2000-PE as an amphipathic polymer could form micelles in an aqueous solution by self-assemble by filming-rehydration method. The encapsulation of BTM did not alter the particle size of PEG-PE micelles and the particle size around 16 nm would allow the particle avoiding kidney filtration or liver metabolization as well as escaping macrophage phagocytosis,25 with increased circulation time than the free drug. The high biocompatibility of the anionic surface of PEG-PE micelles has also been reported.26 The high drug loading efficiency of BTM@PEG-PE micelles could be attributed to the poor water solubility of the drug and the two fatty acid acyls of the micelles that help solubilizing and enclosing such lipophilic chemical,27 and BTM would be remained in the hydrophobic phase during the self-assembling process and be encapsulated efficiently. For the release of BTM, encapsulation of BTM in the core of the micelles may enable a sustained drug release and long circulation time, and the controlled release may help to maintain a sustained plasma concentration.
BTM is a non-water-soluble drug and it has been reported that chemicals with poor water-solubility were insufficiently uptake by cells,28 therefore hydrophobic C6 and BTM may less able to enter the cell in an aqueous medium. However, the entrapment in PEG-PE micelles allowed the cargo to be effectively uptake by cells. Moreover, after 4 h of incubation, a higher co-localization of C6 in cells was observed, suggesting that the PEG-PE micelles could be internalized by CT26 cells. The amphiphilic structure of PEG-PE micelles could insert into the plasma membrane and facilitate cell internalization of the cargo.
For the cell viability assay, we observed a minimum effect of the PEG-PE micelles on CT26 cells but a dose-response cytotoxic effect of BTM@PEG-PE micelles. Besides, L02 cells were not affected by BTM@PEG-PE micelles. Those suggested a potential selective cytotoxic effect of BTM@PEG-PE micelles. While there is no significant difference between the cell viability for free BTM and BTM@PEG-PE micelles treatment, the mean values for free BTM treatment was slightly lower than that of BTM@PEG-PE micelles treatment (Figure 3D). We speculate that the solvent (0.3% DMSO) may have caused a proportion of cell death and contributed to the decreased cell viability.
The results of pharmacokinetic study demonstrated a relatively prolonged circulation of PEG-PE micelles in plasma (Figure 5). It has been reported that the PEG as the shell would reduce the adhesion of the micelles to plasma proteins and phagocytosis by RES and exhibited long retention in circulation and help to improve the therapeutic efficacy of the drug encapsulated into the micelles.29 The prolonged circulation retention combined with controlled drug release may have exhibited synergetic effects on the increased bioavailability.
Considering the poor water solubility of free BTM, we used the comparable low dose of BTM (5 mg/kg) in animal studies, while a higher dose of BTM in PEG-PE micelles may be available. Therefore, no significant side effect was observed for all groups. Also, the slightly enhanced anti-cancer efficacy of BTM@PEG-PE micelles may attribute to the low dose of BTM used. A more profound anti-cancer effect was feasible if higher BTM@PEG-PE micelles were used. The safety problem has been a concern for chemotherapy of cancer. BTM is toxic to tumor cells and may also affect normal cells, the toxicology of BTM are concerns for its clinical usage. Synthetic or engineered drug delivery systems often have biocompatibility and immunogenicity issues.30 While the polymeric micelles have been recognized as biocompatible with non-immunogenicity, the biocompatibility is still concerns for its usage.22,31 Also, little is known on whether BTM would induce an immunological response. Therefore, plasma levels of TNF-α and IL-6 in mice were measured after treatment. In our safety evaluation, mice receiving BTM@PEG-PE exhibited improved index for liver and kidney functions and reduced cytokines, demonstrating that the PEG-PE micelles mediated delivery allowed higher treatment dose and increased the tolerability of BTM.
In the present study, BTM was successfully loaded into PEG-PE micelles. In particular, BTM@PEG-PE micelles showed improved cellular uptake, regulated drug release profile. Injection of BTM@PEG-PE micelles slowed tumor growth in tumor-challenged mice and prolonged survival. Also, the BTM@PEG-PE micelles showed tolerable biocompatibility with less immunogenicity and toxicity to that of free BTM. We demonstrated that PEG-PE micelles are safe and efficient delivery vehicles for improving pharmacokinetic and pharmacodynamic profiles of BTM to treat colon cancer. This delivery strategy may have implications for further exploratory studies of pharmacological potentials of resveratrol derivatives, such as BTM.
