")
Back to Journals » Drug Design, Development and Therapy » Volume 9
High-throughput sequencing of 16S rDNA amplicons characterizes bacterial composition in bronchoalveolar lavage fluid in patients with ventilator-associated pneumonia
Authors Yang X, Wang Y, Zhou Z, Wang G, Wang X , Liu Q, Zhou S, Wang Z
Received 30 April 2015
Accepted for publication 2 June 2015
Published 24 August 2015 Volume 2015:9 Pages 4883—4896
DOI https://doi.org/10.2147/DDDT.S87634
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Xiao-Jun Yang,1,* Yan-Bo Wang,2,3,* Zhi-Wei Zhou,4,* Guo-Wei Wang,2 Xiao-Hong Wang,1 Qing-Fu Liu,1 Shu-Feng Zhou,4 Zhen-Hai Wang2,3
1Department of Intensive Care Unit, 2Neurology Center, General Hospital of Ningxia Medical University, Yinchuan, Ningxia, People’s Republic of China; 3Key Laboratory of Brain Diseases of Ningxia, Yinchuan, Ningxia, People’s Republic of China; 4Department of Pharmaceutical Sciences, College of Pharmacy, University of South Florida, Tampa, FL, USA
*These authors contributed equally to this work
Abstract: Ventilator-associated pneumonia (VAP) is a life-threatening disease that is associated with high rates of morbidity and likely mortality, placing a heavy burden on an individual and society. Currently available diagnostic and therapeutic approaches for VAP treatment are limited, and the prognosis of VAP is poor. The present study aimed to reveal and discriminate the identification of the full spectrum of the pathogens in patients with VAP using high-throughput sequencing approach and analyze the species richness and complexity via alpha and beta diversity analysis. The bronchoalveolar lavage fluid samples were collected from 27 patients with VAP in intensive care unit. The polymerase chain reaction products of the hypervariable regions of 16S rDNA gene in these 27 samples of VAP were sequenced using the 454 GS FLX system. A total of 103,856 pyrosequencing reads and 638 operational taxonomic units were obtained from these 27 samples. There were four dominant phyla, including Proteobacteria, Firmicutes, Actinobacteria, and Bacteroidetes. There were 90 different genera, of which 12 genera occurred in over ten different samples. The top five dominant genera were Streptococcus, Acinetobacter, Limnohabitans, Neisseria, and Corynebacterium, and the most widely distributed genera were Streptococcus, Limnohabitans, and Acinetobacter in these 27 samples. Of note, the mixed profile of causative pathogens was observed. Taken together, the results show that the high-throughput sequencing approach facilitates the characterization of the pathogens in bronchoalveolar lavage fluid samples and the determination of the profile for bacteria in the bronchoalveolar lavage fluid samples of the patients with VAP. This study can provide useful information of pathogens in VAP and assist clinicians to make rational and effective therapeutic decisions.
Keywords: bioinformatics, drug resistance, OTU, PCR, DNA sequencing
Introduction
Increasing evidence shows that ventilator-associated pneumonia (VAP), a type of nosocomial pneumonia, has emerged as an important challenge in the intensive care unit (ICU) that lead to a considerable morbidity and mortality, placing a substantial heavy burden on an individual and society.1 VAP occurs in patients who receive mechanical ventilation and is usually acquired in the hospital setting ~48–72 hours after mechanical ventilation.2 The overall rate of VAP is 13.6 per 1,000 ventilator-day, according to the International Nosocomial Infection Control Consortium (http://www.inicc.org/english/index.php). The incidence of VAP varies within a range from 13–51 per 1,000 ventilation-day according to the patient group and hospital setting; the mean duration of occurrence of VAP is ~5–7 days. The VAP mortality ranges from 24%–76%, and there is even higher mortality rate among critically ill patients.1,3,4 In USA, VAP accounts for over 25% of all ICU-acquired infections, and there are over 100,000 cases annually.5 In addition, VAP represents more than one-half of all antibiotic use in the ICU.6,7
The pathogenesis of VAP is complex but typically relates with colonization of the aerodigestive tract with pathogens, micro-aspiration of contaminated secretions, and formation of biofilms.8 In particular, there are several bacteria including Pseudomonas aeruginosa, Klebsiella pneumoniae, Serratia marcescens, Enterobacter, Citrobacter, Stenotrophomonas maltophilia, Acinetobacter, Burkholderia cepacia, and Methicillin-resistant Staphylococcus aureus, that can cause drug resistance in the treatment of VAP. These pathogens make the VAP treatment more challenging. Recently, the commonly used strategies to reduce the development of drug resistance in the treatment of VAP include de-escalation therapy, truncated courses of antibiotics, dosing regimens that account for patient-antibiotic pharmacokinetics and pharmacodynamics, antibiotic cycling, and surveillance cultures.1,6,7 However, the approaches for pathogen monitoring and to avoid drug resistance are still limited, and VAP still is a life-threatening infectious disease with challenges in the diagnosis and poor prognosis. Thus, it needs advanced methods to detect the full array of pathogens in patients with VAP to avoid the compromise of therapy and achieve maximum therapeutic outcome in clinical practice.
Growing evidence shows that the accurate and comprehensive identification of the causative pathogen can facilitate the VAP therapy in clinical practice.1 16S rDNA gene sequencing approach possesses the capability of fast and accurately revealing the identity of the pathogens, because it can overcome the limitations of conventional culture-based bacterial detection method.9,10 With advances in the sequencing technology, the feasibility of 16S rDNA analysis using 454 GS FLX system has already been proven in the research of microbiota in the oral cavity, wound, urine, and gastrointestinal tract; and the massive data generated by 454 GS FLX system make it possible to analyze the diversity of the bacterial communities.11–17 The employment of the 16S rDNA gene sequencing approach can provide a global spectrum of the composition of the pathogens in samples, which will have a great clinical importance in the optimization of the therapy, leading to the maximum therapeutic outcome.
In the present study, in order to reveal the full array of the pathogens of VAP and help the optimization of VAP therapy, we explored the complexity of the bacterial communities in bronchoalveolar lavage fluid samples of patients with VAP using 16S rDNA amplicon 454 pyrosequencing.
Materials and methods
Patients
A total number of 27 patients from the department of ICU in the General Hospital of Ningxia Medical University were enrolled. All patients were diagnosed with VAP according to the clinical criteria provided by the China Health Ministry Guidelines. Clinical signs and symptoms of VAP included fever or low body temperature, new purulent sputum, and hypoxemia (Tables 1 and 2). Consent forms were obtained from all enrolled patients with VAP. The protocol was approved by the Ethics Committee of the General Hospital of Ningxia Medical University. All procedures were conducted in accordance with the criteria of the Declaration of Helsinki.
