")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 14
Frequency of inborn errors of metabolism screening for children with unexplained acute encephalopathy at an emergency department
Authors Abdel Maksoud M, ELsayed SM, Shatla RH, Imam AA , Elsayed RM , Mosabah AA, Sherif AM
Received 18 February 2018
Accepted for publication 23 March 2018
Published 29 June 2018 Volume 2018:14 Pages 1715—1720
DOI https://doi.org/10.2147/NDT.S165833
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Mamdouh Abdel Maksoud,1 Solaf Mohamed ELsayed,2 Rania H Shatla,1 Abdulbasit Abdulhalim Imam,3 Riad M Elsayed,4 Amira AA Mosabah,5 Ashraf M Sherif5
1Pediatric Department, Ain Shams University, Cairo, Egypt; 2Genetics Department, Ain Shams University, Cairo, Egypt; 3Department of Pediatrics, Al-Azhar Faculty of Medicine-Girls, Cairo, Egypt; 4Pediatric Neurology Unit, Pediatric Department, Mansoura University, Mansoura, Dakahlia, Egypt; 5Department of Pediatrics, Cairo University, Cairo, Egypt
Objective: Our study aimed to estimate the frequency of inborn errors of metabolism (IEMs) in patients presenting with acute encephalopathy-like picture at an emergency department (ED).
Subjects and methods: Our study was a prospective observational study conducted on 30 patients admitted to the pediatric ED with unexplained acute encephalopathy. The study included 30 children with an age ranging from 1 month to 5 years. All patients were subjected to full history taking, thorough clinical examination, and laboratory investigations including serum ammonia, serum lactate, arterial blood gases, tandem mass spectroscopy, organic acid of urine, cerebrospinal fluid examination to exclude central nervous system infection plus the routine laboratory tests (kidney functions, liver functions, random blood glucose, complete blood picture), and brain imaging computed tomography and/or magnetic resonance imaging brain.
Results: Thirty children presented with acute encephalopathy at the ED. All were screened for suspected IEMs. Ten (33.3%) of them was positive in the initial screening test. There were four (13.3%) patients with possible mitochondrial diseases, four (13.3%) patients with possible organic acidemia, one (3.3%) patient with possible urea cycle defect, and one (3.3%) patient with possible nonketotic hyperglycinemia.
Conclusion: Any case of unexplained acute encephalopathy presenting to the ED should be investigated for suspected IEM, especially in high-risk families, as early interventions will lead to improved outcome.
Keywords: inborn errors of metabolism, children, acute encephalopathy, emergency department, comatose child, metabolic screen
Background
Acute encephalopathy is a relatively common problem; one of its causes is metabolic disorders.1 Acute metabolic encephalopathy is a condition of acute global cerebral dysfunction resulting in altered consciousness, behavior changes, or seizures that is not due to primary structural brain disease (eg, tumor or hemorrhage) or infection.2 Acute encephalopathy (regardless of the cause) is a medical emergency. In addition to being a common manifestation of a variety of acquired medical or surgical conditions, it is a presenting feature of number of inherited metabolic diseases, particularly in young children.3,4 We should suspect an inborn error of metabolism (IEM) in patients with neurologic abnormalities (eg, developmental delay, hypotonic baby, or feeding difficulties), especially in those patients with multisystem involvement who appear with acute symptoms.5 The early and specific diagnosis of IEMs and prompt initiation of appropriate therapy are still the best determinants of outcome for these patients.6 Delay in diagnosis may have serious consequences resulting in acute metabolic decompensation, progressive neurologic injury, or death.7
The aim of the study was to estimate the frequency of IEM in children presenting with unexplained acute encephalopathy at the pediatric emergency department (ED).
Subjects and methods
The study was a prospective observational cohort study. It was carried out at the ED in Ghamra Military Hospital, Egypt, during the period from November 2014 to December 2015. The study was conducted on 30 patients (18 males and 12 females), with age ranging from 1 month to 5 years. All patients presented with disturbed level of consciousness defined as a state of unconsciousness lasting >6 hours in which a person cannot be awakened, fails to respond normally on painful stimuli, light, or sound, lacks a normal sleep–wake cycle, and does not initiate voluntary actions.8 Patients with encephalopathy due to head trauma, central nervous system (CNS) infection, renal or hepatic impairment, and those with encephalopathy due to hypoxic–ischemic insult were excluded from the study.
