")
Back to Journals » International Journal of Nanomedicine » Volume 11
From micro- to nanostructured implantable device for local anesthetic delivery
Authors Zorzetto L, Brambilla P, Marcello E, Bloise N , De Gregori M, Cobianchi L , Peloso A , Allegri M , Visai L, Petrini P
Received 24 October 2015
Accepted for publication 3 February 2016
Published 8 June 2016 Volume 2016:11 Pages 2695—2709
DOI https://doi.org/10.2147/IJN.S99028
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Laura Zorzetto,1 Paola Brambilla,1 Elena Marcello,1 Nora Bloise,2 Manuela De Gregori,3 Lorenzo Cobianchi,4,5 Andrea Peloso,4,5 Massimo Allegri,6 Livia Visai,2,7 Paola Petrini1
1Department of Chemistry, Materials and Chemical Engineering ‘G. Natta’, Politecnico di Milano, Milan, 2Department of Molecular Medicine, Centre for Health Technologies (CHT), INSTM UdR of Pavia, University of Pavia, 3Pain Therapy Service, IRCCS Foundation Policlinico San Matteo Pavia, Pavia, 4General Surgery Department, IRCCS Foundation Policlinico San Matteo, Pavia, 5Departments of Clinical, Surgical, Diagnostic and Pediatric Sciences, University of Pavia, Pavia, 6Department of Surgical Sciences, University of Parma, Parma, 7Department of Occupational Medicine, Toxicology and Environmental Risks, S. Maugeri Foundation, IRCCS, Lab of Nanotechnology, Pavia, Italy
Abstract: Local anesthetics block the transmission of painful stimuli to the brain by acting on ion channels of nociceptor fibers, and find application in the management of acute and chronic pain. Despite the key role they play in modern medicine, their cardio and neurotoxicity (together with their short half-life) stress the need for developing implantable devices for tailored local drug release, with the aim of counterbalancing their side effects and prolonging their pharmacological activity. This review discusses the evolution of the physical forms of local anesthetic delivery systems during the past decades. Depending on the use of different biocompatible materials (degradable polyesters, thermosensitive hydrogels, and liposomes and hydrogels from natural polymers) and manufacturing processes, these systems can be classified as films or micro- or nanostructured devices. We analyze and summarize the production techniques according to this classification, focusing on their relative advantages and disadvantages. The most relevant trend reported in this work highlights the effort of moving from microstructured to nanostructured systems, with the aim of reaching a scale comparable to the biological environment. Improved intracellular penetration compared to microstructured systems, indeed, provides specific drug absorption into the targeted tissue and can lead to an enhancement of its bioavailability and retention time. Nanostructured systems are realized by the modification of existing manufacturing processes (interfacial deposition and nanoprecipitation for degradable polyester particles and high- or low-temperature homogenization for liposomes) or development of novel strategies (electrospun matrices and nanogels). The high surface-to-volume ratio that characterizes nanostructured devices often leads to a burst drug release. This drawback needs to be addressed to fully exploit the advantage of the interaction between the target tissues and the drug: possible strategies could involve specific binding between the drug and the material chosen for the device, and a multiscale approach to reach a tailored, prolonged drug release.
Keywords: pain management, microparticle, microencapsulation, nanoparticle production, nanogels, liposomes
Local anesthetics: clinical relevance for their use in implantable delivery systems
Local anesthetics (LAs) temporarily and reversibly prevent nociceptive nervous fibers from sending stimuli to the brain.1 These drugs act by blocking sodium and other ion channels present in the nerve cell membrane, thereby stopping the electrical signal before it can cause feelings of pain.1,2
LAs have been used for acute or chronic pain management, and their principal use is after surgery (eg, thoracotomy, laparotomy, cesarean section, breast cancer surgery, cosmetic breast surgery, limb amputation, and others).2,3 A recent study has highlighted the importance of LA administration after major operations to ward off chronic pain onset after thoracotomies and cesarian sections.3
As nociceptive inputs from the wound play a major role, it is logical to “go back to the periphery”, acting locally, to prevent nociceptive inputs from reaching the central nervous system and triggering endogenous pain processes.4 Peripheral LA administration is a rational approach to control pain.5 Continuous wound infusion (CWI) prolongs the action of LAs, thus improving their efficacy. Experimental models of incisional pain6 have demonstrated that the CWI of LAs modulates peripheral inflammation and sensitization in acute post-operative pain (PP). CWI is effective in controlling pain and reducing postoperative morphine administration after a major surgery.7 CWI through an intralesional catheter presents several limitations such as risk of infection (external device), reduction in patient activity (compliance patient problems), difficulty in administration (need to fill the device many times), and systemic toxicity related to inaccurate administration of LAs or steroids, such as convulsions, arrhythmias, cardiovascular (hypo- or hypertension) and metabolic (eg, hyperglycemia) problems.
Clinically used LAs possess common chemical structural characteristics, in which three chemical moieties can be identified: a hydrophilic amino group, a lipophilic benzene ring, and an intermediate chain8 (Figure 1). The intermediate chain can be an amide or an ester group, which allows classifying the drug as an ester-type or amide-type LA.8 Some of the most used amide types are lidocaine, bupivacaine, dibucaine, mepivacaine, and ropivacaine, whereas the most common ester types are benzocaine, tetracaine, proparacaine, procaine, and chloroprocaine. Some molecules present isomerism: the most diffuse chiral molecules are bupivacaine and prilocaine,1,8 with the (S)-enantiomer being more active and less toxic than the (R)-form.9
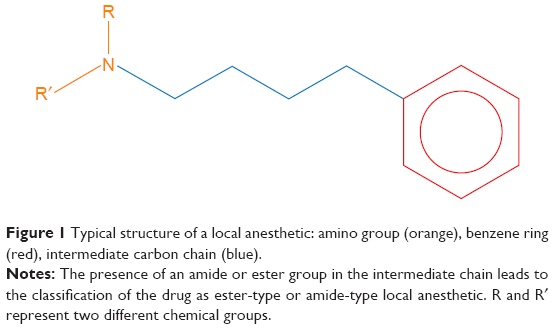 | Figure 1 Typical structure of a local anesthetic: amino group (orange), benzene ring (red), intermediate carbon chain (blue). |
The duration of a drug’s effect is related to its protein-binding ability. Agents that penetrate the nerve membrane and attach more firmly to the membrane proteins show prolonged activity, eg, tetracaine, bupivacaine, and etidocaine. On the contrary, procaine binds poorly to proteins and possesses a short half-life.10 LAs bind also to plasma proteins, albumin (low affinity), and α-1-acid glycoprotein (high affinity), leading to the same effects as tissue proteins.11 The duration of drug’s action is also related to its structure, length, and the groups present.
Amide type is generally more stable and possesses a longer half-life than ester type, which undergoes rapid hydrolysis by nonspecific esterases.11
Most of the clinically useful LAs are available in two forms: as free base or as a salt. The salt form has greater solubility in aqueous media and thus is preferred for a drug delivery system.10,12 The salt, being a weak base, can exist in solution as an un-ionized free base, which is more lipophilic, or as a positively charged cation; both species are involved in the process of conduction blocking.10,13 The base crosses the neuronal bilayer more easily, and thus is preferred for optimal penetration.10 The percentage of drug in the two forms depends on its pKa and the environmental pH. Each drug has a characteristic pKa, with ester type LAs typically having a higher value than the amide type.11 The closer the pKa is to the pH of the tissues, the faster is the onset of action of the drug,13,14 since the percentage of free base formed increases. For this reason, in infected tissues, the LA effect is reduced or even delayed, as the environment tends to be more acidic than usual and the un-ionized form decreases.10
Even though anesthesiologists often prefer them to general anesthetics, LAs present side effects like allergies1,2,8 and stroke15,16 related to their chemical structure (eg, ester types are more problematic than amide types). In particular, lidocaine and bupivacaine have caused strokes leading to death of the patient.15
Chloroprocaine, an ester type LA, has been demonstrated to be less cardiotoxic than most other LAs,15 but it has an extremely short half-life: <10 minutes in vivo.17
Recently, drug delivery systems, according to different user needs obtained by a variety of materials and physical forms, have been developed to minimize severe side effects and to prolong the anesthetic action.1
The state of art: local anesthetic delivery
Implantable systems used for LA delivery
Injectable and implantable systems for LA delivery aim to reduce LA toxicity and prolong the anesthetic effect of drugs with short half-lives, shielding the drug from the environment of the implantation site.1
New delivery platforms are necessary to obtain effective, lasting postoperative peripheral analgesic treatment for several days to block the nociceptive message and to reduce postoperative inflammatory response. This platform has to overcome the limitations of the external inaccurate delivery devices. The development of intraoperative nanogel delivery systems, able to provide anesthetics with planned dosages and timing, is a key step toward attaining the safest and most effective postoperative treatment. Nanotechnology plays a critical role in the development of patient-specific therapies and individualized medicine and offers great opportunities for combating the limitations of postoperative pain management.
One of the simplest systems for LA delivery is represented by the oil depot formulation: lipophilic drugs (eg, lidocaine,18–20 bupivacaine,21 or ropivacaine21,22) are dissolved in different biocompatible oils18–22 and injected intramuscularly, subcutaneously, or intra-articularly.
