")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Fluorescence detection of tramadol in healthy Chinese volunteers by high-performance liquid chromatography and bioequivalence assessment
Received 4 September 2014
Accepted for publication 9 December 2014
Published 26 February 2015 Volume 2015:9 Pages 1225—1231
DOI https://doi.org/10.2147/DDDT.S73723
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Shu-Feng Zhou
Xiao Zhou, Ji Liu
Department of Anesthesia, Shanghai Pulmonary Hospital, Tongji University School of Medicine, Shanghai, People’s Republic of China
Abstract: This study developed a revised high-performance liquid chromatography fluorescence method to determine plasma tramadol concentration, and thereby to examine the bioequivalence of two tramadol formulations among healthy male Chinese volunteers. The study used a double-blind, randomized, 2×2 crossover-design principle. Calculated pharmacokinetic parameters for both formulations were consistent with previous reports. According to the observation of vital signs and laboratory measurement, no subjects had any adverse reactions. The geometric mean ratios (90% confidence interval) of the test drug/reference drug for tramadol were 100.2% (95.3%–103.4%) for the area under the plasma concentration–time curve (AUC) from time zero to the last measurable concentration, 99.6% (94.2%–102.7%) for the AUC from administration to infinite time, and 100.8% (93.1%–106.4%) for maximum concentration. For the 90% confidence intervals of the test/reference AUC ratio and maximum concentration ratio of tramadol, both were in the acceptance range for bioequivalence. According to the two preparations by pharmacokinetic parameter statistics, the half-life, mean residence time, and clearance values showed no significant statistical differences. Therefore, the conclusion of this study was that the two tramadol formulations (tablets and capsules) were bioequivalent.
Keywords: tramadol hydrochloride, in vitro release, pharmacokinetic, bioequivalence, fluorescence detector
Introduction
Tramadol hydrochloride is a synthetic, centrally acting analgesic opioid drug, similar to codeine and morphine in its structure. It works through the inhibition of neuronal reuptake of noradrenaline by weak opioid-receptor agonists, and increases the concentration of serotonin, influencing pain transmission and producing an analgesic effect. In some acute pain conditions, it has proven efficacy and safety, including trauma, renal or biliary colic, and childbirth pains. Chronic pain of malignant or benign origin, in particular neuropathic pain, is a common indication for tramadol.1
Tramadol’s intensity is only an eighth to a tenth that of morphine, with no inhibition of respiration and little dependence. Tramadol is available in various dosage forms for oral administration, such as tablets, capsules, and sustained-release formulations. It is mostly eliminated via biotransformation in the liver in two main pathways: O-demethylation to O-desmethyltramadol (M1) (the pharmacologically active metabolite) by isoenzyme cytochrome P450 2D6 (CYP2D6), and N-demethylation to N-desmethyltramadol (M2) by cytochromes (CYP2B6 and CYP3A4).2 Among these metabolites, only O-desmethyltramadol (M1 metabolite) is pharmacologically active.3 Approximately 10%–30% of the parent drug is excreted unmetabolized in the urine.3,4
The mean absolute bioavailability of tramadol with all oral formulations is approximately 70%. With plasma protein binding of about 20%. Tramadol has linear pharmacokinetic significance in the therapeutic dose range. It is dose-dependent for the relationship between serum concentrations and the analgesic effect, and it varies greatly from individual to individual. The literature has shown that serum concentrations at 100–300 ng/mL were usually an effective concentration.5 The maximum daily recommended dose for clinical use is 400 mg/day.6
Several bioanalytical methods have been reported to determine tramadol alone or in combination with its metabolites in a variety of biological matrices. Methods for estimating tramadol alone have been described, employing high-performance liquid chromatography (HPLC) with ultraviolet light,7,8 fluorescence,9 diode-array detection,10 gas chromatography with flame-ionization detection,11 and mass-spectrometry detection.12–14
In recent years, because of lower prices, generic drugs have been getting more popular among patients. Therefore, bioequivalence study is a commonly accepted method to prove the similar efficacy of two formulations.15 In the People’s Republic of China (PRC), Tramal® (Grünenthal, Germany) is currently a commercially available tramadol capsule, and some generic versions of tramadol tablets have already been developed with lower cost and applied for clinical use. Although the generic and the innovator formulation contain the same active ingredients, they have a different production process and use different accessories. Those factors may affect the release speed and absorption degree in vivo. Therefore, the main purpose of bioequivalence testing is to verify whether the two formulations in humans have the same effect. The aim of this study was to assess bioequivalence for the same dose (100 mg) of the two tramadol formulations.
