")
Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 14
Founder Effects Contribute to the Population Genetic Structure of the Major Dermatophytosis Pathogen Trichophyton rubrum on Hainan Island, China
Authors Geng Y, Wu W , Li R, Xu J, Gu R, Lu J, Zheng W, Zhao F, Zhang J , Gong J
Received 15 July 2021
Accepted for publication 12 October 2021
Published 27 October 2021 Volume 2021:14 Pages 1569—1577
DOI https://doi.org/10.2147/CCID.S329569
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Jeffrey Weinberg
Yuanyuan Geng,1,* Weiwei Wu,2,3,* Rouyu Li,4– 6 Juan Xu,1 Ruixue Gu,1 Jiejie Lu,2 Wenai Zheng,7 Fei Zhao,1 Jianzhong Zhang,1 Jie Gong1
1National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing, People’s Republic of China; 2Department of Dermatology, The Fifth People’s Hospital of Hainan Province, Haikou, People’s Republic of China; 3National Clinical Research Center for Skin and Immune Disease, Beijing, People’s Republic of China; 4Department of Dermatology, Peking University First Hospital, Beijing, People’s Republic of China; 5Beijing Key Laboratory of Molecular Diagnosis on Dermatoses, Beijing, People’s Republic of China; 6Research Center for Medical Mycology, Peking University, Beijing, People’s Republic of China; 7Department of Laboratory Medicine, The Fifth People’s Hospital of Hainan Province, Haikou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jie Gong Tel +86 10 5890 0368
Email [email protected]
Background: Founder events have been observed among numerous plants and animal species living on oceanic islands due to the geographic separation of these islands and the small amount of original life they harbor. However, there has been little research on the ecological characteristics of pathogenic microorganisms on islands. Trichophyton rubrum ranks the most common isolated dermatophyte causing dermatophytosis in clinic and has become an epidemic strain worldwide in recent decades.
Objective: To study the phylogenetic characteristics and the distribution pattern of genetic polymorphism of T. rubrum in China, which further provide theoretical basis for the prevention and control of T. rubrum.
Methods: In the present study, we sequenced and analyzed the genetic characteristics of 204 T. rubrum isolates from Hainan Island and other sites in China. Phylogenetic analysis and genetic polymorphisms were studied based on a total of 41,409 high-quality whole-genome SNPs.
Results: The majority of the isolates from Hainan Island clustered together. Mixed T. rubrum population differentiation was observed among the strains of different geographical origins. In addition, the genetic diversity (π) of the Hainan isolates was low and showed no significant difference from that of isolates from other sites.
Conclusion: This study is the first to discuss general ecological and evolutionary principles related to pathogenic fungi. Our findings reveal a founder effect during the origination of T. rubrum on Hainan Island and provide guidance regarding prevention and treatment strategies.
Keywords: founder effects, Trichophyton rubrum, genetic diversity, population differentiation
Introduction
Founder effects occur mostly when a new population is formed initially by a few individuals (the founders) from the original population.1,2 The founders do not include all the genetics of the original population, resulting in the lack of genetic diversity of newly established population. This is usually the case in groups of many species living on islands, in lakes, and in isolated forests and habitats.2–4
Due to the limited area of oceanic islands and their varying degrees of isolation from nearby continents, they are considered ideal systems for investigating evolutionary patterns such as founder effects.5,6 Founder effects are observable in many plant and animal species; however, few studies have focused on the ecological characteristics of pathogenic microorganisms.2–4,7 Hainan Island is the southernmost tropical island in China. It is a geographically independent island separated from the mainland to the north by the Qiongzhou Strait. The unique geographical features of Hainan Island make it an ideal model for the study of evolutionary processes.8,9
According to a previous report, dermatophytes are the main superficial pathogenic fungi in Hainan Province, among which Trichophyton rubrum is the dominant species.10 Dermatophytes are the most common filamentous pathogenic fungi in the clinic, which tend to infect tissues rich in keratin, such as skin and nails,11 leading to superficial infections, such as tinea cruris, tinea corporis, tinea manuum, etc. Dermatophytes are capable of infecting healthy individuals with competent immune system, which is different from most other pathogenic fungi. Dermatophyte infection is the most common human fungal infection, affecting 20–25% of the global population, and asymptomatic carriers account for 30–70% adult population,12,13 resulting in a great burden of disease and medical burden. According to epidemiological data, T. rubrum is the dominant superficial pathogen, accounting for more than 80% of dermatophyte infections.14 It survives in the environment for at least six months, capable to be universally infected,15 besides, its infections are mostly chronic and easy to relapse.16,17 Due to its biological characteristics and prevalence, T. rubrum has become a model organism for studying human pathogenic filamentous fungi. The identification of T. rubrum included morphological, physiological, and molecular methods, such as microscopy examination, urea hydrolysis, keratinase activity detection, internal transcribed spacer (ITS) polymerase-chain reaction (PCR), matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry (MS), electron spray ionization mass spectrometry (ESI-MS), etc.18,19 ITS PCR was most used method for routine diagnostics.