In esthetic medicine, lidocaine loaded-commercial fillers are commonly employed in patients. For these applications, the release profile is not studied because its characterization is not an intended scope, as they are not designed as drug delivery systems, with LAs added to reduce pain during the filler injection.
Calcium phosphates were loaded with lidocaine for bone defect reconstruction to provide relief from pain after implant.23,24 Scaffolds were formed by sintering ceramic powers and infiltrating them with lidocaine solutions (60 mg/mL23 and 20 mg/mL24). To treat pain after tooth extraction, other implantable devices were developed to fill the bone gap, mixing commercial bone putty (Xybrex; Orthocon, Inc., Irvington, NY, USA) and lidocaine25 or developing sponge-like polycationic and polyanionic hydrogel fillers: lidocaine solution (up to 4 mg/mL) was incorporated into polymer solutions before cross-linking.26
However, none of these systems for LA delivery was developed for a prolonged, tailored release.
Application of micro/nanodevices in the clinical practice
Current achievements and future perspectives in pain therapy raise the question whether hospitalization without pain is still wishful thinking or can became a reality. Micro and then nanodevices are on the horizon of new technologies for pain management, which can overcome the limitations of traditional anesthetic delivery tools, such as drug delivery security and safety of the delivery system, simplicity of setting up, as well as maintenance and management. These systems consist in micro- and nanostructured films, particles, capsules (suspended in a solution or embedded in a gel matrix), and hydrogels, which can be implanted in the site of surgery and release LAs in a tailored manner. Irrespective of the origin of pain, management is necessary in clinical practice leading to essential benefits including shorter hospital stay and improved patient satisfaction. Otherwise, totally implantable devices (according to the US Food and Drug Administration [FDA], ISO 10993-1 micro/nanodevices must be categorized as implant devices with prolonged exposure to the body) with their full integration with the biological milieu of the human body is still a crucial multidisciplinary task to solve. From a clinical perspective, the ideal pain management should cover the entire period necessary with as little discomfort as possible to the patient. The production of devices as biocompatible and small as possible can prevent discomfort. This concept drives this clinically translational research to manufacturing a device characterized by features from micro to nano if considering physical size as the major overriding issue (even if not the only one). LAs, as the main actors of drug delivery through implanted nanodevices, play a pivotal role for the entire system due to their peculiar characteristics: completely reversible action and the relative absence of collateral or side effects compared to other drugs generally used in the clinical scenario. Their use is nowadays limited to intravenous patient-controlled analgesia or patient-controlled epidural systems. Although these analgesic methods are commonly used, they are not free from important disadvantages including their invasiveness and the need for pumping equipment, tubes, or power lines, which could potentially limit patient mobility during, for example, the postoperative period. They also require exhaustive staff-time preparation and resources with the risk of catheter dislodgement or epidural hematoma (increased in case of coadministration of anticoagulant therapy). These limitations show why the development of LA drug delivery system administering therapy from inside the patient’s own body is so crucial and, secondly, why nanotechnology has received a never-seen-before enthusiasm in this field.
Toward tailored, controlled LA delivery with implantable and injectable systems
Drug delivery systems described so far are the widest and simplest methods for LA administration. Among them, it is possible to find also commercial products (eg, EMLA cream27). More advanced ones have been developed to treat the urgent issues represented by chronic and postoperative pains. These can be classified as films, or micro- or nanostructured systems, depending on the final shape and size of the devices constituted by different materials and realized using different techniques (Figure 2). A summary of all the reviewed systems, based on the employed materials, is described in Tables 1–4.
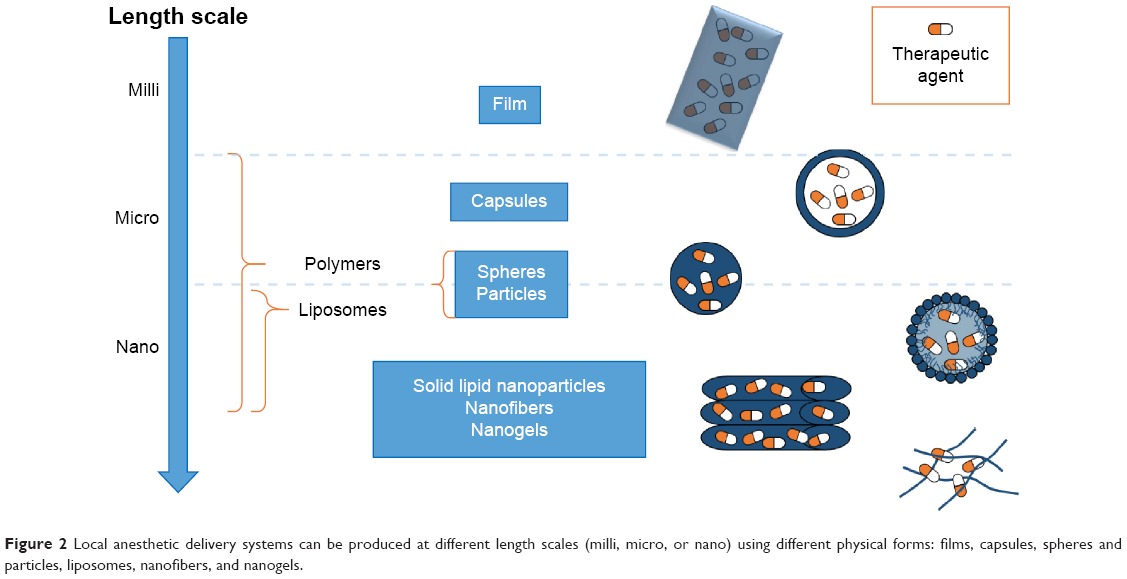 | Figure 2 Local anesthetic delivery systems can be produced at different length scales (milli, micro, or nano) using different physical forms: films, capsules, spheres and particles, liposomes, nanofibers, and nanogels. |
 | Table 1 Summary of polyester-based local anesthetic delivery systems |
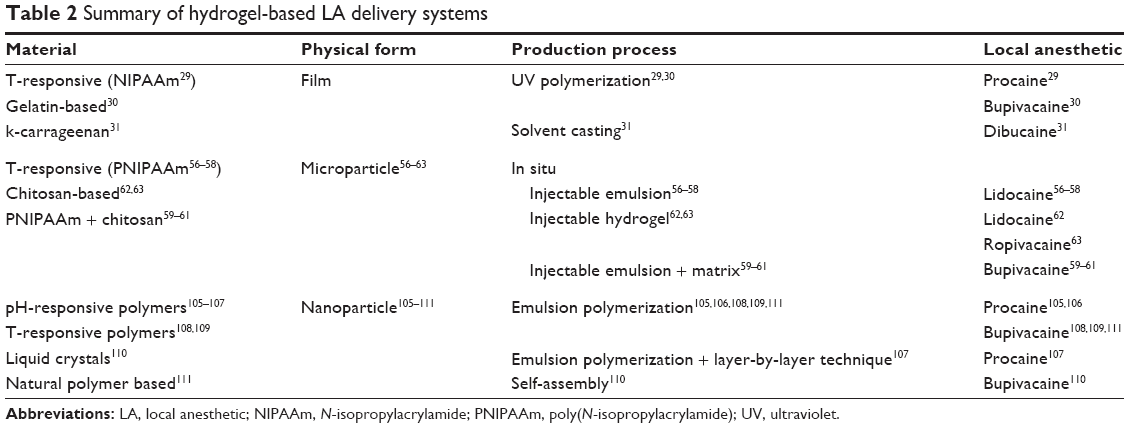 | Table 2 Summary of hydrogel-based LA delivery systems |
 | Table 3 Summary of phospholipid-based LA delivery systems |
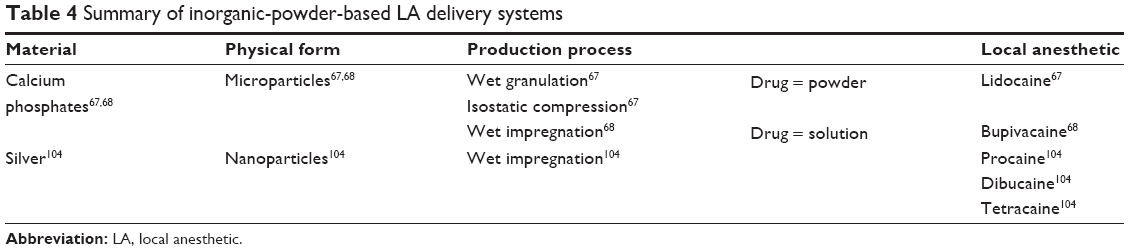 | Table 4 Summary of inorganic-powder-based LA delivery systems |
Films
Drug delivery systems in form of films were developed in particular for wound treatment or as transdermal patch.28 Implantable films were produced by UV polymerization29,30 or solvent casting.31–33 The UV curing technique consists of cross-linking polymer chains using UV radiation and loading the drug29 prior to the cross-linking. Depending on the type of materials, the loading can vary from 10 μg/g of film in the case of a semi-interpenetrated network obtained through UV-curing of poly(ethylene glycol) (PEG) diacrylate in presence of gelatin30 to ~25 mg of drug/g of film using N-isopropylacrylamide (NIPAAm) and 1-vinyl-2-pyrrolidinone.29 Thermoresponsive polymer chains can lead to a more compact hydrogel network, slowing down the drug release.