Materials and methods
Subjects
Twenty healthy male volunteers were recruited from the Department of Pharmacology of our hospital. The criteria for the male subjects were as follows: age 18–35 years, body weight within the normal range (body mass index between 19.6 kg/m2 and 24.8 kg/m2), systolic blood pressure of 110–120 mmHg, and diastolic blood pressure 70–80 mmHg. In addition, the criteria also included current nonsmoking and normal routine clinical examination, chest X-ray, electrocardiogram, and laboratory test items (hematology, blood biochemistry, hepatic function, and urinalysis). Participants needed to have human immunodeficiency virus-, hepatitis B-, and hepatitis C-negative test results. All qualified participants signed an informed consent form (institutional review board, protocol number 93025410).
Drugs and materials
The test formulation of tramadol tablets (50 mg) was produced by CSPC Pharm, Shijiazhuang, PRC. The batch number was 080121. The reference formulation of tramadol capsules (50 mg) was obtained from the local hospital. The batch number was J20110065.
Tramadol and O-desmethyltramadol (purity 99.3% and 99.1%, respectively) were provided by the National Institute for Food and Drug Control (Beijing, PRC). The internal standard, fluconazole (purity 98.7%), was purchased from Sigma-Aldrich (USA). Acetonitrile and ethyl acetate were of analytical grade, and provided by Shanghai Chemical Reagents Company, PRC. Purified water from a Milli-Q system (Millipore, USA) was used throughout the experiment.
In vitro dissolution study
Before performing the in vivo bioequivalence study, the researchers prepared two formulations in a head-to-head in vitro dissolution experiment. The results for in vitro dissolution data were acceptable as per the regulatory guidelines for conducting bioequivalence studies.16 Based on the principles of the Chinese Pharmacopoeia rotating basket method, dissolution testing required that each formulation have six parallel tests, and the results are expressed as means ± standard deviation. Dissolution conditions were an HCl 0.1 M solution of 900 mL as dissolution medium maintained at a basket rotation speed of 50 rotations per minute. The dissolution medium was maintained at 37°C±0.5°C. At 2, 5, 10, 15, 30, and 45 minutes, the samples were drawn. Each sample was analyzed by HPLC to determine the dissolution rate. By using standard mathematical equations on average dissolution value at each interval time point, the dissimilarity factor (f1) and similarity factor (f2) of the two preparations were calculated.
Study design
The study design was a randomized, open-label 2×2 crossover. This study protocol was approved by the ethical committee in the university and the Clinical Trials Research Committee, and followed the principles of the Declaration of Helsinki and its amendments.17
Subjects arrived at the hospital on the day before the study and were put in constant-temperature rooms. A table of random numbers was used to assign subjects in a 1:1 ratio to receive a single 100 mg dose (two capsules or two tablets given with 150 mL of water) of the test or reference formulation of tramadol. Prior to treatment, each subject underwent an overnight fast for at least 10 hours and during each treatment; a standard meal was served 4 hours after dosing. A 2-week washout period followed administration of the initial formulation, after which the alternate formulation was administered.
Blood sampling
Blood samples were collected in heparinized tubes prior to drug-administration zero time, and at 0.5, 1, 2, 3, 4, 6, 8, 10, 12, and 24 hours after dosing. Blood samples (5 mL) were centrifuged (3,000× g for 10 minutes), and the plasma collected was stored at −18°C until analysis. To a 500 μL aliquot of plasma sample in a 10 mL clean glass tube, 20 μL of the internal standard fluconazole (500 μg/mL), and 250 μL of NaOH (0.2 mol/L) were added. The samples were vortexed for 1 minute, and 4 mL ethyl acetate was added. The mixture was vortex-mixed for 5 minutes. After centrifugation (3,000× g for 10 minutes), the upper organic layer was transferred into a clean glass tube and evaporated to dryness at 40°C under a gentle stream of nitrogen. The residue was reconstituted in a 100 μL mobile phase, and transferred to an autosampler vial. An aliquot of 20 μL was injected into the HPLC system for analysis.
