")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Farnesoid X receptor agonist INT-767 attenuates liver steatosis and inflammation in rat model of nonalcoholic steatohepatitis
Received 9 April 2018
Accepted for publication 12 May 2018
Published 16 July 2018 Volume 2018:12 Pages 2213—2221
DOI https://doi.org/10.2147/DDDT.S170518
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
Ying-Bin Hu,1 Xin-Yu Liu,2 Wei Zhan1
1Department of Gastroenterology, Puai Hospital (Wuhan Fourth Hospital), Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei, China; 2Cancer Science Institute, National University of Singapore, Singapore
Introduction: Nonalcoholic steatohepatitis (NASH) is largely driven by the dysregulation of liver metabolism and inflammation. Bile acids and their receptor Farnesoid X receptor (FXR) play a critical role in the disease development. Here, we investigated whether INT-767, the newly-identified dual FXR/TGR5 agonist, can protect rat from liver injury during NASH.
Materials and methods: NASH model was established by feeding the male SD rats with high-fat diet for 16 weeks. INT-767 was given by gavage to NASH rats from week 13 to week 16. At the end of 16 weeks, liver and serum were harvested, and bile acids, glucose and lipid metabolism, liver injury and histological features were evaluated.
Results: INT-767 treatment significantly alleviates high-fat caused liver damage characterized with lipid accumulation and hepatic infiltration of immune cells. INT-767 robustly restores the lipid, glucose metabolism to normal level, attenuates insulin resistance through upregulating FXR level and reverting the dysregulation of its target genes in liver metabolism. Molecularly INT-767 also attenuates the pro-inflammatory response by suppression of TNF-α and NF-κB signaling pathway.
Conclusion: INT-767 may be an attractive candidate for a potential novel strategy on the treatment of NASH.
Keywords: farnesoid X receptor agonist, nonalcoholic steatohepatitis, INT-767, transmembrane G protein-coupled receptor 5
Introduction
Nonalcoholic fatty liver disease (NAFLD) has emerged as the leading cause of chronic liver disease in the world. The clinical-histological spectrum of NAFLD includes two major phenotypes, namely nonalcoholic fatty liver and nonalcoholic steatohepatitis (NASH), the latter being associated with a relatively greater risk of cirrhosis and hepatocellular carcinoma. It is known that insulin resistance, lipotoxicity, oxidative stress and inflammatory factor represent important risk factors for the development of NASH, yet the exact mechanisms controlling the disease pathogenesis remain largely undefined and will require novel therapeutic approaches to prevent the transition from NAFLD to NASH. A promising new approach along these lines is based on recent discoveries of the key role played by bile acids (BAs) in regulating liver and metabolic homeostasis by activating various receptors, including farnesoid X receptor (FXR) and transmembrane G protein-coupled receptor 5 (TGR5).1,2
FXR is a nuclear receptor transcription factor encoded by nuclear receptor subfamily 1, group H, member 4 (NR1H4), which is mainly expressed in the liver, intestine and kidney. It regulates, directly or through the orphan nuclear receptor small heterodimer partner (SHP), a wide variety of target genes critically involved in the control of BAs, lipid, and glucose homeostasis, and in the regulation of immune responses.3–5 TGR5 is a member of the G protein-coupled receptor (GPCR) superfamily and the subsequent elucidation of its potential roles in mammalian physiology greatly supported the concept that BAs are signaling molecules with actions in tissues beyond those of the enterohepatic circulation.6 TGR5 is widely distributed and expressed by adipocytes, sinusoidal endothelial cells, bile duct epithelial cells and Kupffer cells, with broad physiological and pathophysiological implications.7 A recent study showed that pharmacological activation of TGR5 elicited anti-atherogenic effects by reducing macrophage inflammation and lipid uptake.8 Given the impact of FXR and TGR5 signaling on lipid and glucose metabolism, modulators of these receptors and/or agents that influence endogenous BAs levels could have beneficial therapeutic effects in NASH. Although several FXR agonists have been actively tested as therapeutic agents for a variety of chronic and metabolic diseases,9–13 the effect of the newly identified dual FXR/TGR5 agonist, 6α-ethyl-3α,7α,23-trihydroxy-24-nor-5β-cholan-23-sulfate sodium salt (INT-767), in NASH treatment is still elusive.
In this study, we demonstrated that simultaneous activation of FXR/TGR5 with a dual agonist INT-767 improved NASH histological features including alleviation of serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), hyperlipidemia and endotoxin, as well as restoration of normal glucose and lipid metabolism in a rat model of NASH. It was also observed that gene expressions related to BAs export was upregulated, while BAs synthesis, glucose and lipid metabolism, and inflammatory factor were downregulated, which shed new light on the underlying molecular mechanisms in NASH. These findings underscore the critical roles played by the FXR pathways in the pathogenesis of NASH and provide the proof of concept that FXR pathway could be a potential therapeutic target for NASH.
Materials and methods
Ethics statement
Investigation has been conducted in accordance with the ethical standards and according to the Declaration of Helsinki and according to national and international guidelines and has been approved by the Institutional Review Board of Tongji Medical College, Huazhong University of Science and Technology. Animal protocol was approved by the Institutional Animal Care and Use Committee of Tongji Medical College, Huazhong University of Science and Technology and all experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals published by the Association for Assessment and Accreditation of Laboratory Animal Care.