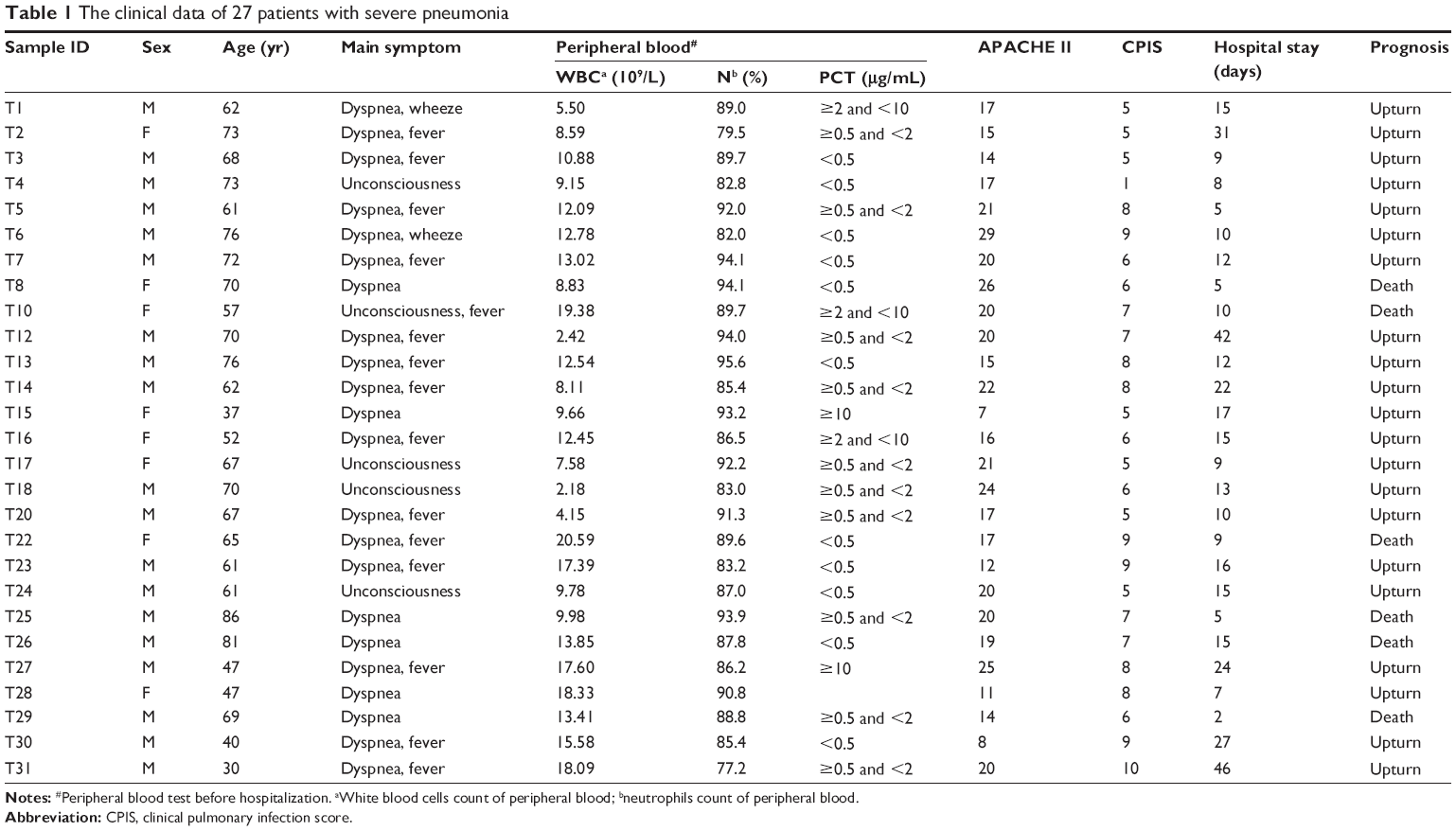 | Table 1 The clinical data of 27 patients with severe pneumonia |
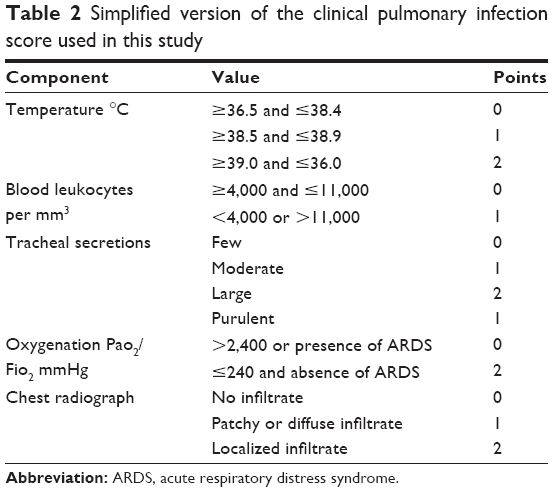 | Table 2 Simplified version of the clinical pulmonary infection score used in this study |
Clinical specimens and clinical laboratory work
The bronchoalveolar lavage fluid samples were collected from the enrolled patients. The collected bronchoalveolar lavage fluid samples were aliquoted into sterile Eppendorf tubes. Following the collection, the bacterial culture was performed. The remaining samples were stored at -80°C for subsequent metagenomic sequencing. The sample collection and transportation were carried out in strict accordance with the sterile operating procedures to avoid contamination.
DNA extraction from the bronchoalveolar lavage fluid samples
The bacterial DNA was isolated from bronchoalveolar lavage fluid samples using QIAamp DNA Micro Kit (Qiagen NV, Venlo, the Netherlands) according to the manufacturer’s instructions. Briefly, 2 mL of bronchoalveolar lavage fluid samples was required for the DNA extraction. The samples were centrifuged at 8,000 rpm for 15 minutes, and the supernatant was removed. The pellet was resuspended and digested using 200 μL of 20 mg/mL lysozyme at 37°C for 1 hour. Following the digestion, the supernatant was collected via centrifugation, and the sample was then resuspended in 200 μL PBS buffer containing 20 μL of proteinase K. Then, a quota of 200 μL AL was added to each diluted sample and incubated at 56°C for 2 hours. The resultant bacterial and human DNA were collected and transferred to sterilized 1.5 mL EP tubes for further extraction using the QIAamp DNA Micro Kit, and the DNA purification was performed according to the manufacturer’s instructions. The DNA concentration was measured by UV spectrophotometer at 260 nm. The average DNA concentration of the samples was 22.8 ng/mL.
The design of barcoded primers
The polymerase chain reaction (PCR) enrichment of the 16S rDNA V3-V5 hypervariable region was performed using the primers as shown in Table 3. The primers contained a ten-base oligonucleotide tag at the 5′ terminal. The sequence after the hyphen was able to pair with the sequences of the end region. The 27 pairs of primers that contained 27 different ten-base oligonucleotides and identical following sequences were used to lead the PCR enrichment (Table 4). The barcoded primers were synthesized by the Shanghai Sangon Biological Engineering Technology & Service Co., Ltd. (Shanghai, People’s Republic of China).
 | Table 3 The sequences of the primers for 16S rDNA V3-V5 hypervariable region |
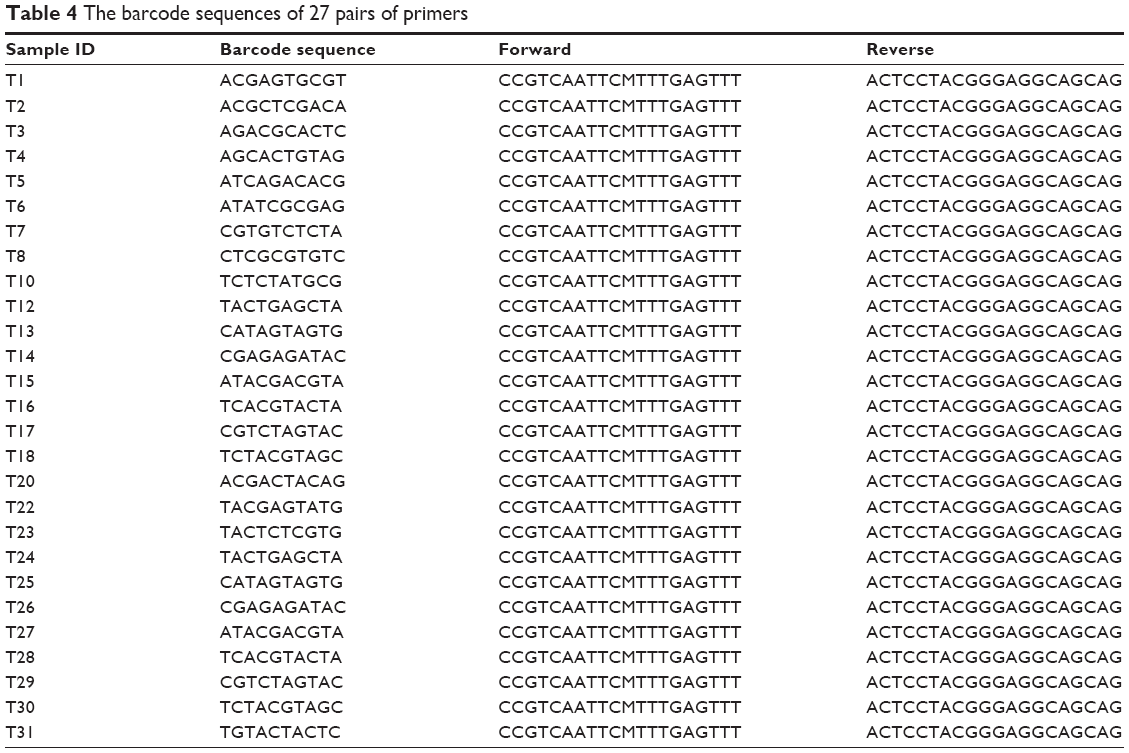 | Table 4 The barcode sequences of 27 pairs of primers |
PCR enrichment of the V3–V5 region
The extracted and purified DNA from 27 bronchoalveolar lavage fluid samples was used for PCR assay. Each PCR reaction system consisted of 12.5 μL PCR master Mix (Dream Taq PCR Master Mix, Fermentas, Burlington, Canada), 0.5 μL forward primer, 0.5 μL reverse primer, 1 μL DNA, and 10.5 μL nuclease-free water. Touchdown PCR conditions were comprised of initial denaturation for 5 minutes at 95°C, denaturation for 30 seconds at 95°C, annealing for 30 seconds at 56°C, and extension for 30 seconds at 72°C. The resultant products were stored at −20°C after the reaction. The pooled tagged single-stranded pyrosequencing library underwent emPCR and pyrosequencing using a Roche 454 GS FLX Pyrosequencer (Hoffman-La Roche Ltd., Basel, Switzerland) according to the manufacturer’s instructions.