Analytical method
Tramadol concentrations were determined using HPLC fluorescence detection based on the method of De Leo et al.18 An Agilent G1321 fluorescence detector was operated at an excitation wavelength of 275 nm and an emission wavelength of 302 nm. Chromatographic separation was achieved on a Gemini C18 column (150×2.0 mm, 5 μm; Phenomenex, USA) with a Security Guard C18 guard column. A mixture of acetonitrile and 0.04 mol/L NaH2PO3 (pH 4.0) =25:75 (v/v) was used as the mobile phase at a flow rate of 1 mL/min. The temperature of the column and autosampler was maintained at 35°C and 4°C, respectively. The chromatographic run time of each sample was 12 minutes.
Method validation
Precision and accuracy
The precision and accuracy of the assay were obtained by comparing the predicted concentration (obtained from the calibration curve), with the actual concentration of tramadol and O-desmethyltramadol spiked in blank plasma. Intraday precision was determined by repeated analysis of each quality-control sample on day 1 (n=5), and interday precision was determined by repeated analysis on 5 consecutive days (n=1 series per day). Precision is expressed as the interday and intraday coefficients of variation ([standard deviation/mean of the recoveries] ×100%). Accuracy was defined as the relative deviation in the computed value (E) of a standard from that of its true value (T), expressed as a percentage (RE%). It was calculated using the formula RE% = (E − T)/T ×100. The limit of quantitation (LOQ) was considered as the concentration of tramadol and O-desmethyltramadol that produced a signal-to-noise (S/N) ratio of 10.
Selectivity and specificity
Tests for selectivity were carried out in six different lots of blank plasma (with heparin sodium as anticoagulant). They were processed by the same extraction protocol and analyzed to determine the extent to which endogenous plasma components may contribute to the interference at the retention time of analytes and the internal standard.
Freeze and thaw stability
The freeze- and thaw-stability study samples were obtained by adding the standard solution in the blank human plasma at three QC levels. These samples were frozen at −20°C for 7 days, and then thawed at room temperature. After being allowed to completely thaw, the samples were refrozen for 24 hours under the same conditions. This freeze–thaw cycle was repeated three times before these samples were analyzed.
Recovery determination
The absolute recovery of tramadol and O-desmethyltramadol was determined by direct comparison of peak areas from extracts versus spiked postextraction samples at 10, 100, and 800 ng/mL and 5, 50, and 500 ng/mL.
Tolerability
Tolerability assessment was primarily completed by monitoring the subject’s vital signs (blood pressure and heart rate). Time points were measured at baseline (predose) and at 0.5, 1, 2, 3, 4, 6, 8, 10, 12, and 24 hours after administration. At baseline and after completion of the study, laboratory tests (hematology, blood biochemistry, hepatic function, and urinalysis) were performed. Subjects were fully informed of adverse events that may occur during the test.
Pharmacokinetic study
The following pharmacokinetic parameters were calculated using noncompartmental methods: maximum plasma concentration (Cmax) and time to Cmax were obtained directly from the experimental data. The area under the plasma concentration–time curve from administration to infinite time (AUC0–∞) was calculated using the trapezoidal method: trapezoidal area from time zero to the last measurable concentration (AUC0–T), extrapolated to infinite time, by addition of the area obtained from the last measurable concentration divided by the terminal elimination rate constant (β); β was estimated from the linear least-squared regression of the terminal phase of the log concentration–time profile. The apparent biological half-life (t½) was calculated as 0.693/β. The formula of AUMC/AUC0–∞ was applied to determine the mean residence time values. The results of the pharmacokinetic calculations are presented as means ± standard deviation.