Solvent casting technique can be applied by exploiting different strategies for drug loading, either during or after the solvent casting. Alternatively, drug loading was achieved by swelling a dried k-carrageenan film in dibucaine solution.31 Simultaneous film casting and drug encapsulation were achieved by loading lidocaine, a lipophilic drug, in a poly(lactic acid) (PLA) solution in chloroform.32 PLA was also employed to produce a composite film.33 During in vivo test in rats, drug release in the plasma lasted up to 10 days.
Although different materials and types of drug were used, when the drug was loaded in the step after film formation,31 the release was faster than when the drug was loaded during the film preparation.32 Drug incorporation in the polymer solution can allow a homogeneous dispersion within the bulk of the material, but it is limited by the solubility of the drug in the solvent used for casting the film.
Microstructured systems
Microspheres, microparticles, and microcapsules are classified as microstructured systems, which can be implanted or injected once suspended in appropriate media, used as injectable matrices, or formed in situ (Figure 3). Among the several proposed solutions, the most widely used materials for microparticle production are synthetic degradable polyesters such as PLA, poly(glycolic acid), poly(lactic-co-glycolic acid) (PLGA), and poly(ε-caprolactone).
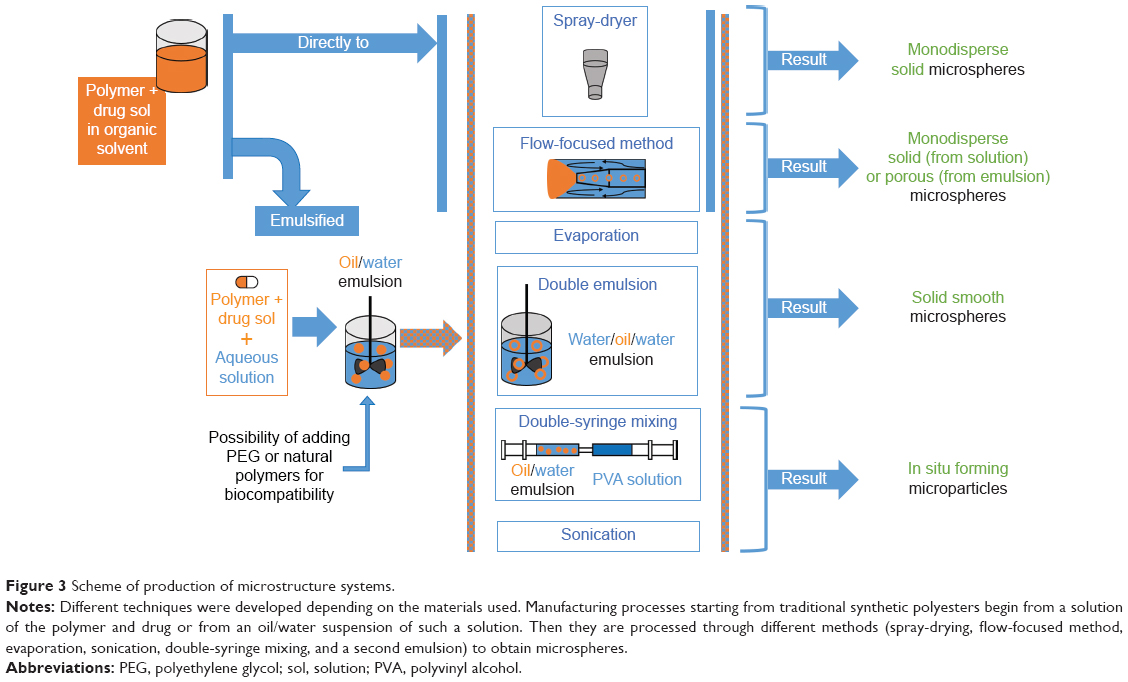 | Figure 3 Scheme of production of microstructure systems. |
The use of these materials to produce microspheres is related to the relative simplicity of microsphere production. In most of the works, microparticles were produced by oil/water (O/W)32,34–42 or water/oil/water (W/O/W) emulsions42,43 and solvent evaporation techniques. Usually, particles produced using those methods are solid and smooth microspheres, with a prolonged drug release lasting from 12 hours to 3–4 days. In some cases, dexamethasone was used as the excipient, enhancing bupivacaine’s in vivo effectiveness without modifying significantly the in vitro release profile.35,36,44
Alternatively, microspheres were produced through a spray-drying technique, which consists of spray-drying an organic drug/polymer solution.45–47 This technique requires higher and thus more expensive technology, but the cost of the spray-dryer is compensated by better results in terms of simplicity of the production process, narrower particle size distribution, and higher drug encapsulation efficiency.45 Spray-drying can be exploited also to produce porous microparticles by using an O/W emulsion instead of a drug–polymer solution.
Porous microparticles were subsequently immersed in an aqueous solution of procaine (hydrophilic drug) and the particle pores were closed by adding an organic solvent in the procaine-loaded bath: in this way a hollow microcapsule was formed. This unconventional method succeeded in reaching drug release up to 9 days and to produce microcapsules that were more easily degradable.48
Another technique to produce monodisperse polymeric microspheres is flow-focusing.49–51 This technique employs a microfluidic device consisting of coaxial capillaries. At the inlet of the capillary with the larger diameter, the drug (lidocaine49,51 or bupivacaine50) and polymer organic solution follows in a certain direction, surrounded by polyvinyl alcohol (PVA) aqueous solution flowing in the same49,50 or opposite direction.51 By tuning the process parameters (eg, viscosity of lipophilic and hydrophilic solutions), it is possible to produce microspheres with final characteristics close to the desired ones and with very narrow particle size distribution.49 Monodisperse microspheres showed slower drug release with reduced initial burst release compared to microspheres produced by traditional methods presenting similar average size but a broader size distribution.50 Moreover, if O/W or W/O/W emulsions were used instead of a drug-loaded polymer solution, it is possible to obtain porous microparticles. This approach was followed by Vladisavljević et al,51 who used numerical simulation to find the most suitable process parameters (eg, flow rate and solution viscosities) to tailor particle characteristics.
Porous microparticles could be produced also using a simpler W/O/W technique. In contrast to the previously described technique, the first emulsion consisted of a water-in-oil emulsion in which the oily phase was constituted by PLGA dissolved in chloromethane in presence of lidocaine, which was subsequently mixed with an aqueous solution of PVA.52
In situ formation of microparticles was achieved by an emulsion of bupivacaine-loaded polymer (PLA with an emulsion stabilizer53 or PLGA54) solution in 2-pyrrolidone with an oily phase:53,54 emulsions were formed both by sonication and by manually mixing the contents of two syringes (one containing the oily phase and the other the polymer phase) previously connected with a double-female luer lock connector.
In spite of prolonged LA release by synthetic polyesters (up to 15–20 days32,47,49), they could also cause significant inflammatory reaction.55 For this reason, alternative strategies were proposed.
