")
Back to Journals » OncoTargets and Therapy » Volume 13
FNDC3B, Targeted by miR-125a-5p and miR-217, Promotes the Proliferation and Invasion of Colorectal Cancer Cells via PI3K/mTOR Signaling
Authors Li Y, Yang J, Wang H, Qiao W, Guo Y, Zhang S, Guo Y
Received 8 August 2019
Accepted for publication 14 January 2020
Published 30 April 2020 Volume 2020:13 Pages 3501—3510
DOI https://doi.org/10.2147/OTT.S226520
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Gaetano Romano
Yilong Li, Jie Yang, Hengyang Wang, Wei Qiao, Yongfeng Guo, Shengtao Zhang, Yajuan Guo
First Department of General Surgery, Ninth Hospital of Xi’an, Xi’an 710054, Shaanxi, People’s Republic of China
Correspondence: Yajuan Guo
The Ninth Hospital of Xi’an, No. 151, Eastern Section of South Second Ring Road, Xi’an 710054, Shaanxi, People’s Republic of China
Email [email protected]
Background: Fibronectin type III domain containing 3B (FNDC3B) acts as an oncogene in various cancers, and abnormal expression of FNDC3B has been found in colorectal cancer (CRC). Our study aimed to illustrate the role of FNDC3B in CRC development.
Methods: Through RT-qPCR and western blotting assays, the mRNA and protein expressions of target genes were measured. CCK-8 and MTT methods were used to detect cell proliferation. Invasion ability was determined using Transwell assay. TargetScan platform and luciferase reporter gene assay were performed to predict and validate the bindings between FNDC3B and miR-125a-5p or miR-217. Besides, the expression correlation was measured by Pearson’s Correlation analysis.
Results: We found that FNDC3B was significantly upregulated in CRC tissues and tumor cell lines, and high expression of FNDC3B predicted a poor survival outcome. The bindings between FNDC3B and miR-125a-5p and miR-217 were respectively at the motifs of CUCAGGG and AUGCAGU. MiR-125a-5p and miR-217 were downregulated in CRC tissues, and both were negatively correlated with FNDC3B expression. Subsequently, the downregulated miR-125a-5p and miR-217 were confirmed as contributors FNDC3B upregulation in CRC. A loss-of-function assay demonstrated that FNDC3B knockdown inhibited the proliferation of CRC cells, while FNDC3B overexpression promoted the proliferation and invasion of tumor cells. Besides, we validated that PI3K/mTOR signaling was involved in the regulation of FNDC3B on the proliferation and invasion of CRC cells.
Conclusion: Generally, our findings demonstrated that FNDC3B facilitated cell proliferation and invasion via PI3K/mTOR signaling, and further promoted CRC progression. The novel miR-125a-5p/FNDC3B and miR-217/FNDC3B axes might be new targets for CRC prognosis and therapy.
Keywords: FNDC3B, colorectal cancer, proliferation, invasion, microRNA, PI3K/mTOR
Introduction
Colorectal cancer (CRC), a disease of the digestive tract, has the fifth high mortality rate in the world, and CRC ranks third in incidence among the most prevalent malignant cancers.1 With development of the economy and lifestyle changes, CRC has seriously threatened the health of people, and placed a heavy economic burden on families and society.2 Although biomarkers for CRC diagnose was increased and chemotherapy combined with surgery improved the prognosis of CRC in recent years. The five-year survival rate of late stage CRC patients was still poor while the recurrence rate was still high.3,4 Therefore, it is urgent to discover the potential biomarkers for CRC diagnosis and prognosis, and clarify the molecular mechanism of CRC development.
Fibronectin type III domain containing 3B (FNDC3B, also FAD104), a member of FNDC3 family, was previously identified as a modulator of osteoblast and adipocyte differentiation.5,6 The fibronectin type III domain is a main structural component of FNDC3B, along with the transmembrane region and proline-rich region.7 As the fibronectin type III domain has the ability to combine with various proteins, FNDC3B plays an important role in multiple cell processes, including cell adhesion, proliferation and growth signaling.8 Recent evidence suggested that FNDC3B was abnormally expressed in a series of human tumors.6 Moreover, Lin et al indicated that FNDC3B was upregulated in hepatocellular carcinoma (HCC), provoking the migration and invasion of HCC cells, and suggested as a diagnostic biomarker in HCC.9 An exploration of CRC mechanism using microarray analysis revealed that FNDC3B was an upregulated gene in CRC.10 However, the biological function of FNDC3B in the growth and metastasis of CRC tumors remains indistinct.
Abundant studies on microRNAs (miRNAs; miRs) have emerged in recent years. MiRNAs are single-stranded structures (about 22 ~ 28 nucleotides long) and cannot encode proteins.11 Generally speaking, miRNAs have complementary target sites within the 3ʹ untranslated region (3ʹ UTR) of their target genes, and suppress mRNA expressions at the post-transcriptional level.12 Increasing evidences presented that miRNAs are crucial regulators of various cancer development, CRC included.13 For example, miR-1271 acted as a tumor suppressor of CRC due to its inhibitory role in regulating metadherin/Wnt signaling and the proliferation and invasion of CRC cells.14 Zhang et al demonstrated that miR-494 was upregulated in CRC and could induce Wnt/β-catenin signaling by targeting adenomatous polyposis coli, thereby boosting CRC cell growth.15 Li and colleagues indicated that miR-140 overexpression inhibited tumor formation and declined the migratory and invasive rates of CRC cells.16 However, little is known about the miRNA which regulates FNDC3B expression in CRC.
In this study, we estimated the expression of FNDC3B in CRC pathological tissues, as well as the relevance between CRC clinical outcomes and ectopic FNDC3B expression. We then identified and validated miRNAs that bond to FNDC3B. In vitro experiments were performed to evaluate the function of FNDC3B in CRC cell proliferation and invasion. Additionally, we considered PI3K/mTOR signaling pathway as a possible target to shed light on the mechanisms of FNDC3B regulating cell proliferation and invasion in CRC.
