")
Back to Journals » OncoTargets and Therapy » Volume 16
FMS-Like Tyrosine Kinase 3 Inhibitors in the Treatment of Acute Myeloid Leukemia: An Update on the Emerging Evidence and Safety Profile
Authors Garciaz S , Hospital MA
Received 12 November 2022
Accepted for publication 11 January 2023
Published 19 January 2023 Volume 2023:16 Pages 31—45
DOI https://doi.org/10.2147/OTT.S236740
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr William C. Cho
Sylvain Garciaz, Marie-Anne Hospital
Department of Hematology, Institut Paoli-Calmettes, Aix-Marseille Université, Centre National de la Recherche Scientifique, UMR7258, Centre de Recherche en Cancérologie de Marseille, INSERM U1068, Marseille, France
Correspondence: Sylvain Garciaz, Institut Paoli-Calmettes, Hematology Department, 232 Bd Sainte-Marguerite, Marseille, 13009, France, Tel + 33 4 91 22 37 54, Fax + 33 4 91 22 30 63, Email [email protected]
Abstract: FMS-like tyrosine kinase 3 (FLT3) is one of the most frequently mutated genes in acute myeloid leukemia (AML). Approximately 30% of the adult cases harbor an internal tandem duplication (FLT3-ITD) and 5– 10% a tyrosine kinase domain (TKD) amino acid substitution (FLT3-TKD). The treatment paradigm of AML patients harboring FLT3 mutations (30%) has been modified by the discovery of tyrosine kinase inhibitors. First- and second-generation inhibitors classify FLT3 inhibitors according to FLT3 specificity: first-generation FLT3 inhibitors include sorafenib and midostaurin and second-generation inhibitors are represented by quizartinib, gilteritinib and crenolanib, among others. Activity of these inhibitors depends on their mechanism of receptor binding (active vs inactive conformation) and efficacy against the FLT3-ITD and -TKD mutations (type 1 inhibitors are active both on FLT3-ITD and TKD, whereas type 2 inhibitors are active only on FLT3-ITD). The FLT3 inhibitors sorafenib, midostaurin, quizartinib and gilteritinib have been tested in monotherapy in several settings including refractory or relapsed AML (R/R AML), post-transplant maintenance as well as in combination with intensive chemotherapy (ICT) or non-intensity regimens. The results of published randomized studies support the use of sorafenib in a post-transplant setting (SORMAIN trial), midostaurin in combination with ICT based (RATIFY trial) and gilteritinib for R/R AML (ADMIRAL trial). Gilteritinib in combination with hypomethylating agent as well as quizartinib are not supported by solid randomized trial results for their use in FLT3-mutated AML patients.
Keywords: tyrosine kinase, resistance, precision medicine, clinical trials, venetoclax
Introduction
Acute myeloid leukemia (AML) is a severe heterogeneous group of neoplastic disorders driven by several molecular alterations. Among the most mutated driver genes in AML is FMS-like tyrosine kinase 3 (FLT3). Approximately 30% of the adult AML cases harbor an internal tandem duplication (FLT3-ITD) and 5–10% a tyrosine kinase domain (TKD) amino acid substitution (FLT3-TKD).1–3 Current treatment cures only a minority of AML patients and less than 20% have a long-term survival. Therapeutic approaches depend on disease subtype and patient’s age, general status, and comorbidities. Intensive cytotoxic therapy (ICT) induction, based on the combination of anthracycline and cytarabine, is given to the youngest patients (<75 years old) followed by cytarabine consolidations and/or allogeneic stem-cell transplantation (ASCT).3,4 Since 2020, older or unfit patients are treated by a non-intensive approach combining the hypomethylating agent azacitidine and the B-cell lymphoma 2 (BCL2) inhibitor venetoclax given orally until progression. This regimen is associated with approximately 65% overall response rate (ORR) including ≈35% complete response (CR) and about 15 months overall survival (OS).5
Although the impact of FLT3-ITD mutation is unclear in the context of venetoclax–azacitidine treatment,6 the negative impact of FLT3-ITD on survival of AML patients treated with ICT is well established.1,7–9 In the 2017 European Leukemia network (ELN) classification, prognosis of FLT3-ITD mutation on the survival of AML patients was dependent on the co-occurrence of the nucleophosmin 1 gene mutation (NPM1mut) and the FLT3-ITD mutation burden.9 In the recently refined ELN 2022 classification, the prognostic impact of FLT3-ITD mutation is independent of other co-occurring mutations as well as mutation burden and is globally associated with an intermediate risk.4 Despite the recent achievements in managing AML patients, the one-fits-all approach is currently challenged by the emergence of targeted therapies. Recently, two new classes of drugs have emerged for selected patients harboring IDH1/2 or FLT3 mutations, the IDH and the FLT3 inhibitors, respectively.10–12 In this review, we will discuss the emerging evidence and safety profiles of the main FLT3 inhibitors that are currently used in clinics or under evaluation in clinical trials.
The results of published randomized studies support the use of sorafenib in a post-transplant setting based on SORMAIN trial, midostaurin in combination with ICT in newly diagnosed (ND) AML based on RATIFY trial and gilteritinib monotherapy for relapsed or refractory (R/R) AML based on ADMIRAL trial. Despite these major advances, several crucial questions remain open. Is there a place for second-generation inhibitors in combination with ICT for patients with ND AML? What is the clinical effect and the safety of combinations of FLT3 inhibitors with non-intensive treatment in particular venetoclax-based regimen in elderly patients ineligible for intensive chemotherapy? What is the best option to tackle relapsed or refractory disease previously treated with FLT3 inhibitors?
Background
FLT3 Mutations
FLT3 belongs to the receptor tyrosine kinase (RTK) family that plays a major role in the regulation of hematopoiesis. FLT3 protein is characterized by the presence of five immunoglobulin-like motifs within their extracellular part that are exclusively expressed in hematopoietic cells. In normal physiology, FLT3 ligand (FLT3L) binding to the extracellular domain induces the dimerization of the receptor and the activation of intracellular signaling pathways such as PI3K/AKT or ERK/MAPK responsible for survival, maturation, and proliferation of hematopoietic cells.2,13,14 Early reports demonstrated an overexpression of FLT3 mRNA in AML and acute B-cell (but not T-cell) leukemia as well as an overexpression of FLT3-L.15,16 However, a breakthrough in our understanding of AML pathophysiology came from the discovery of FLT3 gene mutations located on chromosome 13q12. FLT3-ITD is located within the juxtamembrane (JM) and the TKD and leads to a disrupted JM domain crucial for kinase autoinhibition.17,18 FLT3-ITD produces receptors with a constitutive tyrosine kinase activity due to homodimerization or heterodimerization with the normal FLT3 receptors.19 The most common amino acid substitution within the activation loop of the TKD is an aspartic acid substitution with tyrosine or histidine at residue 835 (D835).20 Other point mutations in the TKD are situated within the activation loop (eg, residues I836, and Y842) of the TKD21,21 and, to a lesser extent, within the TKD1 (eg, residues N676 and F691).22,23 Rarer point mutations and smaller insertions/deletions have also been identified within the TKD and other domains (extracellular and JM domains).21,24–26 All these point mutations result in loss of auto-inhibition and constitutive activation of downstream proliferative signaling cascades (Figure 1).
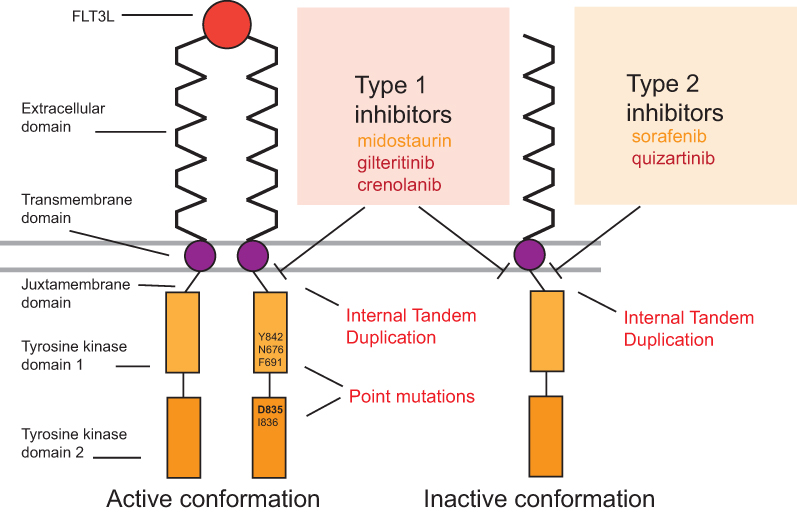 |
Figure 1 Structure of FLT3 receptor in active and inactive conformations, type of mutations and spectrum of activity of the main FLT3 inhibitors. In normal physiology, FLT3l binds to FLT3 receptors that dimerize and activate downstream signaling. Type 1 FLT3 inhibitors are active on FLT3 both on the active and inactive conformations while type 2 inhibitors bind the receptor only in the inactive conformation. Because of this affinity, type 1 inhibitors act on both FLT3-ITD and FLT3 point mutations located on TKD whereas type 2 act only on FLT3-ITD. First-generation inhibitors are highlighted in orange and second-generation inhibitors are highlighted in red. Main TKD mutations are listed. Abbreviation: FT3L, FLT3 ligand. |
FLT3 Inhibitors
Given the prevalence and poor prognosis of FLT3-ITD-mutated AML, targeting FLT3 signaling through small molecule inhibitors is a promising therapeutic strategy. Multiple small-molecule tyrosine kinase inhibitors that target FLT3 are in development for the treatment of patients with AML. First generation of inhibitors having multi-kinase target activity includes lestaurtinib (CEP-701), sunitinib (SU11248), midostaurin (PKC412), and sorafenib (BAY43-9006). These multi-kinase inhibitors have antileukemic effects through the broad inhibition of FLT3 and other pathways but also have multiple off-target effects that increase toxicities. Second-generation FLT3 inhibitors with higher selectivity and inhibitory activity include gilteritinib (ASP2215), quizartinib (AC220), and crenolanib (CP868596). These drugs have greater clinical potential and fewer off-target effects. FLT3 inhibitors are classified into the first or second type according to the binding mode to FLT3 (Figure 1). Type 1 inhibitors, which include midostaurin, gilteritinib, lestaurtinib, and crenolanib, bind the FLT3 receptor in the active conformation near the activation loop or the ATP binding domain. Type 1 inhibitors have the capacity of inhibiting both FLT3-ITD and FLT3-TKD. On the other hand, type 2 inhibitors, including quizartinib and sorafenib bind FLT3 receptor in the inactive conformation in a region adjacent to the ATP-binding domain. As these mutations modify the active kinase conformation of FLT3, type 2 inhibitors are inactive against most FLT3-TKD mutations27 (Figure 1).