Acknowledgment
This research was funded by the Nature Science Foundation of China (grant number NSFC81573718), the Fundamental Research Funds for Central Universities of Central South University (grant number 2019zzts360), and the Hunan Provincial Science and Technology Plan (grant number 2016TP2002).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. doi:10.3322/caac.21551
2. Levin TR, Corley DA, Jensen CD, et al. Effects of organized colorectal cancer screening on cancer incidence and mortality in a large community-based population. Gastroenterology. 2018;155(5):
3. Merchant SJ, Nanji S, Brennan K, et al. Management of stage III colon cancer in the elderly: practice patterns and outcomes in the general population. Cancer Am Cancer Soc. 2017;123(15):2840–2849.
4. Benson AB, Venook AP, Al-Hawary MM, et al. NCCN guidelines insights: colon cancer, version 2.2018. J Natl Compr Canc Netw. 2018;16(4):359–369. doi:10.6004/jnccn.2018.0021
5. Jiang ZD, Chen K, Cheng L, et al. Resveratrol and cancer treatment: updates. Ann N Y Acad Sci. 2017;1403(1):59–69. doi:10.1111/nyas.13466
6. Sinha D, Sarkar N, Biswas J, Bishayee A. Resveratrol for breast cancer prevention and therapy: preclinical evidence and molecular mechanisms. Semin Cancer Biol. 2016;40–41:209–232. doi:10.1016/j.semcancer.2015.11.001
7. Nguyen AV, Martinez M, Stamos MJ, et al. Results of a phase I pilot clinical trial examining the effect of plant-derived resveratrol and grape powder on Wnt pathway target gene expression in colonic mucosa and colon cancer. Cancer Manag Res. 2009;1:25–37. doi:10.2147/CMAR.S4544
8. Patel KR, Brown VA, Jones DJL, et al. Clinical pharmacology of resveratrol and its metabolites in colorectal cancer patients. Cancer Res. 2010;70(19):7392–7399. doi:10.1158/0008-5472.CAN-10-2027
9. San Hipolito-Luengo A, Alcaide A, Ramos-Gonzalez M, et al. Dual effects of resveratrol on cell death and proliferation of colon cancer cells. Nutr Cancer. 2017;69(7):1019–1027. doi:10.1080/01635581.2017.1359309
10. Feng M, Zhong LX, Zhan ZY, Huang ZH, Xiong JP. Resveratrol treatment inhibits proliferation of and induces apoptosis in human colon cancer cells. Med Sci Monit. 2016;22:1101–1108. doi:10.12659/MSM.897905
11. Juan ME, Alfaras I, Planas JM. Colorectal cancer chemoprevention by trans-resveratrol. Pharmacol Res. 2012;65(6):584–591. doi:10.1016/j.phrs.2012.03.010
12. Kapetanovic IM, Muzzio M, Huang ZH, Thompson TN, McCormick DL. Pharmacokinetics, oral bioavailability, and metabolic profile of resveratrol and its dimethylether analog, pterostilbene, in rats. Cancer Chemoth Pharm. 2011;68(3):593–601. doi:10.1007/s00280-010-1525-4
13. Singh G, Pai RS. Optimized PLGA nanoparticle platform for orally dosed trans-resveratrol with enhanced bioavailability potential. Expert Opin Drug Deliv. 2014;11(5):647–659. doi:10.1517/17425247.2014.890588
14. Chabert P, Fougerousse A, Brouillard R. Anti-mitotic properties of resveratrol analog (Z)-3,5,4ʹ-trimethoxystilbene. BioFactors. 2006;27(1–4):37–46. doi:10.1002/biof.5520270104
15. Hsieh TC, Wong C, Bennett DJ, Wu JM. Regulation of p53 and cell proliferation by resveratrol and its derivatives in breast cancer cells: an in silico and biochemical approach targeting integrin alpha v beta 3. Int J Cancer. 2011;129(11):2732–2743. doi:10.1002/ijc.25930
16. Scherzberg MC, Kiehl A, Zivkovic A, et al. Structural modification of resveratrol leads to increased anti-tumor activity, but causes profound changes in the mode of action. Toxicol Appl Pharm. 2015;287(1):67–76. doi:10.1016/j.taap.2015.05.020
17. Hu XB, Kang RR, Tang TT, et al. Topical delivery of 3,5,4-trimethoxy-trans-stilbene-loaded microemulsion-based hydrogel for the treatment of osteoarthritis in a rabbit model. Drug Deliv Transl Res. 2019;9(1):357–365. doi:10.1007/s13346-018-00604-z
18. Sawant RR, Torchilin VP. Polymeric micelles: polyethylene glycol-phosphatidylethanolamine (PEG-PE)-based micelles as an example. Methods Mol Biol. 2010;624:131–149.