Materials and Methods
Patients and Clinical Samples
We recruited 36 cases of CRC patients to obtain the CRC tumor tissues and paired adjacent normal tissues (more than 5 cm from the edge of tumor). The patients were diagnosed and underwent excision surgery in Ninth Hospital of Xi’an from October 2012 to September 2017, and they had not received any kind of treatment, such as chemotherapy and radiotherapy, prior to the surgery. The specimens were immediately frozen using liquid nitrogen after surgery and stored at −80°C. To obtain the overall survival and disease free survival data, CRC patients was the followed up by telephone or e-mail.
Study Approval
The ethic approval of the present study was acquired from the Ethic Committee at the Ninth Hospital of Xi’an. With the accordance of the Declaration of Helsinki, the ethic committee also examined and supervised all experiments and processes involving human specimens in this study. Besides, we had obtained prior written informed consent from the participants and their families.
Cell Culture and Reagent Treatment
The American Type Culture Collection (ATCC, Manassas, USA) was the supplier from which we purchased all the human colorectal cancer cell lines (DLD-1, HCT8, HT29, LS411N, LOVO, SW620) and the normal colonic epithelial cell (NCM460). To culture these cells, we supplemented Dulbecco’s modified Eagle’s medium (DMEM) with 1% nonessential amino acids, 1% penicillin/streptomycin and 10% fetal bovine serum (Invitrogen, Carlsbad, USA). The cells were maintained at 37 °C under a humidified atmosphere with 5% CO2.
Dactolisib (MedChemExpress, NJ, USA) was used to inhibit PI3K/mTOR signaling of the colon cell lines in this study. The cells were treated with Dactolisib solution at the concentration of 5 nM directly or after transfection assay.
RNA Extraction and Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
The total RNA of CRC tissues and cell lines was isolated using MirVana™ miRNA Isolation kit (Ambion, Foster City, USA), and was qualified by 1.5% agarose gel electrophoresis. An Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, USA) was utilized for RNA quality detection. To synthesize first-strand cDNA from the purified RNA, the Revert Aid™ First Strand cDNA Synthesis Kit (Hanover, USA) was purchased from MBI Fermentas. The expressions of target RNAs were detected by qPCR which was performed on an Applied Biosystems 7500 system (Applied Biosystems, Carlsbad, USA) using SYBR-Green Master mix (Takara Bio, Dalian, China). The procedure of qPCR was described as followed: 95°C for 2 min, 40 cycles of 95°C for 5 s and 60°C for 30 s, one cycle of 95°C for 5 s, 60°C for 1 min and 95°C for 15 s, and finally, 50°C for 30 s. The quantification process was performed based on 2−ΔΔCt method,17 and human β-actin and U6 RNA were decided to be the endogenous reference gene in this study.
Western Blotting
To examine the expression of FNDC3B and activation of PI3K/mTOR signaling, we performed Western blotting whose first step was to extract total proteins using an Extraction kit (Nanjing KeyGen Biotech, Nanjing, China). According to the instruction of BCA protein assay kit (Thermo Fisher Scientific, Waltham, USA), the concentration of protein was quantified. SDS-PAGE (15%) was used to resolve the proteins for 2 h. Then, proteins were transferred onto a polyvinylidene fluoride membrane (Bio‑Rad Laboratories, Hercules, USA). The membrane blocking was last for 1 h in 5% non‑fat dried milk at room temperature. After that, we probed the membrane overnight at 4°C with antibodies, anti-FNDC3B, anti-p-PI3K, anti-PI3K, anti-p-mTOR, anti-mTOR (1:500) and anti-β‑actin (1:1000) (Cell Signaling, Danvers, USA). After 3 times’ 10 min-washing with TBST, the horseradish peroxidase-conjugated secondary antibodies (1:1000) (Cell Signaling, Danvers, USA) was used for another 1 h incubation at room temperature. Finally, the visualization of the bands was performed on an ECL Western Blotting Analysis system (GE Healthcare, Chicago, IL, USA).
Cell Transfection Assay
Small interfering RNAs (siRNAs) targeting FNDC3B were designed and generated by RiboBio Co. Ltd. (Guangzhou, China). Invitrogen (Carlsbad, USA) provided miRNA mimic synthesis service. In addition, to construct the overexpression plasmid of FNDC3B, we amplified FNDC3B and cloned it into a pcDNA3.1 (+) vector after identified by 1.5% agarose gel electrophoresis and sequencing (Invitrogen, Carlsbad, USA).
Cells were seeded in 96-well plates at a concentration of 1 × 106 per well, when reaching 80% ~ 90% confluence. The transfection of mimics, siRNA and pcDNA plasmids was performed for 48 h using Lipofectamine® 2000 reagent (Invitrogen, Carlsbad, USA) according to the manufacturers’ protocol.
Cell Proliferation Determination
To determine the cell proliferation of CRC cells under different treatments, we conducted both 3‑(4,5‑dimethyl‑2‑thiazolyl)‑2,5‑diphenyl‑2‑H tetrazolium bromide (MTT) and Cell Counting Kit-8 (CCK-8) assays in line with their manufacturer’s protocols. Briefly, for CCK-8 assay, SW620 and HCT8 cells transfected with negative control (NC) siRNA or siFNDC3B-2 were both divided into four subgroups and were respectively cultured for 24 h, 48 h, 72 h and 96 h after seeded in 96-well plates. Then, each well was supplemented with 10 μL CCK-8 solution and incubated for 1 h at 37 °C. The absorbance of each group was detected at 450 nm using a microplate Reader (Epoch, BioTek Instruments, Winooski, USA).
For MTT assay, HT29 and DLD-1 cells were incubated in a 37°C and 5% CO2 incubator for 96 h after seeded into 96-well plates. While the incubation was complete, 20 μL MTT reagent (5 mg/mL) was added for another 4 h incubation. Then, to dissolve the reaction products, 150 μL dimethyl sulfoxide was added. The absorbance was detected at 570 nm. The final results were normalized to the cellular proliferation at 0 h (100%).