Studies on the T. rubrum population structure suggested two populations, one population contains genotypes originated from Africa, while the other population distributed worldwide excluding Africa area.20,21 T. rubrum has been found to be the dominant epidemic species in recent decades.22,23 In the past few years, the technology of high-throughput sequencing makes great progress, providing information for in-depth understanding of fungi characteristics with the whole-genome data. In dermatophytes, sequencing and analysis of the genome data about some important species have been reported, including Microsporum canis, T. rubrum, T. violaceum, etc., which provide insights into their population differentiation and potential pathogenic genes.24,25 Compared to the genetic variation of other fungal pathogens, the genetic diversity level within T. rubrum is significantly low. Zheng et al resequenced whole genome of 48 T. rubrum strains from China and analyzed their population differentiation and drug resistance based on the sequencing data.17
This study collected and sequenced 204 T. rubrum strains from different regions in China, aiming to describe the genetic characteristics of T. rubrum population in greater coverage, making the results more representative, and providing theoretical foundation for the formulation of treatment strategies in clinic. Besides, there were a large amount of strains from Hainan, which is a good geographic model for studying founder effect, the analysis of related strains can provide information for the evolutionary characteristics of T. rubrum.
Materials and Methods
T. rubrum Isolates
A total of 204 clinical T. rubrum isolates were collected from different cities of China between 2018 and 2019. Samples from patients were isolated in the laboratory using potato dextrose agar (PDA) medium (Land bridge, Beijing, China) containing 0.05% chloramphenicol (Sangon Biotech, Shanghai, China). After 7 days’ cultivation at 28°C, the cultures were separated into monoclonal cultures and identified by the molecular sequencing of their ITS regions.26,27 The primer sequences were as follows:
ITS1 (forward): 5ʹ-TCCGTAGGTGAACCTGCGG-3ʹ
ITS4 (reverse): 5ʹ-TCCTCCGCTTATTGATATGC-3ʹ28
ITS sequence alignment was conducted using national center for biotechnology information (NCBI) nucleotide basic local alignment search tool (BLAST). Strains identified as Trichophyton rubrum were preserved and stored long term at −80°C using 25% glycerol (v/v) at the National Institute for Communicable Disease Control and Prevention (ICDC, Beijing, China).
The sampling sites were recorded as the locations of the hospitals where the isolates were collected. The characteristics of the sampling sites were obtained from data for the cities where these hospitals were located. The population size was obtained from the yearbook published by the statistical bureau of each city. Meteorological information, including the average temperature, humidity and atmospheric pressure, was collected from monthly data from surface meteorological stations in the China National Meteorological Information Center (http://data.cma.cn/data/cdcdetail/dataCode/SURF_CLI_CHN_MUL_MON.html). For cities without a weather station, the meteorological data were replaced by data from the nearest city with a weather station.
DNA Extraction and Whole-Genome Sequencing
All the T. rubrum isolates were cultivated on Sabouraud’s agar (SDA) medium for 10–14 days. T. rubrum cells were first added to centrifuge tubes containing lysis solution (20 mM Tris, 5 mM EDTA, 400 mM NaCl) and zirconium dioxide beads (Φ1 mm and Φ3 mm). The cells were disrupted by shaking using a rapid biological sample preparation system at low temperature (Life Real, Hangzhou, China). After cell disruption, QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) was used to extract genomic DNA in accordance with the operating instructions. Quality of extracted DNA, including concentration and purity, was controlled by sequencing company. Only samples passed the quality inspection will be used for the subsequent library construction and sequencing.
T. rubrum genomes were sequenced by Illumina technology (Novogene, Beijing, China) after library construction. In brief, genomic DNA was randomly fragmented using a Covaris ultrasonic disruptor. A paired-end library was prepared following the steps of end repair and phosphorylation, including A-tailing, index adapter ligation, purification and PCR amplification. After the library was qualified, the Illumina HiSeq X platform was used for high-throughput sequencing, and paired-end reads of 150 bp were obtained, resulting in approximately 3 G of sequencing data for each strain.
De Novo Assembly
Raw reads were first trimmed to improve data reliability. Reads with low-quality bases (Phred score ≤ 5) exceeding 50% of the base number and reads with an Ns content exceeding 10% were discarded. Clean data were employed for genome assembly using SPAdes v3.12.0.29 The parameter “-cov_cutoff” was set to auto. Contigs shorter than 200 bp were filtered to make the assembled data more reliable.30 Sequencing depth was calculated as follows: reads number*150*2/base number in the genome.
SNP Calling
The reference T. rubrum genome CMCC(F)Tli was downloaded from the NCBI Genome dataset. Burrows–Wheeler Alignment tool (version 0.7.17) was used to map the paired-end reads against CMCC(F)Tli genome, choosing the BWA-MEM algorithm. SAMtools (version 1.9) was used to filter, sort and convert files.30
Genome Analysis Toolkit (version 4.1.4.1) was used for SNPs calling as described previously.30,31 Specifically, following conditions were employed to filter SNPs and indels: VariantFiltration, QD < 2.0, LowQD, ReadPosRankSum < −8.0, LowRankSum, FS > 60.0, HightFS, MQRankSum < −12.5, MQRankSum, MQ < 40.0, LowMQ, HaplotypeScore > 13.0, and HaploScore.
