")
Back to Journals » Research and Reports in Urology » Volume 14
Formation of Double Stranded RNA Provokes Smooth Muscle Contractions and Structural Modifications in Bladder Ischemia
Authors Yang JH , Zhao Z , Niu W , Choi HP , Azadzoi KM
Received 2 September 2022
Accepted for publication 11 November 2022
Published 16 November 2022 Volume 2022:14 Pages 399—414
DOI https://doi.org/10.2147/RRU.S388464
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Panagiotis J Vlachostergios
Jing-Hua Yang,1 Zuohui Zhao,2 Wanting Niu,3 Han-Pil Choi,3 Kazem M Azadzoi4
1Department of Surgery, Boston University School of Medicine and Proteomics Laboratory, VA Boston Healthcare System, Boston, MA, USA; 2Department of Urology, Boston University School of Medicine, Boston, MA, USA; 3Research Department, VA Boston Healthcare System, Boston, MA, USA; 4Departments of Urology and Pathology, VA Boston Healthcare System and Boston University School of Medicine, Boston, MA, USA
Correspondence: Kazem M Azadzoi, VA Boston Healthcare System, Building 1A, Room 317 (151), 150 South Huntington Avenue, Boston, MA, 02130, USA, Tel +1 857-364-5602, Email [email protected]
Purpose: Growing evidence suggests that ischemia provokes detrusor overactivity and degenerative responses in the bladder. Underlying mechanisms appear to involve modification of smooth muscle contractile rudiments by hypoxia, redox, cellular stress and cell survival signaling. Downstream pathways of cellular stress and stress response molecules eliciting bladder dysfunction in ischemia remain largely elusive. Our goal was to define the role of double stranded RNA (dsRNA), a stress response molecule provoked by redox, in ischemia mediated bladder dysfunction.
Methods: A rat model of pelvic ischemia along with a cell culture hypoxia model were used to investigate the expression levels, functional consequences, structural aspects, and regulatory mechanisms of dsRNA in the bladder. Gene and protein expression were examined by reverse transcription polymerase chain reaction (RT-PCR), dot blot, and Western blotting, respectively. Tissue structure and function were assessed using histological staining and organ bath. Regulatory mechanisms were analyzed in cultured bladder smooth muscle cells.
Results: The data presented here provide the first evidence of the formation of dsRNA in the overactive bladder. dsRNA is a cellular stress response molecule that sensitizes smooth muscle and regulates inflammatory and degenerative rejoinders. Our data suggest that the production of dsRNA in the bladder is provoked by ischemia. Formation of dsRNA appears to augment bladder smooth muscle contractions and provoke fibrotic and apoptotic responses. Downstream actions of dsRNA in the bladder may involve upregulation of dsRNA-activated protein kinase R (PKR) and caspase-3, the executioner of apoptosis.
Conclusion: Activation of dsRNA/PKR pathway may play a role in sensitization of bladder smooth muscle cells to contractile stimuli, whereas dsRNA and caspase-3 crosstalk appear to modulate cellular stress and instigate degenerative responses in bladder ischemia. These observations suggest the role of dsRNA in bladder dysfunction and may open new perspectives to overcome overactive smooth muscle contractions and structural damage in the bladder.
Keywords: bladder, ischemia, overactivity, dsRNA, contraction, degeneration
Introduction
Ischemia compromises bladder function and leads to structural damage by mechanisms involving metabolic stress, hypoxia, oxidative stress, and activation of the cell survival signaling pathway.1–3 Structural modifications in bladder ischemia include cell shrinkage, condensation and fragmentation of the nucleus, folding and partial disruption of nuclear membrane, degradation of mitochondrial granules, swollen and splintered endoplasmic reticulum, chromatin condensation, accumulation of lysosomes, and increased expression of ribosomes in the nucleus and cytoplasm.2,3 These structural changes result in degeneration of smooth muscle cells and accumulation of connective tissue in the bladder wall.1,2 Functional aberrations in bladder ischemia involve hypersensitivity of smooth muscle cells to contractile stimuli that seems to be mediated by stress response molecules, cellular stress sensors, loss of nerve fibers, differential expression of muscarinic receptors, increased production of eicosanoids and leukotrienes, and post-translational modification of contractile proteins including actin, myosin, caldesmon, calmodulin and tropomyosin.3 These changes were shown to diminish bladder compliance and provoke functional changes consistent with detrusor overactivity.1,2
Recent studies have suggested the involvement of cellular stress and stress response molecules in overactive bladder contractions and structural damage.2,3 However, downstream pathways of cellular stress modulating bladder dysfunction remain largely elusive. Cellular stress in ischemia involves impairment of cellular energy homeostasis due to hypoxia and nutrient deficiency.4,5 Cellular stress upregulates stress response molecules in response to energy deprivation and rejuvenates cellular stress sensors to galvanize protective mechanisms against ischemic insult.6,7 Defensive responses in the stressed cells involve well-coordinated intercommunications between stress sensors and stress response molecules.8,9 Upon sensing stress, cells activate a series of survival signaling cascade to preserve cellular homeostasis and promote survival.8,9 Cellular stress responses are regulated by homeostatic processes that initiate stress signaling and coordinate intercommunication between stress sensors and homeostasis regulating mechanisms.10,11 It was shown that metabolic stress in bladder ischemia interferes with the structural integrity of stress response proteins by mechanisms involving post-translational modifications.3 Such structural alterations in stress response proteins were associated with activation of cell survival signaling via mechanisms involving phosphoinositol 3 kinase (PI3K) and protein kinase B (Akt) survival pathway.1,3
A distinctive feature of cellular stress response appears to be the formation of double-stranded RNA (dsRNA), a unique form of RNA with two complementary strands instead of one single strand in isolation. Formation of dsRNA in response to cellular stress was shown to regulate stress sensors and modulate cellular stress rejoinders.12–16 Formation of dsRNA in the stressed cells has been documented in several organs including brain,12 liver,13 lung,14 heart15 and kidney,16 but its expression and potential role in the overactive bladder has not been previously investigated. Once formed, dsRNA activates stress kinases, namely dsRNA-activated protein kinase R (PKR), which regulates downstream stress signaling pathways and degenerative responses.12–16 Regulation of degenerative processes by dsRNA was shown to involve cleavage of pro-caspases.17 dsRNA interferes with protein transportation and activates proteolysis and protein degradation.16,17 In addition to its role as a translational controlling factor, dsRNA-dependent PKR plays a key role in transcriptional processes and cytoskeleton organization.16,17 Activation of PKR disrupts translocation of transcription factors from cytoplasm to the nucleus and vice versa, resulting in inflammatory responses and structural damage.16,17 In ischemia, stressed cells coordinate endogenous defense mechanisms to counteract hypoxia, nutrient deficiency and redox. dsRNA plays a key role in posttranscriptional conformation of redox responsive genes and transcriptional regulation of the antioxidant response elements.12–15
Accumulation of dsRNA in cells triggers degenerative responses via PKR-mediated provocation of apoptotic molecules.18 When a cell recognizes dsRNA, it is an indication that the cell is stressed and undergoes deterioration.18,19 It was shown that dsRNA-mediated degenerative responses are impelled by disruption of protein synthesis and activation of caspases involved in the execution of apoptosis.19 Caspases have emerged as central mediators linking bladder insults to inflammatory and degenerative responses.20–23 Caspases were shown to interact with inflammatory molecules located intracellularly where they recognize pathologic signals such as pathogen-associated molecular patterns and danger associated molecular patterns.24 Inflammatory molecules interacting with caspases are best known for triggering pyroptosis, a pro-inflammatory process leading to cell lysis.22,23 The activation of these processes has been linked to several events including the production of oxidative radicals, mitochondrial stress, lysosomal damage, cell membrane damage, and cytosolic potassium efflux.25,26 These observations collectively suggest a close link between formation of dsRNA and caspases and the activation of degenerative processes via redox and cellular stress responses.
Formation of dsRNA in the overactive bladder and its potential role in bladder structure and function has not been previously reported. Our goal was to examine the expression levels of Alu RNA elements that are predicted to form a dsRNA structure, determine whether dsRNA is produced in the bladder, define potential structural and functional consequences of dsRNA, and analyze dsRNA intercommunications with degenerative molecules in the bladder.