Invasion Transwell Assay
In order to quantify the invasive ability of cells, we conducted Transwell experiments. Briefly, 1 × 105 cells suspended in 200 μL serum‑free medium were seeded into the upper strata of a Transwell chamber (diameter, 6.5 mm, membrane pore size, 8 μm; Corning, NY, USA) which was pre-coated with 1 mg/mL Matrigel (BD Biosciences, Franklin Lakes, USA), and the lower chamber was supplemented with 600 μL 20% FBS‑DMEM. The incubation was continued for 48 h, and then the non-invaded cells were removed. Subsequently, 4% paraformaldehyde and 0.1% crystal violet (Sigma‑Aldrich, Darmstadt, Germany) were used to fix and stain the cells in the lower chamber for 15 min and 30 min, respectively. All incubation processes took place at 37°C. Finally, the cell number in randomly selected fields were counted under a microscope (Olympus Corporation, Tokyo, Japan).
Bioinformatics Prediction and Mutation Assays
TargetScan Human release 7.2 (http://www.targetscan.org/vert_72/) was the platform we utilized to predicted the combinations between FNDC3B and miR-125a-5p or miR-217. For the validation of the binding relationships between FNDC3B and miR-125a-5p or miR-217, we introduced mutations into the miR-125a-5p or miR-217 binding motifs on 3`UTR of FNDC3B by using fusion PCR, generating two mutant types of FNDC3B 3`UTR.
Luciferase Assay
We inserted the wild and mutant types of FNDC3B 3`UTR into the pMIR-Report Luciferase vector (Ambion, Foster City, USA). Cells were pre-added into 96-well plates 24 h before being co-transfected with miR-125a-5p and miR-217 mimics along with Renilla using Lipofectamine® 2000 reagent (Invitrogen, Carlsbad, USA). The luciferase intensity was measured after 48 h transfection under the guidance of the instruction of dual-luciferase reporter assay (Promega, Madison, USA).
Statistical Analysis
All of the experiments in this study were performed at least three replicates, and the statistical analysis was conducted using GraphPad Prism 7 (GraphPad Software, San Diego, USA). Mean ± standard error of the mean was the manner to present data. Correlations between the expressions of FNDC3B and miR-125a-5p or miR-217 were assessed by Pearson’s correlation analysis. Kaplan‑Meier survival analysis was conducted to determine the overall survival and disease free survival in CRC patients stratified by FNDC3B mRNA expression levels. And the values were expressed as median with interquartile range. The samples were divided into two groups according to the quartile, the lower quartile was the low expression group, and the upper quartile was the high expression group. Difference analysis of data was presented as P values which was calculated by two-tailed Student’s test. And the statistical significance was considered as a P value lower than 0.05.
Results
FNDC3B Was Upregulated in CRC Tissues and Cell Lines