Statistical analysis
In order to test the bioequivalence of the formulations, analysis of variance (ANOVA) for a 2×2 crossover design was performed for log-transformed Cmax, AUC0–T, and AUC0–∞. The log-transformed Cmax ratio, AUC0–T ratio, and AUC0–∞ ratio were obtained for both formulations. ANOVA was performed by using the F-test. Two one-sided t-tests, as described by Schuirmann,19 were used to obtain the probability of exceeding the limits of acceptance for bioequivalence (80%–125%). If the log-transformed ratios of Cmax and AUC values of the two formulations were within a predetermined equivalence range, and their 90% confidence interval (CI) P-value was less than 0.05, the two formulations were considered bioequivalent.
Results
Bioanalytical method validation
This study established a reverse-phase HPLC fluorescence method for the determination of tramadol and O-desmethyltramadol concentrations in human. The study had statistics of human pharmacokinetic parameters. The bioanalytical time per sample was around 10 minutes.
The representative chromatograms of blank plasma (A), and blank plasma added to tramadol, O-desmethyltramadol, the internal standard (B) and sample (C) are shown in Figure 1. The analytical peaks of tramadol, O-desmethyltramadol, and the internal standard were resolved with good symmetry. The retention time of tramadol, O-desmethyltramadol, and the internal standard were 7.2, 6.8, and 6.6 minutes, respectively. No endogenous sources of interference were observed at the retention time of the analyte.
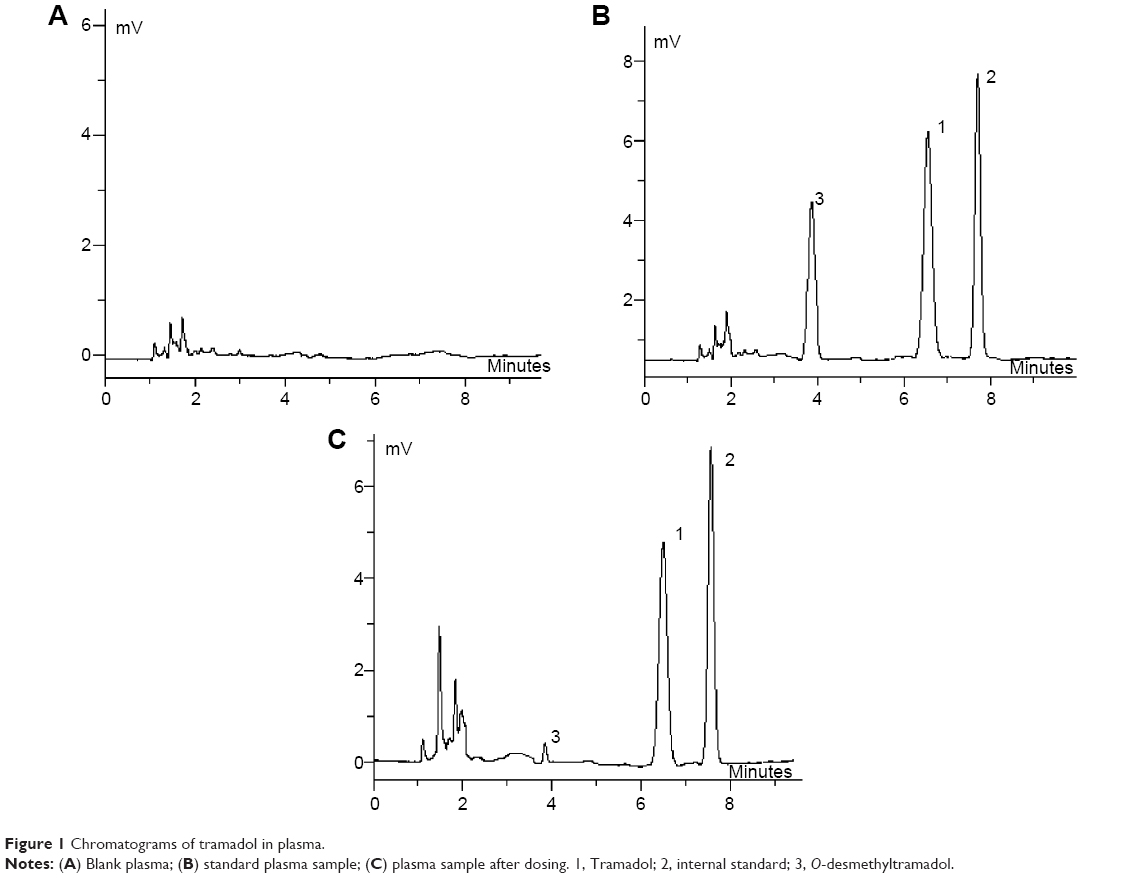 | Figure 1 Chromatograms of tramadol in plasma. |
A sample from a volunteer after intake of tramadol (at 4 hours) is shown in Figure 1C. The chromatograms of LOQ samples were 5 ng/mL for tramadol and 3.5 ng/mL for O-desmethyltramadol (S/N =10). Good linearity was observed over the concentration range of 10–1,000 ng/mL plasma (r=0.9993) for tramadol and 5–600 ng/mL plasma (r=0.9991) for O-desmethyltramadol.