Reagents
INT-767 was from Intercept Pharmaceuticals Inc. (New York, NY, USA). The primary antibodies against FXR, and β-actin were from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). Anti-rabbit IgG HRP-linked antibody was from Cell Signaling Technologies (Danvers, MA, USA). Anti-TGR5 antibody was purchased from LifeSpan BioSciences, Inc. (Seattle, WA, USA). Pre-stained protein molecular weight marker was from Fermentas Life Sciences, (Thermo Fisher Scientific, Waltham, MA, USA). The bicinchoninic acid protein assay kit and SuperSignal West Pico chemiluminescent substrate kit were purchased from Pierce Biotechnology (Thermo Fisher Scientific). Polyvinylidene fluoride (PVDF) membranes and Whatman paper were from EMD Millipore (Billerica, MA, USA). High fat diets (HFD) (60% of calories derived from fat) and chow diets (10% of calories derived from fat) were purchased from Research Diets Inc. (New Brunswick, NJ, USA). The multi-species GLP-1 total ELISA kit and rat insulin ELISA kit were purchased from Merck Millipore (Billerica, MA, USA).
Animal model and experimental design
Male Sprague Dawley rats (200±10 g) were offered by the Experimental Animal Center of Tongji Medical College, Huazhong University of Science and Technology. Rats were housed in a temperature, humidity and ventilation-controlled vivarium, kept on a 12-h light/dark cycle. Forty-eight rats were randomly divided into 3 groups: normal chow diets control group (NC group), HFD model group with DMSO treatment (vehicle group) and HFD model group with INT-767 treatment (INT-767 group). Throughout the 16 weeks, the NC group was fed with normal chow diet while the other two groups were fed with HFDs. In the last 4 weeks (weeks 13–16), NC group and vehicle group were given DMSO by gavage once daily, while INT-767 group was treated with INT-767 (20 mg/kg in DMSO) by gavage once daily. All rats were sacrificed, and samples were collected at the end of study.
Blood and tissue sampling
Heparinized blood samples were collected after puncture of the aorta, spun at 3,500 rpm for 10 min, frozen in liquid nitrogen and stored at −80°C. One part of liver tissue sample was placed in 10% neutral-buffered formalin for histological evaluation and others were frozen in liquid nitrogen and stored at −80°C for Western blot (WB) analysis and quantitative reverse-transcription polymerase chain reaction (RT-qPCR) analysis.
Biochemical measurements in liver tissue and serum
Liver homogenate malondialdehyde (MDA) and free fatty acids (FFAs) were detected with commercial analysis kits obtained from the Jiancheng Institute of Biotechnology (Nanjing, China). The concentrations of ALT, AST, glucose, total cholesterol (TC), triglyceride (TG), low-density lipoprotein (LDL), and high-density lipoprotein (HDL) in the serum were measured by the Abbott Aeroset automatic analyzer (Abbott Laboratories, Abbott Park, IL, USA). Levels of insulin resistance were determined using the homeostasis model assessment of insulin resistance (HOMA-IR) (insulin resistance = fasting glucose mmol/L × fasting insulin μU/mL/22.5).
Histopathology examination
Liver tissues were fixed in 4% paraformaldehyde in 0.1 M phosphate buffer at room temperature overnight. The paraffin-embedded tissue sections (4 μm) were stained with hematoxylin and eosin (H&E) according to the standard protocol. Blinded liver samples were scored by two separate pathologists for NASH severity as previously described.14
Assessment of bacterial translocation
Plasma endotoxin level was measured by the endpoint chromogenic limulus amebocyte lysate (LAL QCL1000) test, per manufacturer’s instruction (Lonza, Basel, Switzerland).
Western blot analysis
Total tissue was prepared in RIPA buffer (Cell Signaling Technology), followed by separation in 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to PVDF membranes. The membranes were probed with antibodies specific for FXR and TGR5. Immunoreactive proteins were visualized using a SuperSignal West Pico (Thermo Fisher Scientific, Waltham, MA, USA) chemiluminescent substrate kit. The relative protein levels were normalized to β-actin.
RNA isolation and RT-qPCR analysis
The relative expression of FXR, SHP, CYP7A1, BSEP, SREBP-1c, PEPCK, G6Pase, TNF-α and NF-κB were determined by RT-qPCR. Total RNA was isolated with TRIzol™ (Thermo Fisher Scientific, Waltham, MA, USA) reagent according to the manufacturer’s instructions. RNA purity and concentration were determined by electrophoresis and BioPhotometer (Eppendorf AG, Hamburg, Germany). One microgram of total RNA was converted to cDNA using the ExScript™ RT reagent kit (TaKaRa, Shiga, Japan). Quantitative real-time PCR was performed on a LightCycler PCR Instrument (Hoffman-La Roche Ltd, Basel, Switzerland) and using SYBR green as the detection fluorophore. The housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control, and gene specific mRNA expression was normalized against GAPDH expression. Primer sequences are summarized in Table 1.
 | Table 1 Primers used in RT-qPCR analysis |
Statistical analysis
Data are expressed as the mean ± SE. Statistical analyses were performed using analysis of variance (ANOVA) or the Kruskal–Wallis test as appropriate. If statistical significance was established, Duncan’s multiple comparison test was used to determine which data sets were significantly different. Statistical analyses were performed using GraphPad Software Inc., (La Jolla, CA, USA).