19. Sarisozen C, Vural I, Levchenko T, Hincal AA, Torchilin VP. Long-circulating PEG-PE micelles co-loaded with paclitaxel and elacridar (GG918) overcome multidrug resistance. Drug Deliv. 2012;19(8):363–370. doi:10.3109/10717544.2012.724473
20. Sawant RR, Torchilin VP. Multifunctionality of lipid-core micelles for drug delivery and tumour targeting. Mol Membr Biol. 2010;27(7):232–246. doi:10.3109/09687688.2010.516276
21. Harada M, Ohuchi M, Hayashi T, Kato Y. Prolonged circulation and in vivo efficacy of recombinant human granulocyte colony-stimulating factor encapsulated in polymeric micelles. J Control Release. 2011;156(1):101–108. doi:10.1016/j.jconrel.2011.06.024
22. Biswas S, Kumari P, Lakhani PM, Ghosh B. Recent advances in polymeric micelles for anti-cancer drug delivery. Eur J Pharm Sci. 2016;83:184–202. doi:10.1016/j.ejps.2015.12.031
23. Li W, Wu J, Zhang J, et al. Puerarin-loaded PEG-PE micelles with enhanced anti-apoptotic effect and better pharmacokinetic profile. Drug Deliv. 2018;25(1):827–837. doi:10.1080/10717544.2018.1455763
24. Porter AG, Janicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6(2):99–104. doi:10.1038/sj.cdd.4400476
25. Wang YZ, Fan W, Dai X, et al. Enhanced tumor delivery of gemcitabine via PEG-DSPE/TPGS mixed micelles. Mol Pharmaceut. 2014;11(4):1140–1150. doi:10.1021/mp4005904
26. Lukyanov AN, Torchilin VP. Micelles from lipid derivatives of water-soluble polymers as delivery systems for poorly soluble drugs. Adv Drug Deliv Rev. 2004;56(9):1273–1289. doi:10.1016/j.addr.2003.12.004
27. Abouzeid AH, Patel NR, Torchilin VP. Polyethylene glycol-phosphatidylethanolamine (PEG-PE)/vitamin E micelles for co-delivery of paclitaxel and curcumin to overcome multi-drug resistance in ovarian cancer. Int J Pharmaceut. 2014;464(1–2):178–184. doi:10.1016/j.ijpharm.2014.01.009
28. Hu X, Yang FF, Liu CY, Ehrhardt C, Liao YH. In vitro uptake and transport studies of PEG-PLGA polymeric micelles in respiratory epithelial cells. Eur J Pharm Biopharm. 2017;114:29–37. doi:10.1016/j.ejpb.2017.01.004
29. Zhang XY, Zhang SH, Kang Y, Huang KQ, Gu ZP, Wu J. Advances in long-circulating drug delivery strategy. Curr Drug Metab. 2018;19(9):750–758. doi:10.2174/1389200219666180511152350
30. Naahidi S, Jafari M, Edalat F, Raymond K, Khademhosseini A, Chen P. Biocompatibility of engineered nanoparticles for drug delivery. J Control Release. 2013;166(2):182–194. doi:10.1016/j.jconrel.2012.12.013
31. Rainbolt EA, Washington KE, Biewer MC, Stefan MC. Recent developments in micellar drug carriers featuring substituted poly(epsilon-caprolactone)s. Polym Chem UK. 2015;6(13):2369–2381. doi:10.1039/C4PY01628A
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.