Intra/interday precision and accuracy were evaluated. The relative standard deviation of tramadol ranged from 5.34% to 7.98% for intra-day and 4.75% to 6.97% for inter-day, respectively. The relative error of tramadol ranged from 3.1% to 5.3% for intraday and 3.6% to 4.7% for interday, respectively. The relative standard deviation of O-desmethyltramadol ranged from 5.54% to 7.63% for intraday and 6.43% to 7.76% for interday, respectively. The relative error of O-desmethyltramadol ranged from 4.2% to 6.5% for intraday and 2.2% to 4.9% for interday, respectively.
Freshly prepared solutions showed no evidence of degradation for either tramadol, O-desmethyltramadol, or the internal standard. No significant degradation was observed for any analyte during sample processing and extraction, including the dry-down procedure. Tramadol and O-desmethyltramadol in sample solution were found to be stable for approximately 24 hours, since the concentrations found were within 93%–99% of the initial concentrations. The results obtained after three freeze–thaw cycles demonstrated that about 82.4%–93.6% of the initial content of tramadol and O-desmethyltramadol were recovered, and that the analytes were stable under these conditions. Plasma samples collected from studies of tramadol and O-desmethyltramadol were evaluated before and after storage at −18°C for stability and found to be stable for at least 3 months. The measured values obtained were in line with the guiding principles of the 2001 edition of the US Food and Drug Administration’s Bioanalytical Method Development.
In vitro release
Figure 2 shows the in vitro release profiles. A dissimilarity factor (f1) of 1.65 (acceptable limit 0–15) and similarity factor (f2) of 92.2 (acceptable limit 50–100) were obtained from the release profiles. To a certain extent, comparative study on the in vitro dissolution can predict whether the two were bioequivalent. The experimental results showed that the two preparations’ in vitro dissolution rates were similar, and the biological behavior in vivo could possibly have been similar too, which led to a positive test result.
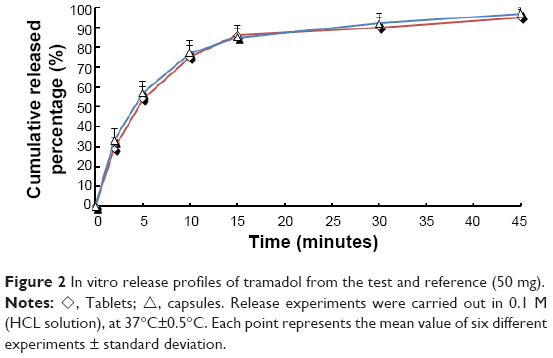 | Figure 2 In vitro release profiles of tramadol from the test and reference (50 mg). |
Pharmacokinetic and bioequivalence studies
A developed bioanalytical method was employed for determining pharmacokinetic parameters after administration of different dosage forms: tramadol tablets and tramadol capsules at a dose of 100 mg in healthy male Chinese volunteers. The plasma concentration versus time profile of tramadol obtained is depicted in Figure 3, and pharmacokinetic parameters obtained are summarized in Table 1. The geometric mean ratios (90% CI) of the test drug/reference drug for tramadol were 100.2% (95.3%–103.4%) for AUC0–T, 99.6% (94.2%–102.7%) for AUC0–∞, and 100.8% (93.1%–106.4%) for Cmax. The 90% CIs of the test/reference AUC ratio and Cmax ratio of tramadol were within the acceptance range for bioequivalence. The differences between the two products for t½, mean residence time, and clearance values were not found to be statistically significant (P>0.05). The geometric mean ratios (90% CI) of AUC0–T, AUC0–∞, and Cmax of tramadol that resulted from the tablet group as well as from the capsule group are presented in Table 2.