First-Generation FLT3 Inhibitors
Sorafenib
One of the most studied FLT3 inhibitors is sorafenib, a type 2 inhibitor targeting multiple serine/threonine and RTK including vascular endothelial growth factor (VEGF) receptor 2, FMS-like tyrosine kinase 3 (FLT3), platelet-derived growth factor (PDGF) receptor, and fibroblast growth factor receptor-1 (FGFR1).28
Monotherapy
Relapsed or Refractory AML
Sorafenib was first evaluated in monotherapy in a Phase 1 study including R/R AML, acute lymphoblastic leukemia, myelodysplastic syndromes (MDS), chronic myelomonocytic leukemia (CMML) or chronic myeloid leukemia in blastic phase.29 Sorafenib was given at 200 mg in two distinct schedules. In schedule “A”, sorafenib was given once or twice daily, 5 days per week, every week for a 21-day cycle, whereas in schedule “B”, the treatment was administered once or twice daily, for 14 days every 21 days. Schedule A was favored first for expansion at maximal tolerated dose (MTD). The study included 50 patients with a median age of 61 years. Twenty-eight (56%) patients had FLT3-ITD only, 5 (10%) had FLT3-TKD D835 only, and 6 (12%) had both mutations. Three (6%) patients had received other prior FLT3 inhibitors (AC220=2, KW 2449=1) and 8 (16%) had prior hematopoietic stem cell transplant (HSCT). Five patients out of the 50 responded (10%), including 3 CR and 2 CR with incomplete platelet recovery (CRp). In addition, 3 patients (all FLT3-ITD) had morphologic leukemia-free state (MLFS). Toxicity was mainly gastrointestinal (GI) and cutaneous including nausea/vomiting (44%) and diarrhea (36%), grade 1–2 skin rash (24%) and grade 1–2 hand-foot syndrome (10%).
Post-Transplant Setting
Given its broad spectrum of kinase inhibition, sorafenib was evaluated in post-transplant setting in a randomized Phase 2 and a Phase 3 study.
Randomized Phase 2 Study
The randomized phase 2 trial SORMAIN included patients with a FLT3-ITD in CR at enrollment after ASCT performed as part of upfront therapy or in the context of R/R AML.30 Patients were randomized to receive either sorafenib at escalated from 400 to 800 mg per day, or placebo. Treatment started between day-60 and day-100 after ASCT and was given continuously for 24 months or until the occurrence of relapse or intolerable toxicity. Between October 2010 and May 2016, 83 patients were enrolled in the study, 43 received sorafenib and 40 received placebo. Median age was 40 years, 71% of the patients were transplanted in first CR (CR1). The study was prematurely stopped because of slow recruitment rate. At the time of data entry, median relapse-free survival (RFS) was not reached in the sorafenib arm and 30.9 months in the placebo arm. The hazard ratio for relapse or death was 0.39 (p=0.013) in the sorafenib group compared with placebo. There was no difference in OS at the time of data entry after a median follow-up of 55 months. Only nine patients were treated upfront with TKI (midostaurin) plus chemotherapy as part of the induction phase. Hence, it is unclear to which extent results apply also to patients undergoing midostaurin. There were more acute or chronic GVHD in the sorafenib group (76.8%) than in the placebo group (59.8%). The other common grade ≥3 adverse events (AE) occurring in ≥10% of sorafenib-treated patients were infections (26.2%), GI toxicity (14.3%), electrolyte alterations (14.3%) and skin toxicity (15.4%).
Phase 3 Study
A phase 3 study (NCT02474290) included FLT3-ITD 18–60-year-old patients, transplanted in CR1 after a myeloablative regimen. Patients were randomly assigned to sorafenib or no treatment in a 1:1 manner.31 Sorafenib maintenance 400 mg twice a day was started between day-30 to day-60 after ASCT and was continued until day-180. Between June 20, 2015, and July 21, 2018, 100 patients were allocated to sorafenib maintenance and 102 to control (no maintenance). Median age was 35, 25–29% harbored NPM1mut and 80% were in the intermediate cytogenetics group. Eleven patients in the sorafenib group and 32 in the control group relapsed. The primary endpoint was 1-year cumulative incidence of relapse (CIR). CIR was 7% in the SOR group and 24.5% in the control group (HR:0.25, p=0.001). Two-year OS and event-free survival (EFS) were 81% versus 68% (p=0.012) and 79% versus 57% (p<0.001) in the sorafenib and control groups, respectively. The most frequent reasons for dose reduction were acute graft-versus-host disease (GVHD) (41%), hematological adverse events (25%) and skin toxicity (11%). The most common grade 3/4 adverse events were infections (≈25%), acute GVHD (≈22%), chronic GVHD (≈18%) well balanced in the two groups. Hematological toxicity was more frequent in the sorafenib group (15%) than in the control (7%). A subsequent comprehensive preclinical study demonstrated that sorafenib increased the IL-15 production by FLT3-ITD leukemic cells leading to the potentiation of the allogeneic CD8+ T cell response and disease eradication in preclinical models.32
Combination with ICT
Sorafenib was also evaluated in combination with ICT in a phase 1–2 as well as in several randomized phase 2 and phase 3 trials.
Phase 1–2 Study
Phase 1–2 study included 18–65-year-old ND AML patients. Intensive treatment was conventional 7+3 regimen. Sorafenib was given at 800 mg/day from day-1 to day-7 during induction and at 800 mg/d for 28 days during consolidations followed by a one-year maintenance. Between 1 February 2008 and 31 March 2010, 62 patients with a median age of 53 years were included. Twenty-three patients had FLT3 mutations, including 17 with FLT3-ITD (10 with low mutation burden), 4 with D835 mutation and 2 with both mutations. Seventy-nine percent of the patients have achieved a CR, including 95% of the patients with FLT3-ITD. With a median follow-up of 52 months (range, 2.3–62.9 months), the OS and RFS were 29 and 13.8 months, respectively. Grade ≥3 adverse events possibly related to sorafenib during induction included hyperbilirubinemia in four patients, elevated transaminases (five), diarrhea (four), rash (two), pancreatitis (one), colitis (one), pericarditis (one), hand and foot syndrome (two), and elevated creatinine.33,34
Randomized Phase 2 Studies
A first randomized phase 2 trial included 18–60-year-old patients with ND AML irrespective of the FLT3 mutational status.35 Patients received 400 mg sorafenib or placebo twice daily from day-10 to day-19 in combination with one or two cycles of 7+3 induction chemotherapy. During consolidations cycles, sorafenib or placebo was given from day-8 until 3 days before the next consolidation cycle. At maintenance, 400 mg sorafenib twice daily or placebo was administered continuously for 12 months after the last consolidation cycle. Between 27 March 2009 and 28 November 2011, 134 patients received sorafenib and 133 received placebo. Median age was 50 years, 46 (17%) patients had FLT3-internal tandem duplication (ITD)-positive AML, and 7% had FLT3-TKD. Response rate was equivalent between the 2 arms (≈60%). Median event-free survival was 21 months in the sorafenib group compared with 9 months in the placebo group. There was no difference for OS. A recent actualization showed that the 5-year EFS in the 2 arms were 41% and 27% (HR=0.60, p=0.011).36 Patients benefit from sorafenib was higher in the subgroup of FLT3-ITD AML (HR =0.55) than FLT3wt patients (HR=0.71). The most frequent grade 3/4 adverse events in the sorafenib group were fever (54%), infection (35%), diarrhea (11%), cardiac events (7%), rash (7%) and hand-foot skin reaction (7%).