Phylogenomics, Structure, and Population Genetics Analyses
The phylogenetic relationships of the 204 T. rubrum isolates were analyzed by RAxML (version 8.2.12) based on the dataset composed of 41,409 filtered SNPs.30,32 A maximum likelihood phylogenetic tree was inferred with the GTRMMA model, and the bootstrap value was set to 1000. iTOL (https://itol.embl.de/) was used for visualization. ADMIXTURE (version 1.3.0) was used to analyze the population structure. K value was examined through the cross-validation (CV) process, and the best fit was selected with the minimum CV error.
Statistics of Population Genetics
Nucleotide diversity (π) was calculated by VCFtools33 based on the data of all 204 isolates classified by geographic origin, respectively, with the site-pi option. Differentiation between isolates from Hainan Island and other collecting sites (Fst) was obtained using VCFtools. All the analysis was done genome-wide along with 5 kb sliding windows. Plots were done using the R package ggplot2.34
Results
Independent Evolution of T. rubrum from Hainan Island
To examine the population genetic characteristics of T. rubrum in China, the genomes of 204 T. rubrum isolates collected from 7 provinces were sequenced and analyzed. To maximize the ecological and geographical breadth of their origins, this collection included 89 strains from Hainan Island that were geographically independent from inland areas and 115 strains from inland cities. Overall, the sampling sites covered different climate zones and areas showing different ecological and social characteristics (Figure 1, Table 1).
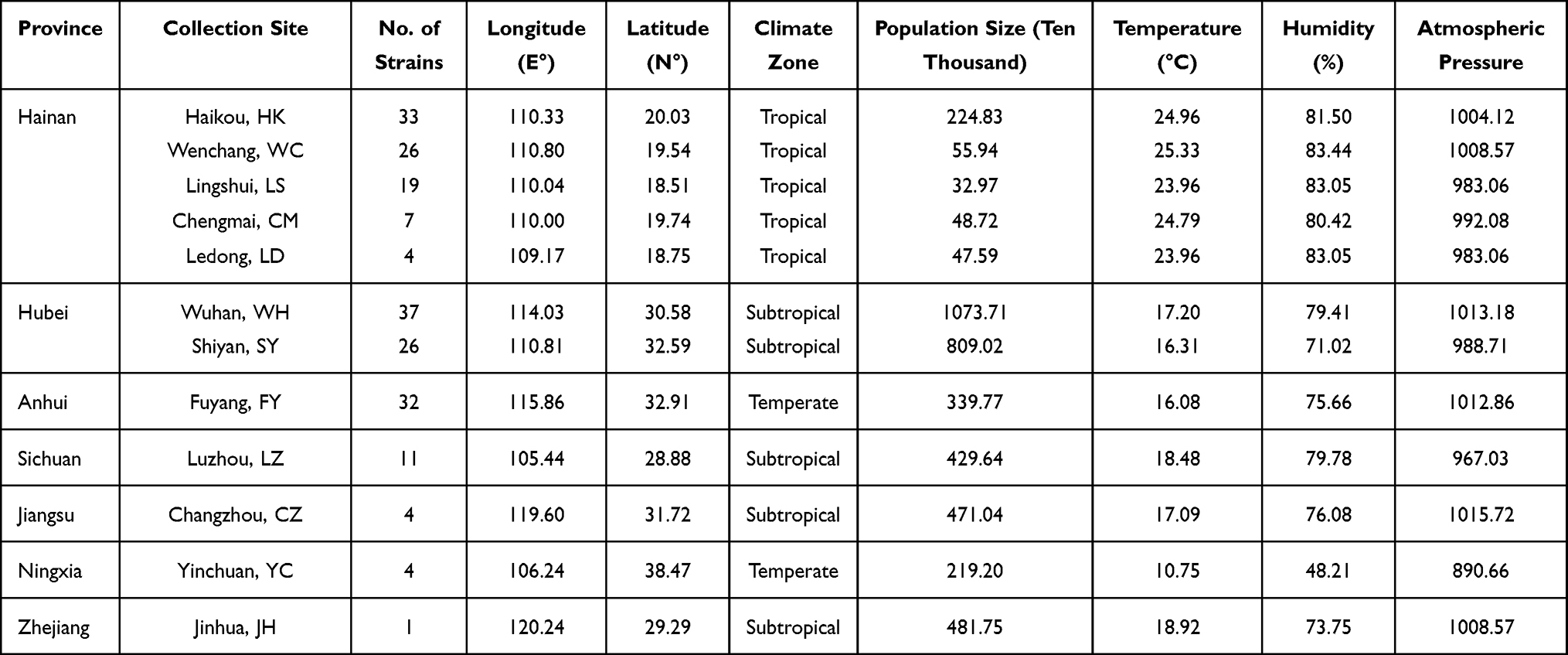 |
Table 1 Ecological and Geographic Information of T. rubrum Isolates in This Study |
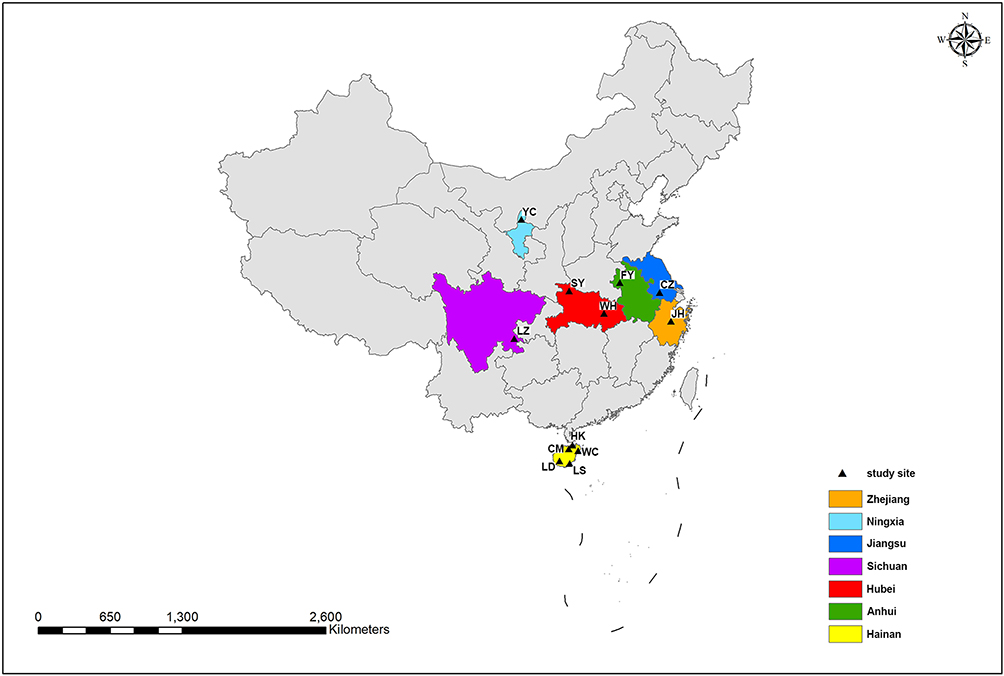 |
Figure 1 Geographical distributions of T. rubrum isolates collected in China. Sampling sites from different provinces were marked with different colors, and the cities were indicated by black triangle. |
The calculated sequence coverage ranged from 70x to 193x (average = 132.9x; median = 134.5x). A total of 41,049 single-nucleotide polymorphisms (SNPs) (Supplementary Data 1) were identified through the 204 genomes. Phylogenetic relationship of the 204 T. rubrum from different geographical regions was shown by a maximum likelihood phylogenetic tree, which resulted from the 41,049 confident SNPs. The evolutionary relationships of the strains from different regions were not completely separated according to the phylogenetic tree, but between the lineages, we observed geographical patterns in which the majority of isolates from Hainan Island clustered together (Figure 2, pink area), suggesting an independent evolutionary trend of these strains.