Results
INT-767 improved NASH histological features in high-fat diet induced rat models
There is a need to create animal models that recapitulate the physiology, histology and manifestation seen in humans with NASH. Current NASH animal models have certain limitations such as requiring specific gene knockout and/or unphysiologically dietary manipulations, failure of reproducing the insulin resistance or liver histology typical of human NASH. In our study, we established a rat model using HFD to induce NASH, assessed by liver section histological analysis. Compared to NC group (the normal chow diets control group), severe cytoplasmic vacuoles in the hepatocytes and inflammation were observed in the vehicle group (HFD model group with DMSO treatment), which was confirmed by H&E staining of the liver sections (Figure 1A, 1B). But no obvious lipid droplets and inflammatory infiltration were seen in the INT-767 group (HFD model group with INT-767 treatment) (Figure 1C). Moreover, based on unbiased pathological assessment, steatosis and inflammation scores in the INT-767 group are significantly lower than those in the vehicle group (χ2 =10.17, P <0.01 and χ2 =21.21, P <0.01, respectively).
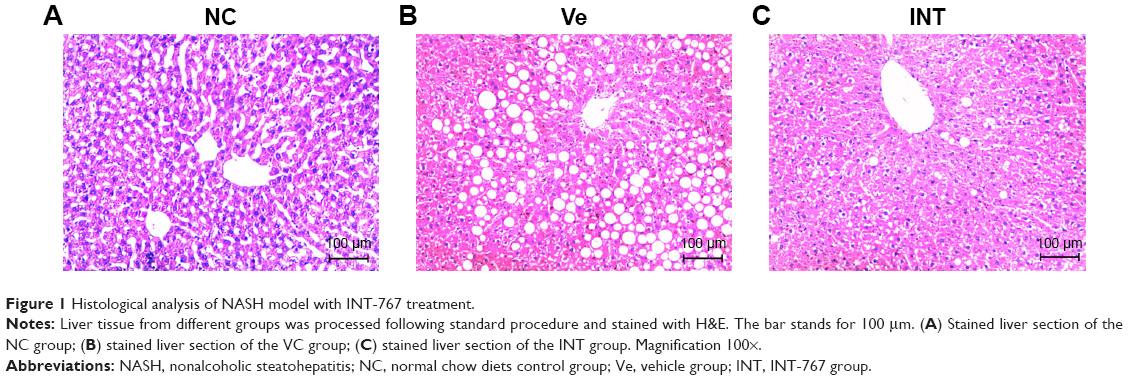 | Figure 1 Histological analysis of NASH model with INT-767 treatment. |
Effect of INT-767 on bile acid metabolism
As BAs’ metabolism has been indicated to play a crucial role in the pathogenesis of NASH. FXR-knockout (FXR-KO) has been reported to accelerate the development of NASH in low-density lipoprotein receptor (LDLR) deficient background, even without HFD,15 suggesting FXR’s important role in pathogenesis of NASH. To examine whether FXR itself was also regulated in response to HFD, liver homogenates were subject to WB analysis. As shown in Figure 2A, compared with the NC group, the vehicle group showed dramatically reduced levels of both FXR and TGR5 protein in the liver homogenates, and the quantification of WB analysis which was included at the right panel (Figure 2A). This result suggested that key regulators of BA metabolism through FXR and TGR5 was suppressed in HFD-induced NASH model. Strikingly, INT-767 treatment completely reverted the suppression of FXR and TGR5 protein levels observed in the vehicle group. We next investigated the change of the downstream target genes of FXR in BAs metabolism. As shown in Figure 2B, FXR mRNA was confirmed to be downregulated in the vehicle group, while INT-767 treatment recovered its level to the extent that it exceeded that of the NC group. The similar pattern was observed for the genes activated by FXR such as SHP and bile salt export pump (BSEP) which promote the transport of BAs. On the contrary, cytochrome P450 family 7 subfamily A member 1 (CYP7A1), the rate-limiting enzyme for cholesterol synthesis, showed the opposite pattern since it was suppressed by SHP. These results indicated that INT-767 treatment led to enhanced BAs transport and reduced BAs synthesis.
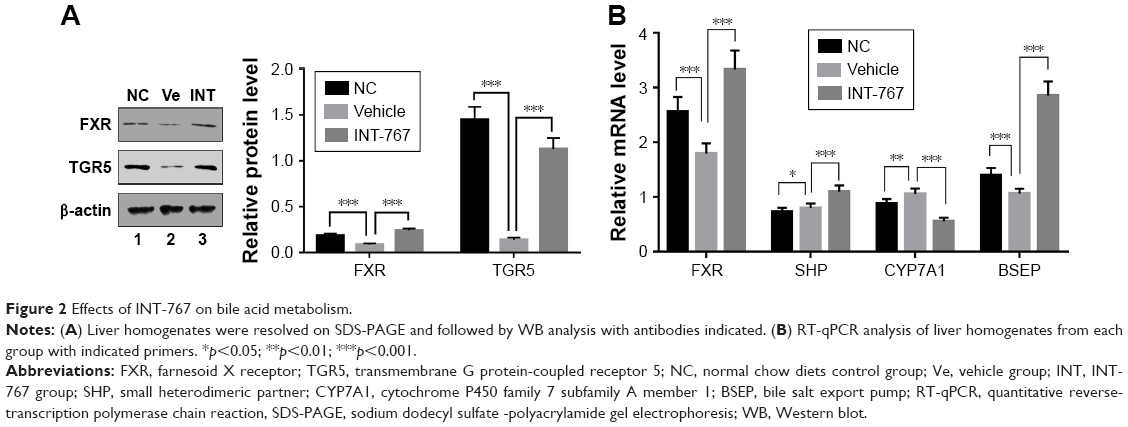 | Figure 2 Effects of INT-767 on bile acid metabolism. |
Effects of INT-767 on insulin resistance and glucose metabolism
We next examined the insulin resistance and glucose metabolism which have been reported as another important aspect of NASH.16 Insulin resistance was clearly observed in the vehicle group compared to the NC group (Figure 3A), and INT-767 could significantly recover the sensitivity of insulin, using the HOMA-IR. Glucagon-like peptide-1 (GLP-1) is an incretin that increases insulin sensitivity, aids glucose metabolism and achieves its roles by binding to its specific receptor (GLP-1R) on human hepatocytes.17 Consistently, lower level of GLP-1 was observed in the vehicle group compared to the NC group, while INT-767 treated group showed increased level of GLP-1 (Figure 3B). From Figure 3C, we also found that two important enzymes of phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase) were upregulated in the vehicle group compared to the NC group. INT-767 treated group showed reduced levels of both enzymes to those found in normal controls.
