")
Back to Journals » International Medical Case Reports Journal » Volume 13
Fibrous Meningioma in a Patient with Encephalocraniocutaneous Lipomatosis: A Rare Case with Unique Features
Authors Al Qawasmeh M , Aldabbour B , Alhayek K , El-Salem K
Received 23 June 2020
Accepted for publication 31 July 2020
Published 17 August 2020 Volume 2020:13 Pages 347—351
DOI https://doi.org/10.2147/IMCRJ.S269007
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Majdi Al Qawasmeh, Belal Aldabbour, Kefah Alhayek, Khalid El-Salem
Department of Neurology, King Abdullah University Hospital (KAUH), Jordan University of Science and Technology (JUST), Irbid, Jordan
Correspondence: Majdi Al Qawasmeh
Neurology Department of KAUH, King Abdullah University Hospital, P.O. Box 630001, Irbid 22110, Jordan
Tel +962 797477731
Email [email protected]
Abstract: Encephalocraniocutaneous lipomatosis “ECCL” is a rare, sporadic neurocutaneous disorder that results from a lethal autosomal mutation surviving by somatic mosaicism. It is characterized by unilateral involvement of skin, eyes and central nervous system in addition to a propensity for mesenchymal tumors. A 30-year-old male with previously controlled epilepsy presented with recurrent seizures. Brain imaging revealed a left parietal parasagittal enhancing tumor, in addition to left sided gyriform calcifications, and bilateral cerebral atrophy and ventricular dilatation more prominent on the left side. He also presented multiple left sided sebaceous nevi and abundant subcutaneous lipomas in addition to left mandibular condylar cysts. The brain tumor was excised, and cytopathology revealed a WHO grade I fibrous meningioma. After a thorough evaluation and exclusion of alternative diagnoses, the patient was diagnosed with definite encephalocraniocutaneous lipomatosis as per Moog’s criteria. Several cases of ECCL recently presented with different intracranial neoplasms. Here we report the first case of ECCL in association with meningioma.
Keywords: neurocutaneous syndromes, neurocutaneous disorders, encephalocraniocutaneous lipomatosis, Haberland syndrome
Introduction
Encephalocraniocutaneous lipomatosis “ECCL” or Haberland Syndrome is a sporadic neurocutaneous disorder first described in 1970 by Haberland and Perou.1 It is characterized by a usually unilateral involvement of skin, eyes, and central nervous system, in addition to a propensity for mesenchymal tumors (mostly lipomas).2 The condition remains rare, with about 100 cases reported in the literature.3 In spite of attempts by Hunter4 and later by Moog2 to establish diagnostic criteria for ECCL, the condition remains heterogenous and clinical presentations in the literature represent a spectrum of manifestations and severity. Here we report the first case of ECCL in association with fibrous meningioma and nevus sebaceous of Jadassohn.
Case Report
A 30-year-old male patient known to have epilepsy was referred to the neurology clinic at our tertiary facility for evaluation and control of recurrent breakthrough seizures. He was born to nonconsanguineous parents, with an uneventful delivery and normal early development. He was of average intelligence and had completed 12 years of school education. Family history was negative for epilepsy, neurological or skin conditions. His epilepsy started at age 16 and has previously been controlled with sodium valproate 500 mg twice daily, with infrequent generalized tonic-clonic seizures over the last few years.
Neurological examination was normal. Electroencephalography “EEG” was unrevealing. Brain CT revealed a hypoattenuating lesion at the left parasagittal parietal area, with sulcal effacement and mass effect. It also revealed gyriform calcifications in the left fronto-temporo-parietal region, with bilateral cerebral atrophy and dilatation of both lateral ventricles which was significantly more prominent on the left side (Figure 1). Incidentally, bone cysts were noted in the left mandibular condyle extending down to the ramus. Brain MRI with contrast (Figure 2) revealed a dura-based mass in the left parasagittal posterior parietal region, with enhancement and extensive surrounding vasogenic edema. It also confirmed the left gyriform calcifications, parenchymal atrophy and ventricular dilatation noted on brain CT. No vascular anomalies were noted in any of the images. The patient was referred to neurosurgery and the intracranial mass was excised (Figure 3). Cytopathology and immunostaining revealed the mass to be due to fibrous meningioma, WHO grade I.