All included patients were subjected to full history talking and clinical examination, and the following laboratory tests.
Laboratory tests
Routine laboratory tests
The following laboratory tests were conducted: complete blood count, blood gas analysis, kidney function and liver functions tests, serum electrolytes, random blood glucose, serum ammonia, serum lactate, and cerebrospinal fluid examination to rule out CNS infection.
Metabolic screen
All children included in the study were further investigated by tandem mass spectrometry (MS/MS). MS/MS technology expands the metabolic disorder screening panel (ie, the number of disorders that can be detected) by incorporating an acylcarnitine profile, which enables detection of fatty acid oxidation disorders (eg, medium-chain acyl-CoA dehydrogenase deficiency) and other organic acid disorders. MS/MS can reliably analyze ~20 metabolites in one short-duration run (ie, ~2 minutes) and provide a comprehensive assessment from a single blood spot specimen.9,10
Urine organic acid analysis was performed using gas chromatography–mass spectrometry in all patients.11
Neuroimaging was done for all cases with either computed tomography and/or magnetic resonance imaging of the brain to rule out intracranial pathology or trauma.
The mothers of the subjects under study were informed about the purpose of the study and the plan of work before they agreed to participate. A written consent was obtained before the study began.
The study was approved by the local ethics committee of Ain Shams University.
Statistical analysis was done using manual methods to calculate the percentage of the obtained data of the patients.
Results
Demographic and clinical data of the patients
In this study, there were 30 patients diagnosed with acute unexplained encephalopathy who presented at the ER of our hospital, with their ages ranging from 1 month to 5 years. Infants of age <1 year numbered 11 (37%) and children aged from 1 to 5 years numbered 19 (63%); the number of males was 60. Patients with positive consanguinity numbered 19 (63%), and 5 (17%) patients had a positive family history. All patients included in our study had disturbed conscious level; seizures were recorded in 63%, delayed motor development was present in 33%, hypotonia in 20%, mental retardation in 13%, hypertonia and spasticity in 10%, muscle weakness in 10%, gait abnormalities in 7%, microcephaly in 7%, and speech abnormalities were present in 3% of the patients. Forty percent of our patients had failure to thrive and chronic vomiting. Hepatomegaly and short stature were detected in 30% of cases. Chronic diarrhea was reported in 20%, hypoglycemia in 10%, and only 7% had cardiac manifestation and ophthalmic manifestation (Tables 1 and 2).
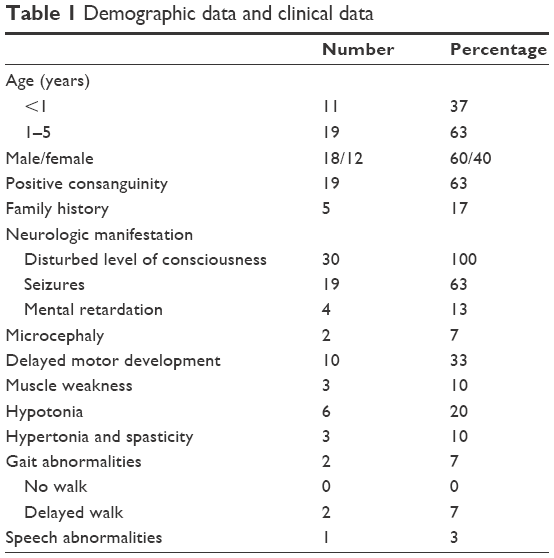 | Table 1 Demographic data and clinical data |
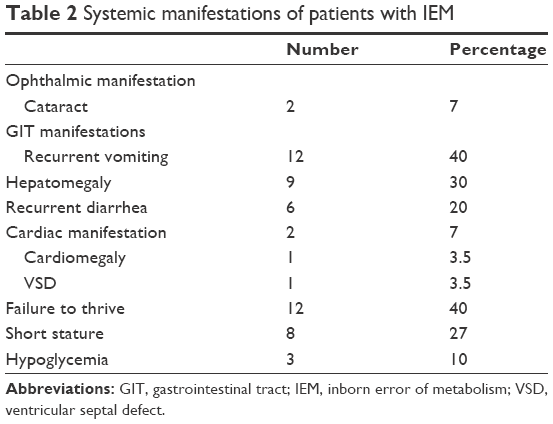 | Table 2 Systemic manifestations of patients with IEM |
Plasma amniography
Plasma amino acid analysis results showed absolute alanine elevation >450 μmol in 12 patients (40%) and co-carnitine abnormalities in 6 patients (20%); 2 of them had a low concentration and 4 had a high concentration. Glycine was elevated in four patients (13%), alanine in two patients (7%), phenylalanine in only one patient (3%), and low borderline citrulline was found in one patient (Table 3).