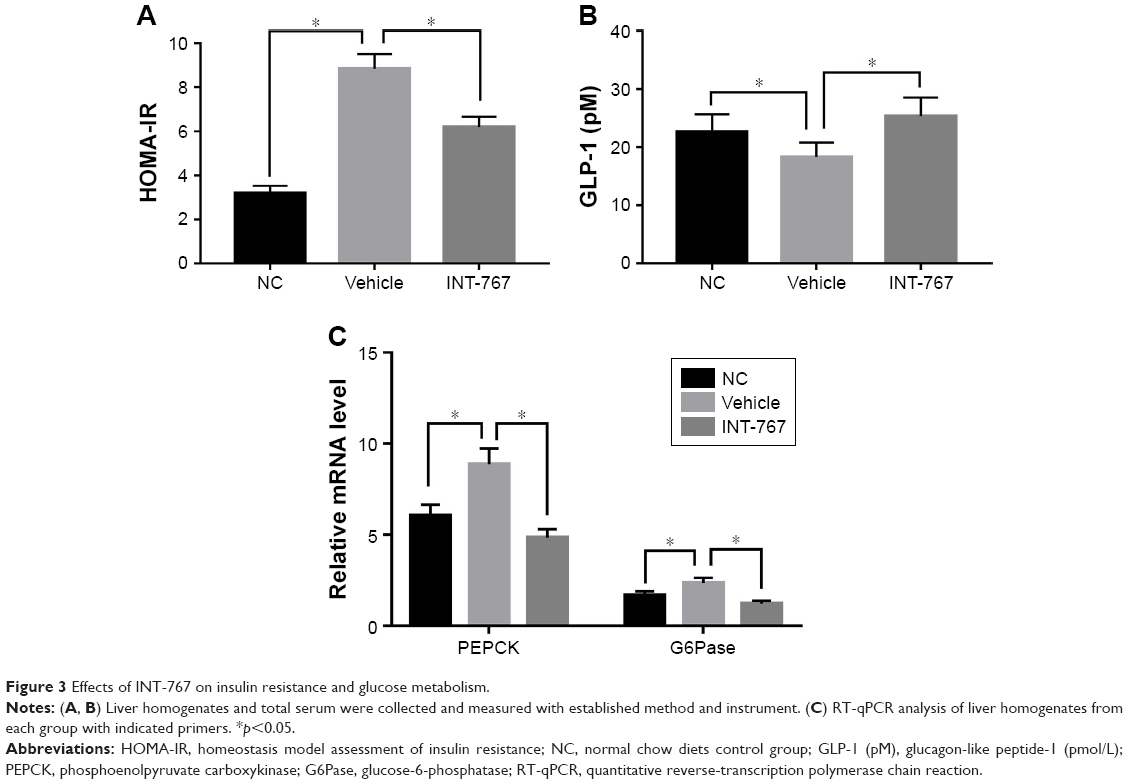 | Figure 3 Effects of INT-767 on insulin resistance and glucose metabolism. |
Effects of INT-767 on lipid metabolism
We next examined the lipid metabolism which is the core program of NASH. As shown in Figure 4 (A–D), hyperlipidemia was observed in the vehicle group compared to the NC group, by measuring TC, TG, LDL and FFAs. Enhanced lipid oxidation and associated oxidative stress as indicated by MDA level was found in the vehicle group compared to the NC group. INT-767 treatment could significantly reduce the hyperlipidemia but had no significant effect on HDL and MDA (Figure 4E and F), suggesting that downregulation of total cholesterol by INT-767 was largely mediated by the reduction of LDL and had little effect on lipid peroxidation. Lipid overflow as well as de novo lipogenesis (DNL) seem to be the key points of hepatocyte accumulation of lipids.18 One key transcription factor in DNL is sterol regulatory element-binding protein-1c (SREBP-1c).19 The result of RT-qPCR showed that SREBP-1c mRNA was induced in the vehicle group compared to the NC group, and downregulated by INT-767 treatment (Figure 4G).