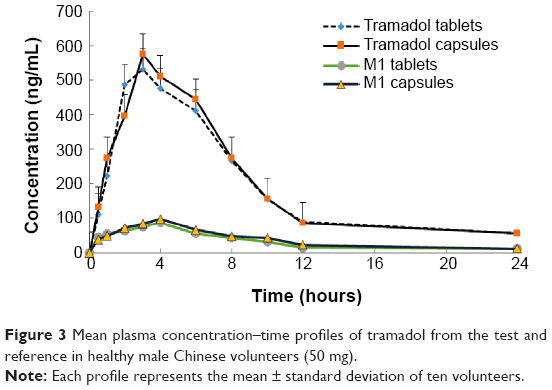 | Figure 3 Mean plasma concentration–time profiles of tramadol from the test and reference in healthy male Chinese volunteers (50 mg). |
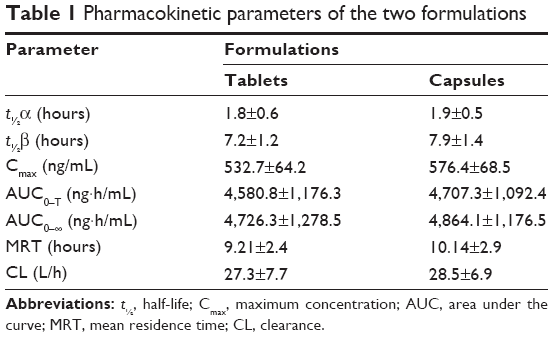 | Table 1 Pharmacokinetic parameters of the two formulations |
 | Table 2 Statistical comparison of the two formulations |
Discussion
This study developed a revised HPLC fluorescence method to determine plasma tramadol concentration, thereby to examine the bioequivalence of two tramadol formulations in healthy male Chinese volunteers. Calculated pharmacokinetic parameters for both formulations were consistent with those reported by García-Quetglas et al.20 No adverse events were reported by subjects or found on analysis of vital signs or laboratory tests.
If the two formulations were bioequivalent (extent and rate), then under the same conditions of dose and administration, they must have similar clinical efficacy.21 Bioequivalence criteria were defined as 90% CI of the geometric mean ratios of the test/reference drug of between 80.0% and 125.0% for AUC0–T, AUC0–∞, and Cmax. Statistical comparison of AUC0–T, AUC0-∞, and Cmax clearly demonstrated no significant differences in the two tramadol formulations. Based on the pharmacokinetic and statistical results of this study, we can conclude that the tramadol capsules were bioequivalent to the tramadol tablets with respect to both their extent and rate of absorption, and that the two products can be considered interchangeable in medical practice.
Disclosure
The authors report no conflicts of interest in this work.