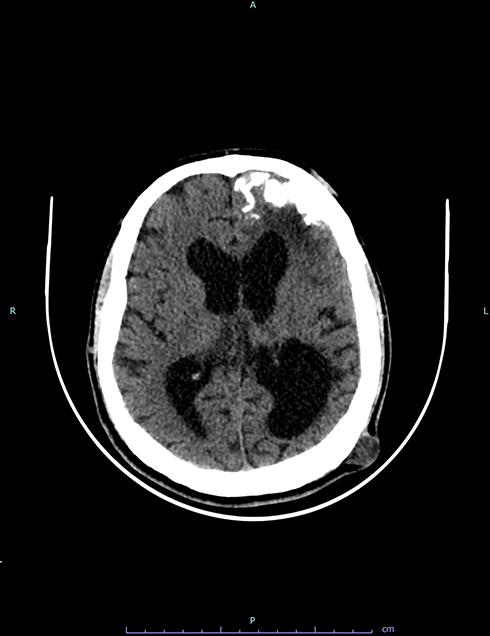 |
Figure 1 Axial brain CT demonstrates left gyriform calcifications as well as bilateral cerebral atrophy and dilatation of both lateral ventricles that is more prominent on the left side. |
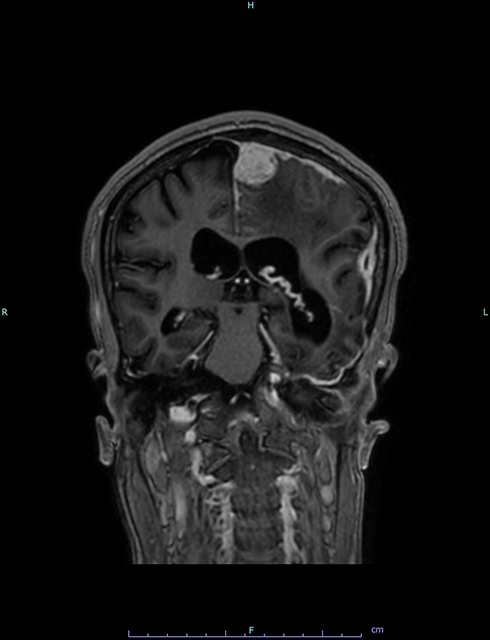 |
Figure 2 Coronal brain MRI (T1 with contrast) demonstrates a dura-based mass in the left parasagittal posterior parietal region, with enhancement and surrounding vasogenic edema. |
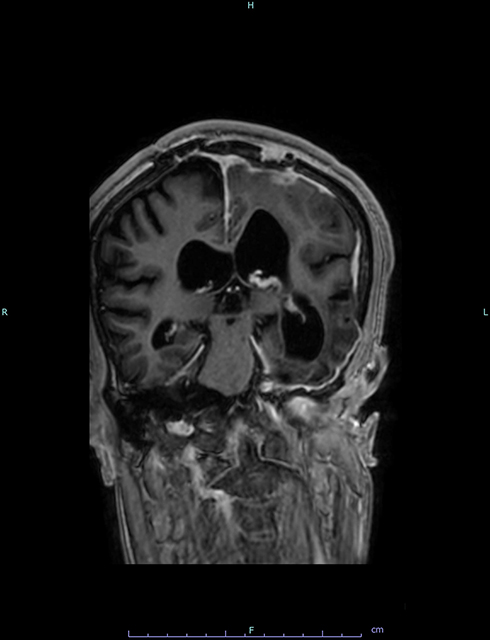 |
Figure 3 Coronal brain MRI (T1 with contrast) shows post-operative changes after resection of the meningioma. It also demonstrates asymmetrical dilatation of the lateral ventricles. |
Also, during evaluation, it was noted that the patient had several left-sided skin lesions over his face (Figure 4), scalp, posterior neck, abdomen, back and the left inguinal area. He also had multiple subcutaneous lipomas of different sizes, which were also restricted to the left side of his body. One of these lipomas was in the left frontotemporal area (Figure 4). The skin lesions and lumps have been present for “as long as he could remember”. The abundance and diversity of lateralized Central Nervous System (CNS) and skin manifestations prompted a thorough evaluation for a neurocutaneous syndrome, and based upon a review of literature, ECCL was suspected. No intraspinal lipomas were revealed on whole-spine MRI. Biopsies from the skin lesions revealed them to be due to nevus sebaceous of Jadassohn, excluding nevus psiloliparis. There was no nonscarring alopecia, skin tags or aplasia. Ophthalmological evaluation was normal except for left melanosis sclerae. No evidence of aortic coarctation was discerned on clinical examination, chest x-ray or Transthoracic Echo. A comprehensive bone survey looking for cysts other than the ones detected in the left mandible was not performed, but none were detected upon revision of the available previous radiographs that were taken for different reasons.