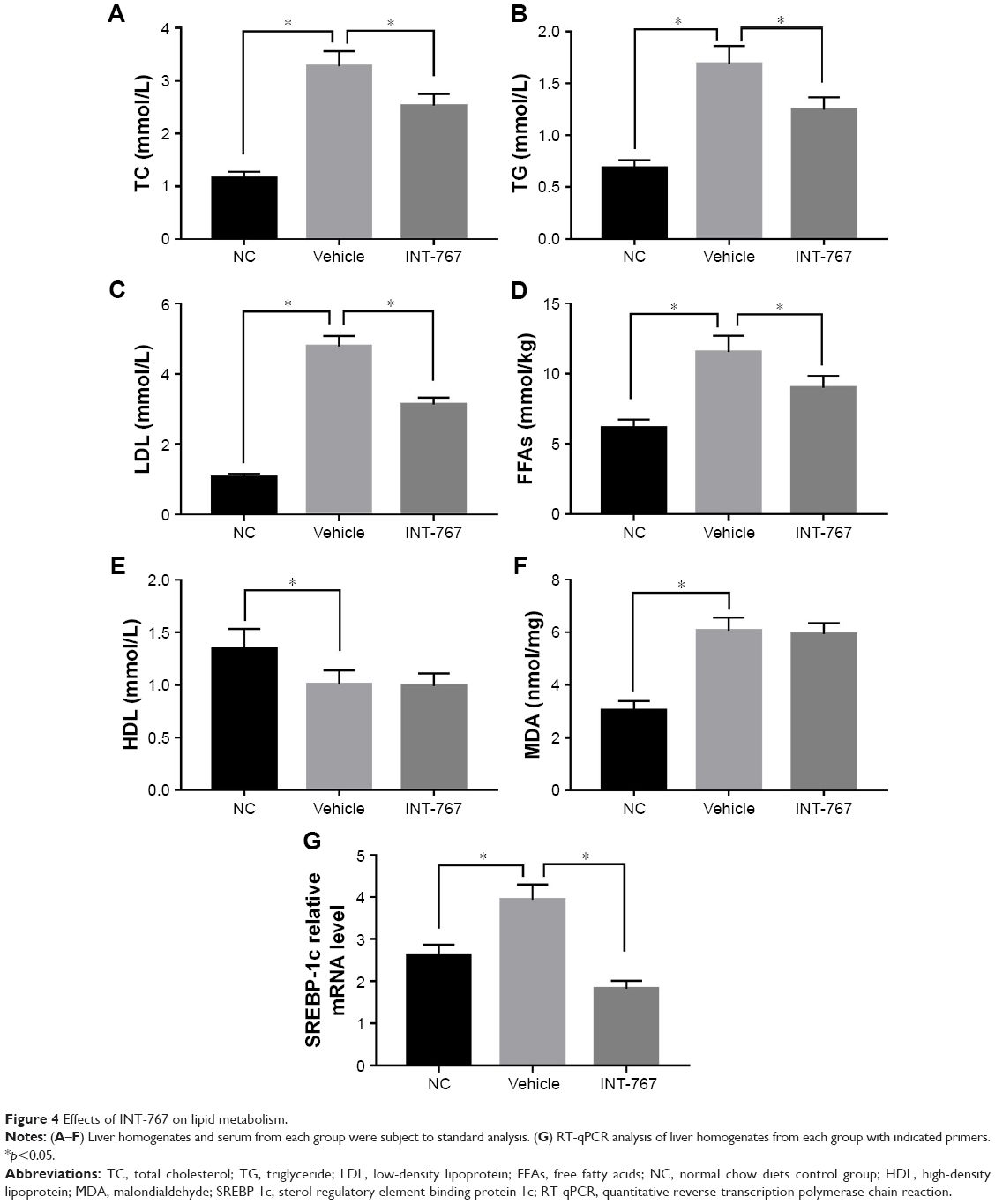 | Figure 4 Effects of INT-767 on lipid metabolism. |
INT-767 alleviates the pro-inflammatory response and liver damage in NASH
To investigate further the mechanism of INT-767, we examined the liver inflammatory response associated with liver damage. Compared with the NC group, the vehicle group showed elevated level of ALT and AST (Figure 5A and B) which represented liver damage, as well as endotoxin level suggesting the leakage of intestine bacteria component into liver (Figure 5C). INT-767 treatment could significantly reverse the increase of these factors. The underneath mechanism could be explained by downregulation of the pro-inflammatory cytokine tumor necrosis factor-α (TNF-α) by INT-767 as shown in Figure 5D. Meanwhile, nuclear factor-κB (NF-κB), the upstream modulator of TNF-α, was also found to be reduced in the INT-767 treatment group.