For the validation of FNDC3B expression in CRC, we performed RT-qPCR on 36 paired clinical CRC samples and corresponding normal tissues. The results showed that the mRNA level of FNDC3B in the 32 collected tumor specimens was significantly upregulated compared with the healthy tissues (Figure 1A). Furthermore, the expression of FNDC3B protein was also remarkably increased in 7 randomly selected CRC tissues (Figure 1B). We also analyzed the clinical characteristics of patients and described the information in Table 1. Similarly, we detected elevated mRNA expression of FNDC3B in six CRC cell lines compared with NCM460 (Figure 1C). As presented in Figure 1D and E, individuals with high FNDC3B expression level exhibited worse overall survival and disease free survival. In general, the upregulation of FNDC3B was validated in CRC specimens and was associated with the poor outcome of patients.
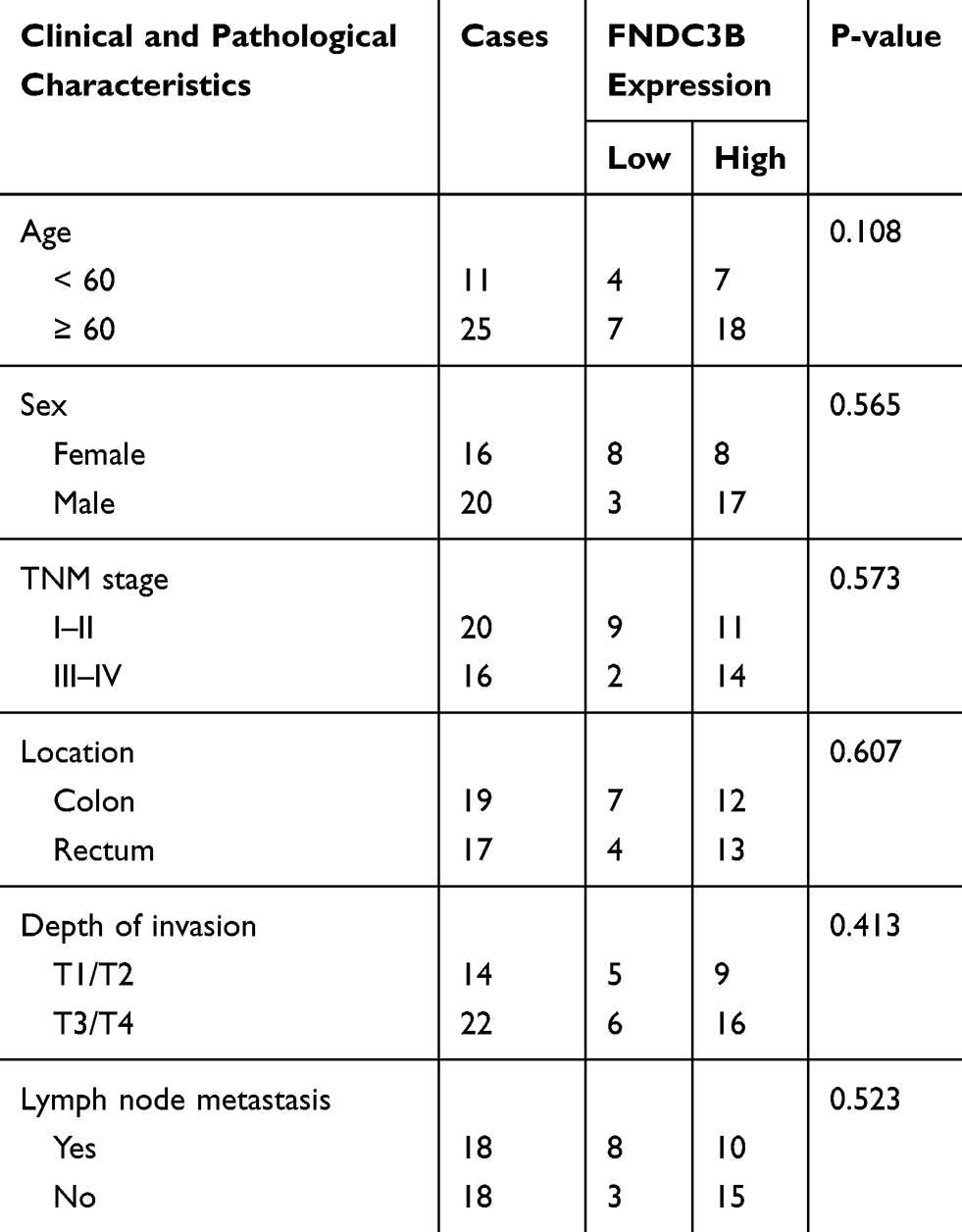 |
Table 1 Relevance Between Clinicopathological Characteristics of CRC Patients and FNDC3B Expressions |
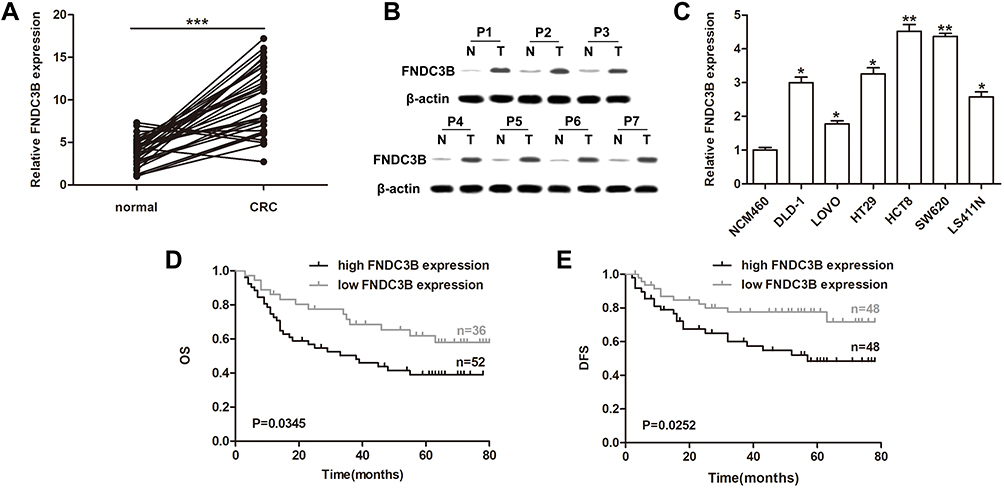 |
Figure 1 High expression of FNDC3B in CRC was associated with poor outcome. Notes: (A) Expressions of FNDC3B in CRC samples and adjacent tissues were detected by RT-qPCR. (B) Western blot assay analyzed the protein levels of FNDC3B in CRC and paired normal tissues. (C) RT-qPCR analysis of FNDC3B expressions in CRC cell lines. (D) The overall survival of CRC patients with different expression levels of FNDC3B was conducted by using Kaplan-Meier. (E) High expression of FNDC3B in patients presented a shorter disease free survival compared with the low FNDC3B expression group. *P < 0.05; **P < 0.01; ***P < 0.001. |
FNDC3B Upregulation Was Modulated by the Decreased Expressions of miR-125a-5p and miR-217 in CRC
TargetScan Human 7.2 was used to investigate miRNAs which could bind to FNDC3B and regulate its expression. We found that miR-125a-5p and miR-217 directly targeted to the 3`UTR of FNDC3B at the sequences from 1425 bp to 1459 bp (CUCAGGG) and 1600 bp ~ 1607 bp (AUGCAGU), respectively (Figure 2A). Also, we detected decreased the expressions of both miR-125a-5p and miR-217 RT-qPCR in CRC tissues using RT-qPCR (Figure 2B). More importantly, Pearson’s correlation analysis demonstrated that there existed negative correlations between the expressions of FNDC3B and these two miRNAs (Figure 2C). Transfections of miR-125a-5p and miR-217 mimics inhibited the protein levels of FNDC3B in all six CRC cell lines (Figure 2D). The seed sequences for miR-125a-5p and miR-217 in the 3`UTR of FNDC3B were respectively mutated to CUCGAUA and CGUUAGU (Figure 2E). Expectedly, the result of luciferase reporter assay indicated that the transfections of miR-125a-5p and miR-217 prominently reduced the luciferase intensity of wild type FNDC3B, while the mutant one was unaffected (Figure 2F). Taken together, these findings showed that miR-125a-5p and miR-217 modulated the expression of FNDC3B through directly binding to the seed sequences.
