")
Back to Journals » International Journal of General Medicine » Volume 14
Four Immune-Related Genes (FN1, UGCG, CHPF2 and THBS2) as Potential Diagnostic and Prognostic Biomarkers for Carbon Nanotube-Induced Mesothelioma
Authors Xie D, Hu J, Wu T, Cao K, Luo X
Received 11 June 2021
Accepted for publication 19 August 2021
Published 29 August 2021 Volume 2021:14 Pages 4987—5003
DOI https://doi.org/10.2147/IJGM.S324365
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Dongli Xie,1,* Jianchen Hu,1,* Tong Wu,2 Kangli Cao,3 Xiaogang Luo1
1College of Textile and Clothing Engineering, Soochow University, Suzhou, 215123, People’s Republic of China; 2Shanghai LEVSON Nanotechnology Co., Ltd, Shanghai, 200444, People’s Republic of China; 3Shanghai Institute of Spacecraft Equipment, Shanghai, 200240, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xiaogang Luo Tel +86 512-67162531
Email [email protected]
Background: Malignant pleural mesothelioma (MPM), a highly aggressive cancer, was mainly attributed to asbestos exposure. Carbon nanotubes (CNTs) share similar negative features to asbestos, provoking concerns about their contribution to MPM. This study was used to identify genes associated with CNT-induced MPM.
Methods: Microarray datasets were available in the Gene Expression Omnibus database. The limma method was used to identify differentially expressed genes (DEGs) in CNT-exposed MeT5A cells (GSE48855) or mice (GSE51636). Weighted correlation network analysis (WGCNA) and protein–protein interaction (PPI) network construction were conducted to screen hub DEGs. The mRNA expression levels of hub DEGs were validated on MPM samples of GSE51024, GSE2549 and GSE42977 datasets, and their diagnostic efficacy was determined by receiver operating characteristic curve analysis. The prognostic values of hub DEGs were assessed using online tools based on The Cancer Genome Atlas data. Their functions were annotated by Database for Annotation, Visualization and Integrated Discovery (DAVID) enrichment and correlation with immune cells and markers.
Results: WGCNA identified that two modules were associated with disease status. Thirty-one common DEGs in the GSE48855 and GSE51636 datasets were overlapped with the genes in these two modules. Twenty of them had a high degree centrality (≥ 4) in the PPI network. Four DEGs (FN1, fibronectin 1; UGCG, UDP-glucose ceramide glucosyltransferase; CHPF2, chondroitin polymerizing factor 2; and THBS2, thrombospondin 2) could predict the overall survival, and they were confirmed to be upregulated in MPM samples compared with controls. Also, they could effectively predict the MPM risk, with an overall accuracy of > 0.9. DAVID analysis revealed FN1, CHPF2 and THBS2 functioned in cell-ECM interactions; UGCG influenced glycosphingolipid metabolism. All genes were positively associated with infiltrating levels of immune cells (macrophages or dendritic cells) and the expression of the dendritic cell marker (NRP1, neuropilin 1).
Conclusion: These four immune-related genes represent potential biomarkers for monitoring CNT-induced MPM and predicting the prognosis.
Keywords: carbon nanotubes, malignant transformation, malignant pleural mesothelioma, tumor-infiltrating immune cells, prognosis, diagnosis
Introduction
Carbon nanotubes (CNTs) are one of the most frequently used engineered nanomaterials for industrial, consumer and biomedical applications1–5 because of their unique properties, such as lightweight, high tensile strength and electrical conductivity.6 However, CNTs share similar negative physical characteristics (eg needle-like structure, high aspect ratio, durability and biopersistence)7,8 and similar routes of exposure (eg inhalation) to asbestos fibers, which account for 88% of the population-attributable risk of malignant pleural mesothelioma (MPM).9,10 These similarities indicate that widespread exposure to CNTs may also have the potential to induce the development of asbestos-like MPM, especially in occupational workers. Furthermore, MPM has been demonstrated to be a highly aggressive cancer, with a 5-year survival rate of <10%.11 Therefore, it is of clinical significance to identify biomarkers for surveillance of occupational workers to prevent the onset of CNT-induced MPM.
Recent studies have used in vitro and in vivo models to explore the molecular mechanisms of CNT exposure for MPM. Lohcharoenkal et al found chronic exposure with single-walled CNTs (SWCNTs) and multi-walled CNTs (MWCNTs) induced a significant increase in cell growth, invasion and migration of non-tumorigenic human lung mesothelial MeT5A cells as compared to Survanta-treated control,12 during which the expression of matrix metallopeptidase 2 (MMP-2) gene was most strikingly upregulated, while knockdown of MMP-2 reduced the aggressive behaviors of CNT-transformed MeT5A cells.12 Huang et al reported that exposure to MWCNTs (0.1 μg/mL) for three months significantly increased the proliferation, migration, invasion and colony-forming abilities of MeT5A cells.13 Transcriptome sequencing, quantitative polymerase chain reaction (qPCR) and Western blot analyses revealed activation of the nuclear factor kappa B (NF-κB)/interleukin (IL)-6/signal transducer and activator of transcription 3 (STAT3) inflammatory pathway may be an important mechanism in MWCNT-induced malignant transformation of MeT5A cells; silencing of these pathway genes reversed the above malignant phenotypes of MeT5A cells.13 Macrophage co-culture13–15 and rat/mouse peritoneal cavity injection14,16 experiments demonstrated MWCNTs may induce the development of MPM by stimulating the macrophage accumulation, which then enhanced the production of NF-κB, IL-6, IL-1β, tumor necrosis factor-α (TNF-α) and STAT3 of mesothelial cells. In the study of Chernova et al, the expression levels of STAT3, IL-6 and phosphatidylinositol 3-kinase (PI3K) genes were identified to be increased, while cyclin dependent kinase inhibitor 2A (CDKN2A) was shown to be decreased in CNT-induced MPM lesions at 6 months post-injection.17 These findings suggest that the above-related genes may represent candidate biomarkers to predict the risk of CNT-induced MPM. However, the key molecules that drive CNT-associated MPM remain not well understood at present.
In this study, we attempted to identify promising biomarkers for CNT-induced MPM by a comprehensive analysis of transcriptome microarray data (GSE48855) of CNT-transformed MeT5A cells.12 Weighted gene co-expression network analysis (WGCNA)18 was used to cluster genes with similar expression patterns in the GSE48855 dataset to the same module and connect modules with the phenotypic trait. The expression levels of genes in crucial modules extracted from the GSE48855 dataset were further validated in the mRNA expression profile (GSE51636) of mice exposed to CNTs or not.17 Furthermore, the expression, predictive and prognostic abilities of hub genes in MPM samples were also explored using the datasets (GSE51024,19 GSE254920 and GSE4297721,22) from the Gene Expression Omnibus (GEO) database and several online tools based on The Cancer Genome Atlas (TCGA) dataset. The functions of hub genes were predicted by linking to the infiltration levels of various immune cells since inflammation seemed to be an important contributor for CNT-associated MPM.13–15
Materials and Methods
Data Source
The mRNA expression profile datasets of GSE4885512 and GSE5163617 were retrieved from the GEO database (http://www.ncbi.nlm.nih.gov/geo/) using “carbon nanotubes” AND “mesothelioma” as the keywords, while GSE51024,19 GSE254920 and GSE4297721,22 microarray datasets were selected from the GEO database because they compared the gene expression profiles between MPM and normal specimens. In the GSE48855 dataset, there were, respectively, three human immortalized pleural mesothelial cells (MeT5A) exposed to subcytotoxic concentration (0.02 μg/cm2) of SWCNTs, MWCNTs or crocidolite asbestos for up to 4 months to induce malignant transformation; Survanta® dispersant (n = 3) or saline (n = 3) was set as the control for CNT or asbestos, respectively.12 The microarray platform of the GSE48855 dataset was NimbleGen Homo sapiens HG18 090828 opt expr HX12 (12x135k) (GPL10191). In the GSE51636 dataset, long carbon nanotubes (5 μg per mouse, n = 4), long asbestos fibers (5 μg per mouse, n = 4) and control (0.5% BSA/saline, n = 4) were injected into the pleural cavity of female C57Bl/6 mice, which induced the inflammatory lesions at 12 weeks and mesothelial hyperplasia at 12–20 months.17 Since microarrays were performed on diaphragm tissues at 12 weeks after CNT administration,17 the GSE51636 dataset was only used as the validation dataset to evaluate the expression levels of crucial genes in precancerous lesions. The microarray platform of the GSE51636 dataset was Agilent-028005 SurePrint G3 Mouse GE 8x60K Microarray (GPL10787). Fifty-five frozen resected MPM tissues and 41 paired normal lung parenchyma tissues were used for the gene expression profiling study in the GSE51024 dataset (platform: [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array, GPL570);19 40 MPM surgical specimens, 5 normal pleura and 5 normal lung specimens, 5 MPM cell lines and 1 non-tumorigenic mesothelial cell-line MeT5A were included in the GSE2549 dataset (platform: [HG-U133A] Affymetrix Human Genome U133A Array, GPL96);20 39 MPM specimens (histological subtype: n = 24 as epithelioid; n = 8 as sarcomatoid; n = 7 as biphasic), 7 normal pleura and 2 normal lung specimens were contained in the GSE42977 dataset (platform: Illumina HumanRef-6 v2.0 expression beadchip, GPL6790).21,22 MPM was believed to arise from mesothelial cells in the pleura and frequently envelopes lung tissues. Thus, both pleura and lung tissues were included as controls.20 The workflow of the study is shown in Figure 1.