Materials and Methods
Bladder Ischemia Model
Animal care and experimental protocols were in accordance with the animal care and use guidelines and approval of our Institutional Animal Care and Use Committee. Bladder ischemia was developed in apolipoprotein E knockout (ApoE−/−) rats (n = 8, Envigo, Indianapolis, IN, USA) displaying spontaneous hypercholesterolemia. To promote the development of atherosclerotic occlusive plaques, we performed partial endothelial denudation of the iliac arteries using a 2F Fogarty arterial embolectomy catheter (Baxter Healthcare Corporation, CA), as previously described.1–3 The sham control rats underwent similar surgical procedures without arterial endothelial denudation (n = 8). After 8 weeks, animals were anesthetized with inhalation of 1–2% isoflurane mixed with oxygen, a midline abdominal incision was made, and bladder ischemia was verified as described below.
Laser Speckle Contrast Imaging of Bladder Blood Flow
Direct measurement of bladder microcirculatory blood flow was achieved using the Moor FLPI-2 Laser Speckle Contrast Imaging system (Moor Instruments, Wilmington, DE, USA). Blood flow imaging of large bladder areas was obtained in real time and contact free manner. Laser Speckle Contrast Imaging is capable of measuring bladder tissue perfusion by detecting microcirculation blood flow up to tissue depth of 3 mm. To account for the effects of filling and distension on bladder perfusion, blood flow was recorded in the empty bladder and then at intravesical volumes of 0.4 mL and 0.8 mL. Bladder blood perfusion at five sites were recorded for 30 seconds by positioning the speckle laser 25 cm above the bladder. Mean bladder blood flow was calculated using the moorFLPI-2 Review Software, v. 4.0 (Moor Instruments, Wilmington, DE, USA). After blood flow recording, the animals were euthanized and then bladder tissue samples from the 8 treated and 8 control animals were processed for analysis as described below.
Dot Blot Analysis of dsRNA
Bladder tissue samples were processed for homogenization, and the total RNA was extracted by adding TRIzol reagent (Invitrogen, Waltham, MA, USA) according to manufacturer’s instructions. Ten µg of the total RNA was treated with RNase free DNase (Ambion, Austin, Texas, USA) to remove single stranded RNA. The RNase-resistant RNAs were precipitated with ethanol, resuspended in water, and spotted onto nitrocellulose membrane (Millipore, Burlington, MA, USA). After dried at 80°C for 2 hours, the membranes were incubated with J2 antibody (1:1000) for 1 hour and then with Horseradish peroxidase-conjugated IgG (1:2000) for 1 hour. After several washes, the signals were visualized by enhanced chemiluminescence (Millipore, Burlington, MA, USA). For quantification, the density of each dot was digitalized using Image J.
RT-PCR for Alu RNA Expression
Using the reverse transcription polymerase chain reaction (RT-PCR), Alu RNA expression of ischemic tissue samples was compared versus control. Total RNA was extracted from bladder tissue samples using TRIzol reagent (Invitrogen). Total RNA (1 µg) was analyzed by RT-PCR using qScript cDNA SuperMix (Quanta Biosciences). The expressions of Alu RNA were statistically analyzed in ischemic versus control samples. The primers for the Alu RNA are CATGGTGAAACCCCGTCTCTA for the forward primer and GCCTCAGCCTCCCGAGTAG for the reverse primer, as previously reported.55
Western Blotting
Ischemic and control bladder tissues were homogenized to prepare total tissue proteins. Equal amounts of proteins were separated on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride filter membranes (Millipore, Bedford, MA, USA), which were incubated overnight with the primary antibodies against PKR, phospho-PKR (Biorbyt, St Louis, MO), caspase-3 (Santa Cruz, Dallas TX), or β-actin (Cell Signaling, Danvers, MA, USA), and then with the fluorescent-labelled secondary antibody. Signals from the fluorescent were scanned with Typhoon 8600 imager (GE Healthcare, Pittsburgh, PA, USA) and protein levels quantified by densitometry using Image J.
Assessment of Tissue Contractile Activity
Contractile reactivity of ischemic and control bladder tissues with intact mucosa were examined in the organ bath, as we have previously described.1–3 In brief, tissues were studied in organ baths containing physiologic solution and aerated with 95% O2, 5% CO2. At isometric tension, contractile reactivity to electrical field stimulation (EFS, 10 volts, 0.8 msec and varying frequencies) was examined before and then after tissue treatment with the dsRNA/toll-like receptor 3 (TLR3) complex inhibitor 614310 (Sigma Aldrich, St Louis, USA). In another set of experiments, control bladder tissues were treated with the PKR activator DHBDC (Sigma Aldrich, St Louis, USA) at resting tension and spontaneous contractile activity after treatment was compared to before treatment.
H&E, Trichrome, and Immunohistochemical Staining
Frozen bladder tissues were sectioned at 8 µm with a cryostat and stained routinely for hematoxylin and eosin (H&E) staining and Masson’s trichrome staining, using the standard protocols. For immunohistochemical staining of caspase-3, the sections were heated in antigen unmasking solution (Vector, Burlingame, CA, USA) according to the manufacturer's instruction. Sections were blocked by 5% bovine serum albumin (BSA) for 30 min before applying the primary antibody against caspase-3 (Novus, Centennial, CO, USA) at 4°C overnight. Protein visualization was achieved by using VisUCyte mouse/rabbit IgG antibody (R&D systems, Minneapolis, MN, USA) and AEC+ substrate chromogen (Dako, Santa Clara, CA, USA). The stained sections were digitally imaged by MoticEasyScan Pro6 (Motic, Hong Kong).
Detection of Apoptotic Cells by TUNEL Staining
Terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) assay kit (Abcam, Boston, MA, USA) was used, according to the manufacturer's protocol with some modifications. In brief, the optimal cutting temperature compound was removed from the cryo sections, and then the sections were permeabilized by proteinase K for 20 min, and the endogenous peroxidase was quenched by 3% H2O2. After equilibration, the sections were incubated with the terminal deoxynucleotidyl transferase labeling reaction mixture for 2 hours at room temperature, then the labeling was stopped, and the sections were blocked. The TUNEL-positive cells were then visualized by a horseradish peroxidase conjugation-diaminobenzidine system, and the nuclei were counter stained by Gill 2 hematoxylin. The sections were mounted in an aqueous-based mounting medium and scanned by the MoticEasyScan Pro-6.
Cell Culture and Transfection
Cell culture dishes of primary human bladder smooth muscle cells (Lonza, Allendale, NJ, USA) were prepared, then randomly divided into treatment and control groups. The treatment group of cells was transfected with the dsRNA analogue polyinosinic-polycytidylic acid (Poly[I]-Poly[C]) sodium salt, namely, dsRNA poly(I:C) using lipofectamine (Sigma Aldrich, St Louis, USA), as we have previously reported.38,39 Control cells were transfected with lipofectamine only containing 0 µg dsRNA poly(I:C). After 12 hours, viable cells were collected and processed for Western blot analysis. Caspase-3 expression in cells transfected with 8 µg and 24 µg dsRNA poly(I:C) was analyzed versus cells transfected with lipofectamine only containing 0 µg dsRNA poly(I:C).
Statistical Analysis
Data are expressed as mean ± standard error of the mean. Data involving two-group comparisons were analyzed by Student’s t-test. Data involving greater than two groups were analyzed by analysis of variance (ANOVA) followed by post hoc comparisons using SigmaPlot statistical software. Significant differences were determined at p < 0.05 level.
Results
Occlusive Disease in Bladder Microcirculation
Our initial efforts were focused on defining the structure of bladder microcirculation and the status of bladder blood perfusion in the rat model of bladder ischemia. Hematoxylin and eosin (H&E) staining of five ischemic bladder samples were compared versus five control samples. Assessment of bladder microcirculation revealed an interesting phenomenon suggesting spreading of arterial occlusive disease from the iliac arteries to the arterioles of the bladder wall. Bladder tissues from apolipoprotein E knockout rats (ApoE−/−) revealed an intriguing structural manifestation that atherosclerotic occlusive disease extends from the site of endothelial balloon denudation of the iliac arteries to smaller arteries, ultimately involving microcirculation of the bladder wall (Figure 1). Marked atherosclerotic occlusive disease characterized by thickening of the intima, swollen subendothelial layer, arterial luminal plaques, and conspicuous decrease in arteriolar luminal size were evident in the ischemic bladder tissues from ApoE−/− rats undergoing iliac artery ballooning described in Methods (Figure 1). In addition, inflammatory changes characterized by swollen epithelium, thickening of lamina propria, irregular nuclear distribution, mucosal hyperemia, loss of mucosal structural integrity, suburothelial hyperplasic denuded areas and dense irregular connective tissue were evident in the ischemic bladder (Figure 1).