A second randomized phase 2 trial sponsored by the Australasian Leukaemia and Lymphoma Group (ALLG) was reported at ASH meeting in 2020. Patients were aged 18–65 years and ND FLT3-ITD positive (allelic ratio (AR) ≥0.05) AML. Patients were randomized in a 2:1 manner to sorafenib or placebo 400 mg twice a day on days 4–10 of induction and each consolidation cycle. Maintenance was 400 mg twice a day on days 1–28 for 12 cycles. Between Jan 2013-May 2018, 99 patients were allocated to induction with either sorafenib (n=65) or placebo (n=33). The CR + CR with incomplete hematologic recovery (CRi) was 91% in the sorafenib arm (CR 80%, CRi 11%) and 94% in the placebo arm (CR 70%, CRi 24%). With a median overall follow-up of 25 months, there was no significant difference in or OS. The frequency of grade 3–4 adverse events (AEs) was similar between the two arms, apart from palmar-plantar rash, reported as drug-related in 15.4% and 6.1% pts in the sorafenib and placebo arms, respectively.37
Phase 3 Study
Finally, a phase 3 trial (NCT00373373) reported the results of sorafenib in combination with ICT in older patients.38 ND AML patients >60 years were enrolled in the study irrespective of FLT3 mutational status. Patients received either sorafenib 400 mg or placebo from day 3 after the end of induction and/or consolidation chemotherapy until 3 days before the first day of the next chemotherapy course. Sorafenib 400 mg or placebo maintenance was given to all patients in CR after consolidation during 1 year after start of induction therapy. One hundred and two patients were randomly assigned to the sorafenib arm, and 95 were assigned to the placebo (control) arm. Median age was 68 years, and 14% of the patients had FLT3-ITD and 20% had adverse cytogenetics. This trial failed to reach its primary end point as intent-to-treat analysis showed no difference in the EFS between both groups (median EFS, 7 months for placebo vs 5 months for sorafenib). Median OS was 15 months for placebo vs 13 months for sorafenib. In the subgroup of patients with FLT3-ITD–positive AML, authors did not observe significant differences in response or survival rates but they found an excess of toxicity in the sorafenib arm, in particular more grade >3 infections and more early death due to combined infection (15 versus 4 patients, p=0.035). This led to more treatment discontinuation in the sorafenib arm than in the placebo arm, possibly explaining the negative results.
Combination with Non-Intensive Regimen
Sorafenib was evaluated with non-intensive treatment in two phase 2 studies. One included younger patients >18 years old relapsing or refractory after ICT or >60 years old patient non candidate for ICT.39 Sorafenib was given at 400 mg BID continuously from day-1 of each cycle concomitantly with azacitidine. Between January 2011 and September 2012, a total of 43 patients with AML were included with a median age of 64 years. FLT-3-ITD was detected prior to the initiation of treatment in 40 of 43 (93%) patients. Among the 37 evaluable patients, 6 had no prior therapy, 12 were primarily refractory to their previous induction regimen, and 19 had relapsed after prior therapy. Nine patients had failed prior therapy with FLT3 kinase inhibitors (5 with quizartinib, 1 with midostaurin), 3 had failed 2 prior FLT3 inhibitors. Response rate was 16% CR, 27% CRi, 3% partial response (PR). The median duration of response was 2.3 months. Median OS was 6.2 months. The most common grade ≥3 adverse events were thrombocytopenia, neutropenia, anemia, and neutropenia with fever or infection.
The second study reported the results of ND AML patients ≥60 years included in the NCT02196857 and NCT01254890 trials.40 The presence of the FLT3-ITD mutation was not a necessary inclusion criterion on the NCT01254890; however, the NCT02196857 required ≥10% FLT3-ITD burden. Treatment consisted in azacitidine 75 mg/m2 daily × 7 days and sorafenib 400 mg twice daily. The study included 27 patients with untreated AML (median age of 74 years). FLT3-ITD was present in 100% of the patients. The overall response rate (ORR) in the 27 evaluable patients was 78% (26% CR, 45% CRi/CRp, and 2 7% PR). The OS was 8.3 months for the entire group and 9.2 months for the 19 responders. The median duration of CR/CRp/CRi was 14.5 months. The most common grade 1/grade 2 AEs were hyperbilirubinemia (22%), diarrhea (22%), fatigue (22%), and nausea (19%). Among the grade 3/4 AEs, infections (26%) and neutropenic fever (26%) were the most common.
Taken together, these results support the utilization of sorafenib in a post-transplant setting. Main side-effects included GI and skin toxicities and liver enzymes elevation. Combination with chemotherapy or non-intensive treatment is not encouraged by the results of the clinical studies.
Midostaurin
Midostaurin is a type 1 FLT3 inhibitor acting both on FLT3-ITD and FLT3-TKD and having an activity against multiple tyrosine kinase receptors including PKC-α, FLT3, c-KIT, VEGFR, and PDGFR.41
Monotherapy
Relapsed or Refractory AML
Phase 2 Study
The first phase 2 proof-of-concept study included advanced MDS or R/R AML with ITD mutation or a D835Y TKD mutation.42 Midostaurin was given at a dose of 75 mg orally 3 times a day until toxicity or progression. Twenty patients (18 ITD, 2 D835Y) were enrolled including 19 AML and 1 MDS with a median age of 62 years CR rate was 0% and one patient had a PR. Fourteen patients (70%) achieved greater than a 50% reduction in the peripheral blast count and 6 patients achieved a more than 50% blast count in the bone marrow. Two patients were removed from the study on day 74 and day 99 to undergo HSCT after having had significant clinical benefit. Main side-effects was GI (grade 1/2 nausea/vomiting, anorexia or diarrhea). Grade 3 hyperbilirubinemia was seen in 1 patient. Three patients had a pulmonary fatal grade 5 events with unclear relation with the drug.
Randomized Phase 2 Study
In an open-label, randomized, Phase II study, midostaurin monotherapy was tested in patients with AML/high-risk MDS with either wild-type or mutated FLT3.43 Patients had to have an R/R AML ineligible to standard chemotherapy or HR-MDS/CMML with FLT3 status available and no previous treatment with FLT3 inhibitors. Patients were randomly assigned to receive midostaurin by continuous oral twice-daily doses of 50 or 100 mg. Ninety-five patients were enrolled including 85 with AML. Thirty-five patients harbored a FLT3 mutations, including 26 ITD. Sixty-one patients were more than 65 years old. Only one patient experienced PR but blast reductions were frequently observed, occurring in 71% of the patients with FLT3-mutant and 42% of the patients with wild-type FLT3. Blasts reduction (reaching >50%) occurred at a median of 29 days. Median time to treatment failure was estimated at 50 days. Median OS was 130 days. Most common grade 1/2 event was GI: nausea (60%), vomiting (48%), diarrhea (38%) and constipation 17%). Most common grade 3/4 events were oedema (20%), febrile neutropenia (18%) and pneumonia (10%).
Post-Transplant Setting
The randomized open-label phase 2 trial RADIUS (NCT01883362) included patients (aged 18–70 years) with documented FLT3-ITD AML who had undergone an ASCT in CR1.44 Patients were randomized 1:1 within 28 to 60 days after ASCT to receive midostaurin (50 mg twice daily in twelve 4-week cycles) or not. Between February 5, 2014, and June 13, 2016, 74 patients were screened, and 60 patients (30 per arm) were randomized. The estimated RFS at 18 months (95% CI) was 89% (69–96%) with midostaurin and 76% (54–88%) with standard of care alone (HR, 0.46, P = 0.27). Most AEs in both arms were grade 1/2. The most common AEs in the midostaurin arm were low-grade gastrointestinal toxicity.
Combination with ICT
Phase 1 Study
The first study reporting the results of midostaurin combined with ICT included 18–60 years old patients with non-previously treated AML. Two treatment schedules were initially planned, a continuous one (starting at day-1) and a sequential one (starting at day-8). Initial dose was 100 mg twice daily. Because of toxicity of the 100 mg BID, the regimen was decreased to 50 mg twice daily for 14 days either continuously (day 1–14) or sequentially (day 1–8 and day 15–22). The chemotherapy regimen consisted of one or two inductions with 7+3 regimen and high-dose cytarabine consolidations in which midostaurin was given according to the schedule assigned during induction. Sixty-nine patients were enrolled, 29 received 100 mg twice daily, 50 mg twice daily. Patients had a median age of 50 years, 27 patients out of 40 harbored FLT3-ITD, 23% had adverse cytogenetics. The overall CR rate was 80% (92% in the FLT3-ITD group, vs 74% in the FLT3wt group). The schedule (sequential vs continuous) did not affect the response rate. One-year OS was 85% and 2-years OS was 62% in patients with FLT3-mutant AML, compared with 78% and 52%, respectively, in patients with FLT3–wild-type AML. In the 100 mg BID cohort, the discontinuation rate was high (23/29) mainly due to grade 3/4 GI AE, whereas the midostaurin was generally well tolerated in combination with chemotherapy at the 50 mg twice-daily dose. Higher rate of discontinuation was noted in the concomitant (55%) compared with the sequential (35%) schedule but overall, the toxicity reported was similar in the two schedules. Side-effects were mainly GI toxicity including nausea (83%), diarrhea (68%) and vomiting (65%). Grade 1–2 liver enzymes increases were seen in about 20% of the patients.