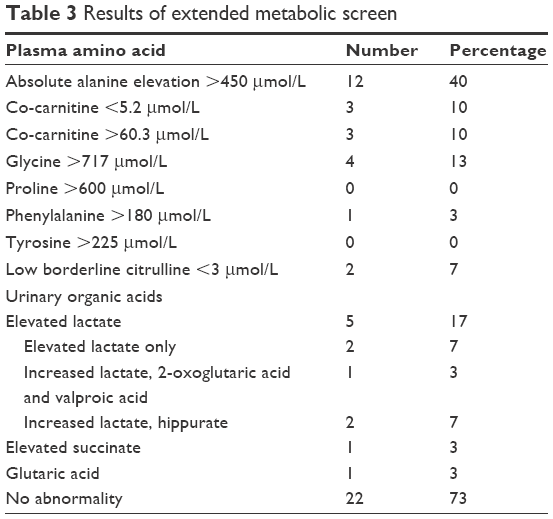 | Table 3 Results of extended metabolic screen |
Urinary organic acid analysis in the studied patients
There was elevated lactate in five patients (17%), increased lactate, 2-oxoglutaric acid, and valproic acid in one patient (3%), elevated lactate alone in two patients (7%), elevated succinate in one patient (3%), increased lactate and hippurate in two patients (7%), and elevated glutaric acid in one patient (3%), as shown in Table 3.
Radiographic abnormalities
Computed tomography brain was available in 25 patients (83%), of which 17 patients (68%) had abnormalities. This included 11 patients with cerebral atrophy, 3 patients with brain edema, and 2 patients with basal ganglia abnormalities. Magnetic resonance imaging was done for 27 patients, revealing atrophic changes in 12 patients, basal ganglia calcification in 2 patients, and white matter abnormalities in 5 patients. It is worth noting that two patients had cataract (7%) and echocardiography abnormalities were detected in two patients (7%); one of them had cardiomegaly and the other had ventricular septal defect (VSD). Abdominal ultrasound abnormalities were observed in 12 patients (40%); 30% had hepatomegaly and 10% had hepatosplenomegaly (Table 4).
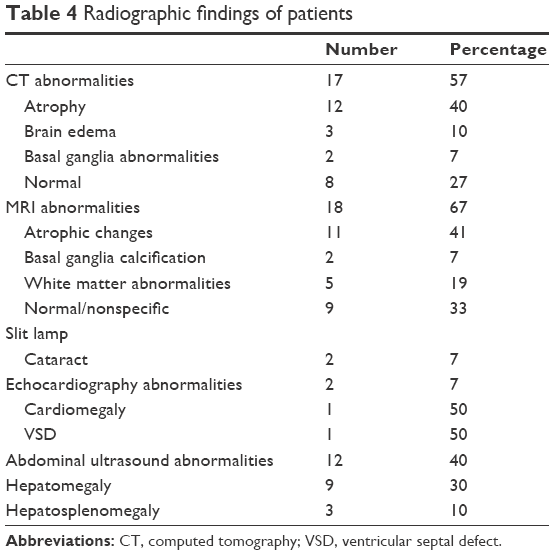 | Table 4 Radiographic findings of patients |
Discussion
Acute encephalopathy is a common and potential medical emergency in patients with IEM.3 Metabolic disorders or IEMs result from a block (partial or complete) in a pathway in the body’s metabolism. There are a large number of conditions included in this group of disorders. Although metabolic disorders are individually rare, collectively, they are not uncommon.11
In the current study, 30 patients presented with unexplained acute encephalopathy at the ED of our hospital, with their ages ranges from 1 month up to 5 years. All children were subjected to metabolic screen for possible IEM, and we found that 13% of patients had possible mitochondrial dysfunction (MD). Recent methods used for establishing diagnosis of MD, such as whole-exome sequencing and next-generation sequencing, based on massively parallel sequencing of the entire 16, mitochondrial genome which allow for diagnosis of MD with high sensitivity.12–14 In our study, the confirmatory tests were not done at the time of study as these tests were not available in our country and due to financial issues. The most common clinical presentation in our patients was seizures in three patients, followed by delayed motor development in three patients. Similarly, previous studies on MD noted that the neurologic manifestations were the most common presenting manifestation (45%–90%) with abnormal tone, seizures, and delayed motor development being the most frequent presentations.15,16