In some studies, PLA or PLGA microsphere were produced in presence of PEG,37,42 which is well known for its nonfouling properties and can reduce inflammatory response. In one case, a mixture of the PLA and PEG was prepared, producing the microsphere by coacervation method, in which hexane (not able to dissolve PLA) was added to a stirred polymer solution (PLA + PEG + lidocaine) in methylene chloride. The coacervation droplets adhered to the suspended drug particles and coalesced around them to form polymer microparticulates.42
In another study, PLA/PEG copolymer was used and particles were realized following the O/W emulsion technique. In this case, particles were coated with gelatin by solvent-casting of a gelatin solution to tailor the drug release, which reached 3-day release during in vitro tests in water at 37°C.37 Combination with natural polymers is a strategy also used in another study, where PLGA microparticles were produced by an O/W emulsion in which alginate was dissolved in the aqueous solution.40 In vitro release of bupivacaine in PBS (37°C, pH 7.4) lasted for up to 24 hours.40
Other studies focused their attention on the use of stimuli-responsive materials (Figure 4). Thermosensitive polymers such as poly(N-isopropylacrylamide) (PNIPAAm)56 and copolymers57,58 were used to produce microspheres by cross-linking preformed polymer chains: an aqueous polymer solution was acidified and glutaraldehyde was added as the cross-linking agent. The solution was then poured into light mineral oil containing lecithin as emulsifying agent. Drug loading was obtained by soaking the microspheres in a lidocaine solution. However, this method did not guarantee prolonged drug release in PBS even up to 100 minutes in vitro.57 PNIPAAm microparticles were also embedded in an injectable chitosan thermogel matrix:59–61 the embedding of microparticles was achieved through an innovative coinjection technique through a double-barreled syringe. In these cases, the chitosan matrix was useful to both prolonging bupivacaine release in PBS (>6 hours in vitro) and avoiding particle dispersion.59–61 Chitosan injectable loaded matrices62,63 were also studied with different results in terms of duration of anesthetic release in PBS: from 40 minutes62 up to 48 hours.63 Other polymeric materials used for loaded microsphere production were tetraethoxysilane64,65 and poly(acrylic acid).66
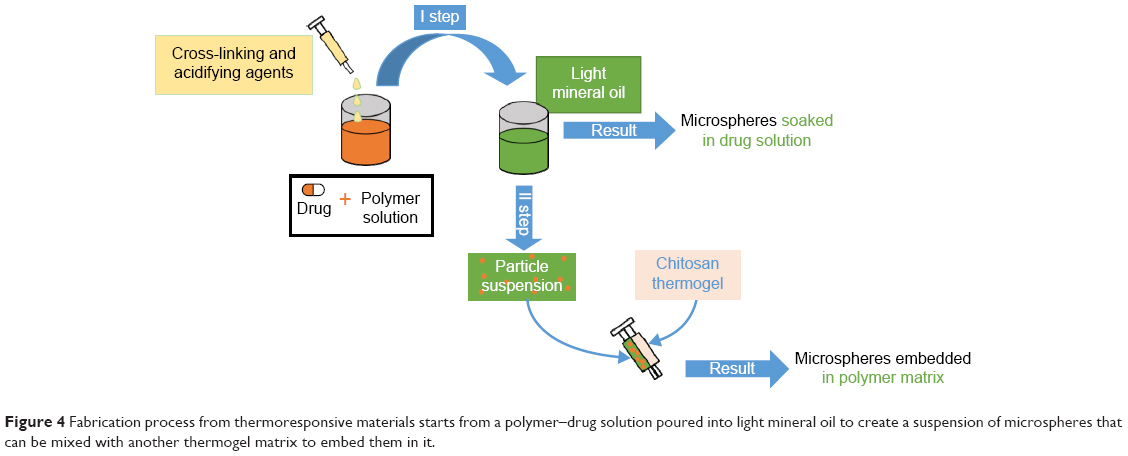 | Figure 4 Fabrication process from thermoresponsive materials starts from a polymer–drug solution poured into light mineral oil to create a suspension of microspheres that can be mixed with another thermogel matrix to embed them in it. |
Inorganic microparticulates can be loaded with LAs (Figure 5). Different methods of loading lidocaine on calcium phosphate powders were tested: wet granulation, isostatic compression, and a combination of both.67,68 These techniques started with mechanically mixing calcium phosphate and lidocaine. In wet granulation, the powder was moistened with water and passed through a sieve. Isostatic granulation involves the application of high pressure to the mixed powder (calcium phosphate and lidocaine).67 Bupivacaine was loaded on calcium phosphate apatite: in this case, wet impregnation of an ethanol solution of the drug resulted in a uniform loading than when mixing bupivacaine powder with calcium phosphate powder.68
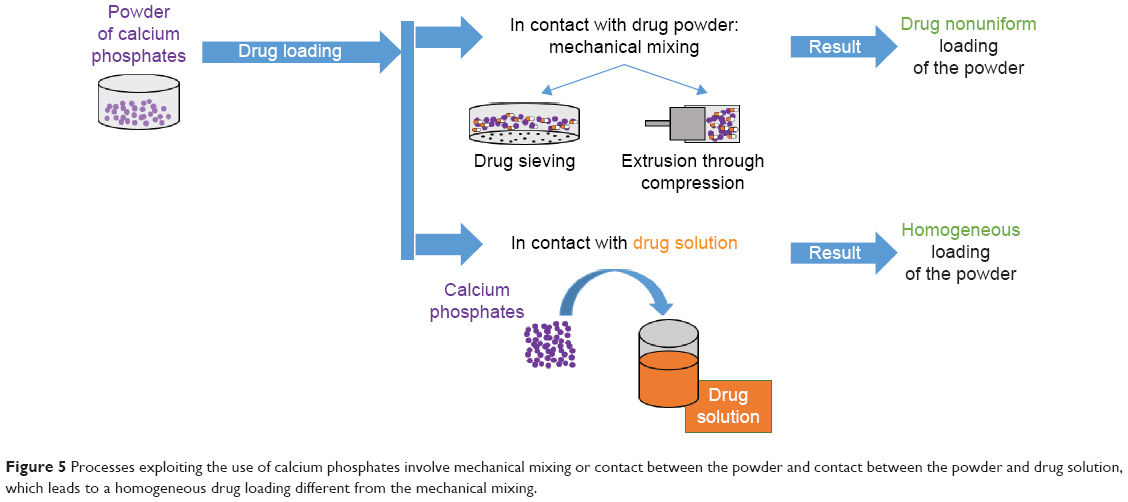 | Figure 5 Processes exploiting the use of calcium phosphates involve mechanical mixing or contact between the powder and contact between the powder and drug solution, which leads to a homogeneous drug loading different from the mechanical mixing. |
From micro to nano
In the past decades, a large number of publications have focused on the advantages resulting from the adoption of nanosystems instead of microsystems and therefore adopting different strategies to obtain nanoscale devices (Figure 6).
 | Figure 6 Scheme of nanostructured device production. |
The aim of the scaling down is to make the size of the drug delivery system more similar to the biological environment, which is nanostructured. In this way, the molecular interactions could be more specific, and the drug could be absorbed into the targeted tissue to attain an enhancement of its bioavailability and retention time. The nanosystems, indeed, were demonstrated to improve intracellular penetration.69–71
In order to have a controlled and reliable LA release, the drug would be encapsulated in a biocompatible and biodegradable nanostructured system, to minimize tissue reaction and to control precisely the rate of drug delivery, which has to be comparable with the rate of generation of nontoxic degradation products. In fact, an initial uncontrolled large dose of drug release could be lethal for the organism and therefore must be avoided.14,72 Moreover, LA nanoencapsulation increases the drug’s efficacy, specificity, tolerability, and therapeutic index.69
The LA release kinetics and the drug loading efficiency are influenced by the size of the system. Larger particles could load a larger amount of drug, leading to a longer block duration and thus a longer effect. On the contrary, smaller particles have the advantage of reducing the release rate of the incorporated drug, thereby improving the drug binding, even though smaller systems show a rapid initial release of the drug, which could be toxic.73 This is due to the fact that on moving from micro to nanosystems the surface-to-volume ratio of the particles increases and therefore also the initial dissolution rate.74,75 Therefore, the development of a nanostructured system has to take into account ad hoc strategies to exploit the possible advantages offered by those systems (in particular the higher encapsulation efficiency and specificity in targeting biological structures) and limit the drawbacks related to their size.
Liposomes to solid lipid nanoparticles
Liposomes are phospholipid-based carriers.27 Because of their multilayered structure, with alternating aqueous compartment and lipid bilayer, they are able to incorporate both lipophilic and hydrophilic drugs, which can pass the lipophilic barrier of cell membranes.
The potential benefits in the use of liposomes as injectable or implantable devices for LA delivery were highlighted when it was found that an injection of soybean phosphatidylcholine nanoemulsion reduced bupivacaine’s cardiotoxicity.76
Liposome synthesis consists in obtaining large multilamellar vesicles (LMVs) from dried phospholipid films by suspending the film in an aqueous solution and vortexing it. Though LMVs were initially used, in also small unilamellar vesicles (SUVs) were also used.77 From an LMV suspension in D2O, SUVs were obtained by freeze-drying the suspension.78,79 Alternatively, SUVs could be obtained by homogenization80 or extrusion81,82 of an organic suspension of LMVs (Figure 7). Different phospholipids alone or mixed have been used in different works: egg phosphatidylcholine;78,81 soy phosphocholine and cholesterol;79,80 egg phosphatidylcholine, cholesterol, and α-tocopherol;82,83 and hydrogenated soy phosphatidylcholine and cholesterol.77
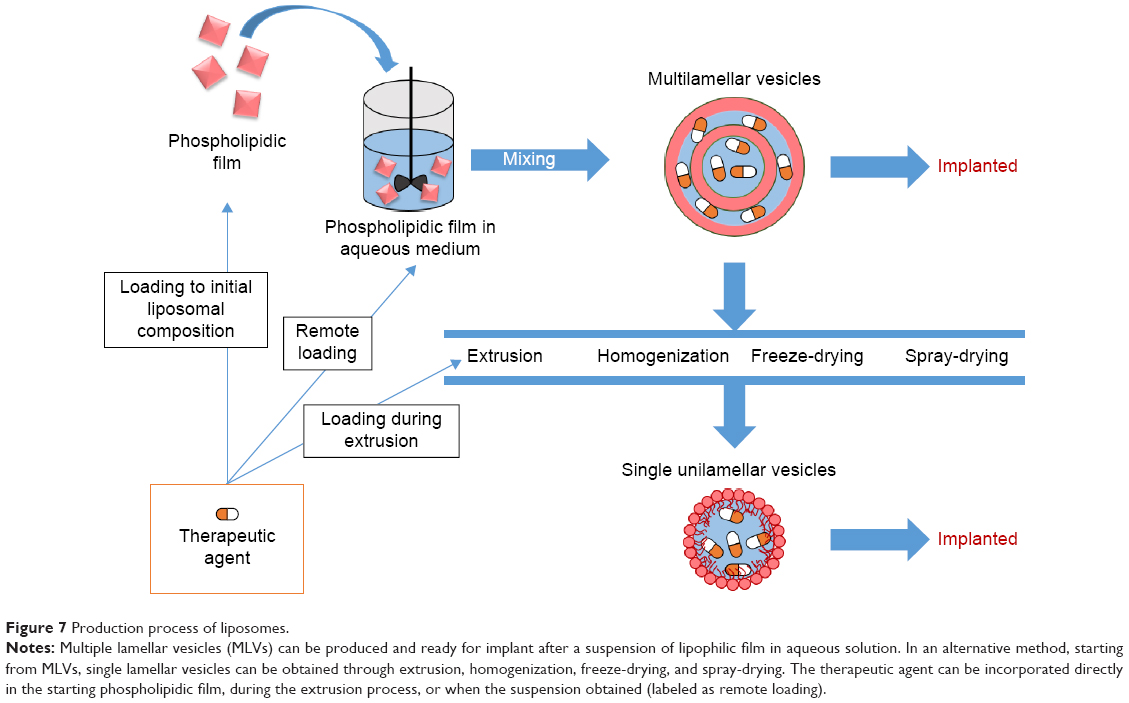 | Figure 7 Production process of liposomes. |
In general, drug loading can be done by simply adding the drug to a solution containing the phospholipids,83,84 or after an extrusion process81,82 through an ammonium sulfate gradient.77,79,80 The latter realizes remote loading of the drug in preformed LMVs produced in presence of (NH4)2SO4. (NH4)2SO4 present in the extraliposomal medium is removed by dialysis against normal saline. In this way, an ammonium sulfate gradient is formed over the liposomal membrane. Bupivacaine was loaded by incubating the liposomal formulations with a concentrated bupivacaine solution at the pH range 5.0–5.5. Remote loading was found to be a more effective encapsulation method, since it led to 24–48 hours release in saline solution in vitro77,80 and 45 hours in vivo lasting of the anesthetic effect,80 which is six to eight times more than the release duration obtained with other loading techniques.