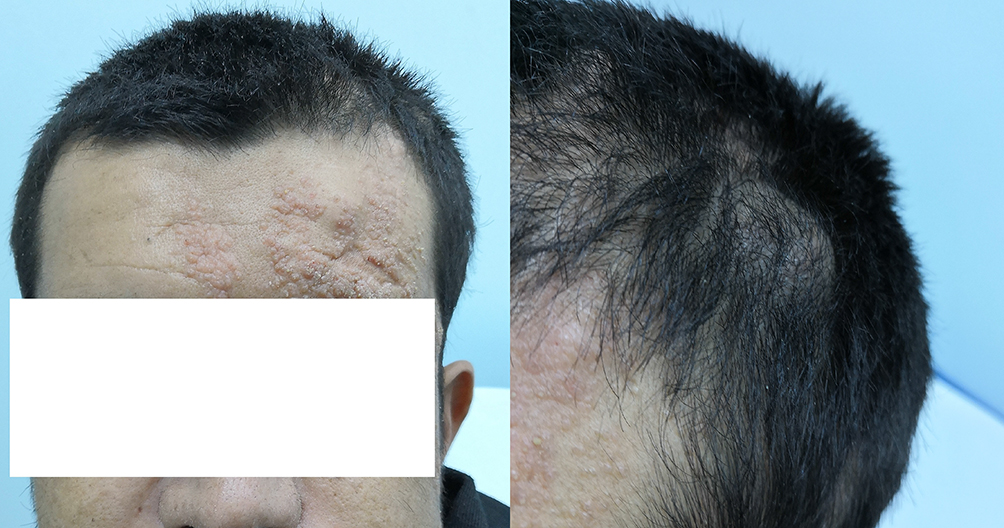 |
Figure 4 Left frontotemporal lipoma (right) and left face lesions due to nevus sebaceous (left). |
During follow up, the patient suffered a left mandibular condylar fracture which was precipitated by a minor trauma and which was later complicated by osteomyelitis. Two years after meningioma excision, the patient’s epilepsy is again well controlled with medical treatment. This case demonstrated involvement of three systems (skin, central nervous system, and other), with major criteria in the latter two. Based on these findings, and the exclusion of alternative diagnoses, it meets Moog’s criteria for a definite case of ECCL.
Discussion
Recent genetic studies have demonstrated mosaic mutations in FGFR1, KRAS and NRAS genes as well as constitutive activation of RAS-MAPK signaling pathway in patients with ECCL.5–7 These findings support earlier hypotheses that ECCL results from a post-zygotic, sporadic, lethal mutation affecting mesenchymal derivatives and surviving due to somatic mosaicism.2,8
The cutaneous hallmark of ECCL is nevus psiloliparis (NP), a hairless fatty nevus that was seen in 44 out of 54 cases reviewed by Moog.2 Other skin manifestations included in the Moog criteria are non-scarring alopecia, frontotemporal subcutaneous lipoma(s), focal skin aplasia or hypoplasia on the scalp, and small nodular skin tags on the eyelids or between the outer canthus and tragus. Our patient had multiple coetaneous lipomas including one in the frontotemporal region, but the other skin criteria were not met. The finding of nevus sebaceous of Jadassohn is unique to our case, and we believe it is in line with the ECCL spectrum as this uncommon hamartomatous skin lesion is known to have a potential to develop a variety of tumors of mesenchymal origin.9
On the other hand, although ocular manifestations were the second most common anomaly observed by Moog (43/54 cases), ophthalmologic examination in our patient was unremarkable except for left melanosis sclerae. This reflects the heterogeneity and diverse presentations of ECCL. Also, Moog’s observations are based on a relatively small number of cases and are likely to change as more cases are aggregated.