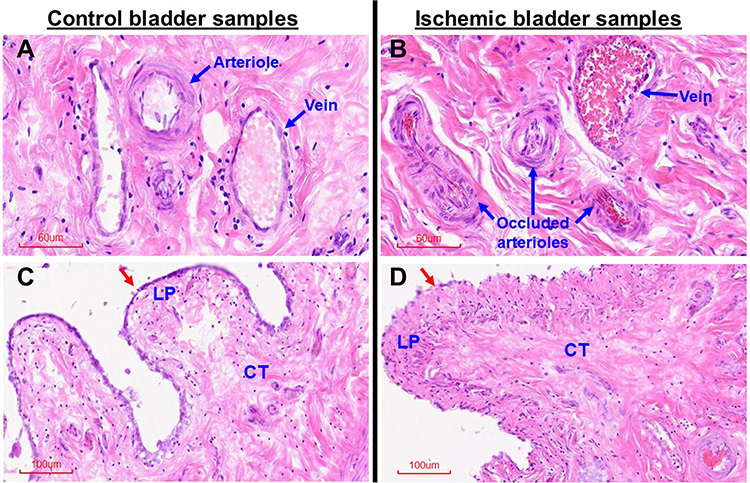 |
Figure 1 H&E staining of bladder tissues. This figure shows morphologic changes in microvasculature (blue arrows), mucosa (red arrows), lamina propria (LP) and suburothelial connective tissue (CT) of the ischemic tissue samples in comparison with control samples. (A) shows normal bladder wall arterioles and vein in the control sample. (B) shows thickening of arteriolar intima and marked decrease in arteriolar luminal size suggesting atherosclerotic occlusive disease in the arterioles of the ischemic bladder. (C) shows normal mucosal lining (red arrow) and normal LP and CT in the control sample. (D) shows structural changes in the mucosa, LP and CT of the ischemic sample in comparison with corresponding structures in the control sample shown in (C). Red arrow in (D) points to irregular mucosal layer surrounding a congested edematous LP in the ischemic sample. Dense CT suggesting accumulation of connective tissue is evident in the ischemic bladder sample (D) in comparison with control sample (C). (A and B) are shown at 20X magnification. (C and D) are shown at 10X magnification. |
Impairment of Bladder Blood Perfusion
Bladder microcirculatory blood flow was assessed directly using the moorFLPI-2 Laser Speckle Contrast Imaging system (Moor Instruments, Wilmington, DE, USA), as described in Methods. The advantage of this system over our previously reported Laser Doppler flowmetry1–3 is that it allows imaging of large bladder areas in a real time and contact free manner. The Laser Speckle Contrast Imaging system is capable of full field laser measurements and determines tissue perfusion by detecting mean microcirculatory blood flow up to tissue depth of 3 mm. Using this measurement system, we compared bladder blood perfusion of eight rats with arterial atherosclerosis versus eight sham controls. Under general anesthesia, a lower abdominal incision was made, and the bladder was exposed, cleaned with saline, and prepared for blood flow imaging. To account for the effects of filling and distension on bladder blood flow, we measured perfusion in the empty bladder and then at intravesical volumes of 0.4 mL and 0.8 mL. The speckle laser was positioned 25 cm above the bladder. For each measurement, bladder blood perfusion was assessed for a 30 second period. Bladder blood flow was recorded, and the mean blood flow was determined using the moorFLPI-2 Review Software, v. 4.0 (Moor Instruments, Wilmington, DE, USA). Our measurements revealed significantly lower bladder blood flow in animals with arterial atherosclerosis in comparison with controls in the empty bladder and at 0.4 and 0.8 mL intravesical volumes (Figure 2). In the control animals, bladder filling at 0.4 and 0.8 mL had no significant effect on bladder blood perfusion in comparison with blood perfusion in the empty state. In animals with arterial disease, filling at 0.8 mL significantly decreased bladder perfusion in comparison with blood perfusion in the empty state, suggesting that filling and distension exacerbate blood perfusion deficit in bladders that were already ischemic (Figure 2).
 |
Figure 2 Bladder blood perfusion. Laser Speckle Contrast Imaging of bladder microcirculatory blood perfusion is shown in an animal with arterial atherosclerosis versus a sham control animal. The average data from eight atherosclerotic and eight control animals are shown on the right. The data shows significantly lower bladder blood perfusion in animals with arterial atherosclerosis in the empty bladder and after filling at 0.4 mL and 0.8 mL intravesical volumes, suggesting bladder ischemia. Filling caused further decrease in bladder perfusion reaching significance at 0.8 mL. *In the ischemia samples indicate significant decrease in bladder perfusion versus blood perfusion at corresponding intravesical volumes in the control group. µ indicates significantly lower bladder perfusion at 0.8 mL intravesical volume versus empty bladder in the ischemia group. |
Ischemia Provoked dsRNA Formation in the Bladder
Formation of dsRNA in the overactive bladder has not been previously reported. The role of dsRNA in the pathophysiology of bladder dysfunction remains virtually unknown. We previously reported that ischemia provokes cellular stress responses in the bladder.1–3 Our present study suggests that bladder ischemia may be an important intervening variable in the formation of dsRNA, a stress signaling molecule. Causative factors contributing to dsRNA formation in bladder ischemia may involve redox, a common phenomenon in ischemia that is strongly linked to the production of dsRNA.27 Our data imply concomitant upregulation of Alu RNA transcription and dsRNA formation in bladder ischemia (Figure 3). Alu RNA represents a family of repetitive sequences, and their sense and antisense transcripts are usually induced prior to dsRNA formation.26–28 In dot blot experiments, equal amounts of total RNAs from the ischemic and control bladder tissues were processed onto nitrocellulose membrane and detected with the J2 dsRNA-specific antibody, as described in Methods. The data revealed significantly greater levels of dsRNA in the ischemic bladder tissues in comparison with control bladder samples (Figure 3A). Concurrently, Alu RNA expression detected by reverse transcription polymerase chain reaction (RT-PCR) showed significant upregulation in the ischemic bladder tissues versus controls (Figure 3B). Because of their repetitive nucleotide sequence, Alu RNA is recognized as a precursor of dsRNA.27 Simultaneous upregulation of Alu RNA and dsRNA may signal activation of stress response in bladder ischemia. Detection of upregulation of dsRNA and Alu RNA in bladder ischemia led us to examine the status of dsRNA-dependent PKR.