Ultimate evolutions in liposome technology involve the addition of sugars in their composition, which leads to lipid–protein–sugar particles (LPSPs),55,85,86 with the aim of accelerating particle degradation in the biological environment. In one study, also dexamethasone was added to the composition to prolong in vivo drug effectiveness.86 In this work, LPSPs were prepared through spray-drying: then di-palmitoylphosphatidylcholine and bupivacaine were dissolved in a given amount of ethanol; sugar (lactose) and albumin were dissolved in water and the solutions were mixed immediately prior to spray-drying. LPSPs provided significantly shorter tissue reaction to foreign body than conventional PLA and PLGA microparticles.55
In one of the latest works, liposomes were coated with chitosan and alginate to overcome the major drawback associated with liposomes: their short shelf-life.77 The coating process succeeded in prolonging liposome conservation, avoiding drug leakage when stored up to 2 years.
An alternative injectable nanocarrier to liposomes87–89 is represented by solid lipid nanoparticles (SLNs), a nanosized colloidal system. SLNs have the advantage of a production process that avoids the use of organic solvents, namely high-pressure homogenization. This process can be performed either at high or low temperatures. In the first case, the process is called hot homogenization:87–89 the drugs (tetracaine87,88 or bupivacaine89) are dissolved in the melted lipid, and the drug-loaded lipid is dispersed in a hot aqueous surfactant solution to form a pre-emulsion, which is subsequently homogenized. The whole process is conducted at 500–1,500 times the atmospheric pressure. In the second variant, called cold homogenization,87 after dissolving the drug in the lipid, solidification is done by pouring the drug-loaded lipid in liquid nitrogen. This leads to the formation of a solid solution (molecular dispersion) of the drug in the lipid matrix. After milling the solid solution, the particles are suspended in a poloxamer solution, and the resulting suspension is homogenized at room temperature and 1,500 times the atmospheric pressure. Even though this carrier can be considered safer than polymeric nanoparticles, through the avoidance of organic toxic solvents, it failed to provide prolonged drug release in phosphate–citrate buffer at 37°C (2 hours).87
Nanostructured systems
Nanostructured systems include nanospheres, nanocapsules, principally designed as injectable systems, and nanofibers. Similar to microstructured systems, synthetic degradable polyesters traditionally play an important role in the production of nanostructured systems.
Nanospheres have been produced by O/W emulsion90–93 and nanoprecipitation.94–99 The first technique consists of making a solution (oily phase) in which the polymer and the drug are dissolved in organic solvents (typically methylene chloride or chloroform). The organic solution is subsequently poured into an aqueous solution (which often contains PVA, which acts as the emulsifier) and the suspension is vortexed. The organic solvent can be eliminated at atmospheric pressure or by rotary evaporation followed by freeze-drying.34 On the other hand, nanoprecipitation consists in dissolving the drug in an organic polymer solution, which is added dropwise to an aqueous phase. The suspension is magnetically stirred until complete solvent evaporation is achieved.
Interfacial deposition (also known as solvent displacement) is an innovative method to produce polyester nanocapsules with hollow oily nuclei.100–102 This method consists in creating an organic phase composed of the drug (benzocaine in all reported works), the polymer, acetone, oil, and the emulsifier. The organic phase is mixed under magnetic stirring with an aqueous solution containing an emulsifier in turn, and acetone is evaporated by rotary evaporation. This technique can be considered an evolution of the O/W suspension, as interfacial deposition differs from it for solvent evaporation from the suspension while mixing it. In vivo duration of nerve blockage could reach 4–5 hours.
An advanced nanostructured device for LA delivery is represented by electrospun bupivacaine-loaded PLGA suture.103 Sutures were produced by adding the drug to the polymeric solution used for the electrospinning process. They released their entire drug payload in PBS at 37°C in 12 days and maintained ~12% of their initial tensile strength after 14 days in vitro.
Silver nanoparticle suspension and LA solutions (procaine, dibucaine, tetracaine) were prepared to study the silver–anesthetic interaction:104 this study, however, is in a preliminary phase and far from any clinical application.
Nanogels
The most advanced research on nanostructured drug delivery devices focuses on nanogels.
Tan et al105,106 have produced pH-responsive nanogels by emulsion polymerization of methacrylic acid–ethyl acrylate cross-linked with di-allyl phthalate. In a more recent study, the same research group succeeded in coating the pH-responsive gels using a layer-by-layer technique. In this case, layers of poly(allylamine hydrochloride) and poly(sodium 4-styrenesulfonate) polyelectrolytes were alternated on the particle surface with the aim of tailoring drug release.107 Interestingly, it was found that beyond a certain thickness of the coating, the drug release had a slow and continuous profile.107 The emulsion polymerization technique is used also to produce functionalized thermoresponsive nanogels based on (PNIPAM). Thanks to the acrylic acid functionalization, the nanogel binds firmly to a cationic drug such as bupivacaine.108,109
A smart production process of nanogels is the self-assembly technique. Self-assembled structures made of bupivacaine and liquid crystalline phases (monoglyceride and medium-chain triglycerides) have been studied.110 Precisely, bupivacaine with appropriate concentrations was mixed with glycerol monooleate or binary glycerol monooleate/medium-chain triglycerides combination, and the mixture was heated at 40°C-50°C. The obtained isotropic solutions were hydrated by adding PBS and then homogenized by vigorous vortexing. The lipid composition led to different crystalline nanostructures, which were shown to influence the drug release, lasting for up to 48 hours.
Also natural polymers such as alginate-based nanogels were investigated for nanogel production111 (Figure 8). Alginate/chitosan nanogels loaded with bupivacaine were produced starting from a dilute (0.0067% w/v) solution of sodium alginate (containing the drug) to which CaCl2 was added under mechanical stirring. To this solution, a dilute chitosan solution was added at a controlled flow rate. The second hydrogel consisted of alginate/anionic surfactant (AOT) nanogels, which were produced starting from an AOT solution in methylene chloride, which was added to an aqueous alginate solution containing bupivacaine, and homogenized. PVA solution was eventually added to the initial solution, creating a double emulsion, to which calcium chloride solution was added to ensure alginate cross-linking. Both compositions showed good stability and cytocompatibility. However, alginate/chitosan nanogels showed a slower drug release in PBS (pH 7.4) than alginate/AOT surfactant (13 hours vs 8 hours).
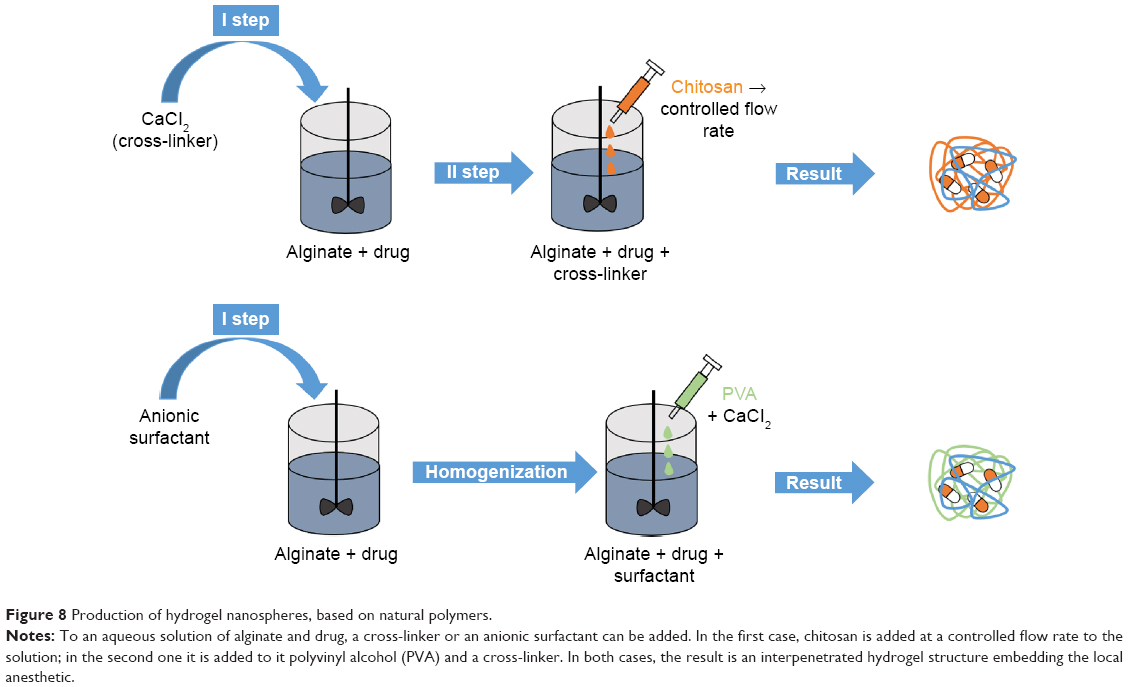 | Figure 8 Production of hydrogel nanospheres, based on natural polymers. |
Concluding remarks and future outlook
LAs are powerful drugs whose use can positively affect modern medical procedures. Together with their utility, they also suffer from safety issues, as most of them show different levels of neurotoxicity and cardiotoxicity. Among the strategies to overcome this drawback, it is possible to develop implantable devices capable of tailored drug release.