Phase 2 Study
The AMLSG 16–10 study compared 18–70-year-old AML patients with FLT3-ITD with historical cohorts of AML patients with FLT3-ITD included in 5 previous AMLSG trials recruiting between 1993 and 2008 at the same centers.45 All patients received 1 or 2 induction cycles according to the 7+3 scheme followed by high or intermediate doses of aracytine consolidations combined with midostaurin administered orally 50 mg twice daily, starting on day 8, until 48 hours before the start of the subsequent chemotherapy cycle or conditioning before ASCT. Midostaurin maintenance was given orally in a dose of 50 mg twice daily for 12 months. Between June 2012 and May 2016, a first cohort of 142 patients was included. To better define the effect of midostaurin in older patients and the relative value of ASCT in the first CR, the study was amended to include a second cohort of 142 patients. Two hundred and eighty-four patients were analyzed in the study. Median age was 54 years, and198 patients were less than 60 and 86 between 61 and 70 years old. Almost 72% had intermediate cytogenetics, 58% were also NPM1mut, 53% had a FLT3 allelic ratio ≥0.5. After induction, 76.4% patients achieved a CR/CRi, 51 (18%) had refractory disease, and 16 (5.6%) died during induction therapy comparable in young and older patients. ASCT was performed in 134 patients. Maintenance therapy was started in 97 (34%) of 284 patients. Median EFS and OS were 13.2 and 26.0 months, respectively. The comparison of EFS in the AMLSG 16–10 study and the historical controls revealed a significant risk reduction for an event in the midostaurin cohort. During induction, the most common adverse events irrespective of the attribution were infection (59%), febrile neutropenia (35%) and gastrointestinal events (25%). Cardiac adverse events were found in 13% of the cases with 6% cardiac arrhythmia and 9% pulmonary events. Cardiac events were more common in older compared with younger patients (22% versus 6%, p=0.004). The most frequent adverse events during maintenance phase were GI toxicity (70%, significantly more frequent after ASCT (80 vs 36%; p=0.0001) followed by infections (51%) and hematological toxicities (46%). Cardiac arrhythmia occurred in 5% of the patients during maintenance phase.
Phase 3 Study
The CALGB 10603 (RATIFY) trial included 18–59-year-old patients with ND AML with FLT3 mutations.46 Induction therapy consisted in 7+3. Midostaurin or placebo was administered in a double-blind fashion, at a dose of 50 mg orally twice daily, on day-8 to day-21 during induction and HDAC consolidations. In maintenance phase, patients received midostaurin or placebo, administered at a dose of 50 mg orally twice daily, for twelve 28-day cycles. From May 2008 through October 2011, a total of 3277 patients were preregistered for the trial, 896 had an FLT3 mutation and 717 were enrolled in the trial, 360 were randomized in the Midostaurin group and 357 in the placebo arm. Median age was 47.9 years, 22.6% had a FLT3-TKD mutation, 77.4% a FLT3-ITD; 47.6% had a low FLT3-ITD allelic ratio (0.05–0.7) and 29.8% a high FLT3-ITD allelic ratio (>0.7). Median duration of trial treatment was 3 months. The primary endpoint was median OS which was 74.5 months in the midostaurin group versus 25.6 months. The hazard ratio for death was 0.78 in the midostaurin group (one-sided p value=0.009). The 4-year OS was 51.4 versus 44.3%, and the 4-year EFS was 28.2% in the midostaurin group and 20.6% in the placebo. The benefit of midostaurin with respect to EFS was consistent across the FLT3 subtypes. Patients harboring NPM1 and Wilms-tumor 1 (WT1) mutations seem to have to higher benefit of midostaurin addition to ICT.47 The rate of grade 3, 4, or 5 anemia was higher in the midostaurin group than in the placebo group (92.7% vs 87.8%, P = 0.03), as was the rate of grade 3, 4, or 5 rash (14.1% vs 7.6%, P = 0.008). Hematological toxicity was the same in the two groups as well as the infection rate (82%) or GI toxicity. Liver enzyme elevation was around 10% in the two groups.
Combination with Hypomethylating Agents
A single-institution study from the MD Anderson phase 1–2 included patients 18 years or older with MDS or AML ineligible to receive standard therapy or with R/R AML regardless of the FLT3 mutational status.48 Midostaurin was administered twice daily for 14 days on each azacitidine cycles (Days 8–21). After the first 23 patients were enrolled, protocol amendment allowed continuous administration of midostaurin unless there were adverse events. The target dose was 50 mg BID. The cycles were repeated every 4 weeks, up to a maximum of 12 cycles. Fifty-four patients were enrolled in this study, 14 in the phase 1 and 40 in the phase 2; 95% of the patients had AML and 5% MDS. FLT3 mutation was FLT3-ITD alone in 68%, with a median ITD ratio of 0.35 and 6% with associated FLT3-TKD D835. The rest of the patients had no FLT3 mutations. Forty-three percent of patients had been previously exposed to a hypomethylating agent, including either azacitidine (n=9) or decitabine (n=11) or both (n=3), and 24% had previously received another FLT3 inhibitor (sorafenib in 10 patients and quizartinib in 3 patients). ORR was 26%, one (2%) patient achieved a CR, 6 (11%) achieved a CRi, 6 (11%) a MLFS, and 1 (2%) patient a PR. Forty (74%) patients were primary refractory to therapy. Toxicity was consistent with other studies with 56% grade 3/4 infections, 11% grade 3/4 decrease in left ventricular fraction and common GI toxicities.
Drug approval of midostaurin in combination with ICT in younger and older patients is based on the results of phase 2 and phase 3 RATIFY, showing a gain in disease control and a tolerable side-effects profile. Special attention must be given to cardiac safety for older patients receiving midostaurin in combination with ICT.
Second-Generation TKI
Quizartinib
Quizartinib was the first drug specifically developed as a potent selective FLT3 inhibitor for AML. The drug shows high selectivity at low concentrations that is almost entirely limited to FLT3, KIT, colony-stimulating factor-1 receptor, platelet-derived growth factor receptor, and RET kinase.49
Monotherapy
Phase 1 Study
Dose-escalation phase 1 study included patients aged ≥18 years with R/R AML, or not a candidate for standard chemotherapy because of age, comorbidity, or other factors.50 Fifty-one patients received quizartinib on an intermittent schedule (at daily doses of 12 to 450 mg) and 25 on a continuous schedule (200 to 300 mg daily). The median age was 60 years. Responses were observed in 23 (30%) of 76 patients (13% CR/CRi, 17% PR). Nine (53%) of 17 FLT3-ITD–positive patients responded. Median response duration was 13 weeks, and median overall survival was 14 weeks. The most frequent treatment-related AEs (observed in >5 patients) were ≤ grade 2 and included nausea (16%), corrected QT interval (QTc) prolongation (12%), dysgeusia (11%), and vomiting (11%). The dose limiting toxicity (DLT) was grade 3 QTc prolongation.
Phase 2 Study
Phase 2 enrolled elderly patients (>60) with or without FLT3-ITD mutations who relapsed within 1 year after first-line ICT or were primary refractory to first-line treatment (cohort 1) and those 18 years or older who were refractory to or relapsed after salvage ICT or after ASCT (cohort 2).51 Quizartinib was given continuously once daily. Seventeen patients initially received 200 mg per day of quizartinib (ie, the maximum tolerated dose in the phase 1 study). Therefore, the protocol was subsequently amended on April 20, 2010, to starting doses of 135 mg per day for men and 90 mg per day for women in all other patients given previous observations of a greater susceptibility to QTc prolongation in women compared with men. Between November 19, 2009 and October 31, 2011, 333 patients: 157 in cohort 1 and 176 in cohort 2 were enrolled. Median ages were 69 years and 51 years in the two cohorts. Seventy-one percent and 77% of the patients had FLT3-ITD in the two cohorts. In cohort 1, the proportion of FLT3-ITD-positive patients who achieved composite CR was 56% including 3% CR, 4% CRp and 50% CR with partial hematologic reconstitution (CRh). Twenty-one percent had PR. In cohort 2, the proportion of FLT3-ITD-positive patients who achieved composite CR was 46% including 4% CR, 1% CRp and 40% CRh. Twenty-nine percent had PR. Responses were also seen in the FLT3wt group (approximately 30% of composite CR). Median OS of FLT3-ITD-positive patients was consistent across both cohorts (25.4 weeks in cohort 1 and 24 weeks in cohort 2). The main treatment-emergent adverse events were nausea (39%) Qtc elongation (29%, 19% grade 1–2 and 10% grade 3). Febrile neutropenia was seen in 23% of the patients.