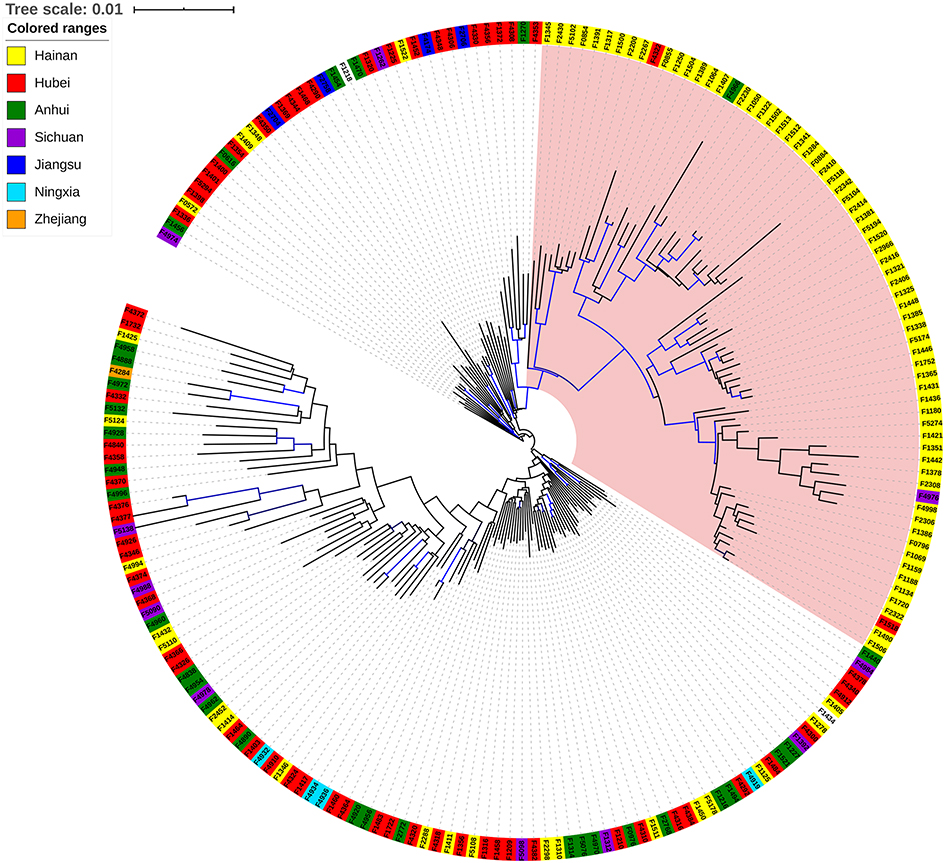 |
Figure 2 Phylogenetic relationships of T. rubrum isolates of China. Phylogeny among the 204 T. rubrum strains sequenced in this study was shown as a maximum likelihood phylogenetic tree. Isolates from different provinces were indicated by different color ranges. Blue Branch lines represented bootstrap supports >90%. |
Genetic Diversity and Population Differentiation
To further verify the classification of T. rubrum strains, we analyzed their population structure. As shown in Figure 3, genetic admixture was observed among T. rubrum from different regions. When K value varied from 2 to 10, the populations were all mixed with no obvious resolution. We further studied genetic diversity among 29,433 polymorphic sites, which were filtered from 41,049 SNPs using VCFtools with the “max missing” parameter set to 1. The average nucleotide diversity (π) calculated from all 204 T. rubrum was 0.22, while the π value of Hainan strains was 0.24. No significant difference was found when nucleotide diversity was compared according to the geographical classification (Figure 4). The low genetic polymorphism observed verified the low levels of variation previously reported among T. rubrum.17,24 In addition, no significant Fst values were obtained between different geographical populations (Figure 4). The results of the genetic polymorphism analysis suggested no clear genetic differentiation, which was consistent with the population structure analysis; thus, the differentiation of isolates from Hainan Island is only reflected in the phylogenetic tree.
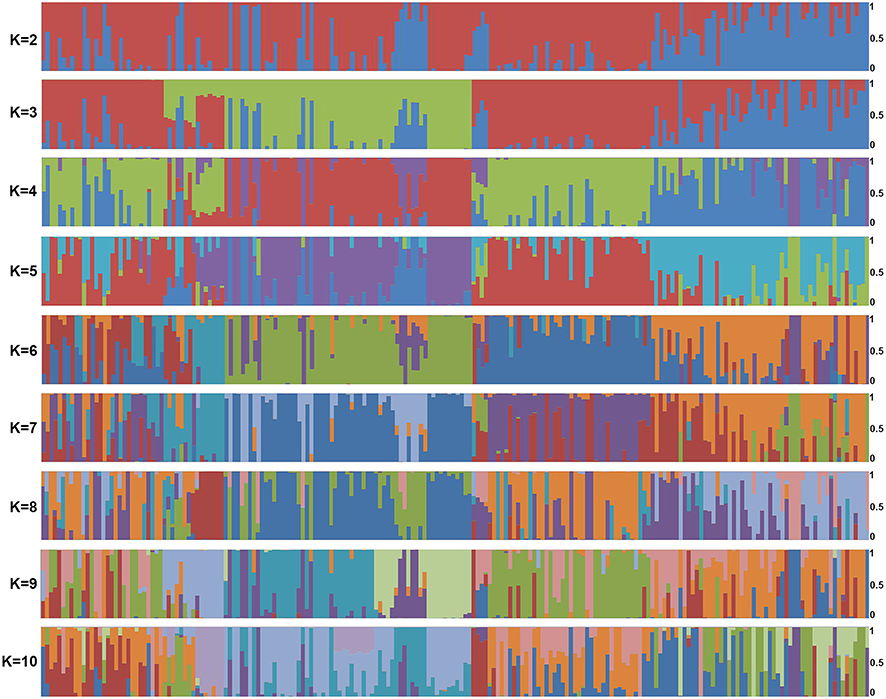 |
Figure 3 Population structure generated by the 204 sequenced T. rubrum strains. Horizontal axis: strains listed in Supplementary Data 1; vertical axis: the fractional representation of populations derived from 204 strains when K = 2–10. |
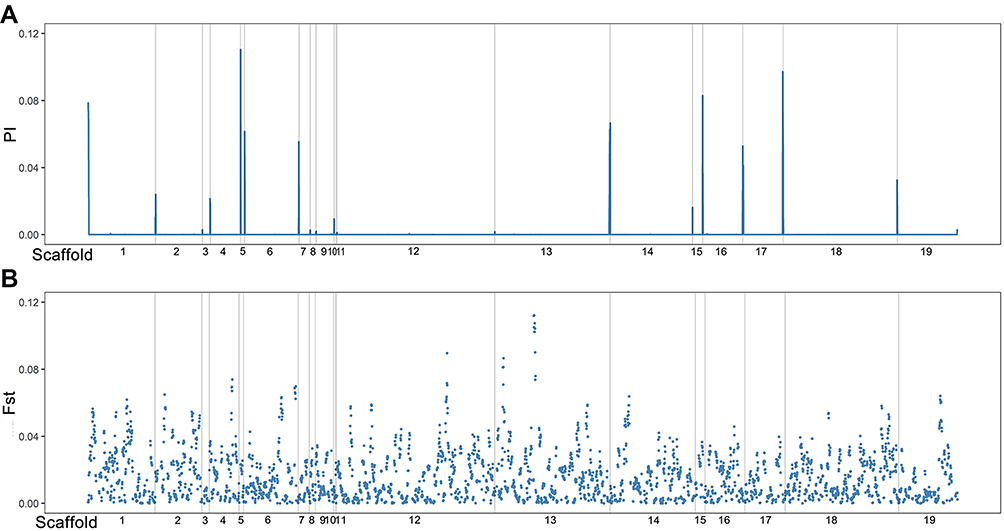 |