Anomalies in the CNS were the third commonest feature noted by Moog. They might occasionally be bilateral, but asymmetry is the rule.2 Intracranial and/or intraspinal lipomas were observed in 33/54 cases, followed by meningeal abnormalities (in 21/54 cases). Other criteria are complete or partial atrophy of a hemisphere, dilated ventricles or hydrocephalus, calcifications in areas other than the basal ganglia, and the presence of porencephalic cysts. Three of these criterions were met. Focal vascular defects are the proposed mechanism underlying the variable CNS features.2
Interestingly, there is an abundance of intracranial tumors other than lipoma in cases with ECCL. Gliomas of different types were reported in 11 different patients (about 10% of reported cases). All of these cases3,10,11 were reported after Moog’s paper was published and hence, they were not included in the cases on which he based his revised criteria. The tumors were mostly low-grade (10/11), and they include: pilomyxoid/pilocytic astrocytoma (5 cases), low-grade astrocytoma (3 cases), glioblastoma (1 case), dysembryoplastic neuroepithelial tumor (1 case), and papillary glioneuronal tumor (1 case). These tumors may be explained by the occurrence of the same FGFR1 mutations recently demonstrated in ECCL in subgroups of sporadic low-grade gliomas.3,10
On the other hand, our case is the first case of ECCL to present with meningioma. Until this presentation is corroborated by similar cases in the future, a coincidental sporadic etiology remains disfavored currently due to several reasons. Firstly, sporadic meningioma is primarily a tumor of the elderly.12 Secondly, mutations in the FGFR gene and constitutive expression and activation of MAPK pathway have been demonstrated in meningioma,13,14 and this shared molecular pathway could explain the co-occurrence of meningioma (and the different other brain tumors) with ECCL. Finally, when grouped together, the prevalence of these different brain tumors in the ECCL cohort far exceeds that in the general community. The authors of this paper believe that this abundance of brain tumors of different types underscores the notion that ECCL is a phenotypic spectrum rather than a single entity and signifies a need to accommodate these findings in any future revisions of current criteria.
The (Other criteria) proposed by Moog include jaw tumors, aortic coarctation, and multiple bone cysts. The latter criterion was demonstrated in our patient. Moreover, our case bears similarity with another case where a bone with developmental abnormalities was easily fractured.15 Neurologically, patient presentations reflect a spectrum ranging from normal development to severe retardation and intractable seizures.2 Our patient lies at the milder end of this clinical spectrum (normal development but having seizures). His neurological examination was normal. He is of average intelligence and good functional status. In addition, his epilepsy started in adolescence and was responsive to medical treatment except in the period surrounding his meningioma diagnosis. Of note, our patient was diagnosed at a later stage and so he is older than the better-performing cases in Moog’s review, whose age range was between 3 months and 24 years.2
The differential diagnosis of ECCL includes several neurocutaneous disorders. Brain atrophy and calcifications may be seen in Sturge-Weber syndrome. However, the condition more commonly presents with a port-wine birthmark and brain vascular anomalies with or without glaucoma, and none of these was present in our patient.16 Proteus syndrome is generally more severe, and a progressive course is essential for the diagnosis.2 Oculoectodermal, oculocerebrocutaneous and Epidermal nevus syndromes were excluded by the absence of their characteristic eye and skin manifestations.2
Conclusions
This case has several unique features and demonstrates the phenotypic heterogeneity of ECCL. Current understanding of this syndrome is incomplete, but it will expand as new genetic and molecular pathways are uncovered and clinical cases with novel presentations are reported.
Ethics Approval and Informed Consent
Ethical approval is not required by the institution for case reports where sufficient anonymization of patient data is guaranteed. The patient has provided informed consent for the case details and accompanying images to be published.