Phase 3 Study
The phase 3 QUANTUM-R trial included patients >18 years with FLT3-ITD R/R AML. Patients were randomly assigned (2:1) to receive single agent quizartinib or the investigator’s choice of one of three preselected salvage chemotherapy regimens.52 Thirty milligram dose of quizartinib given orally once daily was increased to 60 mg in case of the absence of QTc prolongation. Patients receiving concurrent strong CYP3A inhibitors had a reduced starting dose of 20 mg quizartinib once daily which was increased to 30 mg once daily if the same QTc interval criteria were met. Between 2014 and 2017, 367 patients were enrolled, of whom 245 were allocated to quizartinib and 122 to salvage chemotherapy, median age was 55 and 57 years, respectively. Thirty-three percent of the patients had primary refractory AML, 47% a NPM1mut, more than a third of patients in each group had at least 0.5 FLT3-ITD variant allele frequency. Median OS was 6.2 months for quizartinib and 4.7 months for chemotherapy (hazard ratio 0.76, p=0·02). Main side-effects were febrile neutropenia (33%) and QTc prolongation 26% (22% grade 1–2).
Combination with Intensive Chemotherapy
Phase 1 Study
Patients aged between 18 and 60 years with previously untreated AML were eligible in the phase 1 study.53 The first dose level tested (DL1) was quizartinib 60 mg/d administered on day-4 to day-10 10 of induction therapy. A total of 19 patients were enrolled between November 2011 and July 2013. Median age was 43 years, nine patients (47%) were positive for FLT3‐ITD mutation. DLT were observed in 2 of 6 patients in the DL2 cohort. Therefore, dose was reduced to DL‐1 (quizartinib 40 mg/d administered on days 4 through 17 of induction) where only 1 patient had a DLT (grade 3 pericarditis). DL‐1 was identified as the MTD. Sixteen patients (84%) achieved a response to therapy. Fourteen patients (74%) achieved CRc (9 CR, 2 CRp, 3 CRi) and 2 patients (11%) were in MLFS. The most common treatment‐emergent AEs (all grades) reported included nausea (79%), diarrhea (63%), constipation (58%), hypokalemia (53%), hypomagnesemia (53%), neutropenia (53%), febrile neutropenia (47%), and vomiting (47%). Five cardiac events (45%) were considered related to study drug.
On-Going Trials
The phase 2 trial (NCT04107727) and the phase 3 QuANTUM-First study (NCT02668653) are currently recruiting AML patients eligible for ICT.
Although quizartinib demonstrated prolonged OS compared with chemotherapy in the phase 3 study QUANTUM-R, the FDA and the EMA ultimately did not grant approval owing to specific concerns about the design and results of the study.
Gilteritinib
Gilteritinib is a type 1 FLT3 inhibitor which has activity against FLT3, ALK, and AXL (among other kinases). Gilteritinib is multitargeted, but it inhibits FLT3 more than other kinases.54
Monotherapy
Phase 1–2 Study
The phase 1–2 dose-escalation and dose-expansion study included >18-year-old R/R AML patients.55 The presence of an FLT3 mutation was not mandatory, but 10 or more patients with locally confirmed FLT3 mutations (ITD or TKD) were required at each dose level. Gilteritinib was given continuously to participants at seven dose-escalation cohorts starting from 20 to 450 mg/day. Between 2013, and 2015, 265 patients were enrolled in the escalation and expansion phases. FLT3-ITD at screening was confirmed locally in 162 patients. Thirteen individuals had a point mutation in the D835 codon and 16 had both types of FLT3 mutation. Sixty-three (25%) had previously received treatment with a tyrosine kinase inhibitor, most commonly sorafenib. Of the 249 analyzed patients, 100 (40%) achieved a response, with 19 (8%) achieving CR, 10 (4%) CRp, 46 (18%) CRh, and 25 (10%) PR. The median duration of response was 17 weeks. Median OS was 25 weeks. Among 57 FLT3mut patients who had received any previous tyrosine kinase inhibitor, 21 (37%) achieved an ORR, of whom 2 (4%) had a CR, 2 (4%) had CRp, 10 (18%) had CRh, and 6 (11%) had PR. Best responses were achieved at doses ≥80 mg/day or higher. Based on the observed antileukaemic effects and FLT3 inhibition in plasma inhibitory assays, 120 mg/day was chosen as the starting dose for future studies. Grade 1/2 diarrhea was observed in 31% and grade 3 in 5% of the cases. Liver enzyme elevation was seen in ≈20% of the cases, mainly grade 1–2.
Phase 3
The phase 3 ADMIRAL trial included patients >18 years with R/R AML and FLT3mut (FLT3 ITD or TKD D835 or I836 mutations).56 FLT3 mutations were present if the FLT3-ITD allelic ratio was at ≥0.05. Patients were randomized 2:1 between once-daily gilteritinib (120 mg) continuously until treatment failure or the occurrence of toxic effects or salvage chemotherapy. The study included 247 patients in the gilteritinib group and 124 in the chemotherapy group. Median age was 62. Sixty-one percent of the patients had relapsed AML and 39% had primary refractory disease. Only 6.2% previously received midostaurin along with first-line chemotherapy. The most commonly co-mutated genes were NPM1 (46.6%) and DNMT3A (31.0%). Median OS was 10.4 months vs 6.9 months in the gilteritinib and the chemotherapy arms, respectively. Hazard ratio for death was 0.64 in the gilteritinib group (P<0.001). CR with full or partial hematologic recovery was 34.0% in the gilteritinib group and 15.3% in the chemotherapy group. Median duration of remission was 11.0 months. Longer survival was observed with gilteritinib than with chemotherapy across all cohorts of patients with co-mutations, particularly in the cohort of patients with double mutation (DNMT3A and NPM1). The most common serious adverse events considered to be related to gilteritinib therapy were febrile neutropenia (9.3%) and increase in the liver enzyme level (≈5%). Five percent of patients had a QTc elongation. Drug-related adverse events leading to the discontinuation of gilteritinib occurred in 11% of the patients.
Onoing Studies
Gilteritinib has been tested in a phase 1 trial in combination with ICT (NCT02236013). Results have been presented at ASH meeting in 2020.57 A large European phase 3 is currently enrolling patients to be treated with gilteritinib in combination with ICT in comparison with midostaurin (HOVON 156 AML, NCT04027309). Studies testing gilteritinib maintenance post ICT (NCT02927262) and ASCT (NCT02997202) are also currently recruiting.
Combination with Non-Intensive Treatment
The randomized, open-label, phase 3 study LACEWING study (NCT02752035) compared the efficacy and safety of gilteritinib plus azacitidine versus azacitidine alone in patients with ND FLT3mut (ITD and/or TKD) AML ineligible for intensive chemotherapy.58 Patients received azacitidine at standard dose alone or associated with 120 mg/day oral gilteritinib (the gilteritinib alone arm was prematurely stopped). A total of 123 patients were randomized to receive gilteritinib + azacitidine (n = 74) and azacitidine (n = 49). Median age was 77 years, ECOG PS ≥2 was observed in 47.3% in the gilteritinib + azacitidine and 32.7% of the patients in the gilteritinib alone group. Median OS was 9.82 and 8.87 months, in the azacitidine-gilteritinib and azacitidine alone arms, respectively. Due to the failure of reaching primary endpoint, the study was stopped for futility. In the subgroup with FLT3-ITD allelic ratio ≥0.5, gilterinib plus azacitidine was associated with a trend toward a longer OS (10.68 months vs 4.34 months; HR, 0.580 [95% CI, 0.285–1.182]). CRc rates were 58.1% for GIL + AZA and 26.5% for AZA (P < 0.001) but CR rates were similar in the two groups (16.2% vs 14.3%). Patients with a FLT3-ITD allelic ratio ≥0.5 had a higher chance of having CRc in the gilteritinib plus azacitidine group (71.4% vs 20.8%). GI hemorrhage, an AE of special interest identified during animal studies, occurred in 12.3% (n = 9) patients but considered not related to study drug. QTc prolongation, occurred in 13.7% (grade ≥3, 5.5% [n = 4]) of patients receiving gilteritinib and azacitidine.
These data support the use of gilteritinib for patients with R/R AML and FLT3 mutations. Nevertheless, enrollment occurred before widespread use of midostaurin in first-line chemotherapy, which could plausibly generate resistance to FLT3 inhibitors and subsequently alter gilteritinib activity.
Taken together the results of published randomized studies support the use of sorafenib in a post-transplant setting based on SORMAIN trial, midostaurin in combination with ICT in ND AML based on RATIFY trial and gilteritinib monotherapy for R/R AML based on ADMIRAL study. Gilteritinib in combination with hypomethylating agent as well as quizartinib are not supported by solid randomized trial results for their use in FLT3mut AML patients (Table 1).