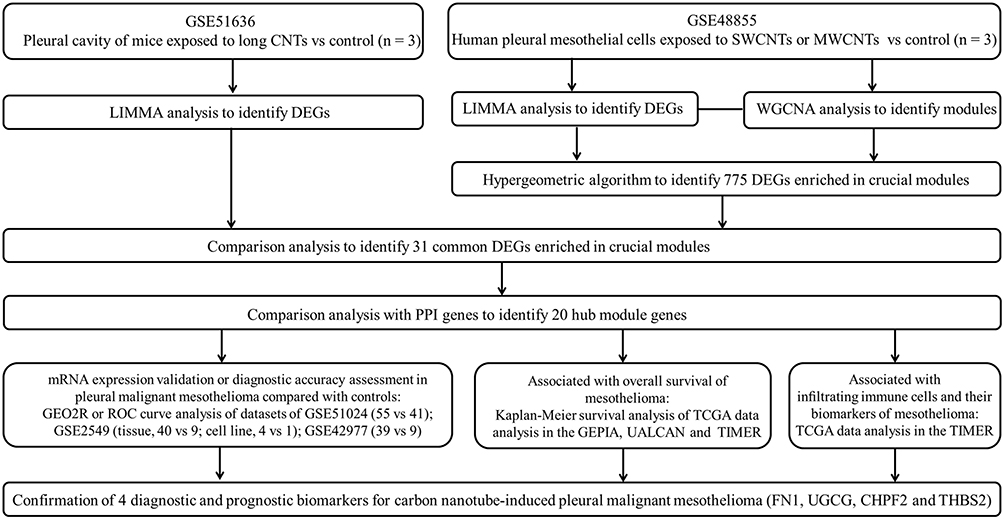 |
Figure 1 Workflow chart of the study. Abbreviations: CNT, carbon nanotubes; SWCNTs, single-walled carbon nanotubes; MWCNTs, carbon nanotubes; DEGs, differentially expressed genes; WGCNA, weighted gene co-expression network analysis; ROC, receiver operating characteristic; PPI, protein–protein interaction; TCGA, The Cancer Genome Atlas; GEPIA, Gene Expression Profiling Interactive Analysis; TIMER, Tumor IMmune Estimation Resource; FN1, fibronectin 1; UGCG, UDP-glucose ceramide glucosyltransferase; CHPF2, chondroitin polymerizing factor 2; THBS2, thrombospondin 2. |
Screening of Differentially Expressed Genes (DEGs)
The matrix and annotation files of GSE48855 and GSE51636 datasets were downloaded from the GEO database. The limma package in R (v3.34.7; https://bioconductor.org/packages/release/bioc/html/limma.html)23 was used to screen DEGs between the CNT (asbestos)-exposed group and the control group in GSE48855 and GSE51636 datasets. The statistical threshold was defined as |log2FC(fold change)| >1 and false discovery rate (FDR) adjusted p-values according to the Benjamini-Hochberg procedure24 <0.05. The heatmap of DEGs identified between SWCNT/MWCNT-exposed and control MeT5A cells in the GSE48855 dataset was plotted using the pheatmap package (v1.0.8; https://cran.r-project.org/web/packages/pheatmap). Furthermore, the DEGs identified in the GSE51636 dataset were converted to human homologous genes using the online database mining tool Biomart (v2.3.6; https://bioconductor.org/packages/release/bioc/html/biomaRt.html)25 and compared with DEGs in the GSE48855 dataset. Venn diagram (http://bioinformatics.psb.ugent.be/webtools/Venn/) was used to identify the overlapped genes. Data of GSE51024, GSE2549 and GSE42977 datasets were directly analyzed using the GEO2R tool (https://www.ncbi.nlm.nih.gov/geo/geo2r/) to confirm the expression levels of crucial genes identified from the GSE48855 and GSE51636 datasets (FDR < 0.05 was considered to be significant) in MPM and controls.
Identification of Co‑expression Modules Associated with CNT-Induced MPM
The WGCNA package in R (v1.61; https://cran.r-project.org/web/packages/WGCNA/index.html)18 was used to construct the co-expression network based on the expression data of all mRNAs in the GSE48855 dataset. As described previously,26 the first step was to determine the soft-thresholding power (β) using the pickSoftThreshold function of WGCNA according to the scale-free topology criterion. Then, the adjacency matrix was converted into a topological overlap matrix (TOM) to construct a dendrogram. Modules were detected using the DynamicTreeCut algorithm of WGCNA with a minimum module size of 40 and a minimum cut height of 0.995. The key modules were screened by: 1) analyzing the correlation of co-expression modules with the disease status of CNT-induced MPM; 2) examining the correlation between gene significance (GS, defined as the correlation between gene expression and each trait) and module membership (MM, defined as the correlation between gene expression and each module eigengene). All correlation analyses were carried out by using the Pearson correlation, with p < 0.05 as the statistical significance measure; and 3) mapping the common DEGs induced by SWCNTs and MWCNTs into the modules by using the hypergeometric algorithm [f(k, N, M, n) = C(k, M)*C(n-k, N-M)/C(n, N)].27 CNT-related modules were identified if their fold enrichment was >1 and p-value was <0.05.
Function Enrichment Analysis for Module Genes
To predict the possible functions of DEGs in crucial modules responsible for MPM, Gene Ontology (GO) terms, Kyoto Encyclopedia of Genes and Genomes (KEGG) and Reactome pathway enrichment analyses were performed using the Database for Annotation, Visualization and Integrated Discovery (DAVID) (v6.8; http://david.abcc.ncifcrf.gov) database. Hyper-geometric test with a p-value <0.05 was considered statistically significant.
Protein–Protein Interaction (PPI) Network Analysis for Module Genes
To further identify hub genes of module DEGs and possible downstream molecular mechanisms, the interactions between DEGs were identified using the Search Tool for the Retrieval of Interacting Genes (STRING; v10.0; https://string-db.org) database. Interaction pairs with a combined score of >0.4 were considered to be reliable and were used to construct the PPI network with the Cytoscape software (v3.6.1; www.cytoscape.org/). Genes in the PPI network were ranked by the CytoNCA plugin in Cytoscape software (http://apps.cytoscape.org/apps/cytonca) according to the degree centrality. Hub DEGs were identified as genes that had a degree centrality of ≥4.
Prognostic Analysis for Module Genes
Gene Expression Profiling Interactive Analysis (GEPIA, http://gepia.cancer-pku.cn/),28 UALCAN (http://ualcan.path.uab.edu/)29 and Tumor IMmune Estimation Resource (TIMER; https://cistrome.shinyapps.io/timer/)30 are interactive web resources for online analyses of levels 3 TCGA RNA-sequencing data and clinical data from 31 cancer types. In the study, we used them to analyze the prognostic performance of CNT-related hub module genes for MPM. Kaplan–Meier curves were plotted to show the differences in overall survival (OS) between the high- and low (or low-medium)-expression groups divided by the median expression value (or 3rd quartile) of each gene. Log-rank p-value <0.05 was set as the cut-off. Only the genes that were demonstrated to be significantly associated with OS in three tools were retained and the OS rate of patients should be consistent with the expected trend according to the expression levels of genes in CNT-MPM and MPM samples (eg, for upregulated genes, patients with a high expression level may have a shorter OS rate).
Diagnostic Analysis for Module Genes
The diagnostic value of hub module genes to differentiate MPM samples from controls was assessed by receiver operating characteristic (ROC) curves. MedCalc (v9.3; MedCalc Software, Mariakerke, Belgium) was used to generate the ROC curves and the diagnostic efficacy of single genes or the gene-combination was evaluated according to the area under each ROC curve (AUC), sensitivity and specificity.
Immune Association Analysis
To further identify the functional mechanisms of hub DEGs, an immune association analysis was performed using the TIMER (https://cistrome.shinyapps.io/timer/) database30 that is a resource for systematic analysis of tumor-infiltrating immune cells across diverse cancer types. In this study, we used the “Gene” module to estimate the correlations between the gene expression of hub genes and the abundance of six immune infiltrates (B cells, CD4+ T cells, CD8+ T cells, neutrophils, macrophages, and dendritic cells). Moreover, the “Correlation” module was also utilized to explore the correlations between the expression of hub genes and gene markers of various immune cells [tumor-associated macrophages (TAMs), dendritic cells and neutrophils].31,32 The gene markers were selected because they were also differentially expressed in CNT-induced MPM. The purity-corrected partial Spearman’s rho value and p-value were provided.
Results
Identification of DEGs Associated with CNT-Induced Malignant Transformation of MeT5A Cells
A total of 9986 genes in the GSE48855 dataset were used for DEG analysis. Under the threshold of FDR < 0.05 and |log2FC| > 1, a total of 1077 genes (including 752 upregulated and 325 downregulated) were identified as DEGs between SWCNT-exposed and dispersant-treated MeT5A cells; a total of 2060 DEGs (including 1493 upregulated and 567 downregulated) were screened from MWCNT-exposed MeT5A cells in comparison with dispersant controls. The heatmap analysis showed that these DEGs could significantly discriminate the transformed MeT5A cells induced by SWCNTs (Figure 2A) or MWCNTs from controls (Figure 2B). Venn diagram displayed there were 565 commonly upregulated (Figure 2C) and 269 commonly downregulated (Figure 2D) between SWCNTs and MWCNTs (Table 1). Thus, these 834 shared genes were used for the following analyses.