Bioinformatic analysis
The high-throughput pyrosequencing reads from 27 bronchoalveolar lavage fluid samples were reassigned to samples according to barcodes. The resultant sequences were clustered into operational taxonomic units (OTUs). The OTUs that reached 97% similarity level were used for alpha diversity analysis that analyzed the species diversity in the single sample by the evaluation of Chao, abundance-based coverage estimators (ACE), Shannon, and Simpson parameters; and the rarefaction curve was also analyzed using the Mothur software v1.27.0 program.18–21 Following the alpha diversity analysis, taxonomy-based analyses were performed through the classification of each sequence using the Naïve Bayesian classifier program in Ribosomal Database Project (RDP) at the Center for Microbial Ecology in Michigan State University (http://rdp.cme.msu.edu/; MI, USA).20,21 The confidence level was of 95%. The sequences were assigned until the genus level in bacteria domain was collected and screened from 27 bronchoalveolar lavage fluid samples.18–21 Each read was assigned a phylum, class, family, and genus. The taxonomic assignment was unambiguous within an 80% confidence threshold, which has been estimated to taxonomically assign reads with over 98% accuracy at genus level. Furthermore, the beta diversity analysis was performed to assess the distribution and content of bacteria and evaluate the total diversity in different samples based on the bacterial profile. Sequences were clustered at 97% nucleotide identity over 90% sequence alignment length using the Mothur software. For this analysis, sequences over 97% identical were considered to correspond to the same OTUs, representing a group of reads that likely belong to the same species.22
Results
High-throughput sequencing reveals 103,856 pyrosequencing reads
In order to identify the pathogens, we first performed the high-throughput sequencing to examine the possible bacterial DNA sequences in 27 bronchoalveolar lavage fluid samples from patients with VAP. There were a total of 103,856 pyrosequencing reads that were identified through 454 pyrosequencing (Table 5). The sequences with insufficient quality or sequences that could not be adequately assigned were not included, such as chimera sequences and a small amount of non-target sequences. For the identified pyrosequencing reads, the average length of the sequences was 550 bp after trimming the primers. Taken together, the high-throughput sequencing approach shows a capability of identifying the bacterial DNA sequences from bronchoalveolar lavage fluid samples from patients with VAP, which may be clinically helpful for the treatment of VAP.
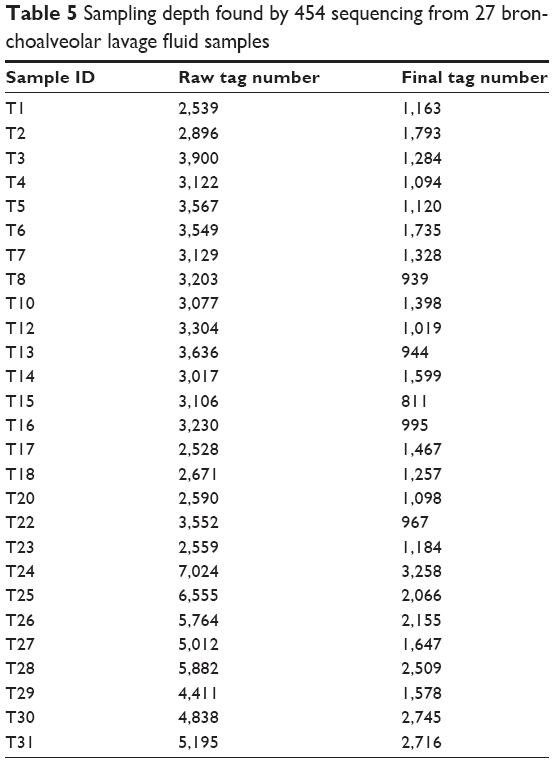 | Table 5 Sampling depth found by 454 sequencing from 27 bronchoalveolar lavage fluid samples |
Classification of the bacteria found in bronchoalveolar lavage fluid samples from patients with VAP
We next classified the obtained pyrosequencing reads using the RDP classifier at a confidence level of 95% and assigned taxonomic classifications to the sequences for biological analysis. The tag sequences of the identified DNA in 27 bronchoalveolar lavage fluid samples from patients with VAP were analyzed using the RDP to annotate species. The data showed that most of the bacterial reads were assigned to genus level and a small number of bacterial reads were assigned to species level (Figures 1 and 2). There were 638 OTUs that were obtained from the 27 bronchoalveolar lavage fluid samples from patients with VAP using Mothur, according to the tag annotation information of species for OTU comments. The Mothur analysis results are able to comment on the classification level under each OTU number (Figure 2), the classification level of each OTU ratio was phylum, class, order, family, genus, and species at 91.4%, 84.3%, 79.0%, 72.7%, 58.2%, and 31.5%, respectively.
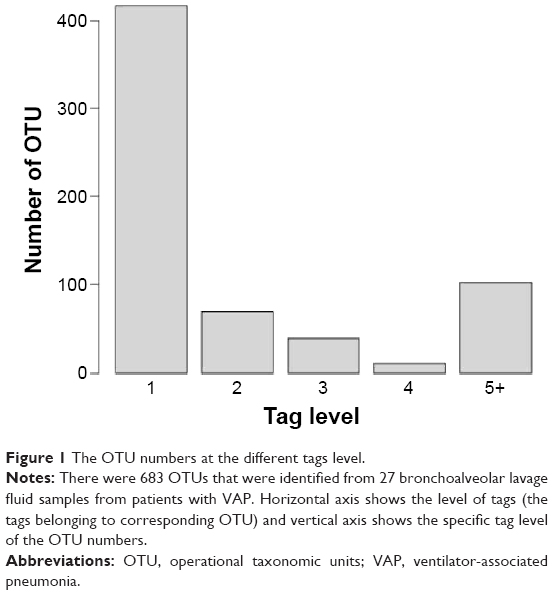 | Figure 1 The OTU numbers at the different tags level. |
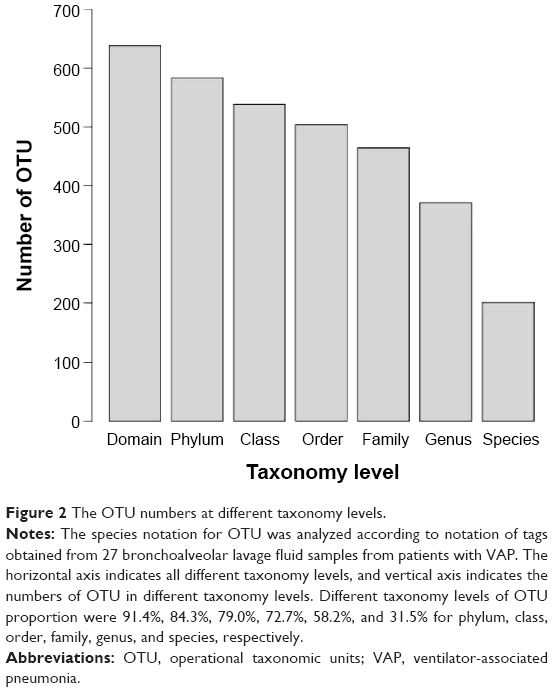 | Figure 2 The OTU numbers at different taxonomy levels. |
Species richness and complexity estimation of microbiota in the bronchoalveolar lavage fluid samples from patients with VAP
Following the classification of the obtained pyrosequencing reads, the species richness complexity were analyzed using alpha diversity analysis. We performed bioinformatic analysis of the large number of pyrosequencing reads to evaluate the species richness and diversity of microbiota in the bronchoalveolar lavage fluid samples from patients with VAP. Based on the OTU data, we calculated alpha diversity. The indices of bacterial richness and diversity of OTUs at a 3% sequence dissimilarity level were summarized in Table 6. The bronchoalveolar lavage fluid sample containing low number of OTU indicated that it had a relatively low diversity in the cerebrospinal fluid sample compared with other environmental species (Figure 3). The Chao and ACE values and the Shannon and Simpson index were calculated, and the results showed that Chao value was ~80, ACE value was ~130, Shannon index was 1.25, and Simpson index was 0.46 (Figure 3). Moreover, the richness of bacterial communities in each bronchoalveolar lavage fluid sample was estimated as presented by the rarefaction curve. The trend of the rarefaction curves also confirmed that there was a low richness in each bronchoalveolar lavage fluid sample, and the saturated shape of the rarefaction curves indicated that bacterial richness of each bronchoalveolar lavage fluid sample was completely sampled (Figure 4).