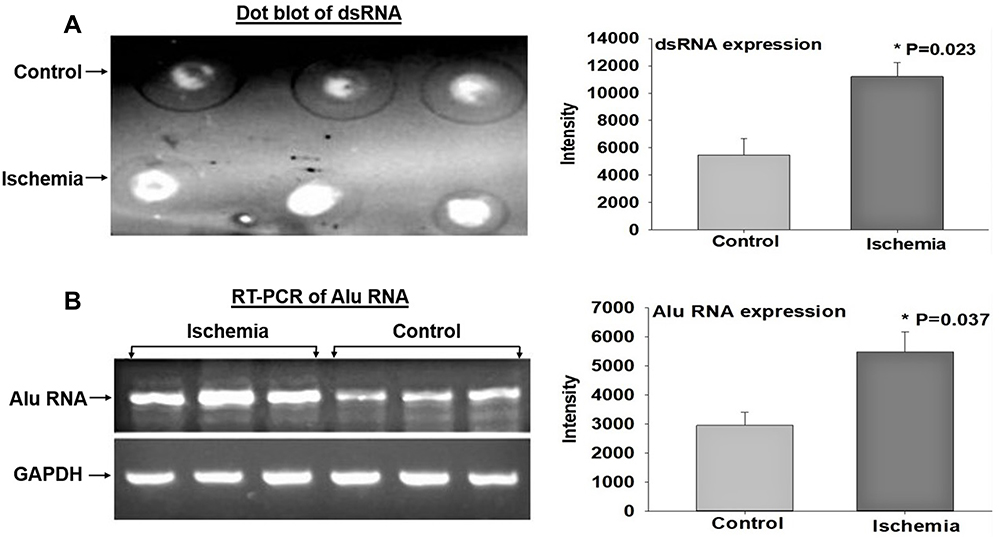 |
Figure 3 Expression of dsRNA and Alu RNA. (A) shows a dot blot sample of dsRNA on the left and average data from three dot blot experiments on the right. (B) shows a RT-PCR sample of Alu RNA on the left and average data from three RT-PCR experiments on the right. The data suggest significant increases in dsRNA and Alu-RNA expression in the ischemic bladder tissues. Total RNAs from the ischemic and sham control bladder tissues were spotted onto nitrocellulose membrane and detected with the J2 antibody or amplified by RT-PCR and analyzed on agarose gel. The intensities were digitalized with Image J and normalized with background or GAPDH. *Indicates significant difference in ischemia versus control samples. |
Expression of dsRNA-Dependent PKR in Bladder Ischemia
The dsRNA-activated protein kinase R (PKR) is a copiously expressed serine/threonine protein kinase that is activated by dsRNA, cytokine, growth factor and cellular stress.28 PKR is an essential kinase for cells to respond adequately to cellular stress conditions including ischemia.28 Our data suggest significant upregulation of both total PKR and phosphorylated PKR (phospho-PKR) in the ischemic bladder (Figure 4), suggesting upregulation and phosphorylation and thus activation of PKR by dsRNA under the ischemic conditions.1–3 Our findings are consistent with previous studies reporting regulation of PKR by ischemia and the involvement of PKR in cellular stress and structural damage.28,29 It was shown that stress granules recruit PKR to the stressed cells to activate cellular stress responses.28 Activation of PKR by dsRNA bridges ischemic insult with cellular stress response molecules to instigate inflammatory and degenerative responses.28,29
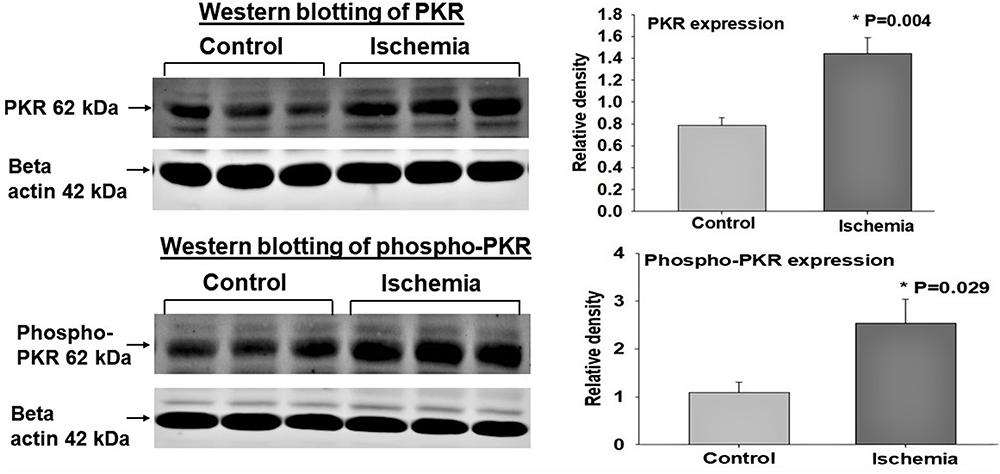 |
Figure 4 Expression of dsRNA-dependent PKR and phospho-PKR. Tissue Western blot samples are shown on the left and analyzed data from three Western blot experiments are shown on the right. The expression levels of dsRNA-dependent PKR and phospho-PKR were significantly greater in the ischemic bladder tissues in comparison with controls, suggesting a link between cellular stress and PKR expression. Upon binding to dsRNA, PKR undergoes autophosphorylation.28–30 It was shown that activated PKR regulates translation and numerous inflammatory and degenerative signaling pathways by catalyzing phosphorylation of substrate targets.29 PKR senses cellular stress and promotes cell death by blocking protein synthesis and activating apoptosis under metabolic stress conditions such as ischemia.30 The induction of apoptosis by PKR was shown to involve the activation of caspase pathways.30 *Indicates significant difference in ischemia versus control samples. |
Liver PKR protein levels were shown to increase in response to stress eliciting conditions.29 PKR actions in cellular stress were shown to depend on the intensity of stressful stimuli. PKR inactivation by gene knockout or pharmacological inhibition was associated with lower levels of inflammatory cytokines and less apoptotic cell death.29 Our observations suggest that formation of dsRNA and subsequent activation of PKR may play a modulating role in smooth muscle dysfunction and degenerative responses in bladder ischemia.28,29
The Role of dsRNA/PKR in Bladder Smooth Muscle Contractions
It has been reported that production of dsRNA alters contractile activity of smooth muscle cells by upregulating stress response molecules in the muscle cells, nerve fibers and receptors of the lung and heart.14,15 Accumulation of dsRNA in the lung was shown to sensitize the airway smooth muscle cells to contractile stimuli by mechanisms involving muscarinic M2 receptors.14 dsRNA-dependent PKR interferes with heart muscle contractility and contributes to the development of heart failure induced by systolic overload.15 We have previously reported that ischemia increases contractile reactivity of bladder smooth muscle cells.1,2 In the present study, contractile reactivity of ischemic and control bladder tissues to increasing frequencies of electrical field stimulation (EFS) was examined before and then after treatment with the dsRNA/toll-like receptor 3 (TLR3) complex inhibitor 614310 (Sigma Aldrich, St Louis, USA), using organ bath. TLR3 binding to dsRNA results in the formation of dsRNA/TLR3 complex and activation of TLR3 leading to functional modifications in the affected tissues.31 Treatment with 614310 decreased contractile reactivity of the ischemic tissues to the control levels in response to all EFS frequencies while having no significant effect on the control tissues (Figure 5A), suggesting the involvement of dsRNA in increased contractile reactivity of the ischemic bladder tissues. In another set of experiments, we found that treatment of control bladder tissues with the PKR activator DHBDC (Sigma Aldrich, St Louis, USA) provokes spontaneous contractile activity at resting baseline tension (Figure 5B), suggesting that PKR promotes bladder smooth muscle contractions.
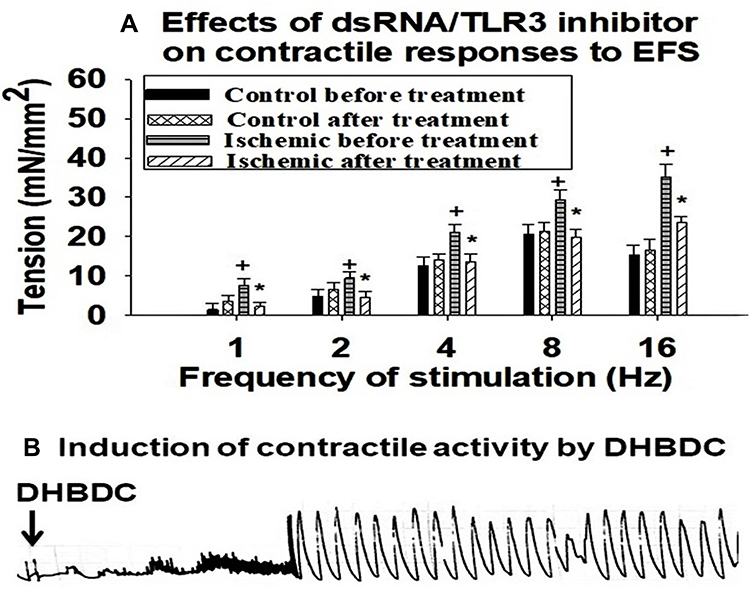 |
Figure 5 Involvement of dsRNA and dsRNA-dependent PKR in increased contractile reactivity of ischemic bladder tissues. (A) is average data from five organ bath experiments suggesting decreased contractile reactivity to EFS after inhibition of the dsRNA/TLR3 complex. The data implies dsRNA involvement in increased reactivity of the ischemic bladder tissues to contractile stimuli. (B) shows incitement of spontaneous contractile activity by DHBDC, suggesting mediating role of PKR in dsRNA provoked contractile activity in the ischemic bladder. +Indicates significantly greater contractions of ischemic tissues versus control samples before treatment with the dsRNA/TLR3 inhibitor. *Indicates significant decrease in contractile reactivity of the ischemic tissues after treatment with the dsRNA/TLR3 inhibitor versus contractions of ischemic tissues before treatment. |
Structural Consequences of dsRNA in the Bladder
To determine whether accumulation of dsRNA plays a role in structural modifications in the ischemic bladder, we carried out structural analysis and immunostaining and Western blotting of caspase-3 to examine the role of dsRNA in structural changes of the ischemic bladder. Structural features of the ischemic bladder were examined using H&E staining as described earlier in Figure 1 and Masson’s trichrome staining as described below. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was used to search for evidence of apoptotic cell death in the ischemic bladder tissues.