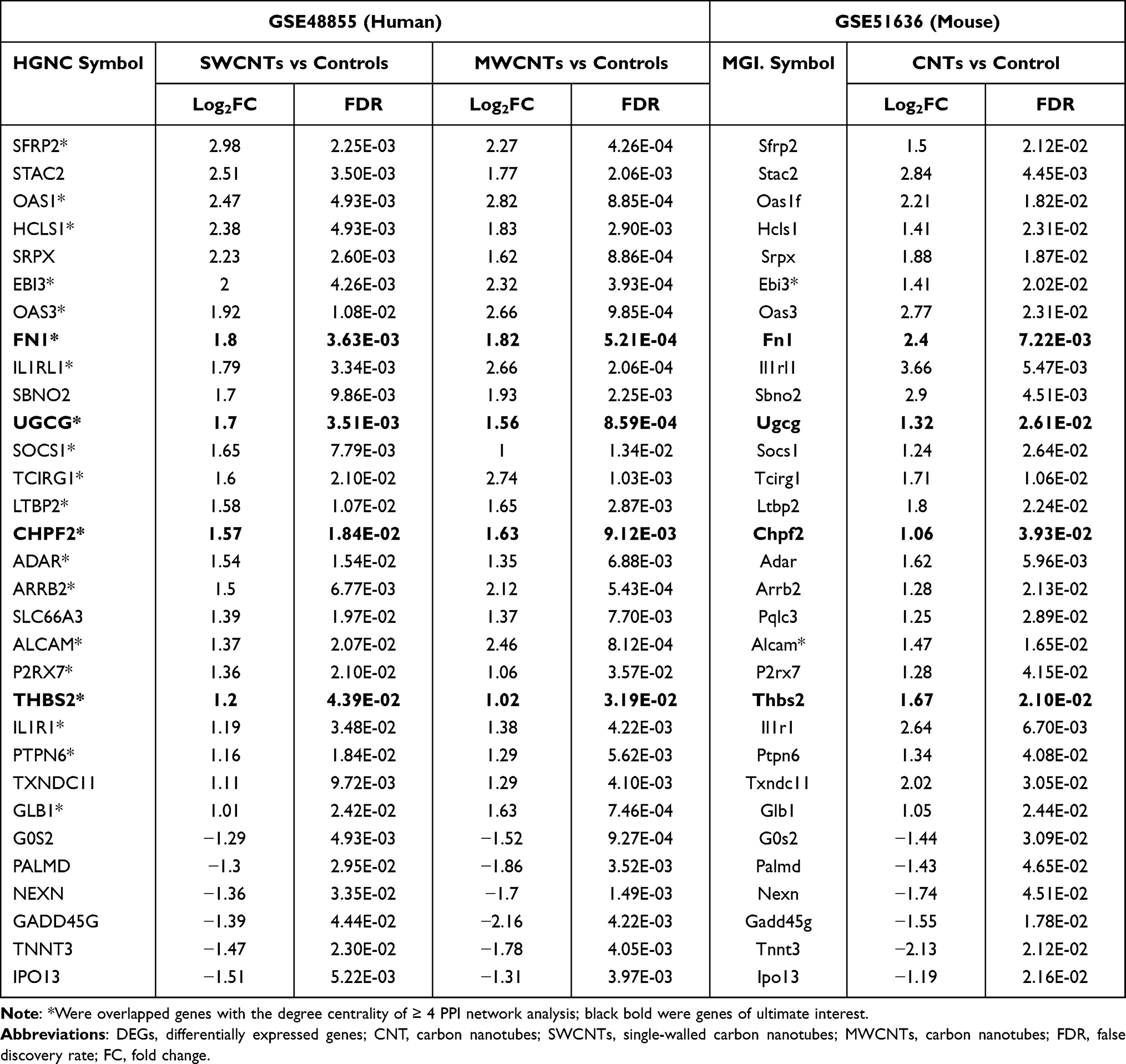 |
Table 1 Homologous Between CNT Exposure and Controls of Human and Mouse |
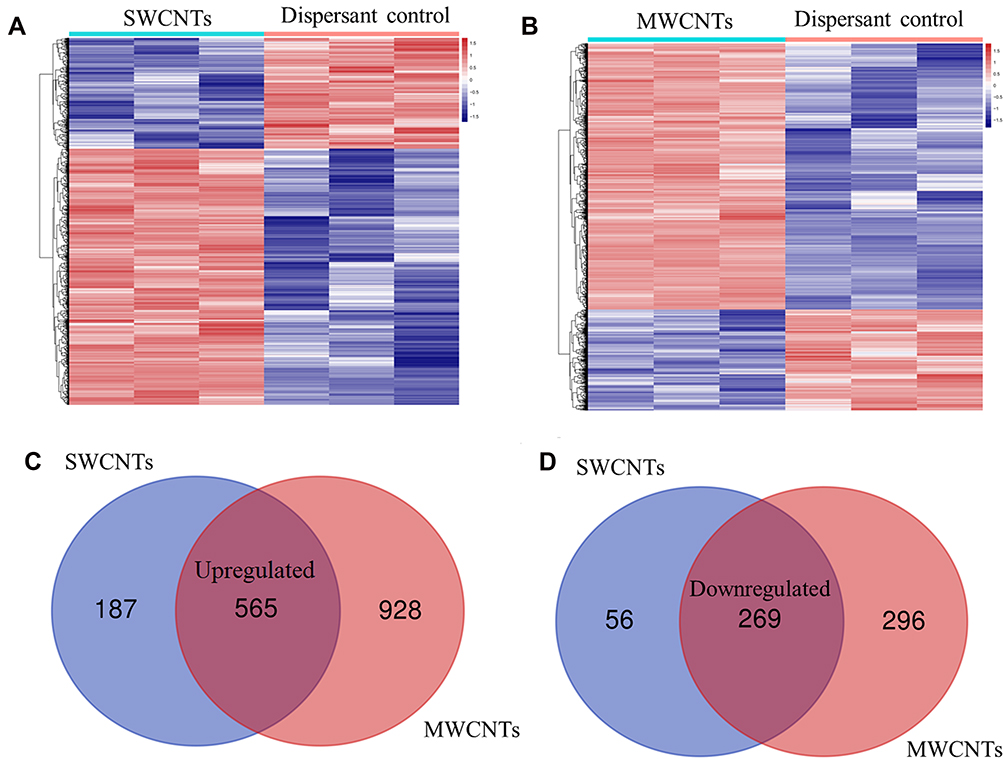 |
Figure 2 Identification of DEGs between CNT-exposed and control human pleural mesothelial cells (MeT5A) in the GSE48855 dataset. (A), the heatmap of DEGs identified by comparing SWCNTs-exposed with dispersant-treated MeT5A cells; (B), the heatmap of DEGs identified by comparing MWCNTs-exposed with dispersant-treated MeT5A cells; (C), Venn diagram to screen the common upregulated genes between SWCNTs and MWCNTs; (D), Venn diagram to the common downregulated genes between SWCNTs and MWCNTs. The heatmap was generated based on the Z-score. Abbreviations: DEGs, differentially expressed genes; SWCNTs, single-walled carbon nanotubes; MWCNTs, multi-walled carbon nanotubes; Red, high expression; blue, low expression. |
Identification of Key Modules Associated with CNT-Induced Malignant Transformation of MeT5A Cells
A WGCNA was used to identify key modules associated with CNT-induced malignant transformation of MeT5A cells based on all genes in the GSE48855 dataset. When the power was 12, the scale-free R2 was 0.9 for the first time (Figure 3A), and the co-expression networks satisfied the requirements of scale-free topology (Figure 3B). Thus, β = 12 was selected as the optimal soft threshold value to generate a TOM dendrogram. After setting the minimum size of 50 genes and the minimum cut height of 0.995, 19 co-expression modules were obtained from the dendrogram and labeled with different colors (Figure 3C). Among them, blue (including 508, all upregulated) and brown (including 267, all downregulated) modules were significantly enriched by DEGs (Table 2). The module–trait relationship analysis illustrated that the blue (r = 0.97; p = 2E-05) and brown (r = −0.98; p = 4E-06) modules were significantly associated with the disease status trait (transformed MeT5A cells or normal controls) (Figure 3D). The scatterplots also suggested that there were significant correlations between GS and MM in these two modules (Figure 3E and F). These findings revealed that blue and brown were key modules and the DEGs in these two modules were especially important for the development of CNT-induced MPM.
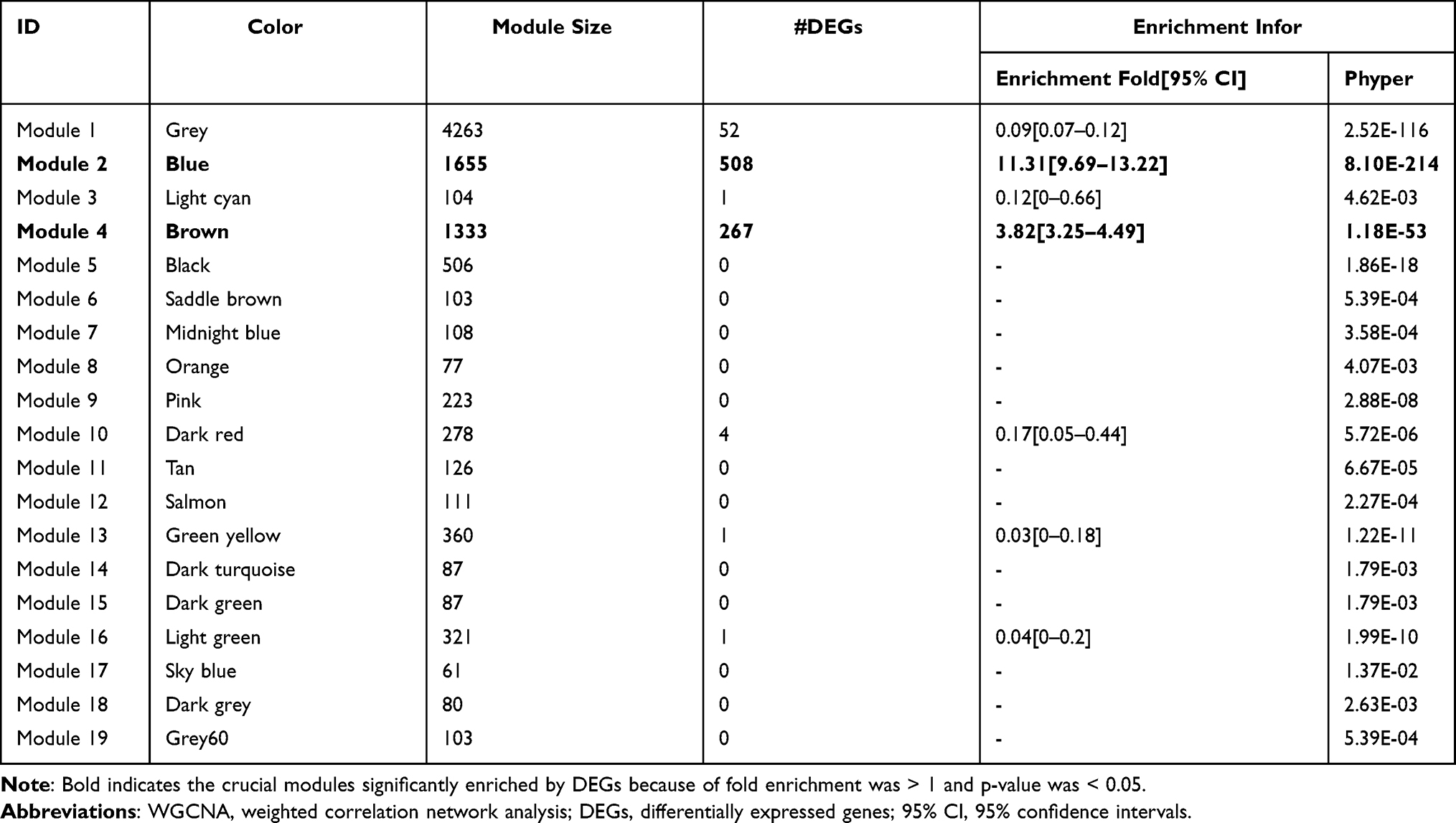 |
Table 2 Modules Based on WGCNA of Genes in the GSE48855 Dataset |
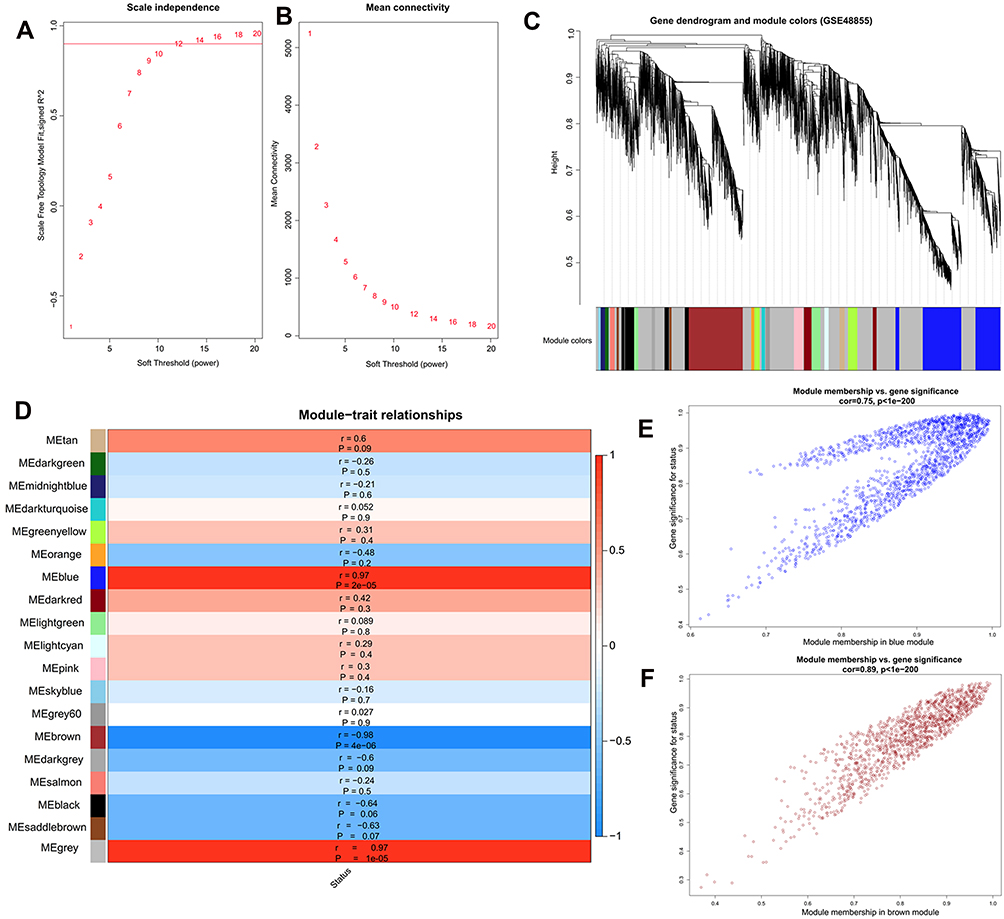 |
Figure 3 Identification of key modules associated with CNT-induced malignant transformation of MeT5A cells by weighted gene co-expression network analysis. (A), calculation of the scale-free topology fit index according to different soft-threshold power values; (B), calculation of mean connectivity according to different soft-threshold power values; (C), a clustering dendrogram of samples; (D), the correlations between modules and disease status (r, correlation coefficient); (E), scatter plot of module eigengenes in the blue module; (F), scatter plot of module eigengenes in the brown module. |
Validation of the Expression of Module Genes in Samples from CNT-Exposed Mice
Under the threshold of FDR < 0.05 and |log2FC| > 1, a total of 1175 genes (including 734 upregulated and 441 downregulated) were identified to be differentially expressed between CNT-exposed mice and controls in the GSE51636 dataset. After Biomart annotation, 693 upregulated and 372 downregulated DEGs were converted to homologous coding genes in humans. Comparing these differentially expressed homologous coding genes in the GSE51636 dataset with all the DEGs in the GSE48855 dataset showed 26 were commonly upregulated (Figure S1A; CHI3L1, chitinase 3 like 1; and all DEGs in Table 1) and 6 were commonly downregulated (Figure S1B; Table 1). Further comparison with the DEGs in blue and brown modules showed that there were 25 commonly upregulated (Figure S1C) and 6 commonly downregulated DEGs (Figure S1D) (all DEGs in Table 1). In addition, 20 of them (indicated as the asterisk in Table 1) were overlapped with the genes with a degree centrality of ≥4 in the PPI network (Figure 4), implying they were hub genes for CNT-induced MPM.