The most varied and studied delivery systems are microstructured PLA or PLGA microparticles. The many manufacturing processes available up to now provide full control of the particle size and dispersion, surface topology, and structure (particle or capsule). Most of the reviewed systems show great stability and prolonged drug release. Various strategies have also been developed to avoid initial burst release, which can worsen safety issues. Despite the maturity of the techniques and release systems, their application potential remains to be demonstrated in reality. Although synthetic polyesters are often chosen for these aims, they show great stability in human body, which is not always required in this specific application and could lead to non-negligible foreign body response, in addition to acidic degradation products. Moreover, an acidic environment is not ideal for LA’s effectiveness, as it could shorten the drug’s half-life. Liposomes and SLNs do not release acids during degradation and can be produced without the use of organic solvents. These materials also have drawbacks, which are now being addressed, as the particles may be too stable in the biological environment but show poor stability on the shelf.
Scaling down to the nanoscale brings indeed several advantages with a device scale comparable to that of the nanostructured biological environment. The strategy to adapt techniques and materials developed for microstructures to produce nanostructures is not straightforward. Production of nanostructured systems could present some peculiar issues, mainly due to the high surface-to-volume ratio, which often leads to burst release of the drug. To this aim, strategies involving specific binding between the drug and the material and a multiscale approach are likely to be the key to reach a tailored, prolonged drug release. According to the progress in the field, the most promising approaches involve the presence of nanosystems in micro or macrodevices, to take full advantage of the different characteristics brought in a unique multiscale device.
Acknowledgments
The authors would like to thank Fondazione Banca del Monte di Lombardia and Fondazione IRCCS Policlinico San Matteo-Pavia for financial support. Massimo Allegri also wishes to thank the Italian Ministry of Health for young researchers, project code GR-2010-2318370. Manuela De Gregori is a member of SIMPAR (Study In Multidisciplinary Pain Research) Group and YAP (Young Against Pain) Group. Massimo Allegri is a member of SIMPAR.
Disclosure
The authors report no conflicts of interest in this work.
References
Edgcombe H, Hocking G, [webpage on the Internet]. Local anaesthetic pharmacology. World Anaesth Tutor Week. 2005:125–127. Available from: http://www.frca.co.uk/documents/la.pdf. Accessed April 8, 2016. | ||
Shipton EA. New formulations of local anaesthetics-part I. Anesthesiol Res Pract. 2012;2012:546409. | ||
Andreae MH, Andreae DA. Local anaesthetics and regional anaesthesia for preventing chronic pain after surgery. Cochrane Database Syst Rev. 2012;10(7):CD007105. | ||
Brennan F, Carr DB, Cousins M. Pain management: a fundamental human right. Anesth Analg. 2007;105(1):205–221. | ||
Sentürk M, Ozcan PE, Talu GK, et al. The effects of three different analgesia techniques on long-term postthoracotomy pain. Anesth Analg. 2002;94(1):11–15. | ||
Kroin JS, Buvanendran A, Watts DE, Saha C, Tuman KJ. Upregulation of cerebrospinal fluid and peripheral prostaglandin E2 in a rat postoperative pain model. Anesth Analg. 2006;103(2):334–343, table of contents. | ||
Gupta A, Favaios S, Perniola A, Magnuson A, Berggren L. A meta-analysis of the efficacy of wound catheters for post-operative pain management. Acta Anaesthesiol Scand. 2011;55(7):785–796. | ||
Galindo Arias M. LEVOBUPIVACAINE – a long acting local anaesthetic, with less cardiac and neurotoxicity. Update Anaesth. 2002;14:23–25. | ||
Tan JPK, Tan MBH, Tam MKC. Application of nanogel systems in the administration of local anesthetics. Local Reg Anesth. 2010;3(1):93–100. | ||
Covino BG, Giddon DB. Pharmacology of local anesthetic agents. J Dent Res. 1981;60(8):1454–1459. | ||
McLure HA, Rubin AP. Review of local anaesthetic agents. Minerva Anestesiol. 2005;71(3):59–74. | ||
Lee KJ, Yang SY, Ryu WH. Controlled release of bupivacaine HCl through microchannels of biodegradable drug delivery device. Biomed Microdevices. 2012;14(3):583–593. | ||
Stevens RA, Chester WL, Schubert A, Brandon D, Grueter JA, Zumrick J. pH-adjustment of 2-chloroprocaine quickens the onset of epidural anaesthesia. Can J Anaesth. 1989;36(5):515–518. | ||
Weiniger CF, Golovanevski L, Domb AJ, Ickowicz D. Extended release formulations for local anaesthetic agents. Anaesthesia. 2012;67(8):906–916. | ||
Guay J. Adverse events associated with intravenous regional anesthesia (Bier block): a systematic review of complications. J Clin Anesth. 2009;21(8):585–594. | ||
Brown DL, Ransom DM, Hall JA, Leicht CH, Schroeder DR, Offord KP. Regional anesthesia and local anesthetic-induced systemic toxicity: seizure frequency and accompanying cardiovascular changes. Anesth Analg. 1995;81(2):321–328. | ||
Kuhnert BR, Kuhnert PM, Philipson EH, Syracuse CD, Kaine CJ, Chang-hyon Y. The half-life of 2-chloroprocaine. Anesth Analg. 1986;65(3):273–278. | ||
Fredholt K, Larsen DH, Larsen C. Modification of in vitro drug release rate from oily parenteral depots using a formulation approach. Eur J Pharm Sci. 2000;11(3):231–237. | ||
Larsen DH, Fredholt K, Larsen C. Assessment of rate of drug release from oil vehicle using a rotating dialysis cell. Eur J Pharm Sci. 2000;11(3):223–229. | ||
Larsen SW, Østergaard J, Friberg-Johansen H, Jessen MNB, Larsen C. In vitro assessment of drug release rates from oil depot formulations intended for intra-articular administration. Eur J Pharm Sci. 2006;29(5):348–354. | ||
Larsen SW, Frost AB, Østergaard J, Marcher H, Larsen C. On the mechanism of drug release from oil suspensions in vitro using local anesthetics as model drug compounds. Eur J Pharm Sci. 2008;34(1):37–44. | ||
Thing M, Larsen C, Østergaard J, Jensen H, Larsen SW. In vitro release from oil injectables for intra-articular administration: importance of interfacial area, diffusivity and partitioning. Eur J Pharm Sci. 2012;45(3):351–357. | ||
Dubnika A, Loca D, Berzina-Cimdina L. Functionalized hydroxyapatite scaffolds coated with sodium alginate and chitosan for controlled drug delivery. Proc Est Acad Sci. 2012;61(3):193. | ||
Salma I, Pilmane M, Skagers A, et al. Early morphofunctional response of contact tissue after intraosal implantation in rabbit jaw of pure synthetic hydroxyapatite (HAp) bioceramic materials and HAp saturated with lidocaine. Stomatologija. 2009;11(4):113–118. | ||
Kumarswamy A, Moretti A, Paquette D, Padilla R, Everett E, Nares S. In vivo assessment of osseous wound healing using a novel bone putty containing lidocaine in the surgical management of tooth extractions. Int J Dent. 2012;2012:1–8. | ||
Liu DZ, Sheu MT, Chen CH, Yang YR, Ho HO. Release characteristics of lidocaine from local implant of polyanionic and polycationic hydrogels. J Control Release. 2007;118(3):333–339. | ||
Sobanko JF, Miller CJ, Alster TS. Topical anesthetics for dermatologic procedures: a review. Dermatologic Surg. 2012;38(5):709–721. | ||
Petrisor G, Ion RM, Brachais CH, Couvercelle J-P, Chambin O. Designing medical devices based on silicon polymeric material with controlled release of local anesthetics. J Macromol Sci Part A. 2012;49(5):439–444. | ||
Geever LM, Cooney CC, Lyons JG, et al. Characterisation and controlled drug release from novel drug-loaded hydrogels. Eur J Pharm Biopharm. 2008;69(3):1147–1159. | ||
Fu Y, Kao WJ. Drug release kinetics and transport mechanisms from semi-interpenetrating networks of gelatin and poly(ethylene glycol) diacrylate. Pharm Res. 2009;26(9):997–1003. | ||