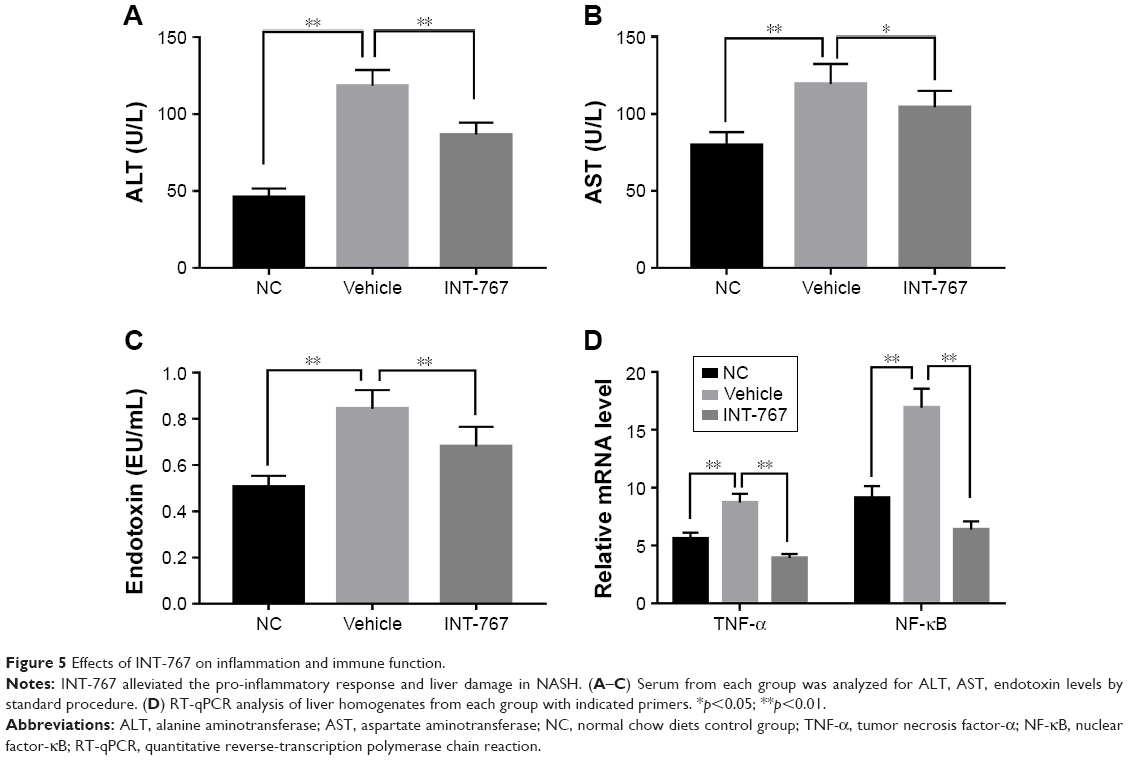 | Figure 5 Effects of INT-767 on inflammation and immune function. |
Discussion
In this study, we demonstrate a significant improvement of liver injury in HFD-induced rat model of NASH treated with the dual FXR/TGR5 agonist INT-767. We also reveal a critical role of FXR/TGR5 activation in regulating hepatic gluconeogenesis (GNG), lipogenesis and hepatic inflammation to maintain metabolic homeostasis in the liver. INT-767 induces FXR and TGR5 gene expression and influences NASH development and progression at multiple levels. Our data support that INT-767 could be a novel FXR agonist with therapeutic effect in NASH.
FXR regulates BA synthesis by cholesterol catabolism. In the liver, the classic BAs synthetic pathway begins with hydrolysis of cholesterol to 7α-hydroxycholesterol by the microsomal enzyme CYP7A1, the rate-limiting step of classic BAs synthesis. This study showed that INT-767 strongly inhibited CYP7A1 expression in the classic BAs synthesis pathway and increases SHP which is a direct FXR target gene in the liver and a suppressor of CYP7A1,20,21 collectively leading to less BA toxicity. FXR-mediated gene suppression occurs through the novel pathway of SHP induction, a member of the nuclear receptor superfamily that binds to and interferes with the positive regulation of gene expression by other nuclear receptors including liver receptor homolog-1 on the target gene such as CYP7A1. INT-767 directly activates the expression of BSEP that transports BAs from the liver to the gall bladder. FXR coordinates the biliary secretion of BAs, cholesterol and phospholipids to form micelles in the canaliculus. This process increases cholesterol solubility and prevents BAs toxicity to the bile duct epithelial cells.22 Furthermore, this process decreases intracellular concentrations of BAs in hepatocytes, thereby alleviating BAs toxicity.23 It is now established that the protective role of FXR against BAs toxicity can be largely attributed to its function in BAs synthesis, transport and excretion.
Treatment with the INT-767 inhibited insulin resistance in a rat NASH model, indicating that FXR activation promotes insulin sensitivity. FXR-KO mice have demonstrated impaired glucose disposal and signs of peripheral insulin resistance.24 Pang et al have shown that impaired FXR signaling pathway results in insulin resistance and elevated hepatic glucose production.25 PEPCK is a well-known vital enzyme that catalyzes a crucial step of GNG in the hepatocytes. Our study has shown that INT-767 decreases expression of the PEPCK. Besides PEPCK, INT-767 also down-regulated the expression of G6Pase, which is a key enzyme responsible for glucose release from hepatocytes to blood in circulation. Altogether, FXR activation plays a key role in inhibiting hepatic GNG and hepatic glucose output. TGR5 signaling is also involved in glucose homeostasis by inducing the release of GLP-1. Treatment with the INT-767 increased the expression of TGR5 and the level of GLP-1. In addition to reducing plasma glucose level, Kumar et al have shown that activation of TGR5 on pancreatic beta cells stimulates insulin release through a cyclic adenosine monophosphate (cAMP) and calcium-dependent mechanism.26 All of these actions of TGR5, are of importance to pharmacology since TGR5 agonists could improve the glucose tolerance as well as liver and pancreatic function.