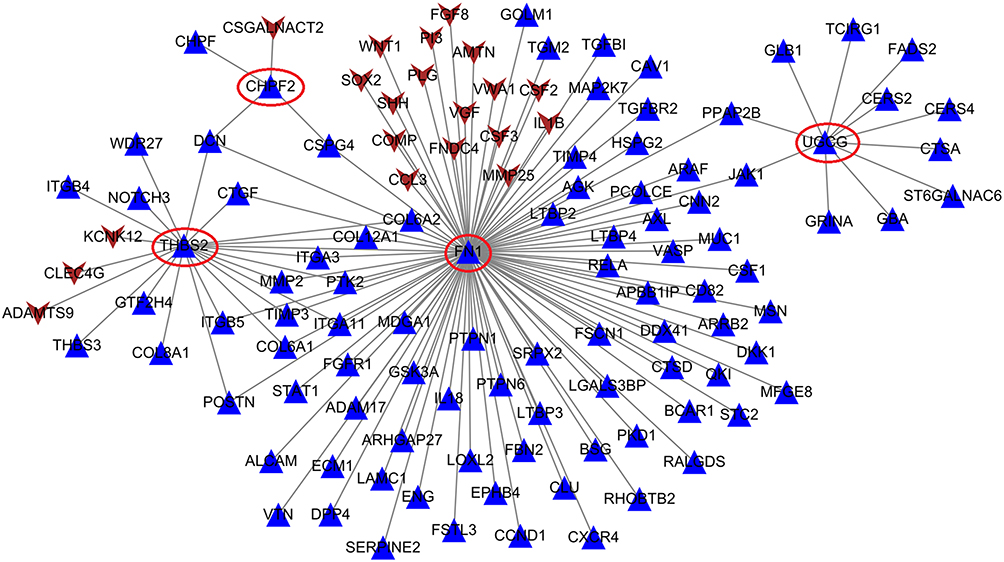 |
Figure 4 The protein–protein interaction network established by interaction pairs of four hub genes. Genes in circular represent hub genes. Gene color is corresponding to module color; regular triangle, upregulated genes; inverted triangle, downregulated genes. |
Analysis of the Prognostic Values of Hub Module Genes in MPM
To verify whether the above 20 hub module genes had the predictive power for the progression of CNT-induced MPM, their associations with OS of MPM patients were explored using GEPIA, UALCAN and TIMER online tools based on the TCGA data. As a result, five genes (FN1, fibronectin 1; ALCAM, activated leukocyte cell adhesion molecule; UGCG, UDP-glucose ceramide glucosyltransferase; CHPF2, chondroitin polymerizing factor 2; THBS2, thrombospondin 2) were found to be significantly associated with OS after GEPIA (Figure 5A–E), TIMER (Figure 5F–J) and UALCAN (Figure S2A–E) online tool analyses. The Kaplan-Meier survival curve analysis showed patients with a high expression level of them had a shorter OS rate than those with a low expression level (Figure 5; Figure S2). These findings were in line with the expected results for these six upregulated genes in CNT-induced MPM, indicating they may be underlying prognostic biomarkers.
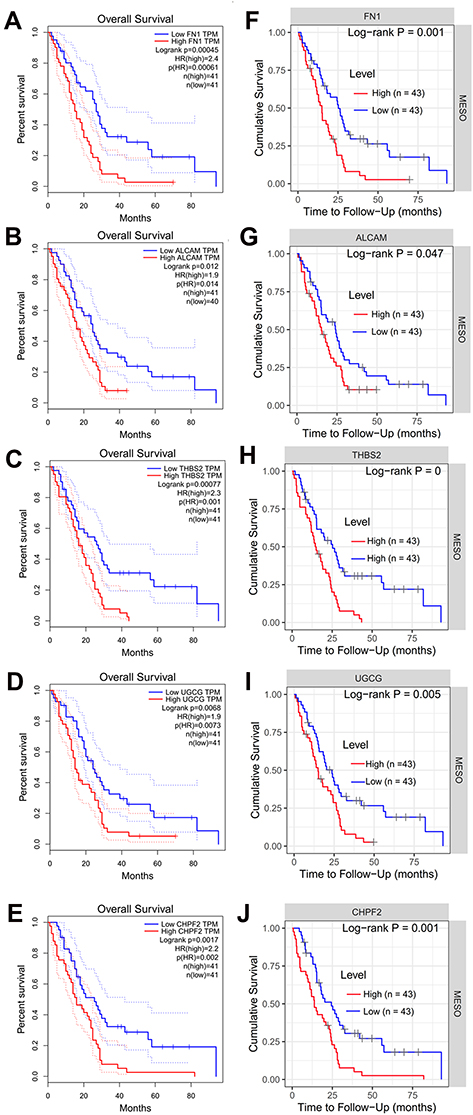 |
Figure 5 Kaplan-Meier survival curves for patients with a high and low expression of five hub module genes in mesothelioma samples. (A–E), GEPIA analysis results; F-J, TIMER analysis results. (A and F) for FN1; (B and G) for ALCAM; (C and H) for THBS2; (D and I) for UGCG; (E and J) for CHPF2. Abbreviations: GEPIA, Gene Expression Profiling Interactive Analysis; TIMER, Tumor IMmune Estimation Resource; HR, hazard ratio; MESO, mesothelioma; FN1, fibronectin 1; ALCAM, activated leukocyte cell adhesion molecule; THBS2, thrombospondin 2; UGCG, UDP-glucose ceramide glucosyltransferase; CHPF2, chondroitin polymerizing factor 2; TPM, transcripts per million. |
Validations of the Expression of Prognosis-Related Hub Module Genes in MPM
Since the TCGA dataset did not contain a control for MPM samples, we collected other microarray datasets (GSE51024, GSE2549 and GSE42977) from the GEO database to validate the expression levels of four prognosis-related hub module genes. After the GEO2R analysis, FN1, UGCG, CHPF2 and THBS2 were validated to be also upregulated (FDR < 0.05) in MPM samples compared to control tissues in the GSE51024 and GSE2549 datasets (Table 3). Comparison of all MPM samples with controls in the GSE42977 dataset only revealed FN1 and THBS2 were upregulated genes, while analysis with sarcomatoid and biphasic histological subtypes showed FN1, UGCG and THBS2 were all significantly differentially expressed. These findings suggested FN1, UGCG, CHPF2 and THBS2 may be potential diagnostic biomarkers to differentiate MPM patients from controls.