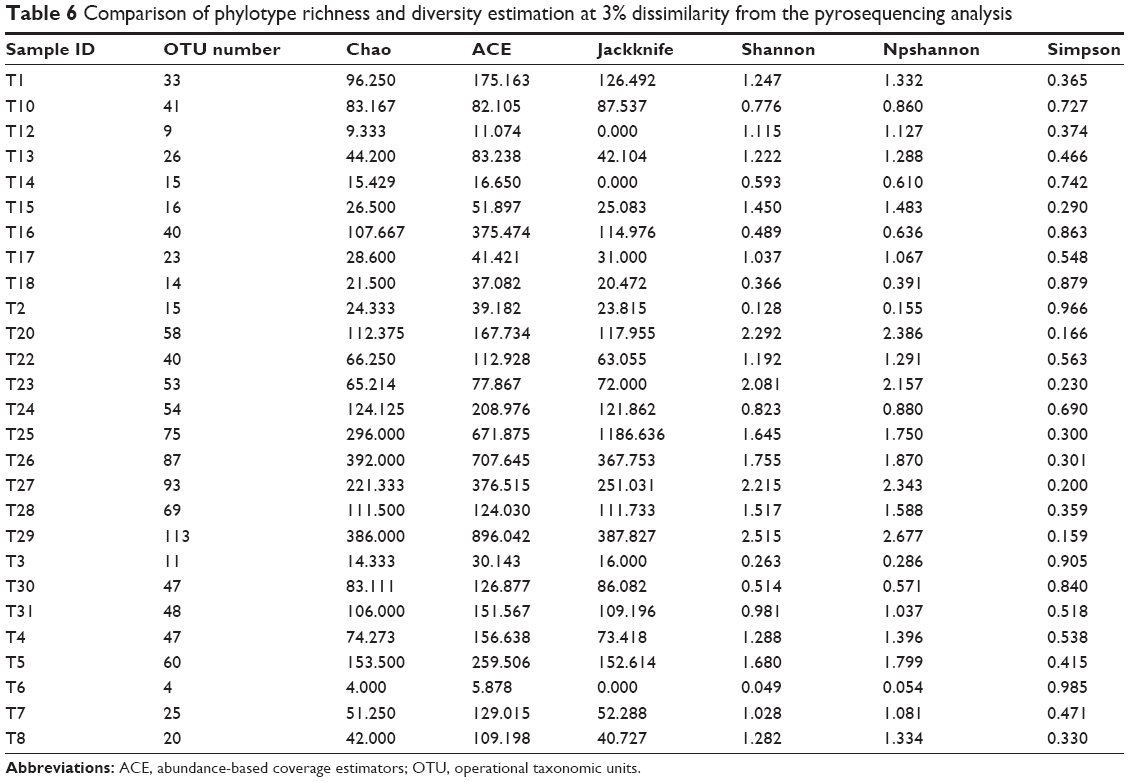 | Table 6 Comparison of phylotype richness and diversity estimation at 3% dissimilarity from the pyrosequencing analysis |
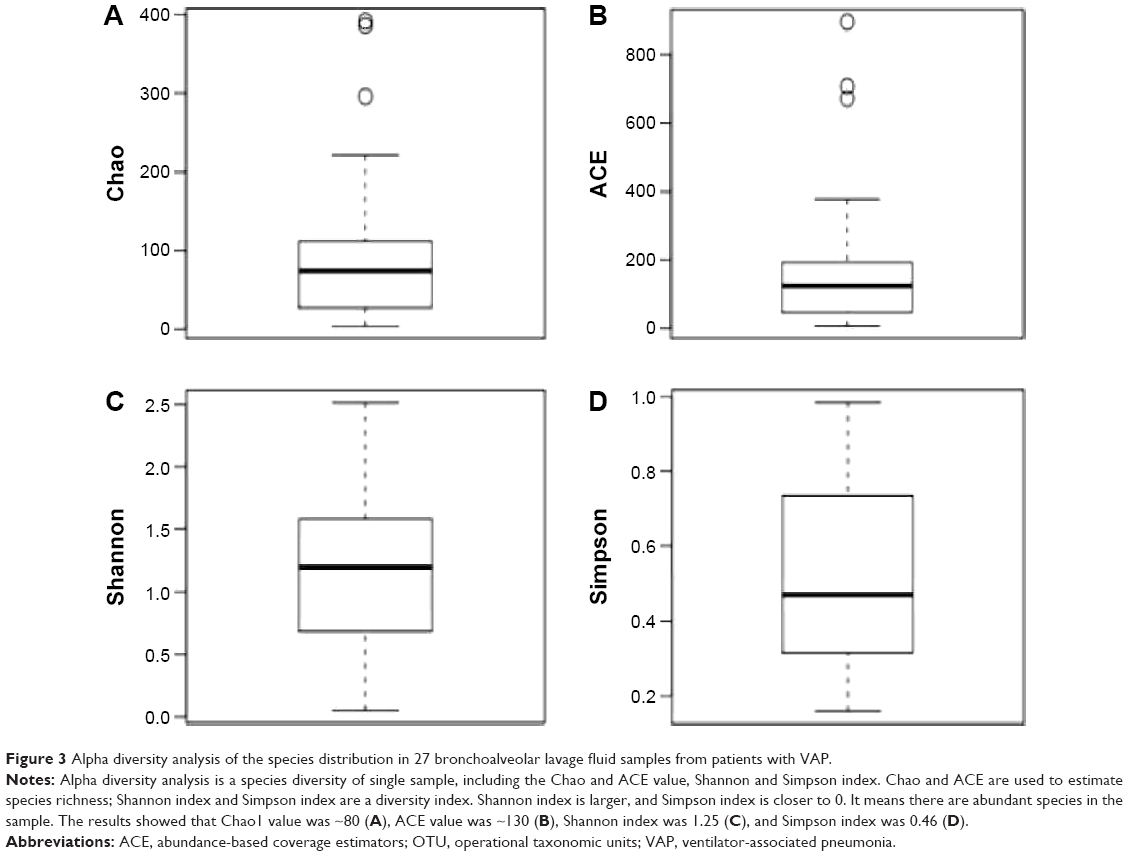 | Figure 3 Alpha diversity analysis of the species distribution in 27 bronchoalveolar lavage fluid samples from patients with VAP. |
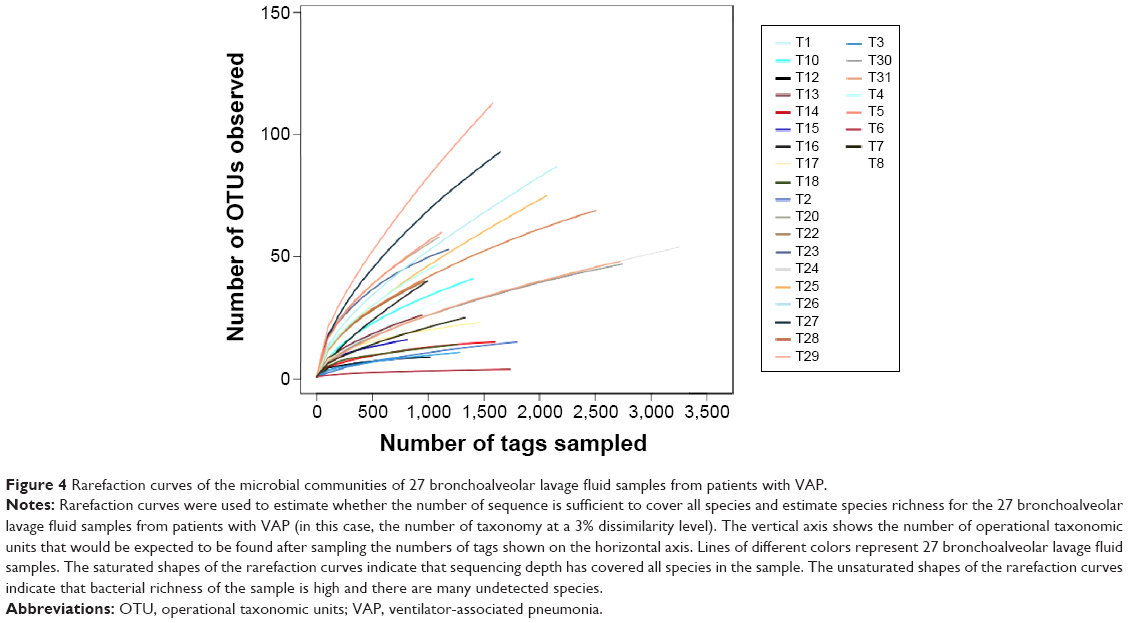 | Figure 4 Rarefaction curves of the microbial communities of 27 bronchoalveolar lavage fluid samples from patients with VAP. |
Comparative analysis of multiple samples
Further, the comparison of the pathogens among the 27 bronchoalveolar lavage fluid samples from patients with VAP was performed. The pathogen profile at phylum and genus level was analyzed (Figures 5 and 6). The relative abundance of each pathogen was determined in the 27 bronchoalveolar lavage fluid samples (Figures 5A and 6A). At phylum level, there were four dominant phyla, including Proteobacteria, Firmicutes, Actinobacteria, and Bacteroidetes (Figure 5B), to a lesser extent, Fusobacteria, Deinococcus-Thermus, Spirochaetes, Nitrospira, Planctomycetes, Synergistetes, and Tenericutes. Of note, Proteobacteria and Firmicutes were widely distributed in all of the samples (Figure 5A); Bacteroidetes and Actinobacteria occurred in half of the samples (Figure 5A). At genus level, these sequences represented 90 different genera (Figure 6A), and there were 12 genera occurring in over ten different samples (Figure 6B). The top five dominant genera, which contained the largest number of sequences, were Streptococcus, Acinetobacter, Limnohabitans, Neisseria, and Corynebacterium (Figure 6A). The most widely distributed genera were Streptococcus, Limnohabitans, and Acinetobacter in the 27 bronchoalveolar lavage fluid samples, and Prevotella, Sphingomonas, Aquabacterium, Corynebacterium, Klebsiella, Pseudomonas, Peptostreptococcus, Porphyromonas, and Tepidimonas were distributed to a lesser extent (Figure 6B; ≥10 samples).
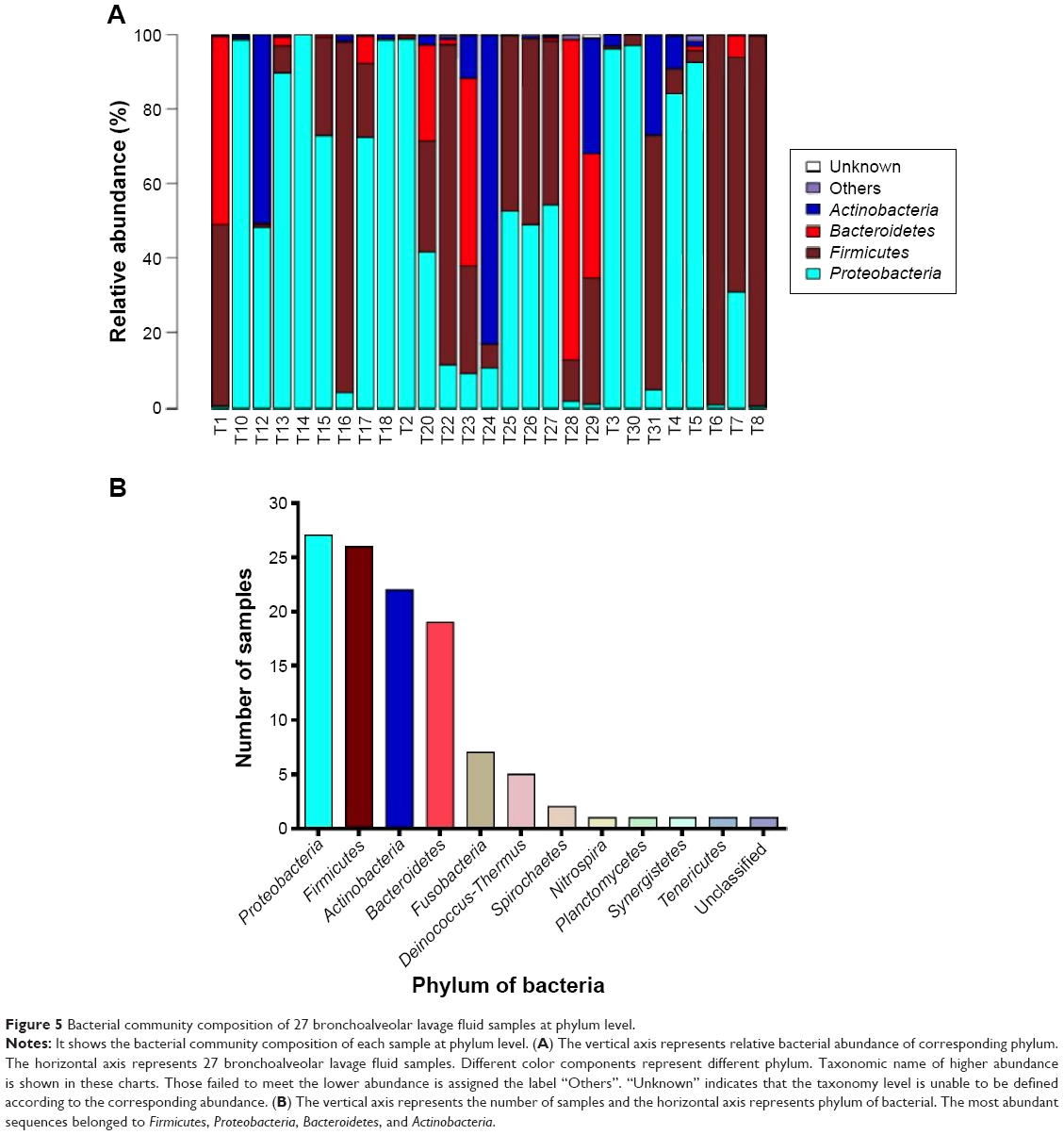 | Figure 5 Bacterial community composition of 27 bronchoalveolar lavage fluid samples at phylum level. |
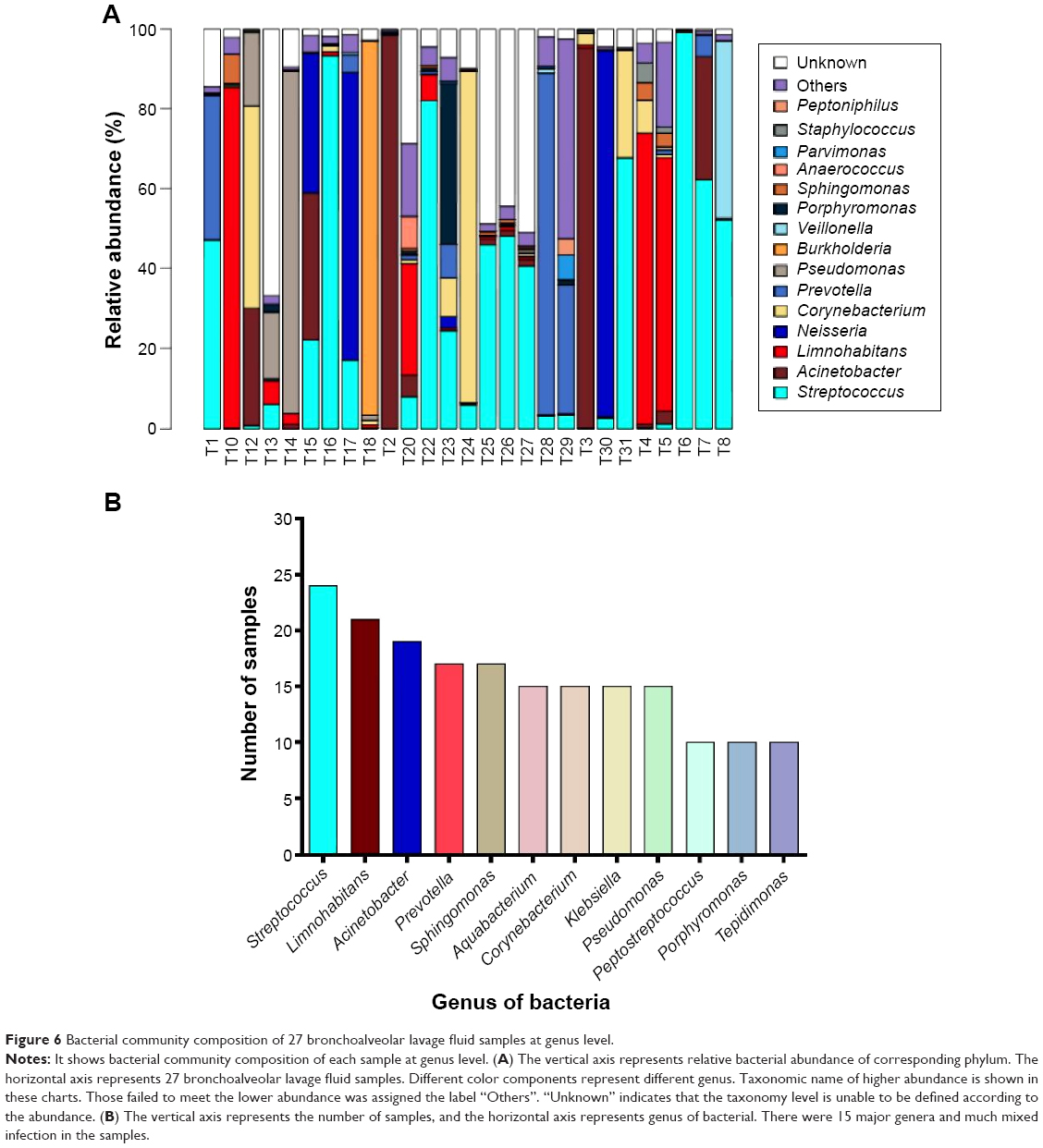 | Figure 6 Bacterial community composition of 27 bronchoalveolar lavage fluid samples at genus level. |