References
Grond S, Sablotzki A. Clinical pharmacology of tramadol. Clin Pharmacokinet. 2004;43(13):879–923. | ||
Wu WN, McKown LA, Liao S. Metabolism of the analgesic drug ULTRAM (tramadol hydrochloride) in humans: API-MS and MS/MS characterization of metabolites. Xenobiotica. 2002;32(5):411–425. | ||
García Quetglas E, Azanza JR, Cardenas E, Sádaba B, Campanero MA. Stereoselective pharmacokinetic analysis of tramadol and its main phase I metabolites in healthy subjects after intravenous and oral administration of racemic tramadol. Biopharm Drug Dispos. 2007;28(1):19–33. | ||
Subrahmanyam V, Renwick AB, Walters DG, et al. Identification of cytochrome P-450 isoforms responsible for cis-tramadol metabolism in human liver microsomes. Drug Metab Dispos. 2001;29(8):1146–1155. | ||
Zydol SR [prescribing information]. Wycombe, UK: Grunenthal; 2010. | ||
Ultram ER [prescribing information]. Steinbach, MB: Biovail; 2010. | ||
Qu L, Feng S, Wu Y, Wu Y. [HPLC method for determination of tramadol hydrochloride in human plasma]. Sichuan Da Xue Xue Bao Yi Xue Ban. 2003;34(3):574–575. Chinese. | ||
Gan SH, Ismail R, Wan Adnan WA, Wan Z. Method development and validation of a high-performance liquid chromatographic method for tramadol in human plasma using liquid-liquid extraction. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;772(1):123–129. | ||
Ebrahimzadeh H, Yamini Y, Sedighi A, Rouini MR. Determination of tramadol in human plasma and urine samples using liquid phase microextraction with back extraction combined with high performance liquid chromatography. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;863(2):229–234. | ||
Küçük A, Kadioğlu Y, Celebi F. Investigation of the pharmacokinetics and determination of tramadol in rabbit plasma by a high-performance liquid chromatography-diode array detector method using liquid-liquid extraction. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;816(1–2):203–208. | ||
Ho ST, Wan JJ, Liaw WJ, Ho CM, Li JH. Determination of tramadol by capillary gas chromatography with flame ionization detection. Application to human and rabbit pharmacokinetic studies. J Chromatogr B Biomed Sci Appl. 1999;736(1–2):89–96. | ||
Gambaro V, Benvenuti C, De Ferrari L, Dell’Acqua L, Farè F. Validation of a GC/MS method for the determination of tramadol in human plasma after intravenous bolus. Farmaco. 2003;58(9):947–950. | ||
Leis HJ, Fauler G, Windischhofer W. Synthesis of d1-N-ethyltramadol as an internal standard for the quantitative determination of tramadol in human plasma by gas chromatography-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;804(2):369–374. | ||
Sha YF, Shen S, Duan GL. Rapid determination of tramadol in human plasma by headspace solid-phase microextraction and capillary gas chromatography-mass spectrometry. J Pharm Biomed Anal. 2005;37(1):143–147. | ||
Center for Drug Evaluation. The Guiding Principle of Bioavailability of China. Beijing: State Food and Drug Administration; 2006. | ||
Prior A, Frutos P, Correa CP. Comparison of dissolution profiles: current guidelines. Available from: http://www.sefig.com/doc/Congreso%20Granada/013_DOC.pdf. Accessed November 1, 2010. | ||
World Medical Association. World Medical Association Declaration of Helsinki: Ethical Principles for Medical Research Involving Human Subjects. Ferney-Voltaire, France: WMA; 2008. Available from: http://www.wma.net/en/30publications/10policies/b3/17c.pdf. Accessed January 7, 2015. | ||
De Leo M, Giorgi M, Saccomanni G, Manera C, Braca A. Evaluation of tramadol and its main metabolites in horse plasma by high-performance liquid chromatography/fluorescence and liquid chromatography/electrospray ionization tandem mass spectrometry techniques. Rapid Commun Mass Spectrom. 2009;23(2):228–236. | ||
Schuirmann DJ. A comparison of the two one-sided tests procedure and the power approach for assessing the equivalence of average bioavailability. J Pharmacokinet Biopharm. 1987;15(6):657–680. | ||
García-Quetglas E, Azanza JR, Sádaba B, Muñoz MJ, Gil I, Campanero MA. Pharmacokinetics of tramadol enantiomers and their respective phase I metabolites in relation to CYP2D6 phenotype. Pharmacol Res. 2007;55(2):122–130. | ||
Kesselheim AS, Misono AS, Lee JL, et al. Clinical equivalence of generic and brand-name drugs used in cardiovascular disease: a systematic review and meta-analysis. JAMA. 2008;300(21):2514–2526. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.