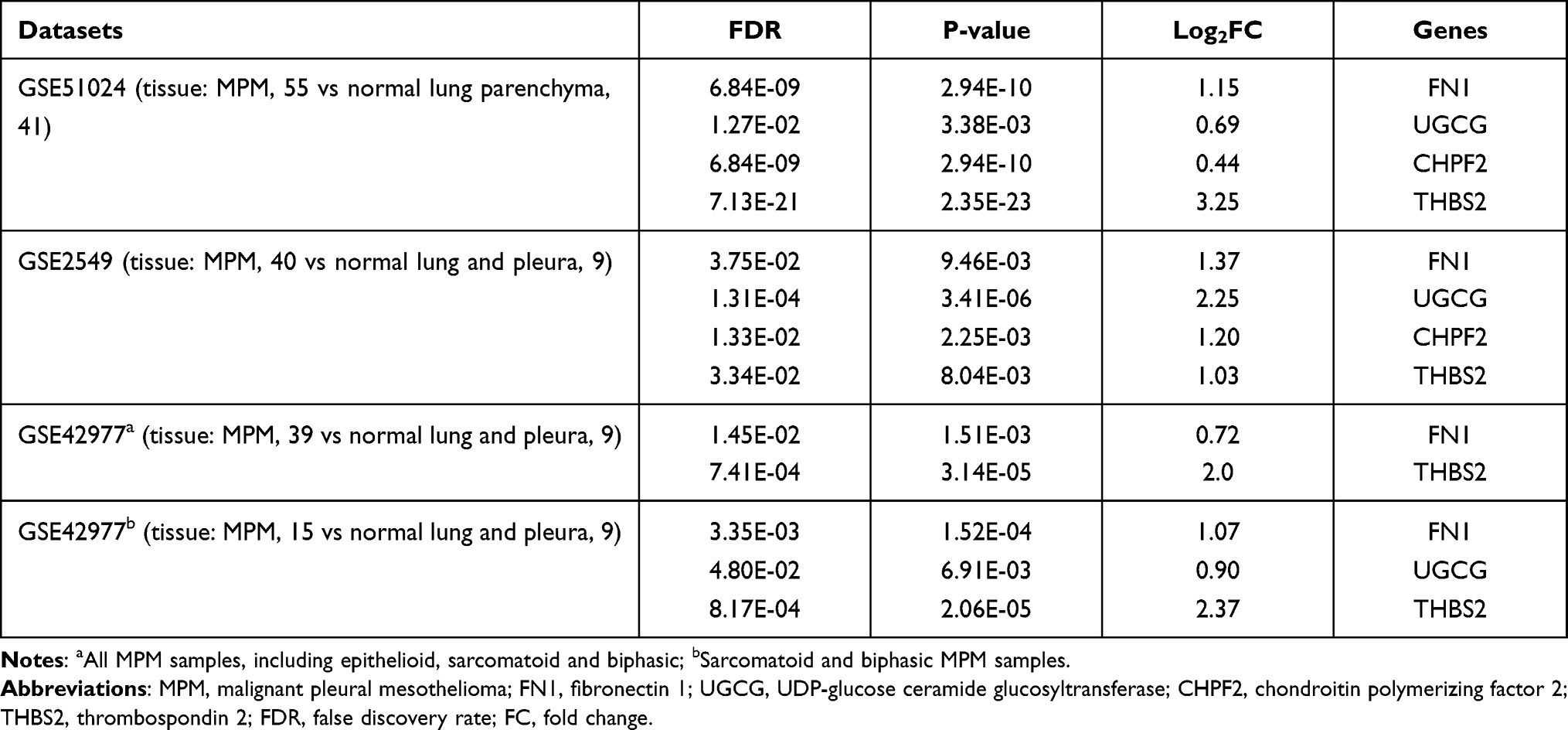 |
Table 3 Validation of the Expression of Hub Genes |
Analysis of the Diagnostic Values of Four Hub Genes for MPM
To explore the diagnostic efficacy of the above four hub genes for MPM, the ROC curve was plotted (Figure 6). The results showed that the AUCs of all these genes were >0.65, indicating that these genes may serve as potential diagnostic markers for MPM (Table 4). Relative to a single gene, the diagnostic efficacy of gene-combination seemed to be higher, with the AUCs > 0.9 in most of the results (Table 4). Furthermore, FN1 and THBS2 were also upregulated genes in asbestos-induced malignant transformation of MeT5A cells or mice (Table S1); in our previous study on CNT-induced malignant transformation of lung cells, FN1 was likewise highly expressed.26 These findings suggest UGCG and/or CHPF2 combination with FN1 and THBS2 may be more specific for the diagnosis of CNT-induced MPM. This conclusion could be demonstrated according to the sensitivity results in GSE2549 analysis (FN1+THBS2, 60%; FN1+UGCG+THBS2, 95%; FN1+UGCG+CHPF2+THBS2, 80%) and specificity results in GSE51042 analysis (FN1+THBS2, 85.37%; FN1+UGCG+THBS2, 97.56%; FN1+UGCG+CHPF2+THBS2, 97.56%) (Table 4).
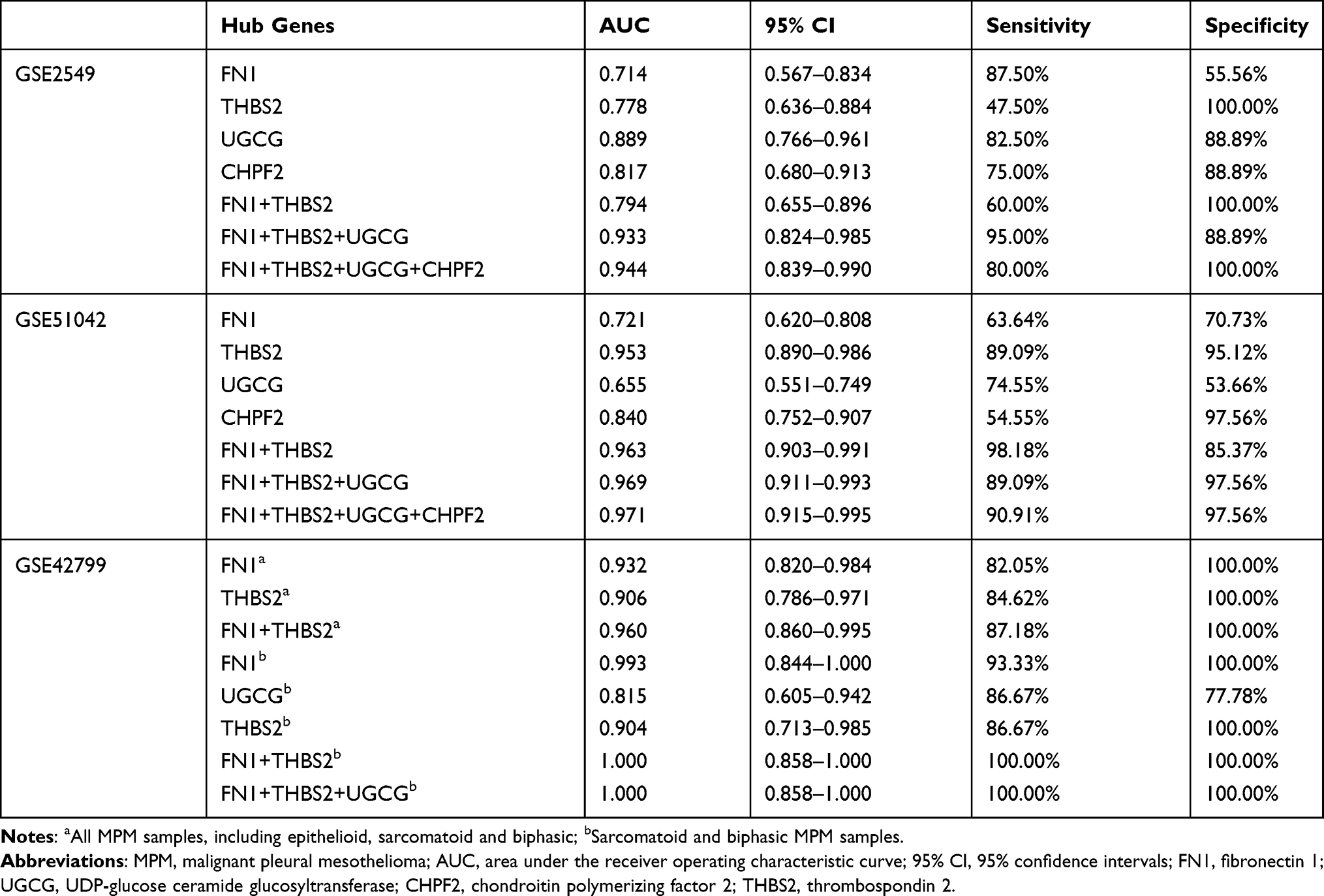 |
Table 4 The Predictive Power of Hub Genes for MPM |
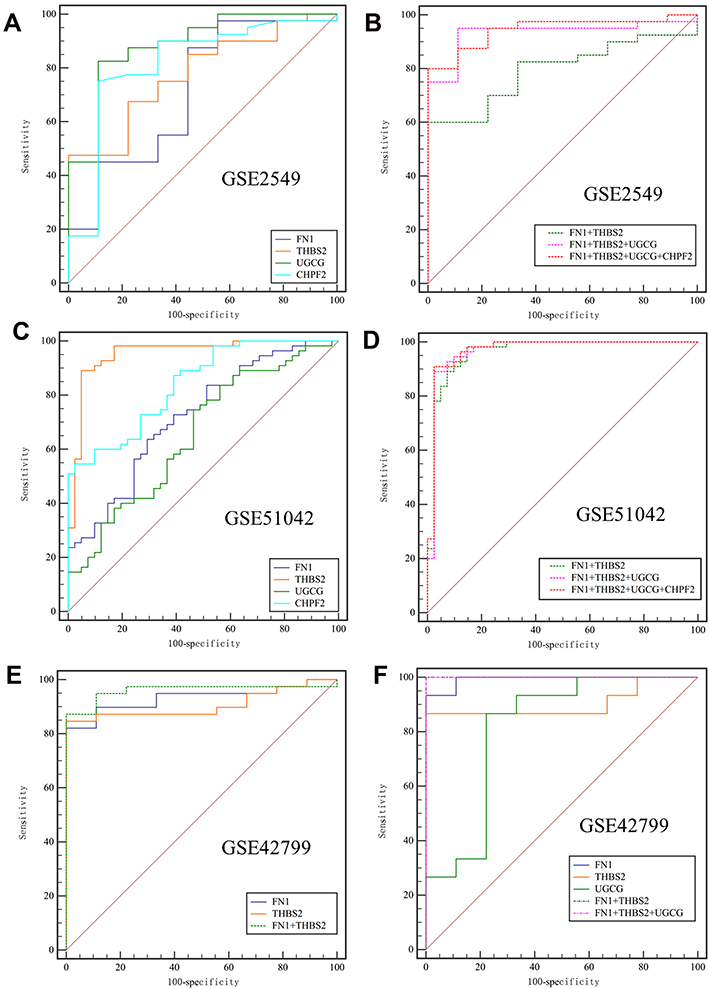 |
Figure 6 ROC curves for evaluating the diagnostic values of four hub genes for distinguishing mesothelioma from normal patients. (A and B), ROC curves were plotted based on the mRNA expression levels of four genes in the GSE2549 dataset. (A), single gene; (B), gene-combination; (C and D), ROC curves were plotted based on the mRNA expression levels of four genes in the GSE51024 dataset. (C), single gene; (D), gene-combination; (E), ROC curve was plotted based on the mRNA expression levels of two genes in all mesothelioma samples of the GSE42977 dataset; (F), ROC curve was plotted based on the mRNA expression levels of three genes in the sarcomatoid and biphasic histological subtypes of the GSE42977 dataset. Abbreviations: ROC, receiver operating characteristic; FN1, fibronectin 1; THBS2, thrombospondin 2; UGCG, UDP-glucose ceramide glucosyltransferase; CHPF2, chondroitin polymerizing factor 2. |
Function Analysis of Four Hub Genes
To reveal the underlying functions of hub genes correlated with MPM, GO, KEGG and Reactome pathway enrichment analyses were conducted for all DEGs in crucial modules. A total of 104 GO-biological process terms were enriched (Table S2), in which FN1 was found to be involved in GO:0007155~cell adhesion, GO:0030198~extracellular matrix organization and GO:0008284~positive regulation of cell proliferation; THBS2 was involved in GO:0007155~cell adhesion; UGCG was involved in GO:0006687~glycosphingolipid metabolic process; CHPF, CSPG4 (chondroitin sulphate proteoglycan 4) and CHPF2 were involved in GO:0030206~chondroitin sulfate biosynthetic process. A total of 25 KEGG pathways were obtained (Table S3), in which FN1, UGCG and THBS2 were also enriched, such as hsa04512:ECM-receptor interaction (FN1, THBS2), hsa04151:PI3K-Akt signaling pathway (FN1, THBS2), hsa04510:Focal adhesion (FN1, THBS2) and hsa00600:Sphingolipid metabolism (UGCG). A total of 27 Reactome pathways were identified, including R-HSA-1474228: Degradation of the extracellular matrix (FN1), R-HSA-186797: Signaling by PDGF (THBS2) and R-HSA-1660662: Glycosphingolipid metabolism (UGCG) (Table S4).