Additionally, in order to evaluate the total diversity and assess the distribution and content of bacteria in these 27 bronchoalveolar lavage fluid samples from patients with VAP, the beta analysis was performed. As shown in Figure 7A and B and Figure 8A–F, there was a substantial difference in the species distribution in the 27 bronchoalveolar lavage fluid samples. The beta diversity of 27 bronchoalveolar lavage fluid samples was indicated by Whittaker index that was used to evaluate the species difference in diversity between different samples. The higher index indicates more difference. The pathogens from number 1, 2, and 12 samples showed the most different diversity from other samples (Figure 7A). According to the Ward analysis data, there were four clusters that can be further divided into ten subclusters (Figure 7B). In addition, the total diversity and distribution of bacteria in these 27 bronchoalveolar lavage fluid samples were evaluated by weighted- and unweighted-UniFrac index (Figure 8A–F), which showed a similar results to that of Whittaker index. Collectively, the results show a comparable difference in the diversity of species distribution and evolution in the 27 bronchoalveolar lavage fluid samples from patients with VAP.
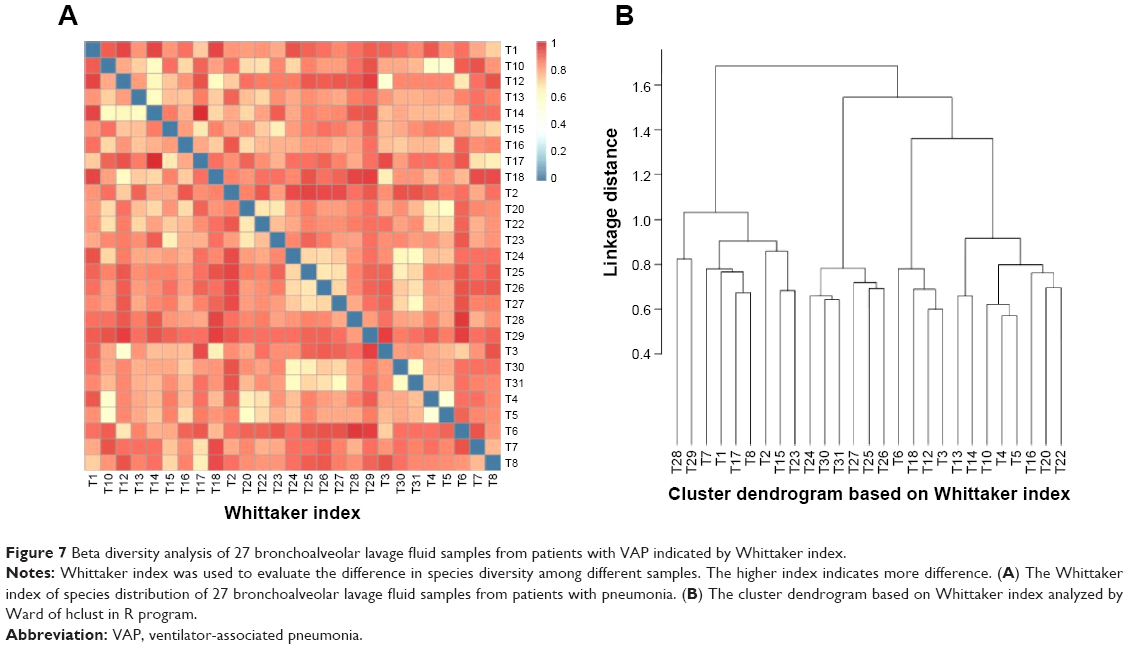 | Figure 7 Beta diversity analysis of 27 bronchoalveolar lavage fluid samples from patients with VAP indicated by Whittaker index. |
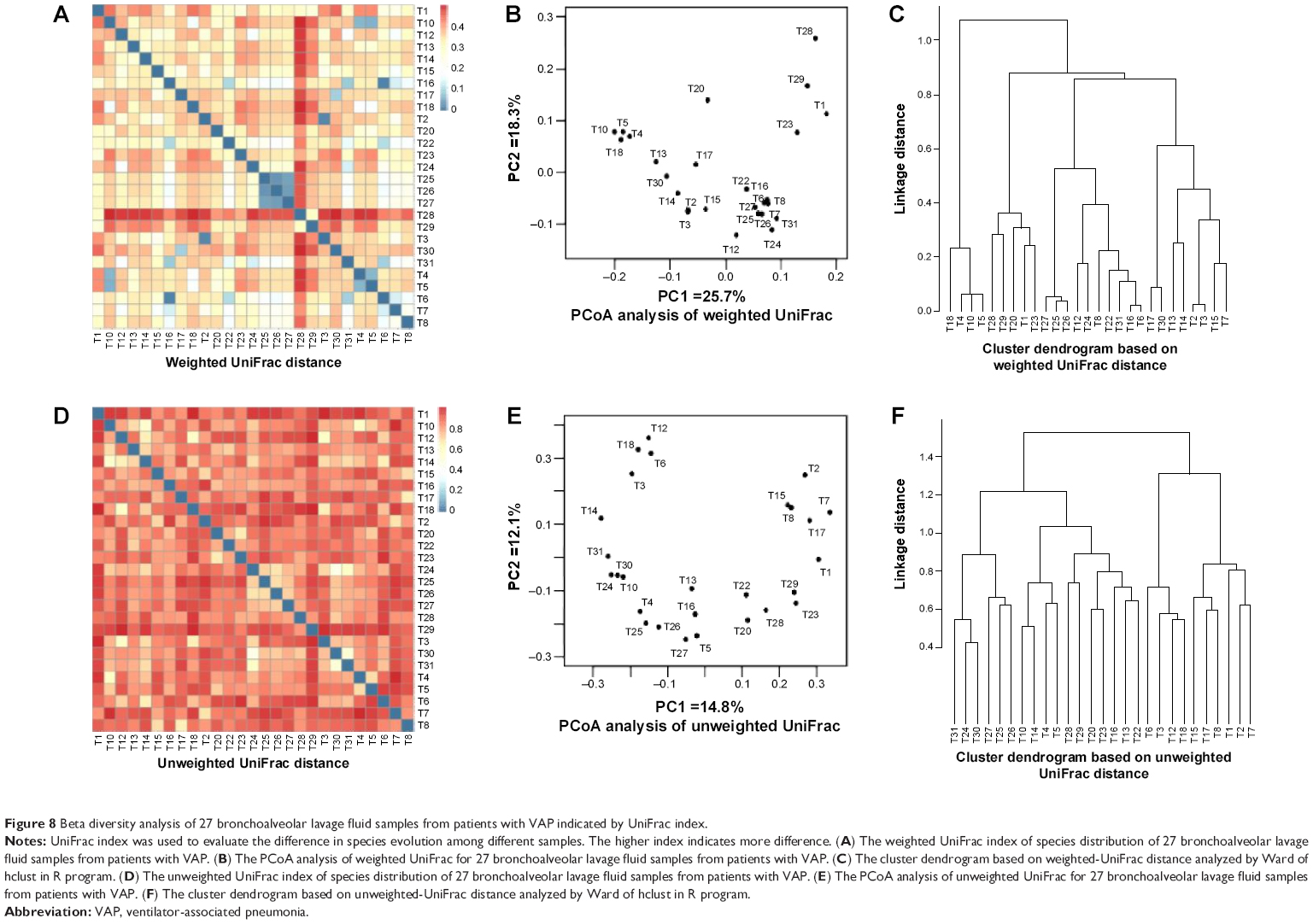 | Figure 8 Beta diversity analysis of 27 bronchoalveolar lavage fluid samples from patients with VAP indicated by UniFrac index. |
Discussion
Due to the substantial VAP-associated morbidity and mortality and the resultant considerable heavy burden on an individual and society, it is of great importance to identify the cause of VAP for the optimization of the treatment of VAP in clinical practice. The complex of pathogen-induced VAP is still a major challenge for the use of current therapeutics. Advances in the identification and profiling of the bacteria have facilitated the revealing of the global view of causative pathogens of VAP. Metagenomic studies have dramatically expanded our understanding of the microbial world without the cultivation of microorganisms and can overcome the shortages of the conventional culture-based approach.17,18,21,23,24 In the present study, we have performed metagenomics high-throughput sequencing to analyze the pathogens that were present in 27 bronchoalveolar lavage fluid samples from patients with VAP. We have identified most of the bacterial pathogens in these 27 samples from patients with VAP. The percentage of sequences belonging to each bacterial genus has been calculated for each patient that may facilitate the optimization of the therapy for VAP treatment and the achievement of maximum therapeutic outcome in clinical practice.