Masson’s Trichrome Staining
Five ischemic bladder samples were compared versus five control samples. Structural assessment of the ischemic bladder tissues by Masson’s trichrome staining revealed loss of smooth muscle cells that appeared to be replaced by dense connective tissue. Tissue samples from the control bladders showed normal distribution of smooth muscle (red stain) and connective tissue (blue stain) with no evidence of aberrant collagen accumulation (Figure 6A). However, dense bundles of collagen were evident in the ischemic tissue samples, suggesting diffuse fibrosis and potential replacement of smooth muscle rudiments by connective tissue (Figure 6B), as detailed in image analysis below.
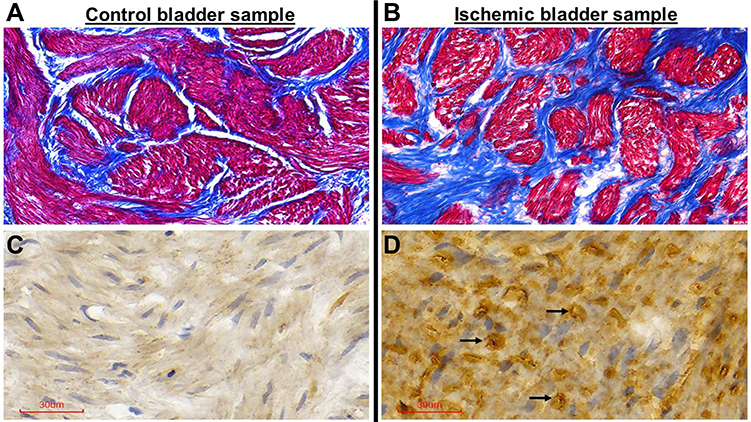 |
Figure 6 Masson’s trichrome and TUNEL staining of bladder tissues. Sections of bladder tissues stained with Masson’s trichrome and TUNEL staining show diffuse fibrosis and widespread apoptotic cells, respectively, in the ischemic samples (B and D) versus control samples (A and C). Trichrome staining of the control sample (A) shows normal distribution of smooth muscle fibers (red stain) and connective tissue (blue stain) while accumulation of dense connective tissue (blue stain) was evident in the ischemic tissues (B). Arrows in the TUNEL stained ischemic sample (D) point to some of the apoptotic cells. All sections are shown at 40X magnification. |
TUNEL Staining
Follow-up experiments were carried out using TUNEL staining to examine whether apoptosis plays a role in loss of smooth muscle cells and accumulation of connective tissue in the ischemic bladder. TUNEL staining, also called the TUNEL assay, is a well-established method to detect cell death. TUNEL staining identifies apoptotic cells undergoing DNA breaks as a consequence of DNA fragmentation in the last phase of apoptosis. In our study, five ischemic samples were compared versus five controls. TUNEL staining suggested evidence of widespread apoptotic cell death in the ischemic bladder tissues (Figure 6D) in comparison with the controls (Figure 6C) as detailed below in image analysis.
Image Analysis
Computerized image analysis of trichrome stained samples revealed a significant decrease in the percentage of smooth muscle in the ischemic bladder tissues in comparison with controls. The percentage of smooth muscle per high power field in samples from five ischemic bladders was 47.83% ± 3.02, which was significantly less than 62.17% ± 3.06 in five control bladder samples (p = 0.008). In histomorphometric image analysis of TUNEL stained samples, the percentage of apoptotic cells per high power field in five ischemic samples was 19.83 ± 2.2, which was significantly greater than 0.3 ± 0.05 in five control samples (p = 0.001). The close link between bladder apoptosis and fibrosis in our study is consistent with reported provocation of fibrosis by apoptosis in other organs.32–34
Immunostaining of Caspase-3
We aimed to examine the expression levels of caspase-3, a well-known apoptotic factor in the bladder2 that interacts with dsRNA.35,36 Caspase-3 acts as a cellular stress intensity sensor with the ability to eliminate damaged cells by activating the execution of apoptosis.2,37 Apoptotic properties of dsRNA were shown to be modulated by the activation of caspases.35 The dsRNA/caspase apoptotic pathway is activated by various cellular stress inducers including ischemia.35–37 Caspase-3 was shown to mediate degenerative responses triggered by ischemia via mechanisms involving DNA damage and protein degradation.36 We initiated caspase-3 assessment using immunostaining and then verified caspase-3 expression by Western blotting, as described below. Our immunostaining suggested marked upregulation of caspase-3 in five ischemic bladder samples versus five controls (Figure 7).
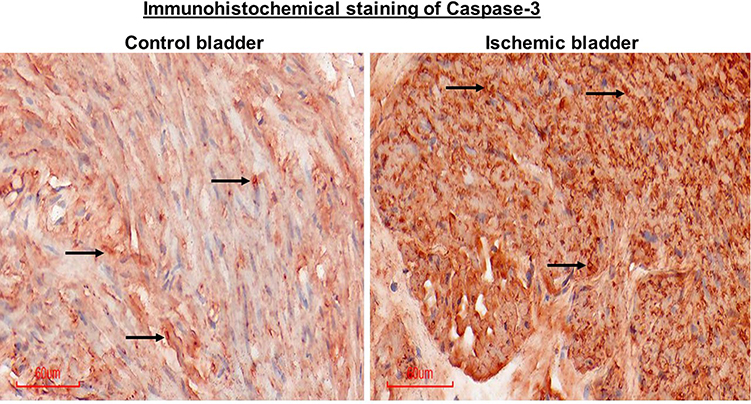 |
Figure 7 Immunohistochemical staining of caspase-3 in bladder tissues. The ischemic bladder tissue shows greater caspase-3 immunopositive cells in comparison with tissue from the control bladder, suggesting apoptotic activity in the ischemic samples. Arrows point to a few immunopositive cells in the two samples. Figures are shown at 20X magnification. |
Regulation of Caspase-3 by dsRNA
Two sets of Western blotting experiments were carried out using bladder tissues and cell culture samples. Tissue Western blotting revealed significant increase in caspase-3 expression in the ischemic bladder tissues versus controls (Figure 8, upper panel), implying upregulation of caspase-3 in bladder ischemia. Western blotting of cultured human bladder smooth muscle cells suggested potential crosstalk mechanisms between dsRNA and caspase-3, as described below. To examine the interrelationship between dsRNA and caspase-3 in the bladder, primary human bladder smooth muscle cells were cultured according to the standard protocols and processed for transfection with dsRNA analogue polyinosinic-polycytidylic acid (Poly[I]-Poly[C]) sodium salt, namely, dsRNA poly(I:C) using lipofectamine, as we have previously reported.38,39 At 12 hours post transfection, viable cells were collected and processed for Western blotting of caspase-3. The expression level of caspase-3 in cells transfected with 8 µg and 24 µg dsRNA poly(I:C) was significantly greater in comparison with cells transfected with lipofectamine only containing 0 µg dsRNA poly(I:C) (Figure 8, lower panel), suggesting regulation of caspase-3 by dsRNA and potential role of dsRNA in caspase-3 upregulation and ensuing apoptotic responses in bladder ischemia.