In our study, we found that four patients had failure to thrive. This is in agreement with Debray et al,15 who reported failure to thrive as a frequent observation in 52% of the cases and a presenting symptom in 10% of the patients. The combination of manifestations such as poor feeding and vomiting associated with persistent lactic acidosis could explain failure to thrive that commonly occurs in mitochondrial diseases.17
In regard to neuroimaging, we had two patients with atrophic changes, one patient with basal ganglia abnormalities, and another one had white matter abnormal signals. Other studies have reported brain atrophic changes in 47% of the studied patients, white matter abnormalities in 40%,14 and basal ganglia hyperintensities in 60% of the patients.18
In our study, four patients had highly suggestive symptoms of organic acidemia; three of them had possibly malonic acidemia and one patient had glutaric aciduria type 1.
Metabolic acidosis and increased anion gap are the characteristic findings of organic acidemias.19,20 This is in agreement with our findings of increased lactate in four patients and increased anion gap in three patients. Plasma lactate is often elevated in organic acidemias as a result of secondary interference with coenzyme A metabolism. In our study, only one patient was diagnosed as glutaric aciduria type 1 patient who presented at age 7 months; there was positive consanguinity, and this is similar to Alkan et al,21 who reported that in most cases of glutaric aciduria type 1, the first episode occurs between the ages of 6 and 18 months. Delayed motor development, persistent vomiting, and failure to thrive were the presenting features, which was similar to those reported by Zafeiriou et al.22
We had a patient possibly diagnosed with nonketotic hyperglycinemia who presented at age 3 months. This is similar to Dinopoulos et al,23 who reported that patients may present in the neonatal period, infancy, or later. Our patient had convulsion and repeated vomiting similar to a study reporting that patients with nonketotic hyperglycinemia develop lethargy, refusal of feeds, hypotonia, hiccups, and rapid progression of the neurologic symptoms leading to seizures and coma.24 In the current study, we found one patient with urea cycle defect, who was aged 2 months, second in the order of birth, and with a positive family history of sibling death. The patient presented with disturbed conscious level (DCL), neurologic manifestation, failure to thrive, and hypoglycemia. This is in agreement with Saudubray et al, who showed 55% of patients with urea cycle defect present in neonatal period and 45% present in infancy; they present with digestive problems and then develop neurologic symptoms.25
In our patients, we observed family history of a similar condition in 67% of our studied cases and positive consanguinity in 72% of our cases. Parental consanguinity increases the likelihood of autosomal recessive IEM because relatives are more likely to carry the same defective gene. Certain IEMs are more prevalent in particular ethnic or religious groups and negative family history does not rule out IEMs because most carriers have no clinical manifestations of the disease.