Makino K, Idenuma R, Murakami T, Ohshima H. Design of a rate- and time-programming drug release device using a hydrogel: pulsatile drug release from kappa-carrageenan hydrogel device by surface erosion of the hydrogel. Colloids Surf B Biointerfaces. 2001;20(4):355–359. | ||
Klose D, Siepmann F, Elkharraz K, Siepmann J. PLGA-based drug delivery systems: importance of the type of drug and device geometry. Int J Pharm. 2008;354(1–2):95–103. | ||
Blanco MD, Bernardo MV, Gómez C, Muñiz E, Teijón JM. Bupivacaine-loaded comatrix formed by albumin microspheres included in a poly(lactide-co-glycolide) film: in vivo biocompatibility and drug release studies. Biomaterials. 1999;20(20):1919–1924. | ||
Chen P-C, Park YJ, Chang L-C, et al. Injectable microparticle-gel system for prolonged and localized lidocaine release. I. In vitro characterization. J Biomed Mater Res A. 2004;704(3):412–419. | ||
Curley J, Castillo J, Hotz J, et al. Prolonged regional nerve blockade. Injectable biodegradable bupivacaine/polyester microspheres. Anesthesiology. 1996;84(6):1401–1410. | ||
Kohane DS, Smith SE, Louis DN, et al. Prolonged duration local anesthesia from tetrodotoxin-enhanced local anesthetic microspheres. Pain. 2003;104(1–2):415–421. | ||
Huang YY, Chung TW, Tzeng TW. A method using biodegradable polylactides/polyethylene glycol for drug release with reduced initial burst. Int J Pharm. 1999;182(1):93–100. | ||
Chen P-C, Kohane DS, Park YJ, Bartlett RH, Langer R, Yang VC. Injectable microparticle-gel system for prolonged and localized lidocaine release. II. In vivo anesthetic effects. J Biomed Mater Res A. 2004;70(3):459–466. | ||
Chung TW, Huang YY, Liu YZ. Effects of the rate of solvent evaporation on the characteristics of drug loaded PLLA and PDLLA microspheres. Int J Pharm. 2001;212(2):161–169. | ||
Zhang H, Lu Y, Zhang G, Gao S, Sun D, Zhong Y. Bupivacaine-loaded biodegradable poly(lactic-co-glycolic) acid microspheres. I. Optimization of the drug incorporation into the polymer matrix and modelling of drug release. Int J Pharm. 2008;351(1–2):244–249. | ||
McAlvin JB, Reznor G, Shankarappa SA, Stefanescu CF, Kohane DS. Local toxicity from local anesthetic polymeric microparticles. Anesth Analg. 2013;116(4):794–803. | ||
Huang YY, Chung TW, Tzeng TW. Drug release from PLA/PEG microparticulates. Int J Pharm. 1997;156(1):9–15. | ||
Bragagni M, Beneitez C, Martín C, De La Ossa DHP, Mura PA, Gil-Alegre ME. Selection of PLA polymers for the development of injectable prilocaine controlled release microparticles: usefulness of thermal analysis. Int J Pharm. 2013;441(1–2):468–475. | ||
Pedersen JL, Lillesø J, Hammer NA, et al. Bupivacaine in microcapsules prolongs analgesia after subcutaneous infiltration in humans: a dose-finding study. Anesth Analg. 2004;99(3):912–918. | ||
Le Corre P, Estèbe JP, Clément R, et al. Spray-dryed bupivacaine-loaded microspheres: in vitro evaluation and biopharmaceutics of bupivacaine following brachial plexus administration in sheep. Int J Pharm. 2002;238(1–2):191–203. | ||
Estebe JP, Le Corre P, Du Plessis L, et al. The pharmacokinetics and pharmacodynamics of bupivacaine-loaded microspheres on a brachial plexus block model in sheep. Anesth Analg. 2001;93(2):447–455, 4th contents page. | ||
Montanari L, Cilurzo F, Selmin F, et al. Poly(lactide-co-glycolide) microspheres containing bupivacaine: comparison between gamma and beta irradiation effects. J Control Release. 2003;90(3):281–290. | ||
Yin W, Yates MZ. Encapsulation and sustained release from biodegradable microcapsules made by emulsification/freeze drying and spray/freeze drying. J Colloid Interface Sci. 2009;336(1):155–161. | ||
Holgado MA, Arias JL, Cózar MJ, Alvarez-Fuentes J, Gañán-Calvo AM, Fernández-Arévalo M. Synthesis of lidocaine-loaded PLGA microparticles by flow focusing. Effects on drug loading and release properties. Int J Pharm. 2008;358(1–2):27–35. | ||
Xu Q, Hashimoto M, Dang TT, et al. Preparation of monodisperse biodegradable polymer microparticles using a microfluidic flow-focusing device for controlled drug delivery. Small. 2009;5(13):1575–1581. | ||
Vladisavljević GT, Shahmohamadi H, Das DB, Ekanem EE, Tauanov Z, Sharma L. Glass capillary microfluidics for production of monodispersed poly (DL-lactic acid) and polycaprolactone microparticles: experiments and numerical simulations. J Colloid Interface Sci. 2014;418:163–170. | ||
Klose D, Siepmann F, Willart JF, Descamps M, Siepmann J. Drug release from PLGA-based microparticles: effects of the “microparticle:bulk fluid” ratio. Int J Pharm. 2010;383(1–2):123–131. | ||
Kranz H, Bodmeier R. Structure formation and characterization of injectable drug loaded biodegradable devices: in situ implants versus in situ microparticles. Eur J Pharm Sci. 2008;34(2–3):164–172. | ||
Kranz H, Yilmaz E, Brazeau GA, Bodmeier R. In vitro and in vivo drug release from a novel in situ forming drug delivery system. Pharm Res. 2008;25(6):1347–1354. | ||
Kohane DS, Lipp M, Kinney RC, et al. Biocompatibility of lipid–protein–sugar particles containing bupivacaine in the epineurium. J Biomed Mater Res. 2002;59(3):450–459. | ||
Fundueanu G, Constantin M, Ascenzi P. Poly(N-isopropylacrylamide-co-acrylamide) cross-linked thermoresponsive microspheres obtained from preformed polymers: influence of the physico-chemical characteristics of drugs on their release profiles. Acta Biomater. 2009;5(1):363–373. | ||
Fundueanu G, Constantin M, Oanea I, Harabagiu V, Ascenzi P, Simionescu BC. Prediction of the appropriate size of drug molecules that could be released by a pulsatile mechanism from pH/thermoresponsive microspheres obtained from preformed polymers. Acta Biomater. 2012;8(3):1281–1289. | ||
Fundueanu G, Constantin M, Asmarandei I, Harabagiu V, Ascenzi P, Simionescu BC. The thermosensitivity of pH/thermoresponsive microspheres activated by the electrostatic interaction of pH-sensitive units with a bioactive compound. J Biomed Mater Res – Part A. 2013;101(6):1661–1669. | ||
Sivakumaran D, Maitland D, Hoare T. Injectable microgel-hydrogel composites for prolonged small-molecule drug delivery. Biomacromolecules. 2011;12(11):4112–4120. | ||
Patenaude M, Hoare T. Injectable, mixed natural-synthetic polymer hydrogels with modular properties. Biomacromolecules. 2012;13(2):369–378. | ||
Sivakumaran D, Maitland D, Oszustowicz T, Hoare T. Tuning drug release from smart microgel-hydrogel composites via cross-linking. J Colloid Interface Sci. 2013;392(1):422–430. | ||
Ganguly S, Dash AK. A novel in situ gel for sustained drug delivery and targeting. Int J Pharm. 2004;276(1–2):83–92. | ||
Foley PL, Ulery BD, Kan HM, et al. A chitosan thermogel for delivery of ropivacaine in regional musculoskeletal anesthesia. Biomaterials. 2013;34(10):2539–2546. | ||
Radin S, Chen T, Ducheyne P. The controlled release of drugs from emulsified, sol gel processed silica microspheres. Biomaterials. 2009;30(5):850–858. | ||
Costache MC, Vaughan AD, Qu H, Ducheyne P, Devore DI. Tyrosine-derived polycarbonate-silica xerogel nanocomposites for controlled drug delivery. Acta Biomater. 2013;9(5):6544–6552. | ||
Cui DC, Lu WL, Sa EA, Gu MJ, Lu XJ, Fan TY. Poly(acrylic acid) microspheres loaded with lidocaine: preparation and characterization for arterial embolization. Int J Pharm. 2012;436(1–2):527–535. | ||
Gautier H, Chamblain V, Weiss P, Merle C, Bouler JM. In vitro characterisation of calcium phosphate biomaterials loaded with lidocaine hydrochloride and morphine hydrochloride. J Mater Sci Mater Med. 2010;21(12):3141–3150. | ||
Verron E, Gauthier O, Janvier P, et al. Analgesic properties of calcium phosphate apatite loaded with bupivacaine on postoperative pain. J Biomed Mater Res – Part B Appl Biomater. 2010;94(1):89–96. | ||