Figure 4 Genetic differentiation in the 204 surveyed T. rubrum strains. (A) Genome-wide (5-kb windows) pairwise Fst between stains from Hainan Island and other collecting sites; (B) genome-wide (5-kb windows) nucleotide diversity (π) of surveyed strains. Gray lines: scaffold boundaries. X-axis: scaffolds exhibit in order. |
Discussion
T. rubrum ranks the most common isolated dermatophyte causing dermatophytosis in clinic and has become an epidemic strain worldwide since the 1950s.14,21,35 From ecological and evolutionary points of view, T. rubrum infection recurrence essentially occurs via the adaptation of a group of T. rubrum to the ecological environment and the host environment. Therefore, in-depth study of the population genetic characteristics of T. rubrum, especially its adaptive evolution, may provide guidance for the prevention and treatment of T. rubrum infection.
In this study, we collected T. rubrum from different geographical regions of China. The phylogenetic characteristics and genetic polymorphism distribution pattern of T. rubrum were studied by analyzing genomic sequences. A total number of 41,049 SNPs were identified among 204 isolates, which was much lower than other fungal genera (2,454,052 SNPs among 157 Saccharomyces cerevisiae isolates;32 589,255 SNPs among 182 Candida albicans isolates,30 or 300,116 SNPs among L- and S-type Aspergillus flavus isolates,36 etc.). However, it’s reasonable for T. rubrum to present less SNPs. Zheng et al detected 23,394 genomic variations through 48 sampled T. rubrum strains;17 in another study, an average of 8092 SNPs were identified for 10 T. rubrum isolates compared to reference genome.24 The low number of SNPs reflected the low degree of variation of T. rubrum, which may result from the clonal reproduction of T. rubrum.37
Based on the 41,049 SNPs, we found that the genetic differentiation structure of T. rubrum in China showed no significant relationship with the geographical regions of the patients and that the genetic diversity of the studied isolates was low, which was consistent with previous studies. Numerous studies have confirmed the low genetic diversity of T. rubrum as it has spread around the world in recent decades, and its genetic differentiation remained low levels.17,20,24