In our study, neurologic manifestations were common. Seizures were observed in 78% of patients, followed by delayed motor development in 28%, hypotonia in 17%, and mental retardation in 6% of patients. This is in accordance with Dott et al,26 who diagnosed 212 patients with IEM. They were generally characterized by convulsion, coma, mental retardation, developmental delay, and abnormal muscular tone. Also, Gulati et al27 reported that most of the IEMs usually present with CNS symptoms. Seizures were a dominant symptom (26%), followed by delayed milestones (18%). Also, Kamate et al28 showed that in patients with IEM, vomiting was observed in 9.4% and organomegaly in 2.3%. We observed cardiac symptoms in one patient in the form of cardiomegaly; this is similar to Han et al, who observed cardiac manifestation in 7%, including heart failure and congenital heart diseases.29
In our study, two patients had cataract. Our findings are nearly similar to Nissenkorn et al, who reported eye involvement was observed in 26% of the patients.30
In our study, we found that about 12 patients had serum ammonia above 48 μmol/L and 7 patients had serum lactate above 20 mg/dL or 2.2 mmol/L. There was an increase in liver enzymes (alanine transaminase and aspartate transaminase) in 5 patients and 11 patients with metabolic acidosis, respectively; this is similar to Debray et al,15 who noted that 72% of the patients had chronic lactic acidemia. Additionally, the study conducted by Touati et al31 reported that 89% of the patients had chronic lactic acidemia and up to 60% had at least one normal lactate concentration, illustrating the variability of this biological parameter and emphasizing that a normal lactate concentration does not exclude IEM. In Cheng et al’s study, 43 patients with IEMs had increased serum ammonia levels and 35% had increased serum lactate; the liver enzymes alanine transaminase and aspartate transaminase were increased in 19% and 42% of patients with metabolic acidosis, respectivley.32
Conclusion
Any case of unexplained acute encephalopathy presenting to the ED should be investigated for IEMs as early diagnosis and treatment will impact the outcome. The challenges to recognize them, as institution of appropriate management can be life-saving. A high index of clinical suspicion (parental history – consanguineous parents, previous unexplained neonatal deaths or Sudden Infant Death Syndrome, relatives with undiagnosed “syndrome”) is required to make the diagnosis as the clinical presentation of most metabolic disorders is nonspecific. However, inconclusive laboratory results do not exclude IEMs, but further investigations are needed to confirm the diagnosis.
Disclosure
The authors report no conflicts of interest in this work.
References
Surtees R, Leonard JV. Acute metabolic encephalopathy: a review of causes, mechanisms and treatment. J Inherit Metab Dis. 1989;12(Suppl 1):42–54. | ||
Parke JT. Acute encephalopathies. In: Mcmillan JA, Feigin RD, DeAngelis C, Jones MD, editors. Oski’s Pediatrics. Principles and Practice, 4th ed. Philadelphia: Lippincott, Williams & Wilkins; 2006:2258. | ||
Leonard JV. Acute metabolic encephalopathy: an introduction. J Inherit Metab Dis. 2005;28:403–406. | ||
Fujinami A, Murayama K, Takayanagi M. [Acute encephalopathy in inherited metabolic diseases]. Nihon Rinsho. 2011;69(3):477–482. Japanese. | ||
Calvo M, Artuch R, Macià E, et al. Diagnostic approach to inborn errors of metabolism in an emergency unit. Pediatr Emerg Care. 2000;16:405–408. | ||
Raghuveer T, Garg U, Graf WD. Inborn errors of metabolism in infancy and early childhood: an update. Am Fam Physician. 2006;73:1981–1990. | ||
CDC. Using tandem mass spectrometry for metabolic disease screening among newborns. A report of a work group. MMWR Recomm Rep. 2001;50(RR-3):1–34. | ||
Weyhenmyeye JA, Gallman EA. Rapid Review Neuroscience. 1st ed. Illinois, USA: Mosby Elsevier; 2007:177–179. | ||
Garg U, Dasouki M. Expanded newborn screening of inherited metabolic disorders by tandem mass spectrometry: clinical and laboratory aspects. Clin Biochem. 2006;39:315–332. | ||