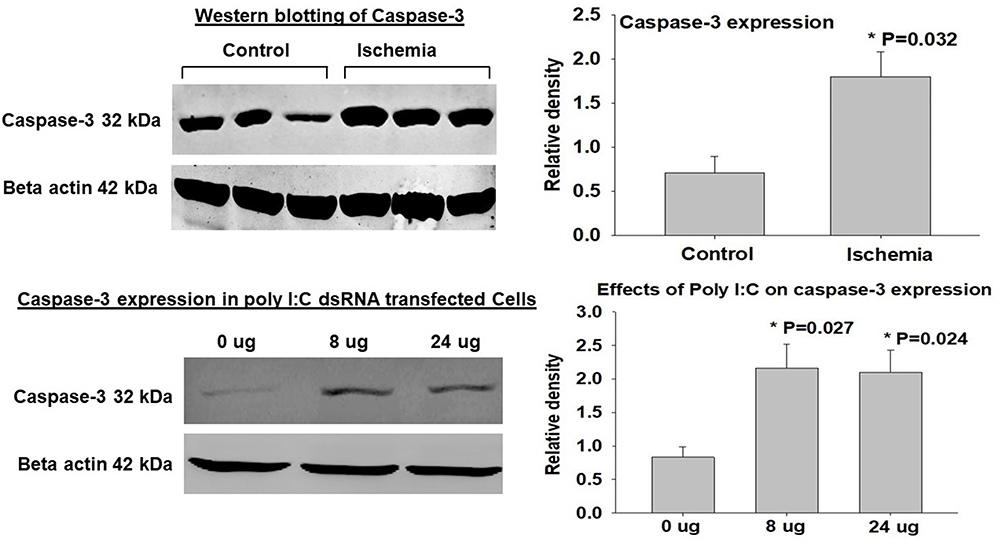 |
Figure 8 Expression of caspase-3. The upper panel shows a tissue Western blotting sample on the left and average data from three Western blot experiments on the right. The data suggest significant upregulation of caspase-3 in samples from the ischemic bladder in comparison with controls. *In the upper panel indicates significance versus control samples. In the lower panel, a sample of caspase-3 expression in poly I:C transfected cells is shown on the left and average data from three transfection experiments are shown on the right. *In the lower panel indicates significant caspase-3 upregulation in the 8 µg and 24 µg samples versus 0 µg samples. The data show significantly greater caspase-3 expression in cultured human bladder smooth muscle cells transfected with dsRNA poly(I:C), suggesting upregulation of caspase-3 by dsRNA in the bladder. |
Discussion
An interesting observation in the rat model of bladder ischemia relates to the spreading of arterial occlusive disease from the site of endothelial denudation in the iliac arteries to smaller pelvic vessels that seems to extend to the bladder wall and involve bladder arterioles. Occlusive disease of the bladder blood supply was verified by measurement of blood flow with Laser Speckle Contrast Imaging system showing a significant decrease in bladder perfusion in ApoE−/− rats undergoing arterial ballooning in comparison with sham controls (Figure 2). Impairment of bladder perfusion in the ApoE−/− rats was exacerbated by bladder filling and distention. Filling at 0.4 and 0.8 mL had no significant effect on bladder perfusion in control rats while causing a significant decrease in bladder perfusion of ApoE−/− rats, suggesting augmentation of preexisting bladder ischemia by filling and distention (Figure 2). Impairment of bladder blood flow in the ApoE−/− rats was associated with morphological changes characterized by irregularity of the mucosal layer, swollen edematous lamina propria and accumulation of connective tissue as shown in Figure 1.
To the best of our knowledge, the data presented here provide the first evidence of dsRNA formation in the overactive bladder. Our data suggest that production of dsRNA and activation of downstream signaling pathway in the overactive bladder may be provoked by ischemia. Upregulation of dsRNA in the ischemic bladder was associated with significant increase in Alu RNA expression, suggesting intercommunication between the two molecules. Alu repeats are recognized as the most common source of dsRNA in human cells.27,40 Because of its repetitive nucleotide sequence, it is widely believed that Alu RNA is a precursor of dsRNA and has the capacity to regulate dsRNA sensors.27 It was shown that upregulation of Alu RNA precedes the formation of dsRNA upon cell exposure to stressful conditions.28 Simultaneous upregulation of Alu RNA and dsRNA in the ischemic bladder may signal cellular stress due to hypoxia and disruption of homeostasis. Major sources of endogenous dsRNA include mitochondrial transcripts, repetitive nuclear sequences, endogenous retroviruses, and natural sense-antisense transcript pairs.40 While cells recognize both exogenous and host-derived dsRNA, underlying mechanisms determining stressed cells fate by dsRNA remain unclear. Our observations suggest that accumulation of dsRNA and subsequent activation of cellular stress responses via dsRNA-dependent protein kinase R (PKR) may contribute to functional changes in the ischemic bladder.
Downstream actions of dsRNA are mediated by activation of PKR. Upon binding to dsRNA, PKR undergoes autophosphorylation at multiple serine, threonine, and tyrosine residues.41 It was shown that activation of PKR by dsRNA requires binding of at least two or more PKR monomers.41 PKR is a member of the family of cellular stress response kinases that inhibits translation by phosphorylation of the stress response proteins. Similar to other protein kinases, catalytic domains of the PKR encompass N-terminal and C-terminal lobes. Stress response kinases including PKR contain a conserved catalytic domain that is linked to different regulatory domains. Regulatory element of PKR consists of an N-terminal dsRNA binding domain consisting of two tandem dsRNA binding motifs.41 The dsRNA binding domain of PKR recognizes dsRNA and distinguishes it from dsDNA or RNA-DNA heteroduplexes.42,43 The ability of dsRNA to act as a PKR activator is correlated with binding of two or more PKR monomers that sequentially attach to a single dsRNA.42,43 These observations suggest that PKR activation involves a mechanism where dsRNA brings two or more PKR monomers in close proximity to enable dimerization by the kinase domain.
Structural and functional analysis of ischemic bladder tissues suggested that activation of dsRNA/PKR and dsRNA/caspase pathways may contribute to overactive contractions and structural damage, respectively. Our observations are consistent with structural and functional consequences of dsRNA reported in other organs including brain, liver, lung, and heart.12–17 Precise mechanisms underlying functional role of dsRNA in the bladder remain elusive. Our data suggest that upregulation of PKR by dsRNA and its subsequent phosphorylation in the ischemic bladder may augment smooth muscle contractile activity. Concomitant upregulation of dsRNA and caspase-3 in the ischemic bladder tissues along with upregulation of caspase-3 by dsRNA in cultured bladder smooth muscle cells suggest the involvement of dsRNA in the development of fibrosis, loss of smooth muscle cells, and apoptotic degenerative responses in the bladder.
Our studies in the organ bath revealed sensitization and over-reactivity of ischemic bladder tissues to electrical field stimulation, as shown in Figure 5. Tissue treatment with the dsRNA/toll-like receptor 3 (TLR-3) complex inhibitor significantly diminished contractile reactivity of ischemic tissues while having no significant effect on control tissues (Figure 5). Furthermore, pharmacological activation of PKR in control tissues by DHBDC provoked spontaneous contractile activity at resting tension in the organ bath. These observations suggest that sensitization of bladder smooth muscle cells to contractile stimuli may be mediated, at least in part, through the formation of dsRNA, recognition of dsRNA by TLR-3, and subsequent activation of PKR. Our findings are consistent with reported sensitization and over-reactivity of pig’s lung airway after exposure to the dsRNA analogue poly(I:C).14 It was shown that treatment with dsRNA poly(I:C) sensitizes the airway smooth muscle to electrical stimulation of the vagal nerve leading to bronchoconstriction.14 This action of dsRNA appears to involve increased acetylcholine release from the vagal nerve and dysfunction of the muscarinic M2 receptors.14 In addition, dsRNA was shown to induce dysfunction of the prejunctional muscarinic receptors on parasympathetic nerves supplying the heart.14 Other studies have suggested a close link between formation of dsRNA and the development of arterial hypertension.44 It is postulated that accumulation of dsRNA and subsequent activation of PKR provokes hypertension by promoting contractile reactivity of vascular smooth muscle cells and increasing vascular tone.44 Studies of mice revealed that treatment with dsRNA poly(I:C) impairs endothelium-dependent relaxation of the blood vessels resulting in increased contractile activity.45 This study suggests that recognition of dsRNA by the endothelial cells alters endothelium-mediated vascular function leading to diminished relaxation and increased contractile activity.45
Structural modifications in bladder ischemia characterized by swollen epithelium, thickening of lamina propria, irregular nuclear distribution, mucosal hyperemia, loss of mucosal structural integrity, suburothelial hyperplasia, loss of smooth muscle cells and fibrosis were associated with markers of apoptosis characterized by positive TUNEL staining and upregulation of caspase-3. Positive TUNEL staining implies DNA breaks as a consequence of DNA fragmentation. Caspase-3 is a sensor of cellular stress intensity and a leading executioner of apoptosis.36,46 This action of caspase-3 was shown to involve DNA damage and protein degradation.36,46 Caspase-3 has the ability to determine the ischemic cell fate by either assembling defensive responses to promote survival or activating the execution of apoptosis.36,46 Regulation of cell fate by caspase-3 is delimited by a series of tightly regulated mechanisms. Caspases may display protective properties by activating cell survival pathway when cells are exposed to mildly stressful conditions.47,48 In contrast, caspases exhibit degenerative apoptotic properties by cleaving a large number of caspase substrates when cells are stressed by abrasive conditions.47 In mild and transient ischemic conditions, caspases are capable of regulating cellular homeostasis to protect cells from metabolic stress.48 Cell culture studies have suggested promotion of cell survival pathway by low caspase activity via mechanisms involving protein kinase B.48 It was shown that activation of protein kinase B in response to low cellular stress was hindered by chemical and genetic inhibition of caspase-3.48 These observations suggest that caspase responses to mild and severe stressful stimuli regulate cell fate upon exposure to stressful conditions.