Emerging evidence shows the advantage of high-throughput sequencing in the identification of the pathogens to potentially improve the specificity of the diagnosis of VAP and the consequent unnecessary antibiotic use and its associated problems, such as drug resistance.9,17,25 The advent of 454 pyrosequencing technology has greatly accelerated studies on the profiling and deciphering the complexity of the human microbiota composition,26,27 leading to an improved understanding in the causative pathogens in the pathogenesis of infectious diseases. The utilization of barcoded primers and 454 pyrosequencing in metagenomics generates a global and comprehensive profiling of the microbiota.28,29 In our study, the findings showed that the high-throughput sequencing approach provided a fast, accurate, and global pyrosequencing reads, which can expand our understanding of the diversity of bacteria present in the respiratory tract of patients with VAP. On the other hand, we detected part of the sequences that belong to unknown species, and there are completely unknown species in 22 of 27 samples in the present study. The observation of these results may be due to the non-homogenous lysis of bacterial cells, primer mismatches, and the presence of mixed bases in PCR process.30–32 These reasons can lead to the failure of the species functional annotation. Therefore, it is critical to eliminate these factors.
Due to the complicated composition of pathogens in the development of VAP, it is of great importance to discriminate the composition of the pathogens at different levels. This will be clinically helpful for the proper therapeutics selection and therapy optimization in the treatment of VAP. The causative bacteria for an early-onset VAP include Streptococcus, Hemophilus influenzae, methicillin-sensitive S. aureus, antibiotic-sensitive enteric Gram-negative bacilli, Escherichia coli, K. pneumoniae, Enterobacter species, Proteus species, and S. marcescens, and the bacteria causing late VAP are typically multiple drug-resistant bacteria, such as methicillin-resistant S. aureus, Acinetobacter, P. aeruginosa, and extended-spectrum beta-lactamase producing bacteria.33,34 Our findings showed that the most common isolates were Streptococcus, Acinetobacter, and Limnohabitans at genus level. In addition, the most prevalently representative sequence belonged to Streptococcus. They belong to opportunistic pathogens, which are widely distributed in nature. They exist in the