Our study demonstrates that INT-767 decreased serum levels of TC, TG and FFAs, revealing the crucial role of FXR in lipid metabolism. FXR deficiency drives pathologic manifestations of NASH, including macrosteatosis, hepatocyte ballooning and inflammation in a mouse model of hypercholesterolemia induced by HFD feeding in LDLR-deficient mice.15 Thus, a combination of FXR deficiency and HFD appears to represent a risk factor for NASH development, further suggesting that FXR activation may be beneficial in the treatment of NASH. Consistent with these data, FXR activation by INT-767 downregulates SREBP-1c via a pathway involving the atypical nuclear receptor SHP thereby reducing expression of several genes involved in DNL. Importantly, we also demonstrated INT-767 could decrease the serum level of LDL in the rat NASH model likely due to downregulation of SREBP-1c. Moreover, lipoprotein profile analysis showed reduced very low density lipoprotein and LDL in LDLR-deficient mice treated with INT-767.27 FXR also acts on reverse cholesterol transport and HDL metabolism by decreasing apolipoprotein A1, increasing scavenger receptor B1 and cholesteryl ester transfer protein expression, thus increasing HDL-C clearance and lowering plasma HDL-C levels in vivo.28,29 In this study, the reduction of HDL observed in INT-767 treatment group was not significant compared with the vehicle group. Although FXR is reported to regulate the clearance of HDL-C, the level of HDL is closely related to HDL synthesis, different subtypes of HDL and their transport in the body. Other pathways may be responsible and detailed mechanism is yet to be elucidated. Similar to HDL, INT-767 does not improve the level of MDA in a rat NASH model. FXR-dependent induction of peroxisome proliferator-activated receptor-α is involved in mitochondrial and peroxisomal fatty acid β-oxidation, thereby shifting hepatic metabolism towards lipid oxidation. MDA is a product of lipid peroxidation through multiple signaling pathways including c-Jun N-terminal kinases (JNK) pathway,30 while FXR could simply be one of the regulators of lipid peroxidation.
In addition to glucose and lipid metabolism, FXR also has anti-inflammatory function in the liver, mainly by the suppression of NF-κB and TNF-α signaling pathway. In murine model of NAFLD, FXR agonist GW4064 treatment reduced hepatic pro-inflammatory cytokine expressions.31 Our study also found activation of FXR by INT-767 alleviated hepatic inflammation induced by endotoxin in a rat NASH model fed with HFD as reflected by reduced serum levels of ALT and AST. Moreover, activation of TGR5 in Kupffer cells appears to induce potent anti-inflammatory effects through inhibition of nuclear translocation of NF-κB and the suppression of TNF-α.32 In addition, this same group showed that pharmacological activation of TGR5 in bone marrow-derived macrophages, reduced lipopolysaccharide-induced chemokine production by a mechanism that required AKT-dependent activation of mTOR complex 1 (mTORC1), which stimulated production of the dominant-negative CCAAT/enhancer binding protein isoform, liver inhibitory protein.33 Yoneno and colleagues demonstrated that TGR5 activation prevented phosphorylation of c-Fos in a cAMP-dependent manner, through which they proposed to contribute to the inhibition of macrophage activation.34 Collectively, INT-767 exerts anti-inflammatory activities and reduces HFD-induced inflammation by downregulation of NF-κB and TNF-α via the possible mechanism of macrophage inhibition.
In conclusion, we demonstrate that dual activation of FXR and TGR5 has a beneficial impact on the development of NASH. The dual FXR/TGR5 agonist INT-767 decreases liver steatosis and inhibits hepatic inflammation in a rat model of NASH. Taken together, this study suggests that INT-767 may be an attractive candidate for a potential novel strategy on the treatment of NASH.
Acknowledgments
This project was guided by the Health and Family Planning Commission of Wuhan municipality (WX17Z07).
Disclosure
The authors report no conflicts of interest in this work.
References
Schaap FG, Trauner M, Jansen PL. Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol. 2014;11(1):55–67. | ||
Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102(6):731–744. | ||
Pozzo L, Vornoli A, Coppola I, Giorgetti L, Gervasi PG, Longo V. Effect of HFD/STZ on expression of genes involved in lipid, cholesterol and glucose metabolism in rats. Life Sci. 2016;166:149–156. | ||
Tanaka N, Aoyama T, Kimura S, Gonzalez FJ. Targeting nuclear receptors for the treatment of fatty liver disease. Pharmacol Ther. 2017;179:142–157. | ||
Yuan L, Bambha K. Bile acid receptors and nonalcoholic fatty liver disease. World J Hepatol. 2015;7(28):2811–2818. | ||
Duboc H, Tache Y, Hofmann AF. The bile acid TGR5 membrane receptor: From basic research to clinical application. Dig Liver Dis. 2014;46(4):302–312. | ||