Kuhara T. Diagnosis of inborn errors of metabolism using filter paper urine, urease treatment, isotope dilution and gas chromatography-mass spectrometry. J Chromatogr B Biomed Sci Appl. 2001;758:3–25. | ||
Fletcher JM. Metabolic emergencies and the emergency physician. J Paediatr Child Health. 2016;52:227–230. | ||
Wong LJ. Next generation molecular diagnosis of mitochondrial disorders. Mitochondrion. 2013;13:379–387. | ||
Pronicka E, Piekutowska-Abramczuk D, Ciara E, et al. New perspective in diagnostics of mitochondrial disorders: two years’ experience with whole-exome sequencing at a national paediatric centre. J Transl Med. 2016;14:174. | ||
Palculict ME, Zhang VW, Wong LJ, Wang J. Comprehensive mitochondrial genome analysis by massively parallel sequencing. Methods Mol Biol. 2016;1351:3–17. | ||
Debray F, Lambert M, Chevalier I, et al. Long-term outcome and clinical spectrum of 73 pediatric patients with mitochondrial diseases. Pediatrics. 2007;119:722–733. | ||
Sharrard M. Clinical presentation of mitochondrial diseases in children with progressive intellectual and neurological deterioration. Dev Med Child Neurol. 2010;52:407–408. | ||
Gibson K, Halliday JL, Kirby DM, Yaplito-Lee J, Thorburn DR, Boneh A. Mitochondrial oxidative phosphorylation disorders presenting in neonates: clinical manifestations and enzymatic and molecular diagnoses. Pediatrics. 2008;122:1003–1008. | ||
Verity CM, Winstone AM, Stellitano L, Krishnakumar D, Will R, McFarland R. The clinical presentation of mitochondrial diseases in children with progressive intellectual and neurological deterioration: a national, prospective, population-based study. Dev Med Child Neurol. 2010;52:434–440. | ||
Wappner RS. Biochemical diagnosis of genetic diseases. Pediatr Ann. 1993;22:282–292, 295–297. | ||
Lindor NM, Karnes PS. Initial assessment of infants and children with suspected inborn errors of metabolism. Mayo Clin Proc. 1995;70:987–988. | ||
Alkan A, Baysal T, Yakinci C, Sigirci A, Kutlu R. Glutaric aciduria type I diagnosed after poliovirus immunization: magnetic resonance findings. Pediatr Neurol. 2002;26:405–407. | ||
Zafeiriou DI, Zschocke J, Augoustidou-Savvopoulou P, et al. Atypical and variable clinical presentation of glutaric aciduria type I. Neuropediatrics. 2000;31:303–306. | ||
Dinopoulos A, Cecil KM, Schapiro MB, et al. Brain MRI and proton MRS findings in infants and children with respiratory chain defects. Neuropediatrics. 2005;36:290–301. | ||
Hoover-Fong JE, Shah S, Van Hove JL, Applegarth D, Toone J, Hamosh A. Natural history of nonketotic hyperglycinemia in 65 patients. Neurology. 2004;63:1847–1853. | ||
Saudubray JM, Nassogne MC, de Lonlay P, Touati G. Clinical approach to inherited metabolic disorders in neonates: an overview. Semin Neonatol. 2002;7:3–15. | ||
Dott M, Chace D, Fierro M, et al. Metabolic disorders detectable by tandem mass spectrometry and unexpected early childhood mortality: a population-based study. Am J Med Genet A. 2006;140:837–842. | ||
Gulati S, Vaswani M, Kalra V, Kabra M, Kaur M. An approach to neurometabolic disorders by a simple metabolic screen. Indian Pediatr. 2000;37:63–69. | ||
Kamate M, Chetal V, Kulgod V, Patil V, Christopher R. Profile of Inborn errors of metabolism in a tertiary care centre PICU. Indian J Pediatr. 2010;77:57–60. | ||
Han L, Ye J, Qiu W, Gao X, Wang Y, Gu X. Selective screening for inborn errors of metabolism on clinical patients using tandem mass spectrometry in China: A four-year report. J Inherit Metab Dis. 2007;30:507–514. | ||
Nissenkorn A, Zeharia A, Lev D, et al. Neurologic presentations of mitochondrial disorders. J Child Neurol. 2000;15:44–48. | ||
Touati G, Rigal O, Lombès A, Frachon P, Giraud M, Ogier de Baulny H. In vivo functional investigations of lactic acid in patients with respiratory chain disorders. Arch Dis Child. 1997;76:16–21. | ||
Cheng KH, Liu MY, Kao CH, et al. Newborn screening for methylmalonic aciduria by tandem mass spectrometry: 7 years’ experience from two centers in Taiwan. J Chin Med Assoc. 2010;73:314–318. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.