Concomitant evidence of apoptotic markers and fibrotic changes in bladder ischemia may imply apoptosis as a provoking factor in the development of fibrosis. This prospect is supported by observations in the lung suggesting that blockade of apoptosis can diminish collagen deposition and prevent the development of fibrosis.32 Studies of tissue samples from human and animals have provided further support that apoptotic processes may be involved in idiopathic fibrotic changes in the lung.33 Furthermore, signaling pathways involved in inflammatory processes were shown to play a role in the regulation of apoptotic and pyroptotic cell death.34 Recruitment of inflammasomes activated apoptotic processes in cultured cells.34 Markers of apoptosis and fibrosis in bladder ischemia were associated with simultaneous upregulation of dsRNA and caspase-3 suggesting potential crosstalk between the two molecules. We examined this perspective in cell culture with the intention to define potential intercommunication between dsRNA and caspase-3. Exposure of human bladder smooth muscle cells to dsRNA poly(I:C) resulted in significant upregulation of caspase-3, suggesting potential regulation of caspase-3 and ensuing apoptotic responses by dsRNA.
Apoptotic action of dsRNA in endothelial progenitor cells was shown to be mediated by activation of caspases through mechanisms involving PKR.18 Upregulation of caspases by PKR appeared to involve phosphorylation of eukaryotic initiation factor 2 and subsequent incitement of apoptotic responses via Fas-associated death domain.18 Caspases were also shown to activate Fas-associated cell domain, cytochrome-c, and apoptosis protease activating factor.49,50 Treatment of cultured cells with the synthetic dsRNA Poly(I:C) was shown to induce apoptotic cell death by mechanisms involving caspases.51 More recently, the endosomal membrane-bound Toll-like receptor 3 (TLR3) was identified as a dsRNA receptor with the capability of triggering apoptosis.52 Components of TLR3 signaling involved in dsRNA-mediated cell death were shown to be mediated by caspases and receptor interacting protein 1.53,54 These observations suggest that apoptotic action of dsRNA requires the formation of a novel complex comprising caspases and TLR3. Recognition of dsRNA by TLR3 and subsequent activation of caspases via TLR3 were shown to be mediated by receptor interacting protein 1.35
Conclusion
We report the first evidence of dsRNA formation in the overactive bladder under the ischemic conditions. Our data suggest that accumulation of dsRNA and activation of dsRNA-dependent PKR may contribute to overactive smooth muscle contractions and provoke structural damage. Recognition of dsRNA by TLR3 and formation of dsRNA/TLR3 complex may contribute to sensitization of smooth muscle cells and increased contractile reactivity. Structural modifications associated with dsRNA appear to be galvanized by upregulation of caspase-3 and activation of apoptotic responses. The precise mechanisms underlying dsRNA actions in the bladder remain as the focus of our future studies.
Abbreviations
dsRNA, double stranded RNA; RT-PCR, reverse transcription polymerase chain reaction; PKR, protein kinase R; PI3K, phosphoinositol 3 kinase; Akt, protein kinase B; ApoE−/−, apolipoprotein E knockout; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; TLR3, toll-like receptor 3; TUNEL, Terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling; Poly[I]-Poly[C], polyinosinic-polycytidylic acid; H&E, Hematoxylin and eosin; EFS, electrical field stimulation.
Institutional Review Board Statement
Animal care and experimental protocols were in accordance with the animal care and use guidelines and approval of the VA Boston Healthcare System Animal Care and Use Committee.
Funding
This work was supported by Merit Review Award Number I01 BX004372 from the United States (US) Department of Veterans Affairs Biomedical Laboratory R&D (BLRD) Service.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Yang JH, Siroky MB, Yalla SV, Azadzoi KM. Mitochondrial stress and activation of PI3K and Akt survival pathway in bladder ischemia. Res Rep Urol. 2017;9:93–100. doi:10.2147/RRU.S132082
2. Yang JH, Li Y, Azad R, Azadzoi KM. Regulation of cellular stress signaling in bladder ischemia. Res Rep Urol. 2020;12:391–402. doi:10.2147/RRU.S271618
3. Yang JH, Choi HP, Yang A, et al. Post-translational modification networks of contractile and cellular stress response proteins in bladder ischemia. Cells. 2021;10(5):1031. doi:10.3390/cells10051031
4. Minhas G, Sharma J, Khan N. Cellular stress response and immune signaling in retinal ischemia-reperfusion injury. Front Immunol. 2016;7:444. doi:10.3389/fimmu.2016.00444
5. Martindale JJ, Metzger JM. Uncoupling of increased cellular oxidative stress and myocardial ischemia reperfusion injury by directed sarcolemma stabilization. J Mol Cell Cardiol. 2014;67:26–37. doi:10.1016/j.yjmcc.2013.12.008
6. Swan CL, Sistonen L. Cellular stress response cross talk maintains protein and energy homeostasis. EMBO J. 2015;34:267–269. doi:10.15252/embj.201490757
7. Mo JS, Meng Z, Kim YC, et al. Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nat Cell Biol. 2015;17:500–510. doi:10.1038/ncb3111
8. Leavy O. Signalling: new roles for cell stress sensor. Nat Rev Immunol. 2014;14:135. doi:10.1038/nri3627
9. Soboloff J, Madesh M, Gill DL. Sensing cellular stress through STIM proteins. Nat Chem Biol. 2011;7:488–492. doi:10.1038/nchembio.619
10. Galluzzi L, Yamazaki T, Kroemer G. Linking cellular stress responses to systemic homeostasis. Nat Rev Mol Cell Biol. 2018;19:731–745. doi:10.1038/s41580-018-0068-0
11. Kultz D. Molecular and evolutionary basis of the cellular stress response. Annu Rev Physiol. 2005;67:225–257. doi:10.1146/annurev.physiol.67.040403.103635
12. Melton LM, Keith AB, Davis S, et al. Chronic glial activation, neurodegeneration, and APP immunoreactive deposits following acute administration of double-stranded RNA. Glia. 2003;44(1):1–12. doi:10.1002/glia.10276
13. Sun R, Park O, Horiguchi N, et al. STAT1 contributes to dsRNA inhibition of liver regeneration after partial hepatectomy in mice. Hepatology. 2006;44(4):955–966. doi:10.1002/hep.21344
14. Bowerfind WM, Fryer AD, Jacoby DB. Double-stranded RNA causes airway hyperreactivity and neuronal M2 muscarinic receptor dysfunction. J Appl Physiol. 2002;92(4):1417–1422. doi:10.1152/japplphysiol.00934.2001
15. Wang H, Xu X, Fassett J, et al. Double-stranded RNA-dependent protein kinase deficiency protects the heart from systolic overload-induced congestive heart failure. Circulation. 2014;129(13):1397. doi:10.1161/CIRCULATIONAHA.113.002209
16. Li G, Scull C, Ozcan L, Tabas I. NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. J Cell Biol. 2010;191(6):1113–1125. doi:10.1083/jcb.201006121
17. Zamanian-Daryoush M, Mogensen TH, DiDonato JA, Williams BR. NF-kappaB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-kappaB-inducing kinase and IkappaB kinase. Mol Cell Biol. 2000;20(4):1278–1290. doi:10.1128/MCB.20.4.1278-1290.2000
18. Xu C, Gamil AAA, Munangandu HM, Evensen Ø. Apoptosis induction by dsRNA-dependent protein kinase R (PKR) in EPC cells via caspase 8 and 9 pathways. Viruses. 2018;10(10):526. doi:10.3390/v10100526
19. Iordanov MS, Ryabinina OP, Schneider P, Magun BE. Two mechanisms of caspase 9 processing in double-stranded RNA- and virus-triggered apoptosis. Apoptosis. 2005;10(1):153–166. doi:10.1007/s10495-005-6070-y
20. Harcourt JL, Offermann MK. Interferon-alpha synergistically enhances induction of interleukin-6 by double stranded RNA in HeLa cells. Eur J Biochem. 2000;267(9):2768–2777. doi:10.1046/j.1432-1327.2000.01300.x
21. Zhan Q, Song R, Li F, et al. Double-stranded RNA upregulates the expression of inflammatory mediators in human aortic valve cells through the TLR3-TRIF-noncanonical NF-κB pathway. Am J Physiol Cell Physiol. 2017;312(4):C407–C417. doi:10.1152/ajpcell.00230.2016
22. Yeh CC, Kuo HM, Li TM, et al. Shikonin-induced apoptosis involves caspase-3 activity in a human bladder cancer cell line (T24). Vivo. 2007;21(6):1011–1019.