Pols TW, Noriega LG, Nomura M, Auwerx J, Schoonjans K. The bile acid membrane receptor TGR5 as an emerging target in metabolism and inflammation. J Hepatol. 2011;54(6):1263–1272. | ||
Pols TW, Nomura M, Harach T, et al. TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell Metab. 2011;14(6):747–757. | ||
Comeglio P, Fillipi S, Sarchielli E, et al. Anti-fibrotic effects of chronic treatment with the selective FXR agonist obeticholic acid in the bleomycin-induced rat model of pulmonary fibrosis. J Steroid Biochem Mol Biol. 2017;168:26–37. | ||
Lian F, Wang Y, Xiao Y, Wu X, Xu H, Liang L, Yang X. Activated farnesoid X receptor attenuates apoptosis and liver injury in autoimmune hepatitis. Mol Med Rep. 2015;12(4):5821–5827. | ||
Mudaliar S, Henry RR, Sanyal AJ, et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology. 2013;145(3):574.e1–582.e1. | ||
Wang XX, Luo Y, Wang D, Adorini L, Pruzanski M, Dobrinskikh E, Levi M. A dual agonist of farnesoid X receptor (FXR) and the G protein-coupled receptor TGR5, INT-767, reverses age-related kidney disease in mice. J Biol Chem. 2017;292(29):12018–12024. | ||
Jadhav K, Xu Yang, Xu Yangong, et al. Reversal of metabolic disorders by pharmacological activation of bile acid receptors TGR5 and FXR. Mol Metab. 2018;9:131–140. | ||
Singh R, Wang Y, Xiang U, Tanaka KE, Gaarde WA, Czaja MJ. Differential effects of JNK1 and JNK2 inhibition on murine steatohepatitis and insulin resistance. Hepatology. 2009;49(1):87–96. | ||
Kong B, Luyendyk JP, Tawfik O, Guo GL. Farnesoid X receptor deficiency induces nonalcoholic steatohepatitis in low-density lipoprotein receptor-knockout mice fed a high-fat diet. J Pharmacol Exp Ther. 2009;328(1):116–122. | ||
Bugianesi E, Moscatiello, Ciaravella MF, Machesini G. Insulin resistance in nonalcoholic fatty liver disease. Curr Pharm Des. 2010;16(17):1941–1951. | ||
Gupta NA, Mells J, Dunham RM, Grakoui A, Handy J, Saxena NK, Anania FA. Glucagon-like peptide-1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology. 2010;51(5):1584–1592. | ||
Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115(5):1343–1351. | ||
Knebel B, Haas J, Hartwig S, et al. Liver-specific expression of transcriptionally active SREBP-1c is associated with fatty liver and increased visceral fat mass. PLoS One. 2012;7(2):e31812. | ||
Zhang Y, Hagedorn CH, Wang L. Role of nuclear receptor SHP in metabolism and cancer. Biochim Biophys Acta. 2011;1812(8):893–908. | ||
Zhou H, Hylemon PB. Bile acids are nutrient signaling hormones. Steroids. 2014;86:62–68. | ||
Moschetta A, Bookout AL, Mangelsdorf DJ. Prevention of cholesterol gallstone disease by FXR agonists in a mouse model. Nat Med. 2004;10(12):1352–1358. | ||
Garzel B, Yang H, Zhang L, Huang S-M, Polli JE, Wang H. The role of bile salt export pump gene repression in drug-induced cholestatic liver toxicity. Drug Metab Dispos. 2014;42(3):318–322. | ||
Bjursell M, Wedin M, Admyre T, et al. Ageing Fxr deficient mice develop increased energy expenditure, improved glucose control and liver damage resembling NASH. PLoS One. 2013;8(5):e64721. | ||
Pang S, Yu L, Lu Y, et al. Farnesoid X receptor expression reduced in obese rat model with insulin resistance. Am J Med Sci. 2015;350(6):467–470. | ||
Kumar DP, Rajagopal S, Mahavadi S, Mirshahi F, Grider JR, Murthy KS, Sanyal AJ. Activation of transmembrane bile acid receptor TGR5 stimulates insulin secretion in pancreatic β cells. Biochem Biophys Res Commun. 2012;427(3):600–605. | ||
Miyazaki-Anzai S, Masuda M, Levi M, Keenan AL, Miyazaki M. Dual activation of the bile acid nuclear receptor FXR and G-protein-coupled receptor TGR5 protects mice against atherosclerosis. PLoS One. 2014;9(9):e108270. | ||
Cipriani S, Mencarelli A, Palladino G, Fiorucci S. FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. J Lipid Res. 2010;51(4):771–784. | ||
Li Y, Wong K, Walsh K, Gao B, Zang M. Retinoic acid receptor β stimulates hepatic induction of fibroblast growth factor 21 to promote fatty acid oxidation and control whole-body energy homeostasis in mice. J Biol Chem. 2013;288(15):10490–10504. | ||
Hu YB, Liu XY. Protective effects of SP600125 in a diet-induced rat model of non-alcoholic steatohepatitis. Scand J Gastroenterol. 2009;44(11):1356–1362. | ||
Yao J, Zhou C-S, Ma X, Fu B-Q, Tao L-S, Chen M, Xu Y-P. FXR agonist GW4064 alleviates endotoxin-induced hepatic inflammation by repressing macrophage activation. World J Gastroenterol. 2014;20(39):14430–14441. | ||
Perino A, Schoonjans K. TGR5 and immunometabolism: Insights from physiology and pharmacology. Trends Pharmacol Sci. 2015;36(12):847–857. | ||
Perino A, Pols TWH, Nomura M, Stein S, Pellicciari R, Schoonjans K. TGR5 reduces macrophage migration through mTOR-induced C/EBPβ differential translation. J Clin Invest. 2014;124(12):5424–5436. | ||
Yoneno K, Hisamatsu T, Shimamura K, et al. TGR5 signalling inhibits the production of pro-inflammatory cytokines by in vitro differentiated inflammatory and intestinal macrophages in Crohn’s disease. Immunology. 2013;139(1):19–29. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.