Acknowledgments
The authors would like to acknowledge the valuable efforts of radiology department at King Abdullah University Hospital.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Haberland C, Perou M. Encephalocraniocutaneous lipomatosis. A new example of ectomesodermal dysgenesis. Arch Neurol. 1970;22(2):144–155. doi:10.1001/archneur.1970.00480200050005
2. Moog U. Encephalocraniocutaneous lipomatosis. J Med Genet. 2009;46(11):721–729. doi:10.1136/jmg.2009.066068
3. Kordacka J, Zakrzewski K, Gruszka R, et al. Sensitive detection of FGFR1 N546K mosaic mutation in patient with encephalocraniocutaneous lipomatosis and pilocytic astrocytoma. Am J Med Genet A. 2019;179(8):1622–1627. doi:10.1002/ajmg.a.61256
4. Hunter AG. Oculocerebrocutaneous and encephalocraniocutaneous lipomatosis syndromes: blind men and an elephant or separate syndromes? Am J Med Genet A. 2006;140(7):709–726. doi:10.1002/ajmg.a.31149
5. Bennett JT, Tan TY, Alcantara D, et al. Mosaic activating mutations in FGFR1 cause encephalocraniocutaneous lipomatosis. Am J Hum Genet. 2016;98(3):579–587. doi:10.1016/j.ajhg.2016.02.006
6. Chacon-Camacho OF, Lopez-Moreno D, Morales-Sanchez MA, et al. Expansion of the phenotypic spectrum and description of molecular findings in a cohort of patients with oculocutaneous mosaic RASopathies. Mol Genet Genomic Med. 2019;7(5):e625. doi:10.1002/mgg3.625
7. Richters RJH, Seyger MMB, Meeuwis KAP, Rinne T, Eijkelenboom A, Willemsen MA. Oculoectodermal syndrome - encephalocraniocutaneous lipomatosis associated with NRAS mutation. Acta Derm Venereol. 2020;100(8):adv00103. doi:10.2340/00015555-3358
8. Happle R. Lethal genes surviving by mosaicism: a possible explanation for sporadic birth defects involving the skin. J Am Acad Dermatol. 1987;16(4):899–906. doi:10.1016/S0190-9622(87)80249-9
9. Gupta SK, Gupta V. Basal cell carcinoma and syringocystadenoma papilliferum arising in nevus sebaceous on face-a rare entity. Indian J Dermatol. 2015;60(6):637. doi:10.4103/0019-5154.169159
10. Valera ET, McConechy MK, Gayden T, et al. Methylome analysis and whole-exome sequencing reveal that brain tumors associated with encephalocraniocutaneous lipomatosis are midline pilocytic astrocytomas. Acta Neuropathol. 2018;136(4):657–660. doi:10.1007/s00401-018-1898-8
11. Bavle A, Shah R, Gross N, et al. Encephalocraniocutaneous Lipomatosis. J Pediatr Hematol Oncol. 2018;40(7):553–554. doi:10.1097/MPH.0000000000001170
12. Holleczek B, Zampella D, Urbschat S, et al. Incidence, mortality and outcome of meningiomas: a population-based study from Germany. Cancer Epidemiol. 2019;62:101562. doi:10.1016/j.canep.2019.07.001
13. Johnson M, Toms S. Mitogenic signal transduction pathways in meningiomas: novel targets for meningioma chemotherapy? J Neuropathol Exp Neurol. 2005;64(12):1029–1036. doi:10.1097/01.jnen.0000189834.63951.81
14. AlSahlawi A, Aljelaify R, Magrashi A, et al. New insights into the genomic landscape of meningiomas identified FGFR3 in a subset of patients with favorable prognoses. Oncotarget. 2019;10(53):5549–5559. doi:10.18632/oncotarget.27178
15. Culleton S, Barras CD, Asadi H, Looby S, Brennan P, Kok HK. Tip of an iceberg: skull fracture as an adult presentation of encephalocraniocutaneous lipomatosis. Case Rep Neurol Med. 2016;2016:3292654.
16. Comi AM. Sturge-Weber syndrome. Handb Clin Neurol. 2015;132:157–168.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.