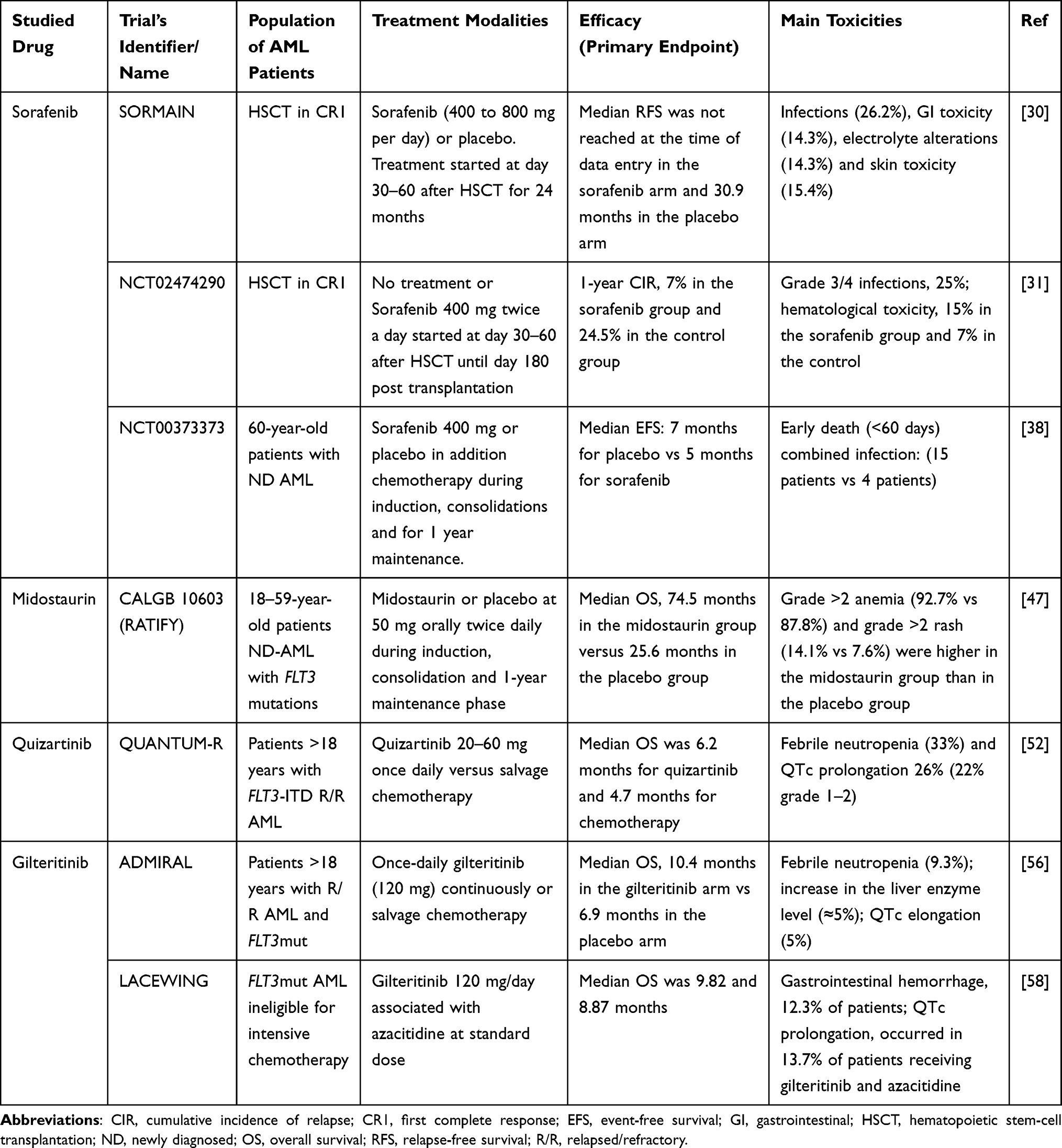 |
Table 1 Main Randomized Studies Evaluating FLT3 Inhibitors in Acute Myeloid Leukemia |
Perspective
Tremendous progress has been made in deciphering the molecular pathogenesis and the tumor heterogeneity in AML. This allowed to move from a one-fits-all to a targeted approach based on a better knowledge of altered molecular pathways.10 Successful development of precision medicines includes the introduction of small-molecule inhibitors of FLT3, IDH1/IDH2 and BCL-2. When administered as single agents, these inhibitors exert substantial activity in patients with R/R AML. Combining FLT3 inhibitors with intensive chemotherapies augments clinical activity and partially overcome the poor prognosis of FLT3mut AML.
Despite these important achievements, some questions remain open Is there a place for second-generation inhibitors in combination with ICT for patients with ND AML? What is the clinical effect and the safety of combinations of FLT3 inhibitors with non-intensive treatment in particular venetoclax-based regimen in elderly patients who are not candidates for intensive chemotherapy? What is the best option to tackle relapsed or refractory disease previously treated with FLT3 inhibitors?
What is the place for second-generation FLT3 inhibitors in combination with ICT? As discussed above, the large European phase 3 (HOVON 156 AML, NCT04027309) is currently enrolling patients to be treated with gilteritinib in combination with ICT in comparison with midostaurin. Other FLT3 inhibitors are currently studied, for instance, the second-generation type I FLT3 inhibitor crenolanib showed evidence of clinical efficacy both in monotherapy and in combination with ICT.59–61 Two phase 3 trials are currently enrolling patients to compare the effect of crenolanib, one investigating ICT in combination with crenolanib or midostaurin in ND FLT3-mutated AML (NCT03258931), the other crenolanib with chemotherapy versus chemotherapy alone in R/R FLT3-mutated AML (NCT03250338).
Is the clinical effect and the safety of combinations of FLT3 inhibitors with non-intensive treatment? Combination of FLT3 inhibitor with the BCL2 inhibitor venetoclax is currently under evaluation. This approach is very promising for FLT3mut AML patients who are not candidates for intensive chemotherapy given the intermediate prognosis of patients treated with venetoclax–azacitidine.5,6 A recently published phase 1 study reported the tolerability and efficacy of gilteritinib with venetoclax-based therapy.62 In this trial enrolling 61 patients, including 56 with FLT3-ITD and 36 patients previously treated with FLT3 inhibitors, venetoclax dose was 400 mg and gilteritinib 120 mg, given once daily continuously. The modified CRc rate (=ORR + MLFS) for FLT3mut patients was 75% (including 18% CR, 22% CRh/CRp and 36% MLFS). Tolerance was manageable with expected grade 3–4 cytopenias. Triplet therapies including FLT3 inhibitors, venetoclax and azacitidine have been retrospectively reported.63 Several prospective early-phase clinical trials are ongoing, testing gilteritinib (NCT04140487) or quizartinib (NCT03661307) among others. Of note, combination of venetoclax and gilterinib seems to be highly synergistic in vitro and in mice xenografts through the inhibition of MCL1 and the kinase AXL.64 This combination will probably be game-changing in the management of FLT3mut AML in the future.
Resistance to FLT3 inhibitors remains a major concern as most of the patients will relapse upon treatment. Several recent reviews cover the topic of resistance mechanisms and propose strategies to overcome them.65,66 An important notion is the oncogene addiction that can explain how broad-spectrum inhibitors like midostaurin may be more effective for ND AML and more targeted inhibitors like quizartinib may be more effective for more R/R AML that are marked by greater dependency on mutant FLT3 as a driver oncogene. Hence, the broad spectrum of activity of the more multi-targeted inhibitors has utility and can offer clinical benefit that more narrow spectrum inhibitors cannot. Drug resistance emerges differently depending on the selectivity of the inhibitor. For instance, studies based on next-generation-sequencing and/or single-cell analyses performed on pre-treatment and relapse pairs of samples discovered that a larger part of patients treated with type 1 inhibitors developed RAS/MAPK mutations contrary with those treated with type 2 inhibitors.67–69 A novel FLT3 inhibitor, FF-10101, in clinical development has shown a high level of efficacy against AML cells harbouring FLT3 mutations including those located in the activation loop acquired upon crenolanib treatment.70,71 Research perspectives in the field of AML resistance, in particular in the setting of FLT3 oncogene addiction, will probably pinpoint the interplay between alternative cell death mechanisms and metabolic deregulations. Recent studies have shown that deregulation of the autophagic pathway is frequent and targetable in FLT3mut AML.72,73 SAR405, a highly potent small-molecule inhibitor of the phosphatidylinositol 3-kinase catalytic subunit type 3 (PIK3C3)/Vps34 induces a blockage at the late endosome-lysosome step autophagy flux and shows interesting preclinical efficacy in FLT3-ITD AML.74 Cell metabolism and mitochondrial respiration are more and more described Achille’s heels of FLT3-ITD AML cells that also can be targeted by therapeutic intervention.75–77 It is likely that repeated molecular and metabolic profiling alongside with in vitro drug sensitivity profiling will provide crucial results for future precision medicine trials.78
Disclosure
The authors report no conflicts of interest in this work.