In addition, several immune and inflammation-related GO biological processes and pathways were found, such as GO:0006955~immune response (IL1B), hsa04060:Cytokine-cytokine receptor interaction (IL1B), R-HSA-448706: Interleukin-1 processing (IL1B), R-HSA-2132295: MHC class II antigen presentation (CTSA, cathepsin A); while FN1 could interact with IL1B; UGCG could interact with CTSA in the PPI prediction analysis (Figure 4), implying our hub genes were also immune-associated in MPM. To further confirm their immune associations, TIMER analysis was performed to explore the correlation between the expression levels of hub genes and the abundance of six immune infiltrates as well as biomarkers of immune cells based on the TCGA data. The results showed FN1, UGCG and THBS2 were positively correlated with the level of immune infiltration of macrophages; FN1, UGCG and CHPF2 had significant correlations with infiltrating levels of dendritic cells (Figure 7). All genes were positively correlated with NRP1 (neuropilin 1), a marker of dendritic cells (Table 5).
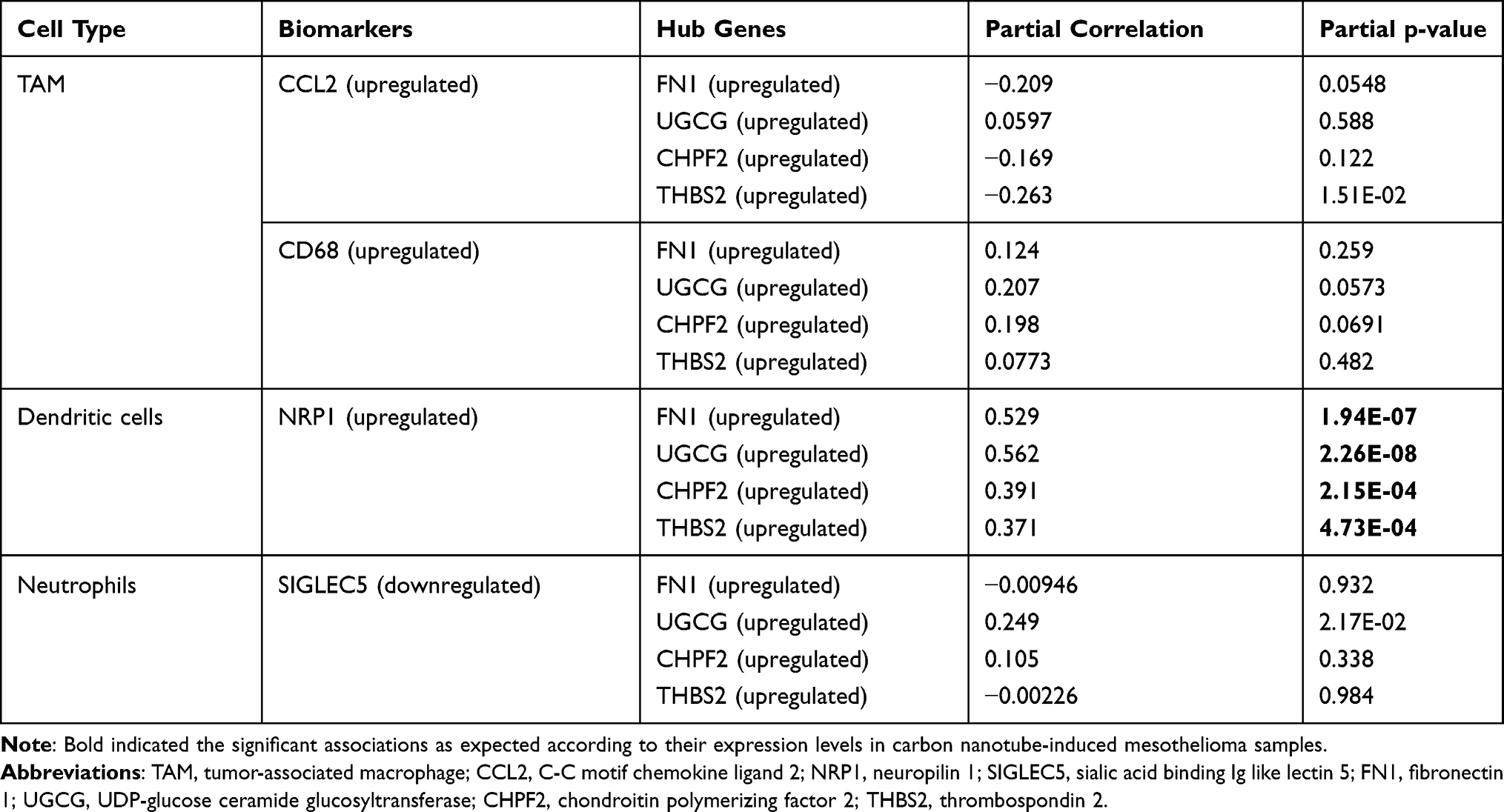 |
Table 5 Correlation Analysis Between Hub Genes and Markers of Immune Cells |
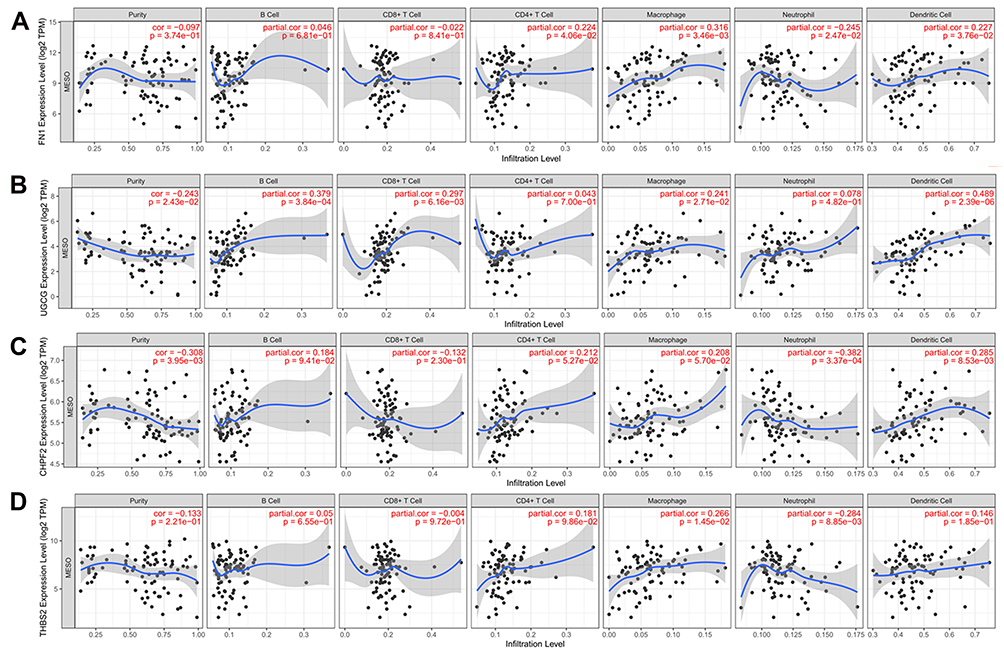 |
Figure 7 Correlations between the expression of hub genes and the abundance of six immune infiltration cells of mesothelioma. (A), FN1; (B), UGCG; (C), CHPF2; (D), THBS2. Abbreviations: FN1, fibronectin 1; THBS2, thrombospondin 2; UGCG, UDP-glucose ceramide glucosyltransferase; CHPF2, chondroitin polymerizing factor 2; cor, correlation. |
Discussion
Relative to asbestos that had been prohibited in several countries,33,34 CNTs are a novel material class and being widely applied in various fields,1–5 indicating CNT exposure may be the main contributor for the development of MPM in the future. Thus, identification of biomarkers for surveillance of occupational workers to prevent the onset of CNT-induced MPM may be of great clinical significance. However, current studies on the molecular mechanisms of CNT-induced MPM remain rare12,13,17 and no studies linked CNT-exposed in vitro and in vivo models with MPM patient samples as reports for lung cancer.26,35 In the present study, we, for the first time, attempted to screen concordant genes differentially expressed both in CNT-exposed MeT5A cells and mice compared with controls and validate the expression, diagnostic and prognostic abilities of hub genes in specimens from MPM patients. Based on WGCNA and PPI network analyses, 20 genes were considered as hub module genes associated with the development of CNT-induced MPM. Four of them (FN1, UGCG, CHPF2 and THBS2) were found to be significantly associated with OS; significantly upregulated in MPM specimens compared to control tissues; and they could effectively predict the MPM risk with an overall accuracy >0.9 using the gene-combination. These findings suggest that these four genes may serve as potential diagnostic and prognostic biomarkers for monitoring MPM in CNT workers.
The extracellular matrix (ECM) is a natural substrate to provide a support structure and an attachment site for cells. Also, cell-ECM interactions have been generally recognized as an important mechanism for tumor cell proliferation, migration and invasion.36 Thus, the components (including fibronectin, thrombospondin and chondroitin sulfate proteoglycans) of the ECM and related genes may be crucial targets for the development and progression of cancers, including MPM.37 This hypothesis had been demonstrated by previous studies. Siri et al observed a significantly higher concentration of fibronectin in pleural fluids and plasma of patients with MPM as compared with that in patients with pleurisies.38 Kanaji et al found that exogenous addition of fibronectin enhanced the migration and invasion abilities of three MPM cells (H28, H226 and H2052).39 Decreasing the synthesis of fibronectin by retinoic acid inhibited the migration of MPM cells.40 FN1 gene was also identified to have a significant abundance in MPM cell models (JU77, LO68 and OLD1612).41 After GSE2549 data analysis and qPCR experiments, Kim et al validated the expression level of THBS2 was significantly higher in the MPM tissues compared with normal tissues.42 Patients with elevated mRNA levels of THBS2 in MPM tissues had a statistically significantly shorter OS rate than those with a low level.43 siRNA-mediated silencing of THBS2 gene significantly decreased proliferation and colony formation activities of MPM cell line IST-Mes2.44 CSPG4 was expressed in 6 out of 8 MPM cell lines and in 25 out of 41 MPM biopsies, but was not detectable in surrounding healthy cells. Treatment with a monoclonal antibody against CSPG4 significantly reduced MPM cell adhesion, motility, migration, invasiveness, growth and prevented the formation of MPM xenografts in severely combined immune-deficient mice to improve their survival rates.45 CHPF is a member of glycosyltransferases to participate in the biosynthesis of chondroitin sulfate, which recognizes proteoglycan core proteins to form CSPG. CHPF was detected to be highly expressed in lung cancer tissues and cell lines.46 Knockdown of CHPF in lung cancer cells can effectively inhibit the proliferation and promote the apoptosis of cancer cells.46,47 High expression of CHPF was significantly associated with pathologic stage, metastasis and worse prognosis of breast carcinoma patients.48 In line with these studies, our results also showed FN1, THBS2, CSPG4 and CHPF were upregulated in CNT-induced malignant transformed MeT5A cells, mice and MPM tissues, associated with OS and enriched in extracellular matrix-related biological process and pathways. Although no study demonstrated the roles of CHPF2 in cancer, CHPF2 was predicted to interact with CSPG4 and CHPF in our PPI network (Figure 4), indirectly providing an evidence to show the oncogenic functions of CHPF2 in MPM.