However, it is worth noting that the strains from Hainan Island clustered together in the phylogenetic tree, whereas no genetic differentiation was observed in relation to strains outside the island. Previous reports have indicated that T. rubrum is highly clonal. Its reproduction may be asexual, or its sexual reproduction occurs infrequently,24,38 which could be responsible for the low frequency of genetic differentiation observed. Due to the unique geographical location of Hainan Island, which was separated from the mainland, we inferred that the current distribution pattern of T. rubrum on Hainan Island was a result of a founder effect.
Founder effects occur mostly when a new population is formed initially and developed by a few individuals (the founders) from the original population, leading to a low genetic diversity of the new population.6,8,39 The T. rubrum population of Hainan Island experienced a founder effect when it was established. Due to personnel exchanges, people from outside of Hainan enter the island carrying T. rubrum, leading to its spreading and reproduction. Local transmission may occur among populations from contaminated skin flakes, either directly from humans to humans or via environment. The subsequent T. rubrum infections on the island were all caused by descendants of one or several ancestors.
The genetic variability level of island populations is usually lower than mainland populations of the same species.4 As the genetic diversity of T. rubrum is low overall within the species, the T. rubrum population on Hainan Island has maintained low genetic diversity during its evolution and shows no significant differentiation from populations at sites other than Hainan Island. The ecological influence of genetic diversity can be reflected at different levels, from the population to the whole ecosystem.40 High genetic diversity means that populations can better adapt to the environment under selective pressure. In this case, since stains from Hainan Island presented no differentiation from other sites, we suggested no specific modification for the prevention and treatment strategies against T. rubrum.
Based on our findings, we proposed that the T. rubrum population on Hainan Island originated from a specific historical event and reproduced after being introduced to the island. With the increase in cross-strait exchanges, a small number of T. rubrum from other regions have spread and survived on the island. Due to the short evolutionary time (being the epidemic strain worldwide since the 1950s), only a differentiation trend was observed between the population of Hainan Island and those of other regions.