References
1. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209–2221. doi:10.1056/NEJMoa1516192
2. Hospital MA, Green AS, Maciel TT, et al. FLT3 inhibitors: clinical potential in acute myeloid leukemia. Onco Targets Ther. 2017;10:607–615. doi:10.2147/OTT.S103790
3. Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373(12):1136–1152. doi:10.1056/NEJMra1406184
4. Döhner H, Wei AH, Appelbaum FR, et al. Diagnosis and management of AML in adults: 2022 ELN recommendations from an international expert panel. Blood. 2022:2022016867. doi:10.1182/blood.2022016867
5. DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med. 2020;383(7):617–629. doi:10.1056/NEJMoa2012971
6. Konopleva M, Thirman MJ, Pratz KW, et al. Impact of FLT3 mutation on outcomes after venetoclax and azacitidine for patients with treatment-naïve acute myeloid leukemia. Clin Cancer Res. 2022;28(13):2744–2752. doi:10.1158/1078-0432.CCR-21-3405
7. Patel JP, Gönen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366(12):1079–1089. doi:10.1056/NEJMoa1112304
8. Kottaridis PD, Gale RE, Frew ME, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood. 2001;98(6):1752–1759. doi:10.1182/blood.v98.6.1752
9. Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–447. doi:10.1182/blood-2016-08-733196
10. Döhner H, Wei AH, Löwenberg B. Towards precision medicine for AML. Nat Rev Clin Oncol. 2021;18(9):577–590. doi:10.1038/s41571-021-00509-w
11. Récher C. The beginning of a new therapeutic era in acute myeloid leukemia. EJHaem. 2021;2(4):823–833. doi:10.1002/jha2.252
12. Sutamtewagul G, Vigil CE. Clinical use of FLT3 inhibitors in acute myeloid leukemia. Onco Targets Ther. 2018;11:7041–7052. doi:10.2147/OTT.S171640
13. Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100(5):1532–1542. doi:10.1182/blood-2002-02-0492
14. Levis M. FLT3/ITD AML and the law of unintended consequences. Blood. 2011;117(26):6987–6990. doi:10.1182/blood-2011-03-340273
15. Birg F, Courcoul M, Rosnet O, et al. Expression of the FMS/KIT-like gene FLT3 in human acute leukemias of the myeloid and lymphoid lineages. Blood. 1992;80(10):2584–2593. doi:10.1182/blood.V80.10.2584.2584
16. Zheng R, Levis M, Piloto O, et al. FLT3 ligand causes autocrine signaling in acute myeloid leukemia cells. Blood. 2004;103(1):267–274. doi:10.1182/blood-2003-06-1969
17. Meshinchi S, Appelbaum FR. Structural and functional alterations of FLT3 in acute myeloid leukemia. Clin Cancer Res. 2009;15(13):4263–4269. doi:10.1158/1078-0432.CCR-08-1123
18. Nakao M, Yokota S, Iwai T, et al. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia. 1996;10(12):1911–1918.
19. Kiyoi H, Towatari M, Yokota S, et al. Internal tandem duplication of the FLT3 gene is a novel modality of elongation mutation which causes constitutive activation of the product. Leukemia. 1998;12(9):1333–1337. doi:10.1038/sj.leu.2401130
20. Yamamoto Y, Kiyoi H, Nakano Y, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. 2001;97(8):2434–2439. doi:10.1182/blood.v97.8.2434
21. Bacher U, Haferlach C, Kern W, Haferlach T, Schnittger S. Prognostic relevance of FLT3-TKD mutations in AML: the combination matters--an analysis of 3082 patients. Blood. 2008;111(5):2527–2537. doi:10.1182/blood-2007-05-091215
22. Opatz S, Polzer H, Herold T, et al. Exome sequencing identifies recurring FLT3 N676K mutations in core-binding factor leukemia. Blood. 2013;122(10):1761–1769. doi:10.1182/blood-2013-01-476473
23. von Bubnoff N, Engh RA, Aberg E, Sänger J, Peschel C, Duyster J. FMS-like tyrosine kinase 3-internal tandem duplication tyrosine kinase inhibitors display a nonoverlapping profile of resistance mutations in vitro. Cancer Res. 2009;69(7):3032–3041. doi:10.1158/0008-5472.CAN-08-2923
24. Chatain N, Perera RC, Rossetti G, et al. Rare FLT3 deletion mutants may provide additional treatment options to patients with AML: an approach to individualized medicine. Leukemia. 2015;29(12):2434–2438. doi:10.1038/leu.2015.131
25. Fröhling S, Schlenk RF, Breitruck J, et al. Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: a study of the AML Study Group Ulm. Blood. 2002;100(13):4372–4380. doi:10.1182/blood-2002-05-1440
26. Reindl C, Bagrintseva K, Vempati S, et al. Point mutations in the juxtamembrane domain of FLT3 define a new class of activating mutations in AML. Blood. 2006;107(9):3700–3707. doi:10.1182/blood-2005-06-2596
27. Daver N, Schlenk RF, Russell NH, Levis MJ. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia. 2019;33(2):299–312. doi:10.1038/s41375-018-0357-9
28. Hasskarl J. Sorafenib: targeting multiple tyrosine kinases in cancer. Recent Results Cancer Res. 2014;201:145–164. doi:10.1007/978-3-642-54490-3_8
29. Borthakur G, Kantarjian H, Ravandi F, et al. Phase I study of sorafenib in patients with refractory or relapsed acute leukemias. Haematologica. 2011;96(1):62–68. doi:10.3324/haematol.2010.030452
30. Burchert A, Bug G, Fritz LV, et al. Sorafenib maintenance after allogeneic hematopoietic stem cell transplantation for acute myeloid leukemia with FLT3-internal tandem duplication mutation (SORMAIN). J Clin Oncol. 2020;38(26):2993–3002. doi:10.1200/JCO.19.03345
31. Xuan L, Wang Y, Huang F, et al. Sorafenib maintenance in patients with FLT3-ITD acute myeloid leukaemia undergoing allogeneic haematopoietic stem-cell transplantation: an open-label, multicentre, randomised phase 3 trial. Lancet Oncol. 2020;21(9):1201–1212. doi:10.1016/S1470-2045(20)30455-1
32. Mathew NR, Baumgartner F, Braun L, et al. Sorafenib promotes graft-versus-leukemia activity in mice and humans through IL-15 production in FLT3-ITD-mutant leukemia cells. Nat Med. 2018;24(3):282–291. doi:10.1038/nm.4484
33. Ravandi F, Arana YC, Cortes JE, et al. Final report of phase II study of sorafenib, cytarabine and idarubicin for initial therapy in younger patients with acute myeloid leukemia. Leukemia. 2014;28(7):1543–1545. doi:10.1038/leu.2014.54
34. Ravandi F, Cortes JE, Jones D, et al. Phase I/II study of combination therapy with sorafenib, idarubicin, and cytarabine in younger patients with acute myeloid leukemia. J Clin Oncol. 2010;28(11):1856–1862. doi:10.1200/JCO.2009.25.4888
35. Röllig C, Serve H, Hüttmann A, et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): a multicentre, phase 2, randomised controlled trial. Lancet Oncol. 2015;16(16):1691–1699. doi:10.1016/S1470-2045(15)00362-9
36. Röllig C, Serve H, Noppeney R, et al. Sorafenib or placebo in patients with newly diagnosed acute myeloid leukaemia: long-term follow-up of the randomized controlled SORAML trial. Leukemia. 2021;35(9):2517–2525. doi:10.1038/s41375-021-01148-x
37. Wei A. Results of a phase 2, randomized, double-blind study of sorafenib versus placebo in combination with intensive chemotherapy in previously untreated patients with FLT3-ITD acute myeloid leukemia (ALLG AMLM16). ASH; 2020. Available from: https://ash.confex.com/ash/2020/webprogram/Paper137334.html.