UGCG is the only enzyme for the synthesis of glucosylceramide via transferring a UDP-glucose molecule to ceramide. Glucosylceramide is the precursor for complex glycosphingolipids which are not only important components of cellular membrane but also second messengers in signal transduction to regulate various physiological and pathophysiological processes, such as excessive cell proliferation.49 Therefore, UGCG may also be an underlying gene associated with carcinogenesis. This hypothesis had been proved by several studies. For example, Salustiano et al found UGCG was highly expressed in leukemias with a multidrug resistance. Pharmacological inhibition of UGCG greatly improved the chemosensitivity and promoted cell apoptosis.50 The study team of Schömel et al suggested overexpressed UGCG may maintain breast cancer cell proliferation and doxorubicin resistance by increasing glutamine, glycolysis and oxidative glucose metabolisms.51–53 UGCG deficiency suppressed hepatocellular tumor initiation and growth in mice.54 The cisplatin-resistant MPM cells also expressed increased Gb3 glycosphingolipids,55 which indirectly reflected the upregulation of UGCG. In accordance with these studies, UGCG was upregulated in CNT-induced malignant transformed MeT5A cells, mice and MPM tissues, associated with OS and enriched in glycosphingolipid metabolism pathways.
Increasing evidence had highlighted CNT exposure induced the malignant transformation by promoting the accumulation of immune cells (macrophages13–15 and dendritic cells56). T cell immunoglobulin mucin 4 (Tim4) was reported to mediate the recognition of foreign pathogen MWCNTs by peritoneal macrophages.57 Phagocytosed MWCNTs activated the NLR family pyrin domain containing 3 inflammasome in macrophages and caused IL-1β release to the adjacent pleural mesothelial cells.15,57,58 An amplified inflammatory response was subsequently stimulated to promote cell proliferation, migration, invasion, colony formation of mesothelial cells.13 Tkach et al observed that exposure to SWCNTs for one day and seven days facilitated the recruitment of dendritic cells to the lung.56 Beyeler et al detected that the frequencies of pulmonary dendritic cells were increased following the MWCNT treatment.59 The CNT-induced migration of dendritic cells to the lung was driven by C-C motif chemokine ligand 5 secreted from macrophages.60 Macrophage-dendritic cell-human bronchial epithelial cell co-culture model triggered the release of more pro-inflammatory cytokines (TNF-α and IL-8) than the monoculture model,61,62 which may subsequently induce the formation of lung cancer lesion. Furthermore, the NRP1 biomarker of dendritic cells was also proved to be an oncogenic gene in lung cancer;63,64 inhibition of its expression significantly reduced the proliferation, migratory and invasive capabilities of cancer cells.64,65 Furthermore, FN1 was found to an associating protein to involve Tim4-mediated efferocytosis.66 CSTA (cystatin A) activated the phagocytosis in macrophages and dendritic cells by cell-surface neuraminidase 1.67 FN1 could interact with all other hub genes (THBS2 and CHPF2), while UGCG could interact with CSTA (Figure 4). Accordingly, we speculated our hub genes may participate in macrophage activation-induced dendritic cell recruitment to contribute to CNT-associated MPM. Consistent with this theory, we found the expression of FN1, UGCG and THBS2 (CHPF2) were positively correlated with the levels of macrophages (dendritic cells) and NRP1.
This research has some limitations. First, this study preliminarily screened four genes concordantly expressed in CNT-exposed in vitro and in vivo models with MPM patient samples as potential biomarkers to predict the development risk and prognosis of CNTs-induced MPM. Whether they were also concordantly expressed in blood or pleural effusions samples needs to be confirmed in new clinical samples or animal models in order to be applied in clinic more conveniently. Second, the expression of hub genes was only validated at mRNA levels, not protein levels. Proteomics, immunohistochemistry and Western blotting should be designed to explore the protein expression. Third, although most of the genes were revealed to be associated with MPM, they were not linked with CNT exposure. Also, whether these genes are truly mesothelial cancer driver genes or genes overexpressed in an adaptive response to the tumoral progression are also unclear. In vitro and in vivo studies (overexpression and silencing) should be performed to investigate the function mechanisms of four hub genes in the malignant transformation of CNTs-induced MPM.44 Fourth, their immune associations needed to be verified by co-culture of macrophages (dendritic cells) with mesothelial cells, the use of endocytosis inhibitors and deletion of hub genes.57,60
Conclusion
The present study reveals that four genes (FN1, UGCG, CHPF2 and THBS2) may be candidate biomarkers for monitoring CNT-induced MPM and predicting the prognosis. They may function by involving cell-ECM interactions, glycosphingolipid metabolism pathways and influencing the immune response.
Data Sharing Statement
All data were downloaded from GEO (GSE48855, GSE51636, GSE51024, GSE2549 and GSE42977; http://www.ncbi.nlm.nih.gov/geo/).
Acknowledgments
Financial support from the National Key Research and Development Program of China (2017YFA0204600), National Natural Science Foundation of China (51703027) and Project funded by China Postdoctoral Science Foundation (2017M621322, 2018T110324) is greatly acknowledged.
Disclosure
Tong Wu is an employee of Shanghai LEVSON Nanotechnology Co., Ltd. Kangli Cao is an employee of Shanghai Institute of Spacecraft Equipment. The authors declare no other potential conflicts of interest for this work.
References
1. Luo X, Liang Y, Weng W, Hu Z, Zhu M. Polypyrrole-coated carbon nanotube/cotton hybrid fabric with high areal capacitance for flexible quasi-solid-state supercapacitors. Energy Storage Mater. 2020;33:11–17. doi:10.1016/j.ensm.2020.07.036
2. Luo X, Weng W, Liang Y, et al. Multifunctional fabrics of carbon nanotube fibers. J Mater Chem A. 2019;7:8790–8797. doi:10.1039/C9TA01474H
3. Abazari S, Shamsipur A, Bakhsheshi-Rad HR, Ismail AF, Sharif S, Razzaghi M. Carbon nanotubes (CNTs)-reinforced magnesium-based matrix composites: a comprehensive review. Materials. 2020;13(19):4421. doi:10.3390/ma13194421
4. Yin Z, Cui C, Chen H, Duoni YX, Qian W. The application of carbon nanotube/graphene-based nanomaterials in wastewater treatment. Small. 2020;16(15):e1902301. doi:10.1002/smll.201902301
5. Negri V, Pacheco-Torres J. Carbon nanotubes in biomedicine. Top Curr Chem. 2020;378(1):15. doi:10.1007/s41061-019-0278-8
6. Wang JN, Luo XG, Wu T, Chen Y. High-strength carbon nanotube fibre-like ribbon with high ductility and high electrical conductivity. Nat Commun. 2014;5:3848. doi:10.1038/ncomms4848
7. Kane AB, Hurt RH, Gao H. The asbestos-carbon nanotube analogy: an update. Toxicol Appl Pharmacol. 2018;361:68–80. doi:10.1016/j.taap.2018.06.027
8. Nagai H, Toyokuni S. Differences and similarities between carbon nanotubes and asbestos fibers during mesothelial carcinogenesis: shedding light on fiber entry mechanism. Cancer Sci. 2012;103(8):1378–1390. doi:10.1111/j.1349-7006.2012.02326.x
9. Spirtas R, Heineman EF, Bernstein L, et al. Malignant mesothelioma: attributable risk of asbestos exposure. Occup Environ Med. 1994;51(12):804–811. doi:10.1136/oem.51.12.804
10. Lacourt A, Gramond C, Rolland P, et al. Occupational and non-occupational attributable risk of asbestos exposure for malignant pleural mesothelioma. Thorax. 2014;69(6):532–539. doi:10.1136/thoraxjnl-2013-203744
11. Domen A, De Laet C, Vanderbruggen W, et al. Malignant pleural mesothelioma: single-institution experience of 101 patients over a 15-year period. Acta Chir Belg. 2017;117(3):157–163. doi:10.1080/00015458.2016.1272253
12. Lohcharoenkal W, Wang L, Stueckle TA, et al. Chronic exposure to carbon nanotubes induces invasion of human mesothelial cells through matrix metalloproteinase-2. ACS Nano. 2013;7(9):7711–7723. doi:10.1021/nn402241b
13. Huang X, Tian Y, Shi W, et al. Role of inflammation in the malignant transformation of pleural mesothelial cells induced by multi-walled carbon nanotubes. Nanotoxicology. 2020;14(7):947–967. doi:10.1080/17435390.2020.1777477
14. Xu J, Futakuchi M, Shimizu H, et al. Multi-walled carbon nanotubes translocate into the pleural cavity and induce visceral mesothelial proliferation in rats. Cancer Sci. 2012;103(12):2045–2050. doi:10.1111/cas.12005
15. Murphy FA, Schinwald A, Poland CA, Donaldson K. The mechanism of pleural inflammation by long carbon nanotubes: interaction of long fibres with macrophages stimulates them to amplify pro-inflammatory responses in mesothelial cells. Part Fibre Toxicol. 2012;9:8. doi:10.1186/1743-8977-9-8
16. Takagi A, Hirose A, Futakuchi M, Tsuda H, Kanno J. Dose-dependent mesothelioma induction by intraperitoneal administration of multi-wall carbon nanotubes in p53 heterozygous mice. Cancer Sci. 2012;103(8):1440–1444. doi:10.1111/j.1349-7006.2012.02318.x
17. Chernova T, Murphy FA, Galavotti S, et al. Long-fiber carbon nanotubes replicate asbestos-induced mesothelioma with disruption of the tumor suppressor gene Cdkn2a (Ink4a/Arf). Curr Biol. 2017;27(21):3302–3314.e3306. doi:10.1016/j.cub.2017.09.007
18. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 2008;9:559. doi:10.1186/1471-2105-9-559
19. Suraokar MB, Nunez MI, Diao L, et al. Expression profiling stratifies mesothelioma tumors and signifies deregulation of spindle checkpoint pathway and microtubule network with therapeutic implications. Ann Oncol. 2014;25(6):1184–1192. doi:10.1093/annonc/mdu127
20. Gordon GJ, Rockwell GN, Jensen RV, et al. Identification of novel candidate oncogenes and tumor suppressors in malignant pleural mesothelioma using large-scale transcriptional profiling. Am J Pathol. 2005;166(6):1827–1840. doi:10.1016/S0002-9440(10)62492-3
21. De Rienzo A, Richards WG, Yeap BY, et al. Sequential binary gene ratio tests define a novel molecular diagnostic strategy for malignant pleural mesothelioma. Clin Cancer Res. 2013;19(9):2493–2502. doi:10.1158/1078-0432.CCR-12-2117
22. De Rienzo A, Coleman MH, Yeap BY, Severson DT. Association of RERG expression with female survival advantage in malignant pleural mesothelioma. Cancers. 2021;13(3):565. doi:10.3390/cancers13030565
23. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi:10.1093/nar/gkv007
24. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. 1995;57:289–300.