Taken together, the results of this research showed that T. rubrum was introduced into Hainan from off of the island, followed by its evolution for a short time, and there was a trend of differentiation observed in comparison with strains from sites other than Hainan Island. This study discussed general ecological and evolutionary principles with regard to pathogenic fungi. Due to the epidemiological characteristics of T. rubrum (high prevalence, worldwide dominant epidermal pathogens), it can be compared with the genome data of other dermatophytes, and reveals candidate genes related to its pathogenicity or invasion of the host. Besides, association studies with phenotype information such as drug resistance will provide a theoretical basis for development of new antifungal drug and further benefits clinical treatment.
Data and Code Availability
Assembled genomes have been uploaded to NCBI GenBank with BioProject ID PRJNA673795.
Ethics Statement
The authors confirm that the ethical policies of the journal, as noted on the journal’s author guidelines page, have been adhered to and the appropriate ethical review committee approval has been received. This study was approved by the ethics committee of the National Institute for Communicable Disease Control and Prevention (ICDC) and is consistent with the guidelines of the Helsinki Declaration.
Acknowledgments
This work was supported by Youth Program of National Natural Science Foundation of China (Grant Number 81701983), the Foundation for Young Scholars from China CDC (2018A102), Hainan Provincial Natural Science Foundation of China (Grant Number 817314), and Construction Project of Hainan Province Clinical Medical Center.
Disclosure
The authors declare no conflicts of interest.
References
1. Templeton AR. The reality and importance of founder speciation in evolution. BioEssays. 2008;30(5):470–479. doi:10.1002/bies.20745
2. Broders HG, Mahoney SP, Montevecchi WA, Davidson WS. Population genetic structure and the effect of founder events on the genetic variability of moose (Alces alces) in Canada. Mol Ecol. 1999;8(8):1309–1315. doi:10.1046/j.1365-294X.1999.00695.x
3. Alsos IG, Ehrich D, Eidesen PB, et al. Long-distance plant dispersal to North Atlantic islands: colonization routes and founder effect. AoB Plants. 2015;7. doi:10.1093/aobpla/plv036
4. Jones ME, Paetkau D, Geffen E, Moritz C. Genetic diversity and population structure of Tasmanian devils, the largest marsupial carnivore. Mol Ecol. 2004;13(8):2197–2209. doi:10.1111/j.1365-294X.2004.02239.x
5. Fernández-Mazuecos M, Vargas P. Genetically depauperate in the continent but rich in oceanic islands: cistus monspeliensis (Cistaceae) in the Canary Islands. PLoS One. 2011;6(2):e17172. doi:10.1371/journal.pone.0017172
6. Matzke NJ. Model selection in historical biogeography reveals that founder-event speciation is a crucial process in Island Clades. Syst Biol. 2014;63(6):951–970. doi:10.1093/sysbio/syu056
7. Hedrick PW, Gutierrez-Espeleta GA, Lee RN. Founder effect in an island population of bighorn sheep. Mol Ecol. 2001;10(4):851–857. doi:10.1046/j.1365-294X.2001.01243.x
8. Yan W, Hou B, Xue Q, Geng L, Ding X. Different evolutionary processes in shaping the genetic composition of Dendrobium nobile in southwest China. Genetica. 2015;143(3):361–371. doi:10.1007/s10709-015-9835-4
9. Liu ZY, Wang ZL, Yan WY, Wu XB, Zeng ZJ, Huang ZY. The sex determination gene shows no founder effect in the giant honey bee, Apis dorsata. PLoS One. 2012;7(4):e34436. doi:10.1371/journal.pone.0034436
10. Juan X, Weiwei W, Mengshi Z, et al. Molecular epidemiological study on human superficial fungal infections in Hainan Province. J Parasitic Biol. 2019;14(9):1015–1018,1023. in Chinese.
11. Zhan P, Liu W. The changing face of dermatophytic infections worldwide. Mycopathologia. 2017;182(1–2):77–86. doi:10.1007/s11046-016-0082-8
12. Havlickova B, Czaika VA, Friedrich M. Epidemiological trends in skin mycoses worldwide. Mycoses. 2008;51(Suppl 4):2–15. doi:10.1111/j.1439-0507.2008.01606.x
13. Petrucelli MF, Abreu MH, Cantelli BAM, et al. Epidemiology and diagnostic perspectives of dermatophytoses. J Fungi. 2020;6(4):310.
14. Seebacher C, Bouchara JP, Mignon B. Updates on the epidemiology of dermatophyte infections. Mycopathologia. 2008;166(5–6):335–352. doi:10.1007/s11046-008-9100-9
15. Wang L, Ma L, Leng W, et al. Analysis of the dermatophyte Trichophyton rubrum expressed sequence tags. BMC Genomics. 2006;7(1):255. doi:10.1186/1471-2164-7-255
16. Blutfield MS, Lohre JM, Pawich DA, Vlahovic TC. The immunologic response to Trichophyton rubrum in lower extremity fungal infections. J Fungi. 2015;1(2):130–137. doi:10.3390/jof1020130
17. Zheng H, Blechert O, Mei H, et al. Whole-genome resequencing of Trichophyton rubrum provides insights into population differentiation and drug resistance. Mycopathologia. 2020;185(1):103–112.