Mendes LP, Delgado JMF, Costa AD, et al. Biodegradable nanoparticles designed for drug delivery: the number of nanoparticles impacts on cytotoxicity. Toxicol Vitr. 2015;29:1268–1274. | ||
Shang L, Nienhaus K, Nienhaus GU. Engineered nanoparticles interacting with cells: size matters. J Nanobiotechnology. 2014;12(1):5. | ||
Verma A, Stellacci F. Effect of surface properties on nanoparticle-cell interactions. Small. 2010;6(1):12–21. | ||
De Paula E, Cereda CMS, Tofoli GR, Franz-Montan M, Fraceto LF, de Araújo DR. Drug delivery systems for local anesthetics. Recent Pat Drug Deliv Formul. 2010;4(1):23–34. | ||
Weiniger CF, Golovanevski M, Sokolsky-Papkov M, Domb AJ. Review of prolonged local anesthetic action. Expert Opin Drug Deliv. 2010;7(6):737–752. | ||
Junghanns JUAH, Müller RH. Nanocrystal technology, drug delivery and clinical applications. Int J Nanomedicine. 2008;3(3):295–309. | ||
Rangasamy M. Nano technology : a review. J Appl Pharm Sci. 2011;01(02):8–16. | ||
Morey TE, Varshney M, Flint JA, Rajasekaran S, Shah DO, Dennis DM. Treatment of local anesthetic-induced cardiotoxicity using drug scavenging nanoparticles. Nano Lett. 2004;4(4):757–759. | ||
Cohen R, Kanaan H, Grant GJ, Barenholz Y. Prolonged analgesia from Bupisome and Bupigel formulations: from design and fabrication to improved stability. J Control Release. 2012;160(2):346–352. | ||
Fernandes Fraceto L, De Matos Alves Pinto L, Franzoni L, et al. Spectroscopic evidence for a preferential location of lidocaine inside phospholipid bilayers. Biophys Chem. 2002;99(3):229–243. | ||
Grant GJ, Piskoun B, Bansinath M. Analgesic duration and kinetics of liposomal bupivacaine after subcutaneous injection in mice. Clin Exp Pharmacol Physiol. 2003;30(12):966–968. | ||
Grant GJ, Barenholz Y, Bolotin EM, et al. A novel liposomal bupivacaine formulation to produce ultralong-acting analgesia. Anesthesiology. 2004;101(1):133–137. | ||
Cabeça LF, Fernandes SA, de Paula E, Marsaioli AJ. Topology of a ternary complex (proparacaine-beta-cyclodextrin-liposome) by STD NMR. Magn Reson Chem. 2008;46(9):832–837. | ||
De Araujo DR, Cereda CMS, Brunetto GB, et al. Pharmacological and local toxicity studies of a liposomal formulation for the novel local anaesthetic ropivacaine. J Pharm Pharmacol. 2008;60(11):1449–1457. | ||
De Araujo DR, Cereda CMS, Brunetto GB, Pinto LM, Santana MH, de Paula E. Encapsulation of mepivacaine prolongs the analgesia provided by sciatic nerve blockade in mice. Can J Anaesth. 2004;51(6):566–572. | ||
Toongsuwan S, Li LC, Erickson BK, Chang HC. Formulation and characterization of bupivacaine lipospheres. Int J Pharm. 2004;280(1–2):57–65. | ||
Kohane DS, Lipp M, Kinney RC, Lotan N, Langer R. Sciatic nerve blockade with lipid-protein-sugar particles containing bupivacaine. Pharm Res. 2000;17(10):1243–1249. | ||
Colombo G, Padera R, Langer R, Kohane DS. Prolonged duration local anesthesia with lipid-protein-sugar particles containing bupivacaine and dexamethasone. J Biomed Mater Res – Part A. 2005;75(2):458–464. | ||
Zur Mühlen A, Schwarz C, Mehnert W. Solid lipid nanoparticles (SLN) for controlled drug delivery – drug release and release mechanism. Eur J Pharm Biopharm. 1998;45(2):149–155. | ||
Schwarz C, Mehnert W. Freeze-drying of drug-free and drug-loaded solid lipid nanoparticles (SLN). Int J Pharm. 1997;157(2):171–179. | ||
Pietkiewicz J, Sznitowska M, Placzek M. The expulsion of lipophilic drugs from the cores of solid lipid microspheres in diluted suspensions and in concentrates. Int J Pharm. 2006;310(1–2):64–71. | ||
Görner T, Gref R, Michenot D, Sommer F, Tran MN, Dellacherie E. Lidocaine-loaded biodegradable nanospheres. I. Optimization of the drug incorporation into the polymer matrix. J Control Release. 1999;57(3):259–268. | ||
Polakovic M, Gorner T, Gref R, Dellacherie E. Lidocaine-loaded biodegradable nanospheres. II. Modeling of drug release. J Control Release. 1999;60(2–3):169–177. | ||
Moraes CM, De Matos AP, De Lima R, Rosa AH, De Paula E, Fraceto LF. Initial development and characterization of PLGA nanospheres containing ropivacaine. J Biol Phys. 2007;33(5–6):455–461. | ||
Ramos Campos EV, Silva de Melo NF, Guilherme VA, et al. Preparation and characterization of poly(ε-caprolactone) nanospheres containing the local anesthetic lidocaine. J Pharm Sci. 2013;102(1):215–226. | ||
Govender T, Riley T, Ehtezazi T, et al. Defining the drug incorporation properties of PLA-PEG nanoparticles. Int J Pharm. 2000;199(1):95–110. | ||
Bilati U, Allémann E, Doelker E. Development of a nanoprecipitation method intended for the entrapment of hydrophilic drugs into nanoparticles. Eur J Pharm Sci. 2005;24(1):67–75. | ||
Govender T, Stolnik S, Garnett MC, Illum L, Davis SS. PLGA nanoparticles prepared by nanoprecipitation: drug loading and release studies of a water soluble drug. J Control Release. 1999;57(2):171–185. | ||
Moraes CM, De Lima R, Rosa AH, De Paula E, Fraceto LF. Encapsulation of local anesthetic bupivacaine in biodegradable poly(DL-lactide-co-glycolide) nanospheres: factorial design, characterization and cytotoxicity studies. Macromol Symp. 2009;281(1):106–112. | ||
Moraes CM, De Paula E, Rosa AH, Fraceto LF. Physicochemical stability of poly(lactide-co-glycolide) nanocapsules containing the local anesthetic bupivacaine. J Braz Chem Soc. 2010;21(6):995–1000. | ||
Garcia X, Escribano E, Domenech J, Queralt J, Freixes J. In vitro characterization and in vivo analgesic and anti-allodynic activity of PLGA-bupivacaine nanoparticles. J Nanoparticle Res. 2011;13(5):2213–2223. | ||
Moraes CM, de Matos AP, de Paula E, Rosa AH, Fraceto LF. Benzocaine loaded biodegradable poly-(d,l-lactide-co-glycolide) nanocapsules: factorial design and characterization. Mater Sci Eng B Solid-State Mater Adv Technol. 2009;165(3):243–246. | ||
Silva de Melo NF, de Araújo DR, Grillo R, et al. Benzocaine-loaded polymeric nanocapsules: study of the anesthetic activities. J Pharm Sci. 2012;101(3):1157–1165. | ||
De Melo NFS, Grillo R, Guilherme VA, et al. Poly(lactide-co-glycolide) nanocapsules containing benzocaine: influence of the composition of the oily nucleus on physico-chemical properties and anesthetic activity. Pharm Res. 2011;28(8):1984–1994. | ||
Weldon CB, Tsui JH, Shankarappa SA, et al. Electrospun drug-eluting sutures for local anesthesia. J Control Release. 2012;161(3):903–909. | ||
Mocanu A, Pasca RD, Tomoaia G, et al. New procedure to synthesize silver nanoparticles and their interaction with local anesthetics. Int J Nanomedicine. 2013;8:3867–3874. | ||
Tan JPK, Goh CH, Tam KC. Comparative drug release studies of two cationic drugs from pH-responsive nanogels. Eur J Pharm Sci. 2007;32(4–5):340–348. | ||
Tan JPK, Zeng AQ, Chang CC, Tam KC. Release kinetics of procaine hydrochloride (PrHy) from pH-responsive nanogels: theory and experiments. Int J Pharm. 2008;357(1–2):305–313. | ||
Tan JPK, Wang Q, Tam KC. Control of burst release from nanogels via layer by layer assembly. J Control Release. 2008;128(3):248–254. | ||
Hoare T, Young S, Lawlor MW, Kohane DS. Thermoresponsive nanogels for prolonged duration local anesthesia. Acta Biomater. 2012;8(10):3596–3605. | ||
Hoare T, Sivakumaran D, Stefanescu CF, Lawlor MW, Kohane DS. Nanogel scavengers for drugs: local anesthetic uptake by thermoresponsive nanogels. Acta Biomater. 2012;8(4):1450–1458. | ||
Yaghmur A, Rappolt M, Oøstergaard J, Larsen C, Larsen SW. Characterization of bupivacaine-loaded formulations based on liquid crystalline phases and microemulsions: the effect of lipid composition. Langmuir. 2012;28(5):2881–2889. | ||
Grillo R, de Melo NFS, de Araújo DR, de Paula E, Rosa AH, Fraceto LF. Polymeric alginate nanoparticles containing the local anesthetic bupivacaine. J Drug Target. 2010;18(9):688–699. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.