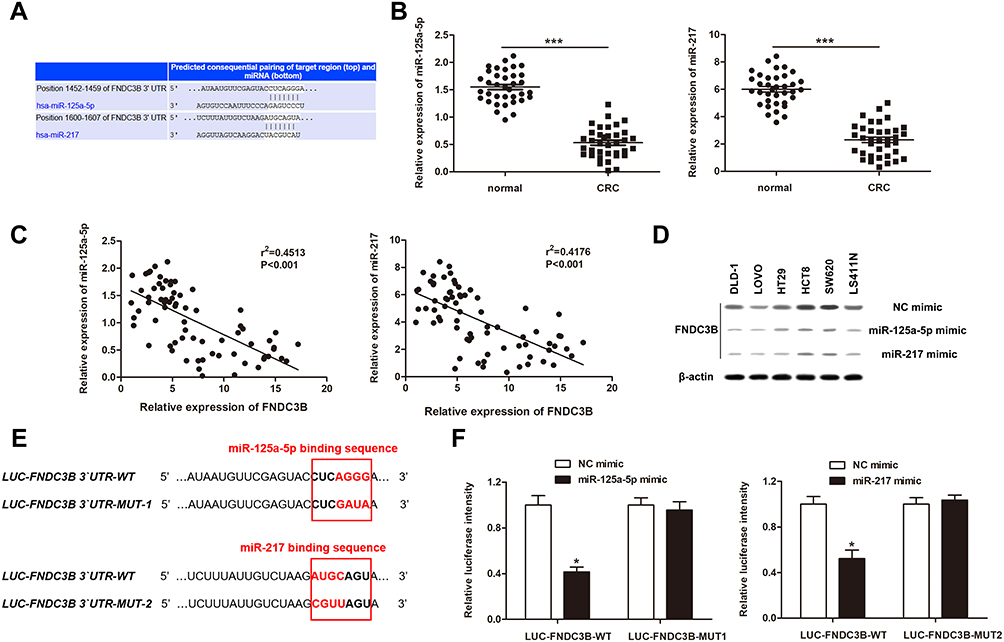 |
Figure 2 FNDC3B was the direct target of miR-125a-5p and miR-217. Notes: (A) TargetScan predicted the binding sites of miR-125a-5p and miR-217 on FNDC3B 3`UTR, respectively. (B) The expressions of miR-125a-5p and miR-217 in CRC samples were analyzed by RT-qPCR. (C) Pearson’s correlation analysis exhibited the negative correlations between the expressions of FNDC3B and miR-125a-5p or miR-217. (D) Western blot showed the protein levels of FNDC3B in CRC cells after transfected with miRNA mimics. (E) The mutant nucleotides in the binding sites between FNDC3B 3`UTR and miR-125a-5p or miR-217. (F) Luciferase intensities of reporter vectors contained FNDC3B 3`UTR of WT or MUT type were measured after miRNA mimic transfections. *P < 0.05 and ***P < 0.001 represented the significant differences from the control value, respectively. |
Regulation of FNDC3B on Cell Proliferation and Invasion in CRC Was Influenced by miR-125a-5p and miR-217
To further explore the role of FNDC3B in CRC, we synthesized 2 siRNAs of FNDC3B and constructed a plasmid carrying FNDC3B. And the cell lines, SW620 and HCT8, with highest upregulation level of FNDC3B were used for the loss-of-function assay, while DLD-1 and HT29 cells with lower FNDC3B upregulation level were utilized for overexpression experiment. The results of Western blotting presented that siFNDC3B-1 and siFNDC3B-2 effectively reduced the protein levels of FNDC3B in SW620 and HCT8 cells, while pcDNA-FNDC3B enhanced the expressions of FNDC3B protein in DLD-1 and HT29 cells in a dose-dependent manner (Figure 3A). We determined a significantly declined proliferation in FNDC3B silenced SW620 and HCT8 cells (Figure 3B and C). In DLD-1 and HT29 cells, the transfections of miR-125a-5p and miR-217 had an inhibitory effect on proliferation and invasion; however, the proliferation and invasion rates in both mimics and pcDNA-FND3CB transfected CRC cells were significantly higher than mimics transfected cells (Figure 3D–G). All in all, our findings indicated that upregulated FNDC3B promoted the proliferation and invasion of CRC tumor cells in vitro, which was modulated by miR-125a-5p and miR-217.
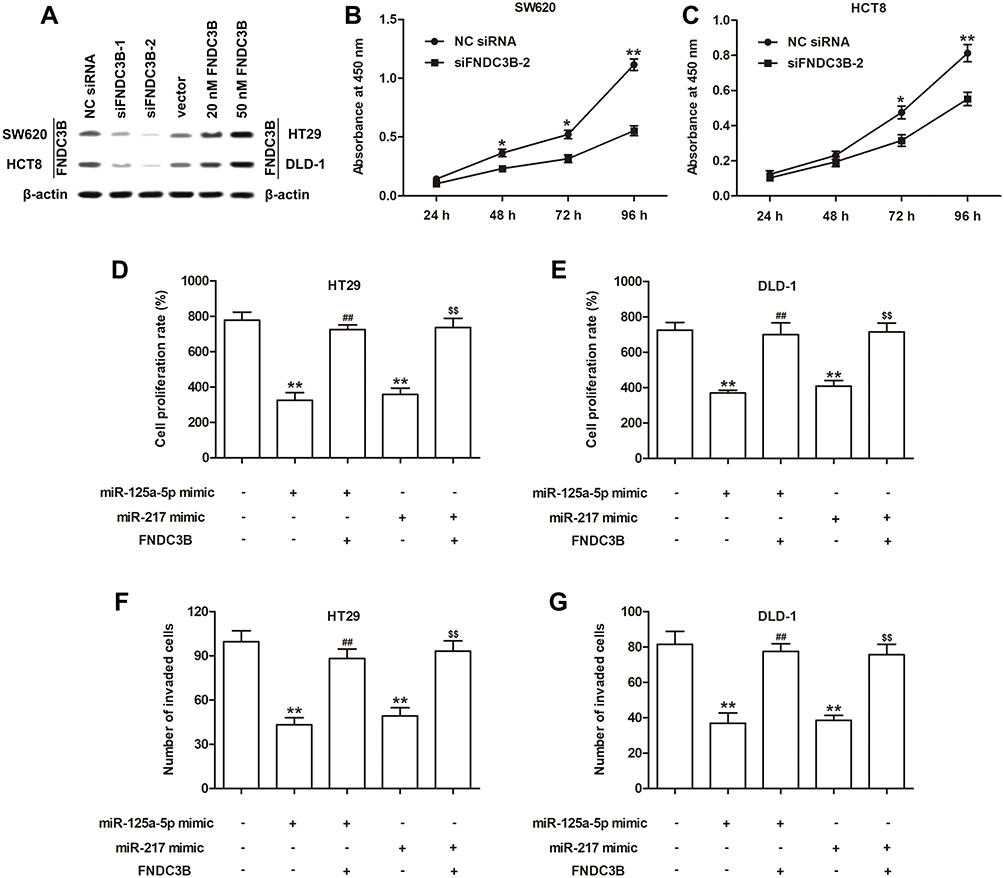 |
Figure 3 Regulation of FNDC3B on cell proliferation and invasion in CRC was influenced by miRNAs. Notes: (A) Western blotting exhibited the transfection efficiencies of siRNAs and overexpression vector for FNDC3B. (B and C) The proliferation curves of SW620 and HCT8 cells with FNDC3B knockdown were detected by CCK-8. (D and E) MTT assay showed the proliferation abilities of HT29 and DLD-1 cells after miRNA mimic or/and pcDNA transfections. (F and G) The invaded rates of HT29 and DLD-1 cells were analyzed using Transwell method. Significant differences versus control group were indicated by *P < 0.05 and **P < 0.01. ##P < 0.01 and $$P < 0.01 were respectively versus miR-125a-5p or miR-217 group. |
FNDC3B Promoted Cell Proliferation and Invasion in CRC via PI3K/mTOR Signaling
By detecting the phosphorylation levels of PI3K and mTOR in CRC and healthy tissues, we concluded that the PI3K/mTOR signaling pathway was strongly activated in CRC (Figure 4A). Besides, knockdown of FNDC3B significantly decreased the protein levels of p-PI3K and p-mTOR in SW620 and HCT8 cells, while pcDNA-FNDC3B enhanced the expressions of p-PI3K and p-mTOR in DLD-1 and HT29 cells (Figure 4B and C). The results of MTT assay showed that dactolisib, a PI3K/mTOR inhibitor, inhibited the proliferation of DLD-1 and HT29 cells, while the overexpression of FNDC3B accelerated cell proliferation; except that, we found that dactolisib reversed the adverse effect of FNDC3B overexpression on cell proliferation (Figure 4D). The aggravated effect of FNDC3B overexpression on the invasion rates of DLD-1 and HT29 cells was reversed by Dactolisib (Figure 4E). Collectively, these results suggested that FNDC3B accelerated CRC cell proliferation and invasion via activating PI3K/mTOR signaling pathway.