18. Lipner SR, Scher RK. Onychomycosis: clinical overview and diagnosis. J Am Acad Dermatol. 2019;80(4):835–851. doi:10.1016/j.jaad.2018.03.062
19. Su H, Packeu A, Ahmed SA, et al. Species distinction in the Trichophyton rubrum complex. J Clin Microbiol. 2019;57(9):e00352–e00319. doi:10.1128/JCM.00352-19
20. Ohst T, de Hoog S, Presber W, Stavrakieva V, Graser Y. Origins of microsatellite diversity in the Trichophyton rubrum-T. violaceum clade (Dermatophytes). J Clin Microbiol. 2004;42(10):4444–4448. doi:10.1128/JCM.42.10.4444-4448.2004
21. Gräser Y, Fröhlich J, Presber W, de Hoog S. Microsatellite markers reveal geographic population differentiation in Trichophyton rubrum. J Med Microbiol. 2007;56(Pt 8):1058–1065. doi:10.1099/jmm.0.47138-0
22. Liang G, Zheng X, Song G, et al. Adult tinea capitis in China: a retrospective analysis from 2000 to 2019. Mycoses. 2020;63(8):876–888. doi:10.1111/myc.13102
23. Yu J, Liu WD, Tong ZS, et al. Aetiology of superficial fungal infections of the foot in urban outpatients in mainland China: a multicentre, prospective case study. Mycoses. 2020;63(11):1235–1243. doi:10.1111/myc.13168
24. Persinoti GF, Martinez DA, Li W, et al. Whole-genome analysis illustrates global clonal population structure of the ubiquitous dermatophyte pathogen Trichophyton rubrum. Genetics. 2018;208(4):1657–1669. doi:10.1534/genetics.117.300573
25. Martinez DA, Oliver BG, Gräser Y, et al. Comparative genome analysis of Trichophyton rubrum and related dermatophytes reveals candidate genes involved in infection. mBio. 2012;3(5):e00259–00212. doi:10.1128/mBio.00259-12
26. Ansari S, Hedayati MT, Zomorodian K, et al. Molecular characterization and in vitro antifungal susceptibility of 316 clinical isolates of dermatophytes in Iran. Mycopathologia. 2016;181(1–2):89–95. doi:10.1007/s11046-015-9941-y
27. Badali H, Mohammadi R, Mashedi O, de Hoog GS, Meis JF. In vitro susceptibility patterns of clinically important Trichophyton and Epidermophyton species against nine antifungal drugs. Mycoses. 2015;58(5):303–307. doi:10.1111/myc.12315
28. White TJ, Bruns T, Lee S, Taylor J. PCR Protocols: A Guide to Methods and Applications. San Diego: Academic Press; 1990.
29. Garriss G, Nannapaneni P, Simões AS, et al. Genomic characterization of the emerging pathogen Streptococcus pseudopneumoniae. mBio. 2019;10(3):e01286–e01319. doi:10.1128/mBio.01286-19.
30. Ropars J, Maufrais C, Diogo D, et al. Gene flow contributes to diversification of the major fungal pathogen Candida albicans. Nat Commun. 2018;9(1):2253. doi:10.1038/s41467-018-04787-4
31. Peter J, De Chiara M, Friedrich A, et al. Genome evolution across 1011 Saccharomyces cerevisiae isolates. Nature. 2018;556(7701):339–344. doi:10.1038/s41586-018-0030-5
32. Gallone B, Steensels J, Prahl T, et al. Domestication and divergence of Saccharomyces cerevisiae beer yeasts. Cell. 2016;166(6):1397–1410.e1316. doi:10.1016/j.cell.2016.08.020
33. Danecek P, Auton A, Abecasis G, et al. The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–2158. doi:10.1093/bioinformatics/btr330
34. Wickham H. Ggplot2: Elegant Graphics for Data Analysis. New York: Springer; 2009.
35. White TC, Findley K, Dawson TL
36. Chang PK. Genome-wide nucleotide variation distinguishes Aspergillus flavus from Aspergillus oryzae and helps to reveal origins of atoxigenic A. flavus biocontrol strains. J Appl Microbiol. 2019;127(5):1511–1520. doi:10.1111/jam.14419
37. Graser Y, Kuhnisch J, Presber W. Molecular markers reveal exclusively clonal reproduction in Trichophyton rubrum. J Clin Microbiol. 1999;37(11):3713–3717. doi:10.1128/JCM.37.11.3713-3717.1999
38. Kano R, Isizuka M, Hiruma M, Mochizuki T, Kamata H, Hasegawa A. Mating type gene (MAT1-1) in Japanese isolates of Trichophyton rubrum. Mycopathologia. 2013;175(1–2):171–173. doi:10.1007/s11046-012-9603-2
39. Templeton AR. The theory of speciation via the founder principle. Genetics. 1980;94(4):1011–1038. doi:10.1093/genetics/94.4.1011
40. Hughes AR, Inouye BD, Johnson MT, Underwood N, Vellend M. Ecological consequences of genetic diversity. Ecol Lett. 2008;11(6):609–623. doi:10.1111/j.1461-0248.2008.01179.x
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.