23. Hughes FM, Turner DP, Todd Purves J. The potential repertoire of the innate immune system in the bladder: expression of pattern recognition receptors in the rat bladder and a rat urothelial cell line (MYP3 cells). Int Urol Nephrol. 2015;47(12):1953–1964. doi:10.1007/s11255-015-1126-6
24. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–820. doi:10.1016/j.cell.2010.01.022
25. Franchi L, Eigenbrod T, Muñoz-Planillo R, et al. Cytosolic double-stranded RNA activates the NLRP3 inflammasome via MAVS-induced membrane permeabilization and K + efflux. J Immunol. 2014;193(8):4214–4222. doi:10.4049/jimmunol.1400582
26. Shin JM, Choi D-K, Sohn K-C, et al. Double-stranded RNA induces inflammation via the NF-κB pathway and inflammasome activation in the outer root sheath cells of hair follicles. Sci Rep. 2017;7(7):44127. doi:10.1038/srep44127
27. Arnaiz E, Miar A, Dias Junior AG, et al. Hypoxia regulates endogenous double-stranded RNA production via reduced mitochondrial DNA transcription. Front Oncol. 2021;24(11):779739. doi:10.3389/fonc.2021.779739
28. Reineke LC, Kedersha N, Langereis MA, van Kuppeveld FJ, Lloyd RE, Racaniello VR. Stress granules regulate double-stranded RNA-dependent protein kinase activation through a complex containing G3BP1 and Caprin1. mBio. 2015;6(2):e02486. doi:10.1128/mBio.02486-14
29. Terada T, Ueyama J, Ukita Y, Ohta T. Protein expression of double-stranded RNA-activated protein kinase (PKR) in intrahepatic bile ducts in normal adult livers, fetal livers, primary biliary cirrhosis, hepatolithiasis and intrahepatic cholangiocarcinoma. Liver. 2000;20(6):450–457. doi:10.1034/j.1600-0676.2000.020006450.x
30. Gil J, Esteban M. Induction of apoptosis by the dsRNA-dependent protein kinase (PKR): mechanism of action. Apoptosis. 2000;5(2):107–114. doi:10.1023/A:1009664109241
31. Chattopadhyay S, Sen GC. dsRNA-activation of TLR3 and RLR signaling: gene induction-dependent and independent effects. J Interferon Cytokine Res. 2014;34(6):427–436. doi:10.1089/jir.2014.0034
32. Kuwano K, Hagimoto N, Nakanishi Y. The role of apoptosis in pulmonary fibrosis. Histol Histopathol. 2004;19(3):867–881. doi:10.14670/HH-19.867
33. Thannickal VJ, Horowitz JC. Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc Am Thorac Soc. 2006;3(4):350–356. doi:10.1513/pats.200601-001TK
34. Sagulenko V, Thygesen SJ, Sester DP, et al. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 2013;20(9):1149–1160. doi:10.1038/cdd.2013.37
35. Estornes Y, Toscano F, Virard F, et al. dsRNA induces apoptosis through an atypical death complex associating TLR3 to caspase-8. Cell Death Differ. 2012;19(9):1482–1494. doi:10.1038/cdd.2012.22
36. Gill R, Soriano M, Blomgren K, et al. Role of caspase-3 activation in cerebral ischemia-induced neurodegeneration in adult and neonatal brain. J Cereb Blood Flow Metab. 2002;22(4):420–430. doi:10.1097/00004647-200204000-00006
37. Yang JH, Choi HP, Niu W, Azadzoi KM. Cellular stress and molecular responses in bladder ischemia. Int J Mol Sci. 2021;22(21):11862. doi:10.3390/ijms222111862
38. Wang W, Wang W-H, Azadzoi KM, et al. Alu RNA accumulation in hyperglycemia augments oxidative stress and impairs eNOS and SOD2 expression in endothelial cells. Mol Cell Endocrinol. 2016;426:91–100. doi:10.1016/j.mce.2016.02.008
39. Wang W, Wang WH, Azadzoi KM, et al. Activation of innate antiviral immune response via double-stranded RNA-dependent RLR receptor-mediated necroptosis. Sci Rep. 2016;Mar(6):22550. doi:10.1038/srep22550
40. Sáenz JB, Vargas N, Cho CJ, Mills JC. Regulation of the double-stranded RNA response through ADAR1 licenses metaplastic reprogramming in gastric epithelium. JCI Insight. 2022;7(3):e153511. doi:10.1172/jci.insight.153511
41. Lemaire PA, Anderson E, Lary J, Cole JL. Mechanism of PKR Activation by dsRNA. J Mol Biol. 2008;381(2):351–360. doi:10.1016/j.jmb.2008.05.056
42. Nanduri S, Carpick BW, Yang Y, Williams BR, Qin J. Structure of the double-stranded RNA-binding domain of the protein kinase PKR reveals the molecular basis of its dsRNA-mediated activation. EMBO J. 1998;17(18):5458–5465. doi:10.1093/emboj/17.18.5458
43. Nanduri S, Rahman F, Williams BR, Qin J. A dynamically tuned double-stranded RNA binding mechanism for the activation of antiviral kinase PKR. EMBO J. 2000;19(20):5567–5574. doi:10.1093/emboj/19.20.5567
44. Dela Justina V, Giachini FR, Priviero F, Webb RC, Double-stranded RNA. Toll-like receptor activation: a novel mechanism for blood pressure regulation. Clin Sci. 2020;134(2):303–313. doi:10.1042/CS20190913
45. Zimmer S, Steinmetz M, Asdonk T, et al. Activation of endothelial toll-like receptor 3 impairs endothelial function. Circ Res. 2011;108(11):1358–1366. doi:10.1161/CIRCRESAHA.111.243246
46. Carvour M, Song C, Kaul S, Anantharam V, Kanthasamy A, Kanthasamy A. Chronic low-dose oxidative stress induces caspase-3-dependent PKCdelta proteolytic activation and apoptosis in a cell culture model of dopaminergic neurodegeneration. Ann N Y Acad Sci. 2008;1139:197–205. doi:10.1196/annals.1432.020
47. Slee EA, Adrain C, Martin SJ. Executioner caspase-3, −6, and −7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J Biol Chem. 2001;276(10):7320–7326. doi:10.1074/jbc.M008363200
48. Kuranaga E, Miura M. Nonapoptotic functions of caspases: caspases as regulatory molecules for immunity and cell-fate determination. Trends Cell Biol. 2007;17(3):135–144. doi:10.1016/j.tcb.2007.01.001
49. Gil J, Alcamí J, Esteban M. Induction of apoptosis by double-stranded-RNA-dependent protein kinase (PKR) involves the alpha subunit of eukaryotic translation initiation factor 2 and NF-kappaB. Mol Cell Biol. 1999;19(7):4653–4663. doi:10.1128/MCB.19.7.4653
50. Gil J, García MA, Esteban M. Caspase 9 activation by the dsRNA-dependent protein kinase, PKR: molecular mechanism and relevance. FEBS Lett. 2002;529(2–3):249–255. doi:10.1016/S0014-5793(02)03348-3
51. Kalai M, Van Loo G, Vanden Berghe T. Tipping the balance between necrosis and apoptosis in human and murine cells treated with interferon and dsRNA. Cell Death Differ. 2002;Sep(9):981–994. doi:10.1038/sj.cdd.4401051
52. Funami K, Sasai M, Ohba Y, Oshiumi H, Seya T, Matsumoto M. Spatiotemporal mobilization of Toll/IL-1 receptor domain-containing adaptor molecule-1 in response to dsRNA. J Immunol. 2007;179(10):6867–6872. doi:10.4049/jimmunol.179.10.6867
53. Salaun B, Coste I, Rissoan MC, Lebecque SJ, Renno T. TLR3 can directly trigger apoptosis in human cancer cells. J Immunol. 2006;176(8):4894–4901. doi:10.4049/jimmunol.176.8.4894
54. Salaun B, Lebecque S, Matikainen S, Rimoldi D, Romero P. Toll-like receptor 3 expressed by melanoma cells as a target for therapy? Clin Cancer Res. 2007;13(15 Pt 1):4565–4574. doi:10.1158/1078-0432.CCR-07-0274
55. Rihani A, Van Maerken T, Pattyn F. Effective Alu repeat based RT-Qpcr normalization in cancer cell perturbation experiments. PLoS One. 2013;8(8):e71776. doi:10.1371/journal.pone.0071776
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.