form of carrier state, generally not pathogenic, but can cause disease when immunity decreased.35–37 The Streptococcus encompasses both commensal and pathogenic Gram-positive bacteria that inhabit various human body sites, such as Streptococcus pneumoniae, a well-known pathogen associated with pneumonia.38 In recent years, Acinetobacter baumannii has become increasingly prevalent. It has been the main pathogenic bacteria in hospital-acquired infection, particularly in VAP39,40 Thus, the global identification of the causative pathogens in VAP is important for therapeutics selection.
In the treatment of VAP in the absence of risk factors for multidrug-resistant bacteria, the empirical therapy can be selected for the causative pathogens of Streptococcus pneumoniae, Haemophilus influenzae, methicillin-sensitive S. aureus, and antibiotic-sensitive gram-negative enteric organisms. The antibiotics used to treat drug-sensitive VAP include ceftriaxone, quinolones (levofloxacin, moxifloxacin, or ciprofloxacin), ampicillin/sulbactam, or ertapenem. However, the mixed infections with drug-resistant bacteria represent a major challenge in the treatment of VAP, which often occurs. Therefore, in the treatment of VAP in the presence of risk factors for multidrug-resistant organisms, the clinician must consider not only empirical therapies but also the therapies for the drug-resistance causative pathogens that include P. aeruginosa, Klebsiella, Enterobacter, Serratia, Acinetobacter, Stenotrophomonas maltophilia, B. cepacia, and methicillin-resistant S. aureus. The therapeutics should be broadened to achieve the therapeutic effect. This includes 1) either an antipseudomonal cephalosporin (cefepime or ceftazidime), an antipseudomonal carbapenem (imipenem or meropenem), or a β-lactam/ β-lactamase inhibitor (piperacillin-tazobactam), and 2) an antipseudomonal fluoroquinolone (ciprofloxacin or levofloxacin) or an aminoglycoside (amikacin, gentamicin, or tobramycin) plus linezolid or vancomycin.
Conclusion
In summary, the present study shows that the high-throughput sequencing is a clinically valuable approach to analyze the pathogens of VAP with the advantage of overcoming the limitations of convention approaches. The global view of the bacterial composition can provide a better understanding of pathogens in VAP, which can assist clinicians to make rational and effective therapeutic decisions to treat drug-sensitive or resistant bacteria, and achieve maximum therapeutic effect in the treatment of VAP in clinical practice.