38. Serve H, Krug U, Wagner R, et al. Sorafenib in combination with intensive chemotherapy in elderly patients with acute myeloid leukemia: results from a randomized, placebo-controlled trial. J Clin Oncol. 2013;31(25):3110–3118. doi:10.1200/JCO.2012.46.4990
39. Ravandi F, Alattar ML, Grunwald MR, et al. Phase 2 study of azacytidine plus sorafenib in patients with acute myeloid leukemia and FLT-3 internal tandem duplication mutation. Blood. 2013;121(23):4655–4662. doi:10.1182/blood-2013-01-480228
40. Ohanian M, Garcia-Manero G, Levis M, et al. Sorafenib combined with 5-azacytidine in older patients with untreated FLT3-ITD mutated acute myeloid leukemia. Am J Hematol. 2018;93(9):1136–1141. doi:10.1002/ajh.25198
41. Zhao JC, Agarwal S, Ahmad H, Amin K, Bewersdorf JP, Zeidan AM. A review of FLT3 inhibitors in acute myeloid leukemia. Blood Rev. 2022;52:100905. doi:10.1016/j.blre.2021.100905
42. Stone RM, DeAngelo DJ, Klimek V, et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. 2005;105(1):54–60. doi:10.1182/blood-2004-03-0891
43. Fischer T, Stone RM, Deangelo DJ, et al. Phase IIB trial of oral Midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J Clin Oncol. 2010;28(28):4339–4345. doi:10.1200/JCO.2010.28.9678
44. Maziarz RT, Levis M, Patnaik MM, et al. Midostaurin after allogeneic stem cell transplant in patients with FLT3-internal tandem duplication-positive acute myeloid leukemia. Bone Marrow Transplant. 2021;56(5):1180–1189. doi:10.1038/s41409-020-01153-1
45. Schlenk RF, Weber D, Fiedler W, et al. Midostaurin added to chemotherapy and continued single-agent maintenance therapy in acute myeloid leukemia with FLT3-ITD. Blood. 2019;133(8):840–851. doi:10.1182/blood-2018-08-869453
46. Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med. 2017;377(5):454–464. doi:10.1056/NEJMoa1614359
47. Jahn N, Jahn E, Saadati M, et al. Genomic landscape of patients with FLT3-mutated acute myeloid leukemia (AML) treated within the CALGB 10603/RATIFY trial. Leukemia. 2022;36(9):2218–2227. doi:10.1038/s41375-022-01650-w
48. Strati P, Kantarjian H, Ravandi F, et al. Phase I/II trial of the combination of midostaurin (PKC412) and 5-azacytidine for patients with acute myeloid leukemia and myelodysplastic syndrome. Am J Hematol. 2015;90(4):276–281. doi:10.1002/ajh.23924
49. Zarrinkar PP, Gunawardane RN, Cramer MD, et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood. 2009;114(14):2984–2992. doi:10.1182/blood-2009-05-222034
50. Cortes JE, Kantarjian H, Foran JM, et al. Phase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS-like tyrosine kinase 3-internal tandem duplication status. J Clin Oncol. 2013;31(29):3681–3687. doi:10.1200/JCO.2013.48.8783
51. Cortes J, Perl AE, Döhner H, et al. Quizartinib, an FLT3 inhibitor, as monotherapy in patients with relapsed or refractory acute myeloid leukaemia: an open-label, multicentre, single-arm, phase 2 trial. Lancet Oncol. 2018;19(7):889–903. doi:10.1016/S1470-2045(18)30240-7
52. Cortes JE, Khaled S, Martinelli G, et al. Quizartinib versus salvage chemotherapy in relapsed or refractory FLT3-ITD acute myeloid leukaemia (QuANTUM-R): a multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2019;20(7):984–997. doi:10.1016/S1470-2045(19)30150-0
53. Altman JK, Foran JM, Pratz KW, Trone D, Cortes JE, Tallman MS. Phase 1 study of quizartinib in combination with induction and consolidation chemotherapy in patients with newly diagnosed acute myeloid leukemia. Am J Hematol. 2018;93(2):213–221. doi:10.1002/ajh.24974
54. Levis M, Perl AE. Gilteritinib: potent targeting of FLT3 mutations in AML. Blood Adv. 2020;4(6):1178–1191. doi:10.1182/bloodadvances.2019000174
55. Perl AE, Altman JK, Cortes J, et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-label, phase 1–2 study. Lancet Oncol. 2017;18(8):1061–1075. doi:10.1016/S1470-2045(17)30416-3
56. Perl AE, Martinelli G, Cortes JE, et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N Engl J Med. 2019;381(18):1728–1740. doi:10.1056/NEJMoa1902688
57. Pratz K A. Phase 1 study of gilteritinib in combination with induction and consolidation chemotherapy in patients with newly diagnosed AML: final results. ASH; 2020. Available from: https://ash.confex.com/ash/2020/webprogram/Paper137685.html.
58. Wang ES, Montesinos P, Minden MD, et al. Phase 3 trial of gilteritinib plus azacitidine vs azacitidine for newly diagnosed FLT3mut+ AML ineligible for intensive chemotherapy. Blood. 2022;140(17):1845–1857. doi:10.1182/blood.2021014586
59. Cortes JE, Kantarjian HM, Kadia TM, et al. Crenolanib besylate, a type I pan-FLT3 inhibitor, to demonstrate clinical activity in multiply relapsed FLT3-ITD and D835 AML. JCO. 2016;34(15_suppl):7008. doi:10.1200/JCO.2016.34.15_suppl.7008
60. Randhawa JK, Kantarjian HM, Borthakur G, et al. Results of a phase II study of crenolanib in relapsed/refractory acute myeloid leukemia patients (pts) with activating FLT3 mutations. Blood. 2014;124(21):389. doi:10.1182/blood.V124.21.389.389
61. Wang ES, Griffiths EA, Walter RB, et al. Tolerability and efficacy of crenolanib and cytarabine/anthracycline chemotherapy in older patients (aged 61 to 75) with newly diagnosed FLT3-mutated Acute Myeloid Leukemia (AML). Blood. 2019;134:3829. doi:10.1182/blood-2019-130536
62. Daver N, Perl AE, Maly J, et al. Venetoclax plus gilteritinib for FLT3-mutated relapsed/refractory acute myeloid leukemia. J Clin Oncol. 2022:JCO2200602. doi:10.1200/JCO.22.00602
63. Maiti A, DiNardo CD, Daver NG, et al. Triplet therapy with venetoclax, FLT3 inhibitor and decitabine for FLT3-mutated acute myeloid leukemia. Blood Cancer J. 2021;11(2):25. doi:10.1038/s41408-021-00410-w
64. Janssen M, Schmidt C, Bruch PM, et al. Venetoclax synergizes with gilteritinib in FLT3 wild-type high-risk acute myeloid leukemia by suppressing MCL-1. Blood. 2022;140(24):2594–2610. doi:10.1182/blood.2021014241
65. Tecik M, Adan A. Therapeutic targeting of FLT3 in acute myeloid leukemia: current status and novel approaches. Onco Targets Ther. 2022;15:1449–1478. doi:10.2147/OTT.S384293
66. Desikan SP, Daver N, DiNardo C, Kadia T, Konopleva M, Ravandi F. Resistance to targeted therapies: delving into FLT3 and IDH. Blood Cancer J. 2022;12(6):91. doi:10.1038/s41408-022-00687-5
67. Alotaibi AS, Yilmaz M, Kanagal-Shamanna R, et al. Patterns of resistance differ in patients with acute myeloid leukemia treated with type I versus type II FLT3 inhibitors. Blood Cancer Discov. 2021;2(2):125–134. doi:10.1158/2643-3230.BCD-20-0143
68. Schmalbrock LK, Dolnik A, Cocciardi S, et al. Clonal evolution of acute myeloid leukemia with FLT3-ITD mutation under treatment with midostaurin. Blood. 2021;137(22):3093–3104. doi:10.1182/blood.2020007626
69. McMahon CM, Ferng T, Canaani J, et al. Clonal selection with RAS pathway activation mediates secondary clinical resistance to selective FLT3 inhibition in acute myeloid leukemia. Cancer Discov. 2019;9(8):1050–1063. doi:10.1158/2159-8290.CD-18-1453
70. Yamaura T, Nakatani T, Uda K, et al. A novel irreversible FLT3 inhibitor, FF-10101, shows excellent efficacy against AML cells with FLT3 mutations. Blood. 2018;131(4):426–438. doi:10.1182/blood-2017-05-786657
71. Ferng TT, Terada D, Ando M, et al. The irreversible FLT3 inhibitor FF-10101 is active against a diversity of FLT3 inhibitor resistance mechanisms. Mol Cancer Ther. 2022;21(5):844–854. doi:10.1158/1535-7163.MCT-21-0317
72. Seo W, Silwal P, Song IC, Jo EK. The dual role of autophagy in acute myeloid leukemia. J Hematol Oncol. 2022;15(1):51. doi:10.1186/s13045-022-01262-y
73. Dupont M, Huart M, Lauvinerie C, et al. Autophagy targeting and hematological mobilization in FLT3-ITD acute myeloid leukemia decrease repopulating capacity and relapse by inducing apoptosis of committed leukemic cells. Cancers. 2022;14(2):453. doi:10.3390/cancers14020453
74. Ronan B, Flamand O, Vescovi L, et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat Chem Biol. 2014;10(12):1013–1019. doi:10.1038/nchembio.1681
75. Park HJ, Gregory MA, Zaberezhnyy V, et al. Therapeutic resistance in acute myeloid leukemia cells is mediated by a novel ATM/mTOR pathway regulating oxidative phosphorylation. Elife. 2022;11:e79940. doi:10.7554/eLife.79940
76. Bjelosevic S, Gruber E, Newbold A, et al. Serine biosynthesis is a metabolic vulnerability in FLT3-ITD-driven acute myeloid leukemia. Cancer Discov. 2021;11(6):1582–1599. doi:10.1158/2159-8290.CD-20-0738
77. Garciaz S, Guirguis AA, Müller S, et al. Pharmacologic reduction of mitochondrial iron triggers a noncanonical BAX/BAK-dependent cell death. Cancer Discov. 2022;12(3):774–791. doi:10.1158/2159-8290.CD-21-0522
78. Burd A, Levine RL, Ruppert AS, et al. Precision medicine treatment in acute myeloid leukemia using prospective genomic profiling: feasibility and preliminary efficacy of the Beat AML Master Trial. Nat Med. 2020;26(12):1852–1858. doi:10.1038/s41591-020-1089-8
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.