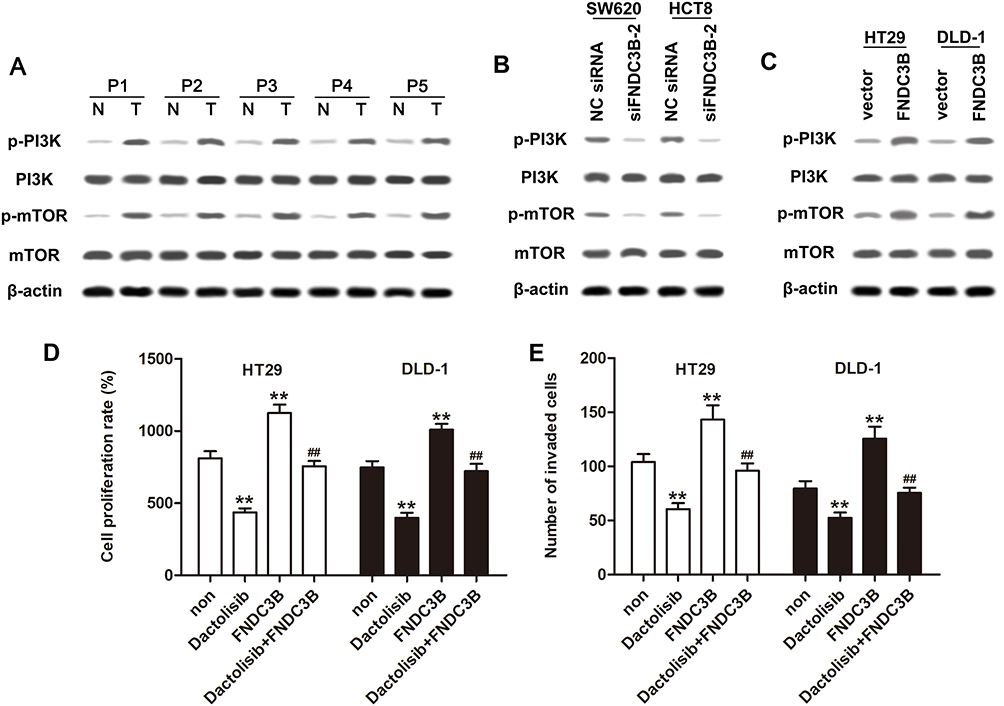 |
Figure 4 Overexpressed FNDC3B promoted cell proliferation and invasion in CRC through activating PI3K/mTOR signaling. Notes: (A) The activation of PI3K/mTOR pathway in tumor and normal tissues of CRC patients was confirmed by Western blotting. (B and C) Western blot analysis of the expressions of p-PI3K and p-mTOR in SW620 and HCT8 cells transfected with siFNDC3B-2 or HT29 and DLD-1 cells transfected with pcDNA-FNDC3B. (D) The proliferation of HT29 and DLD-1 cells treated with Dactolisib along with FNDC3B overexpression was detected by MTT assay. (E) Transwell method was conducted for the detection of the invaded rates in HT29 and DLD-1 cells. P value less than 0.01 was expressed as **.##P value less than 0.01 versus FNDC3B group. |
Discussion
In recent years, CRC has become a primary contributor to the high morbidity and mortality of cancer-related diseases, and more than half of patients succumbs CRC in worldwide every year.18 Additionally, CRC was proved as a multiple-stage carcinoma which arose multiple-step process during its development and carcinogenesis.19 The genetic or epigenetic alterations of multiple genes and interactive factors were involved in CRC progress.20 To explore more effective therapies for CRC, a further detailed understanding of the mechanism which could underlie CRC progression and development is urgent. In this study, we discovered the significant upregulation of FNDC3B in CRC tissues and cell lines and that the patients with high FNDC3B expression turned to poor overall survival and disease free survival compared with those had low FNDC3B expression. Similarly, Zhong et al previously identified the increased expression of FNDC3B in tongue squamous cell carcinoma (TSCC) in both clinical samples and in vitro cell lines.6 FNDC3B was also an upregulated gene in HCC, and was demonstrated to have negative correlation with HCC patient survival both in vivo and in vitro.9 In addition, a recent microarray analysis indicated that FNDC3B was highly expressed in CRC and was suggested as a prognostic and therapeutic target of CRC, which was further verified in our study.10
FNDC3B is initially known as a modulator of differentiation of osteoblast and adipocyte.21 Recently, the role of FNDC3B in cancer biology was emerged as the accumulating evidence indicated that FNDC3B regulated the growth and development of diversified human tumors. For instance, an in vitro investigation showed that overexpressed FNDC3B promoted migration and invasion in Mahlavu and Huh7 cells while FNDC3B knockdown inhibited cell migration, invasion and tumor nodule formation.9 The increased FNDC3B expression was also validated to induce the epithelial-mesenchymal transition of TSCC cells; however, knocking down FNDC3B would result in depression of the migratory and invasive abilities of TSCC cells.6 Xu and colleagues revealed that interfering FNDC3B expression effectively suppressed the glioblastoma cells to proliferate, migrate and invade.22 Consistently, our study also proved that the transfection of siRNA for FNDC3B remarkably decreased both the proliferation and invasion rates of CRC cells, while FNDC3B overexpression enhanced these processes of CRC cells.