Acknowledgments
The authors appreciate the technical support from BGI Inc. (Shenzhen, People’s Republic of China). This work was supported by grants from the Natural Science Foundation of China (Grant numbers: 81450031, 81160151) and the Natural Science Foundation of NingXia (Grant number: NZ14142).
Disclosure
The authors declare that there are no conflicts of interest in this work.
References
Koenig SM, Truwit JD. Ventilator-associated pneumonia: diagnosis, treatment, and prevention. Clin Microbiol Rev. 2006;19(4):637–657. | ||
Vallés J, Martin-Loeches I, Torres A, et al. Epidemiology, antibiotic therapy and clinical outcomes of healthcare-associated pneumonia in critically ill patients: a Spanish cohort study. Intensive Care Med. 2014;40(4):572–581. | ||
Zilberberg MD, Shorr AF. Ventilator-associated pneumonia: the clinical pulmonary infection score as a surrogate for diagnostics and outcome. Clin Infect Dis. 2010;51(suppl 1):S131–S135. | ||
Morrow BM, Argent AC, Jeena PM, Green RJ. Guideline for the diagnosis, prevention and treatment of paediatric ventilator-associated pneumonia. S Afr Med J. 2009;99(4 pt 2):255–267. | ||
Klevens RM, Edwards JR, Richards CL Jr, et al. Estimating health care-associated infections and deaths in U.S. hospitals, 2002. Public Health Rep. 2007;122(2):160–166. | ||
American Thoracic Society; Infectious Diseases Society of America. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med. 2005;171(4):388–416. | ||
Gupta D, Agarwal R, Aggarwal AN, et al; Pneumonia Guidelines Working Group. Guidelines for diagnosis and management of community- and hospital-acquired pneumonia in adults: joint ICS/NCCP(I) recommendations. Lung India. 2012;29(suppl 2):S27–S62. | ||
Kollef MH. What is ventilator-associated pneumonia and why is it important? Respir Care. 2005;50(6):714–721; discussion 721–724. | ||
Woo PC, Tsoi HW, Leung KW, et al. Identification of Mycobacterium neoaurum isolated from a neutropenic patient with catheter-related bacteremia by 16S rRNA sequencing. J Clin Microbiol. 2000; 38(9): 3515–3517. | ||
Woo PC, Lau SK, Teng JL, Tse H, Yuen KY. Then and now: use of 16S rDNA gene sequencing for bacterial identification and discovery of novel bacteria in clinical microbiology laboratories. Clin Microbiol Infect. 2008;14(10):908–934. | ||
Ahmadian A, Gharizadeh B, Gustafsson AC, et al. Single-nucleotide polymorphism analysis by pyrosequencing. Anal Biochem. 2000;280(1):103–110. | ||
Sakamoto M, Umeda M, Ishikawa I, Benno Y. Comparison of the oral bacterial flora in saliva from a healthy subject and two periodontitis patients by sequence analysis of 16S rDNA libraries. Microbiol Immunol. 2000;44(8):643–652. | ||
Price LB, Liu CM, Melendez JH, et al. Community analysis of chronic wound bacteria using 16S rRNA gene-based pyrosequencing: impact of diabetes and antibiotics on chronic wound microbiota. PLoS One. 2009;4(7):e6462. | ||
Dethlefsen L, Huse S, Sogin ML, Relman DA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008;6(11):e280. | ||
Lyra A, Rinttilä T, Nikkilä J, et al. Diarrhoea-predominant irritable bowel syndrome distinguishable by 16S rRNA gene phylotype quantification. World J Gastroenterol. 2009;15(47):5936–5945. | ||
Fournier PE, Raoult D. Prospects for the future using genomics and proteomics in clinical microbiology. Annu Rev Microbiol. 2011;65:169–188. | ||
Siddiqui H, Nederbragt AJ, Lagesen K, Jeansson SL, Jakobsen KS. Assessing diversity of the female urine microbiota by high throughput sequencing of 16S rDNA amplicons. BMC Microbiol. 2011;11:244. | ||
Cole JR, Wang Q, Cardenas E, et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37(Database issue):D141–D145. | ||
Schloss PD, Westcott SL, Ryabin T, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75(23):7537–7541. | ||
Chen CY, Wen TY, Wang GS, Cheng HW, Lin YH, Lien GW. Determining estrogenic steroids in Taipei waters and removal in drinking water treatment using high-flow solid-phase extraction and liquid chromatography/tandem mass spectrometry. Sci Total Environ. 2007;378(3):352–365. | ||
Rosen GL, Reichenberger ER, Rosenfeld AM. NBC: the naive Bayes classification tool webserver for taxonomic classification of metagenomic reads. Bioinformatics. 2011;27(1):127–129. | ||
Sogin ML, Morrison HG, Huber JA, et al. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc Natl Acad Sci U S A. 2006;103(32):12115–12120. | ||
McKenna P, Hoffmann C, Minkah N, et al. The macaque gut microbiome in health, lentiviral infection, and chronic enterocolitis. PLoS Pathog. 2008;4(2):e20. | ||
Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73(16):5261–5267. | ||
Clarridge JE 3rd. Impact of 16S rRNA gene sequence analysis for identification of bacteria on clinical microbiology and infectious diseases. Clin Microbiol Rev. 2004;17(4):840–862, table of contents. | ||
Cho I, Blaser MJ. The human microbiome: at the interface of health and disease. Nat Rev Genet. 2012;13(4):260–270. | ||
Beck JM, Young VB, Huffnagle GB. The microbiome of the lung. Transl Res. 2012;160(4):258–266. | ||
Han MK, Huang YJ, Lipuma JJ, et al. Significance of the microbiome in obstructive lung disease. Thorax. 2012;67(5):456–463. | ||
Zhou Y, Lin P, Li Q, et al. Analysis of the microbiota of sputum samples from patients with lower respiratory tract infections. Acta Biochim Biophys Sin (Shanghai). 2010;42(10):754–761. | ||
Fredricks DN, Smith C, Meier A. Comparison of six DNA extraction methods for recovery of fungal DNA as assessed by quantitative PCR. J Clin Microbiol. 2005;43(10):5122–5128. | ||
Kuczynski J, Lauber CL, Walters WA, et al. Experimental and analytical tools for studying the human microbiome. Nat Rev Genet. 2012;13(1):47–58. | ||
Shin S, Lee TK, Han JM, Park J. Regional effects on chimera formation in 454 pyrosequenced amplicons from a mock community. J Microbiol. 2014;52(7):566–573. | ||
Guillamet CV, Kollef MH. Ventilator associated pneumonia in the ICU: where has it gone? Curr Opin Pulm Med. 2015;21(3):226–231. | ||
Kalanuria AA, Zai W, Mirski M. Ventilator-associated pneumonia in the ICU. Crit Care. 2014;18(2):208. | ||
Johanson WG, Pierce AK, Sanford JP. Changing pharyngeal bacterial flora of hospitalized patients. Emergence of Gram-negative bacilli. N Engl J Med. 1969;281(21):1137–1140. | ||
Johanson WG Jr, Pierce AK, Sanford JP, Thomas GD. Nosocomial respiratory infections with Gram-negative bacilli. The significance of colonization of the respiratory tract. Ann Intern Med. 1972;77(5):701–706. | ||
Bonten MJ, Bergmans DC, Ambergen AW, et al. Risk factors for pneumonia, and colonization of respiratory tract and stomach in mechanically ventilated ICU patients. Am J Respir Crit Care Med. 1996;154(5):1339–1346. | ||
Nobbs AH, Lamont RJ, Jenkinson HF. Streptococcus adherence and colonization. Microbiol Mol Biol Rev. 2009;73(3):407–450, table of contents. | ||
Magnotti LJ, Croce MA, Zarzaur BL, et al. Causative pathogen dictates optimal duration of antimicrobial therapy for ventilator-associated pneumonia in trauma patients. J Am Coll Surg. 2011;212(4):476–484; discussion 484–486. | ||
Memish ZA, Shibl AM, Kambal AM, Ohaly YA, Ishaq A, Livermore DM. Antimicrobial resistance among non-fermenting Gram-negative bacteria in Saudi Arabia. J Antimicrob Chemother. 2012;67(7):1701–1705. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.