25. Durinck S, Spellman PT, Birney E, Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc. 2009;4(8):1184–1191. doi:10.1038/nprot.2009.97
26. Xie D, Luo X. Identification of four methylation-driven genes as candidate biomarkers for monitoring single-walled carbon nanotube-induced malignant transformation of the lung. Toxicol Appl Pharmacol. 2021;412:115391. doi:10.1016/j.taap.2020.115391
27. Cao J, Zhang S. A Bayesian extension of the hypergeometric test for functional enrichment analysis. Biometrics. 2014;70(1):84–94. doi:10.1111/biom.12122
28. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98–w102. doi:10.1093/nar/gkx247
29. Chandrashekar DS, Bashel B, Balasubramanya SAH, et al. UALCAN: a portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia. 2017;19(8):649–658. doi:10.1016/j.neo.2017.05.002
30. Li T, Fan J, Wang B, et al. TIMER: a web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 2017;77(21):e108–e110. doi:10.1158/0008-5472.CAN-17-0307
31. Yang J, Li H, Hu S, Zhou Y. ACE2 correlated with immune infiltration serves as a prognostic biomarker in endometrial carcinoma and renal papillary cell carcinoma: implication for COVID-19. Aging. 2020;12(8):6518–6535. doi:10.18632/aging.103100
32. Huo Q, Li Z, Cheng L, Yang F, Xie N. SIRT7 is a prognostic biomarker associated with immune infiltration in luminal breast cancer. Front Oncol. 2020;10:621. doi:10.3389/fonc.2020.00621
33. Glass WI, Armstrong R, Chen G. Banning asbestos in New Zealand, 1936–2016, an 80-year long saga. Int J Environ Res Public Health. 2017;14(12):1457. doi:10.3390/ijerph14121457
34. Westerholm P, Remaéus B, Svartengren M. The tale of asbestos in Sweden 1972–1986-the pathway to a near-total ban. Int J Environ Res Public Health. 2017;14(11):1433. doi:10.3390/ijerph14111433
35. Guo NL, Wan YW, Denvir J, et al. Multiwalled carbon nanotube-induced gene signatures in the mouse lung: potential predictive value for human lung cancer risk and prognosis. J Toxicol Environ Health A. 2012;75(18):1129–1153. doi:10.1080/15287394.2012.699852
36. Jagirdar RM, Papazoglou ED, Pitaraki E, et al. Cell and extracellular matrix interaction models in benign mesothelial and malignant pleural mesothelioma cells in 2D and 3D in-vitro. Clin Exp Pharmacol Physiol. 2021;48(4):543–552. doi:10.1111/1440-1681.13446
37. Turini S, Bergandi L, Gazzano E, Prato M, Aldieri E. Epithelial to mesenchymal transition in human mesothelial cells exposed to asbestos fibers: role of TGF-β as mediator of malignant mesothelioma development or metastasis via EMT event. Int J Mol Sci. 2019;20(1):150. doi:10.3390/ijms20010150
38. Siri A, Carnemolla B, Raffanti S, Castellani P, Balzano E, Zardi L. Fibronectin concentrations in pleural effusions of patients with malignant and non-malignant diseases. Cancer Lett. 1984;22(1):1–9. doi:10.1016/0304-3835(84)90037-5
39. Kanaji N, Kita N, Kadowaki N, Bandoh S. Fibronectin and hepatocyte growth factor produced by lung fibroblasts augment migration and invasion of malignant pleural mesothelioma cells. Anticancer Res. 2017;37(5):2393–2400. doi:10.21873/anticanres.11578
40. Scarpa S, Giuffrida A, Palumbo C, et al. Retinoic acid inhibits fibronectin and laminin synthesis and cell migration of human pleural mesothelioma in vitro. Oncol Rep. 2002;9(1):205–209.
41. Greening DW, Ji H, Chen M, et al. Secreted primary human malignant mesothelioma exosome signature reflects oncogenic cargo. Sci Rep. 2016;6:32643. doi:10.1038/srep32643
42. Kim MK, Kim HW, Jang M, et al. LOX family and ZFPM2 as novel diagnostic biomarkers for malignant pleural mesothelioma. Biomark Res. 2020;8:1. doi:10.1186/s40364-019-0180-0
43. Melaiu O, Melissari E, Mutti L, et al. Expression status of candidate genes in mesothelioma tissues and cell lines. Mutat Res. 2015;771:6–12. doi:10.1016/j.mrfmmm.2014.11.002
44. Dell’Anno I, Barbarino M. EIF4G1 and RAN as possible drivers for malignant pleural mesothelioma. Int J Mol Sci. 2020;21(14):4856. doi:10.3390/ijms21144856
45. Rivera Z, Ferrone S, Wang X, et al. CSPG4 as a target of antibody-based immunotherapy for malignant mesothelioma. Clin Cancer Res. 2012;18(19):5352–5363. doi:10.1158/1078-0432.CCR-12-0628
46. Hou XM, Zhang T, Da Z, Wu XA. CHPF promotes lung adenocarcinoma proliferation and anti-apoptosis via the MAPK pathway. Pathol Res Pract. 2019;215(5):988–994. doi:10.1016/j.prp.2019.02.005
47. Hou XM, Baloch Z, Zheng ZH, et al. Knockdown of CHPF suppresses cell progression of non-small-cell lung cancer. Cancer Manag Res. 2019;11:3275–3283. doi:10.2147/CMAR.S192036
48. Li Y, Gong H, Feng L, Mao D, Xiao Y, Wang Y. Chondroitin polymerizing factor promotes breast carcinoma cell proliferation, invasion and migration and affects expression of epithelial-mesenchymal transition-related markers. FEBS Open Bio. 2021;11(2):423–434. doi:10.1002/2211-5463.13062
49. Furukawa K, Ohmi Y, Ohkawa Y, et al. New era of research on cancer-associated glycosphingolipids. Cancer Sci. 2019;110(5):1544–1551. doi:10.1111/cas.14005
50. Salustiano EJ, da Costa KM, Freire-de-lima L, Mendonça-Previato L, Previato JO. Inhibition of glycosphingolipid biosynthesis reverts multidrug resistance by differentially modulating ABC transporters in chronic myeloid leukemias. J Biol Chem. 2020;295(19):6457–6471. doi:10.1074/jbc.RA120.013090
51. Schömel N, Gruber L, Alexopoulos SJ, et al. UGCG overexpression leads to increased glycolysis and increased oxidative phosphorylation of breast cancer cells. Sci Rep. 2020;10(1):8182. doi:10.1038/s41598-020-65182-y
52. Schömel N, Hancock SE. UGCG influences glutamine metabolism of breast cancer cells. Sci Rep. 2019;9(1):15665. doi:10.1038/s41598-019-52169-7
53. Wegner MS, Schömel N, Gruber L, et al. UDP-glucose ceramide glucosyltransferase activates AKT, promoted proliferation, and doxorubicin resistance in breast cancer cells. Cell Mol Life Sci. 2018;75(18):3393–3410. doi:10.1007/s00018-018-2799-7
54. Jennemann R, Federico G, Mathow D, et al. Inhibition of hepatocellular carcinoma growth by blockade of glycosphingolipid synthesis. Oncotarget. 2017;8(65):109201–109216. doi:10.18632/oncotarget.22648
55. Tyler A, Johansson A, Karlsson T, et al. Targeting glucosylceramide synthase induction of cell surface globotriaosylceramide (Gb3) in acquired cisplatin-resistance of lung cancer and malignant pleural mesothelioma cells. Exp Cell Res. 2015;336(1):23–32. doi:10.1016/j.yexcr.2015.05.012
56. Tkach AV, Shurin GV, Shurin MR, et al. Direct effects of carbon nanotubes on dendritic cells induce immune suppression upon pulmonary exposure. ACS Nano. 2011;5(7):5755–5762. doi:10.1021/nn2014479
57. Omori S, Tsugita M, Hoshikawa Y, et al. Tim4 recognizes carbon nanotubes and mediates phagocytosis leading to granuloma formation. Cell Rep. 2021;34(6):108734. doi:10.1016/j.celrep.2021.108734
58. Fukai E, Sato H, Watanabe M, Nakae D, Totsuka Y. Establishment of an in vivo simulating co-culture assay platform for genotoxicity of multi-walled carbon nanotubes. Cancer Sci. 2018;109(4):1024–1031. doi:10.1111/cas.13534
59. Beyeler S, Steiner S, Wotzkow C, et al. Multi-walled carbon nanotubes activate and shift polarization of pulmonary macrophages and dendritic cells in an in vivo model of chronic obstructive lung disease. Nanotoxicology. 2020;14(1):77–96. doi:10.1080/17435390.2019.1663954
60. Mukherjee SP, Bondarenko O, Kohonen P. Macrophage sensing of single-walled carbon nanotubes via Toll-like receptors. Sci Rep. 2018;8(1):1115. doi:10.1038/s41598-018-19521-9
61. Clift MJ, Endes C, Vanhecke D, et al. A comparative study of different in vitro lung cell culture systems to assess the most beneficial tool for screening the potential adverse effects of carbon nanotubes. Toxicol Sci. 2014;137(1):55–64. doi:10.1093/toxsci/kft216
62. Müller L, Riediker M, Wick P, Mohr M, Gehr P, Rothen-Rutishauser B. Oxidative stress and inflammation response after nanoparticle exposure: differences between human lung cell monocultures and an advanced three-dimensional model of the human epithelial airways. J R Soc Interface. 2010;7(Suppl 1):S27–40. doi:10.1098/rsif.2009.0161.focus
63. Ding Z, Zhu J, Zeng Y, et al. The regulation of Neuropilin 1 expression by miR-338-3p promotes non-small cell lung cancer via changes in EGFR signaling. Mol Carcinog. 2019;58(6):1019–1032. doi:10.1002/mc.22990
64. Pérez-Lozano ML, Sandoval P, Rynne-Vidal A, et al. Functional relevance of the switch of VEGF receptors/co-receptors during peritoneal dialysis-induced mesothelial to mesenchymal transition. PLoS One. 2013;8(4):e60776. doi:10.1371/journal.pone.0060776
65. Ding Z, Du W, Lei Z, et al. Neuropilin 1 modulates TGF‑β1‑induced epithelial‑mesenchymal transition in non‑small cell lung cancer. Int J Oncol. 2020;56(2):531–543.
66. Lee J, Park B, Moon B, et al. A scaffold for signaling of Tim-4-mediated efferocytosis is formed by fibronectin. Cell Death Differ. 2019;26(9):1646–1655. doi:10.1038/s41418-018-0238-9
67. Seyrantepe V, Iannello A, Liang F, et al. Regulation of phagocytosis in macrophages by neuraminidase 1. J Biol Chem. 2010;285(1):206–215. doi:10.1074/jbc.M109.055475
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.