Desirably, our present study further identified that the expressions of miR-125a-5p and miR-217 were decreased in CRC, and were both negatively correlated with FNDC3B expression. Meanwhile, miR-125a-5p and miR-217 were proved to regulate FNDC3B expression by directly binding to the 3`UTR of FNDC3B, thus to involve in cell proliferation and invasion of CRC. Although evidences about the combinations of miR-125a-5p and miR-217 with FNDC3B was deficient in CRC regulation studies, the roles of these two miRNAs in regulating CRC cell processes had been investigated. Similar to our study, miR-125a-5p was verified to act as a tumor suppressor of colon cancer via binding to anti-apoptotic genes and inhibiting cell proliferation.23 Previous study documented that miR-217 could target to MAPK signaling and was involved in regulating tumor growth and apoptosis of CRC.24 The study of Bian et al validated that inhibiting miR-217 enhanced the proliferation and migration rates of CRC cell in vitro.25 Except that, in a research based on glioblastoma, miR-129-5p was found to directly target FNDC3B, thus inhibiting the cell viability, proliferation and invasion of tumor cells.22
PI3K/mTOR signaling pathway has been reported to be a crucial signal transduction cascade which is hyper-activated in various tumors.26 Moreover, there was research indicated that PI3K/mTOR signaling not only participated in the regulation of cell proliferation, but also metastasis in CRC.27–29 Zhang and colleagues demonstrated that PI3K/mTOR signaling was activated in colon cancer tissues, and the activation of PI3K/mTOR signaling was depressed by miR-218 and thus to inhibit cell proliferation and migration.27 Additionally, PI3K/mTOR activation was regulated by acetylshikonin which examined as a potential anti-CRC drug because its ability to inhibit the proliferation of CRC cells.30 Studies also proved that the phosphorylation of PI3K and mTOR promoted by IMPDH2, induced G1/S phase cell cycle transition, and accelerated the migration and invasion of tumor cells.31 Our present study also addressed the evidence that FNDC3B exerted positive effects on the activation of PI3K/mTOR signaling during promoting CRC cell processes. Contrarily, there was evidence suggested that mTOR inactivation could enhance cellular autophagy, which may be a protective measure for the survival of tumor cells.32 However, the regulatory network of autophagy is very complex, and excessive autophagy will also affect the growth ability of tumor cells themselves.33 Therefore, the role of FDNC3B in autophagy of CRC cells might need further explorations.
Conclusion
In conclusion, we confirmed the oncogenic role of FNDC3B in CRC development, and also we evaluated the correlation between FNDC3B expression and CRC clinical outcome. Our findings underlined that miR-125a-5p and miR-217 were associated with the dysregulation of FNDC3B. Additionally, modulating the activation of PI3K/mTOR signaling was indicated as the mechanism of FNDC3B regulating cell proliferation and invasion in CRC. These evidence provided new insights for the molecular basis of CRC prognosis and therapy.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Fedewa SA, et al. Colorectal cancer statistics, 2017. CA Cancer J Clin. 2017;67(3):177–193.
2. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. doi:10.3322/caac.21338
3. Dienstmann R, Vermeulen L, Guinney J, Kopetz S, Tejpar S, Tabernero J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat Rev Cancer. 2017;17(2):79–92. doi:10.1038/nrc.2016.126
4. Verhoeff SR, van Erning FN, Lemmens VE, de Wilt JH, Pruijt JF. Adjuvant chemotherapy is not associated with improved survival for all high risk factors in stage II colon cancer. Int J Cancer. 2016;139(1):187–193. doi:10.1002/ijc.30053
5. Keishi K, Ayumi K, Shigehiro O, Makoto N, Masayoshi I. Fad104, a positive regulator of adipogenesis, negatively regulates osteoblast differentiation. Biochem Biophys Res Commun. 2010;397(2):187–191. doi:10.1016/j.bbrc.2010.05.077
6. Zhong Z, Zhang H, Hong M, et al. FNDC3B promotes epithelial-mesenchymal transition in tongue squamous cell carcinoma cells in a hypoxic microenvironment. Oncol Rep. 2018;39(4):1853–1859.
7. Wang HY, McMahon C, Ali SM, et al. Novel FNDC3B and MECOM fusion and WT1 L378fs* 7 frameshift mutation in an acute myeloid leukaemia patient with cytomorphological and immunophenotypic features reminiscent of acute promyelocytic leukaemia. Br J Haematol. 2016;172(6):987–990. doi:10.1111/bjh.13552
8. Nishizuka M, Kishimoto K, Kato A, et al. Disruption of the novel gene fad104 causes rapid postnatal death and attenuation of cell proliferation, adhesion, spreading and migration. Exp Cell Res. 2009;315(5):809–819. doi:10.1016/j.yexcr.2008.12.013
9. Lin CH, Lin YW, Chen YC, et al. FNDC3B promotes cell migration and tumor metastasis in hepatocellular carcinoma. Oncotarget. 2016;7(31):49498–49508. doi:10.18632/oncotarget.10374
10. Chen S, Wang Y, Zhang L, et al. Exploration of the mechanism of colorectal cancer metastasis using microarray analysis. Oncol Lett. 2017;14(6):6671–6677. doi:10.3892/ol.2017.7044
11. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. doi:10.1016/j.cell.2009.01.002
12. Gao Y, Liu Y, Du L, et al. Down-regulation of miR-24-3p in colorectal cancer is associated with malignant behavior. Med Oncol. 2015;32(1):362. doi:10.1007/s12032-014-0362-4
13. Yin Y, Zhong J, Li SW, et al. TRIM11, a direct target of miR-24-3p, promotes cell proliferation and inhibits apoptosis in colon cancer. Oncotarget. 2016;7(52):86755. doi:10.18632/oncotarget.13550
14. Sun X, Zhai H, Chen X, Kong R, Zhang X. MicroRNA-1271 suppresses the proliferation and invasion of colorectal cancer cells by regulating metadherin/Wnt signaling. J Biochem Mol Toxicol. 2018;32(2):e22028. doi:10.1002/jbt.2018.32.issue-2
15. Zhang Y, Guo L, Li Y, et al. MicroRNA-494 promotes cancer progression and targets adenomatous polyposis coli in colorectal cancer. Mol Cancer. 2018;17(1):1. doi:10.1186/s12943-017-0753-1
16. Li J, Zou K, Yu L, et al. Microrna-140 inhibits the epithelial-mesenchymal transition and metastasis in colorectal cancer. Mol Ther Nucleic Acids. 2018;10(C):426. doi:10.1016/j.omtn.2017.12.022
17. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi:10.1006/meth.2001.1262
18. Zhang T, Wang F, Wu JY, et al. Clinical correlation of B7-H3 and B3GALT4 with the prognosis of colorectal cancer. World J Gastroenterol. 2018;24(31):3538.
19. Tang J, Wang G, Zhang M, et al. Paradoxical role of CBX8 in proliferation and metastasis of colorectal cancer. Oncotarget. 2014;5(21):10778–10790. doi:10.18632/oncotarget.v5i21
20. Liu HY, Zhang CJ. Identification of differentially expressed genes and their upstream regulators in colorectal cancer. Cancer Gene Ther. 2017;24(6):244–250. doi:10.1038/cgt.2017.8
21. Kishimoto K, Nishizuka M, Ueda T, et al. Indispensable role of factor for adipocyte differentiation 104 (fad104) in lung maturation. Exp Cell Res. 2011;317(15):2110–2123. doi:10.1016/j.yexcr.2011.06.003
22. Xu H, Hu Y, Qiu W. Potential mechanisms of microRNA-129-5p in inhibiting cell processes including viability, proliferation, migration and invasiveness of glioblastoma cells U87 through targeting FNDC3B. Biomed Pharmacother. 2017;87:405–411. doi:10.1016/j.biopha.2016.12.100
23. Tong Z, Liu N, Lin L, Guo X, Yang D, Zhang Q. miR-125a-5p inhibits cell proliferation and induces apoptosis in colon cancer via targeting BCL2, BCL2L12 and MCL1. Biomed Pharmacother. 2015;75:129–136. doi:10.1016/j.biopha.2015.07.036
24. Zhang N, Lu C, Chen L. miR-217 regulates tumor growth and apoptosis by targeting the MAPK signaling pathway in colorectal cancer. Oncol Lett. 2016;12(6):4589–4597. doi:10.3892/ol.2016.5249
25. Bian Y, Gao G, Zhang Q, et al. KCNQ1OT1/miR-217/ZEB1 feedback loop facilitates cell migration and epithelial-mesenchymal transition in colorectal cancer. Cancer Biol Ther. 2019;20(6):886–896. doi:10.1080/15384047.2019.1579959
26. Cao ZX, Yang YT, Yu S, et al. Pogostone induces autophagy and apoptosis involving PI3K/Akt/mTOR axis in human colorectal carcinoma HCT116 cells. J Ethnopharmacol. 2017;202:20–27. doi:10.1016/j.jep.2016.07.028
27. Zhang X, Shi H, Tang H, Fang Z, Wang J, Cui S. miR-218 inhibits the invasion and migration of colon cancer cells by targeting the PI3K/Akt/mTOR signaling pathway. Int J Mol Med. 2015;35(5):1301–1308. doi:10.3892/ijmm.2015.2126
28. Jiang QG, Tai YL, Dong NL, Hai TZ. PI3K/Akt pathway involving into apoptosis and invasion in human colon cancer cells LoVo. Mol Biol Rep. 2014;41(5):3359–3367. doi:10.1007/s11033-014-3198-2
29. Ying-Zi L, Ke W, Jun H, et al. The PTEN/PI3K/Akt and Wnt/β-catenin signaling pathways are involved in the inhibitory effect of resveratrol on human colon cancer cell proliferation. Int J Oncol. 2014;45(1):104–112. doi:10.3892/ijo.2014.2392
30. Ye H, Zhu Y, Huang Q, et al. Acetylshikonin inhibits colorectal cancer growth via PI3K/Akt/mTOR signaling pathway. Chin Med. 2018;9(3):720–726.
31. Duan S, Huang W, Liu X, et al. IMPDH2 promotes colorectal cancer progression through activation of the PI3K/AKT/mTOR and PI3K/AKT/FOXO1 signaling pathways. J Exp Clin Cancer Res. 2018;37(1). doi:10.1186/s13046-018-0980-3.
32. Conza GD, Cafarello ST, Loroch S, et al. The mTOR and PP2A pathways regulate PHD2 phosphorylation to fine-tune HIF1α levels and colorectal cancer cell survival under hypoxia. Cell Rep. 2017;18(7):1699–1712. doi:10.1016/j.celrep.2017.01.051
33. Singh SS, Vats S, Chia AY-Q, et al. Dual role of autophagy in hallmarks of cancer. Oncogene. 2018;37(9):1142. doi:10.1038/s41388-017-0046-6
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.