")
Back to Journals » Vascular Health and Risk Management » Volume 19
Familial Hypertrophic Cardiomyopathy: Diagnosis and Management
Authors Litt MJ, Ali A, Reza N
Received 22 November 2022
Accepted for publication 28 March 2023
Published 6 April 2023 Volume 2023:19 Pages 211—221
DOI https://doi.org/10.2147/VHRM.S365001
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Konstantinos Tziomalos
Michael J Litt,1 Ayan Ali,2 Nosheen Reza1
1Division of Cardiovascular Medicine, Department of Medicine, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, USA; 2Department of Medicine, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, USA
Correspondence: Nosheen Reza, Perelman School of Medicine at the University of Pennsylvania, Department of Medicine, 3400 Civic Center Boulevard, 11th Floor South Pavilion, Philadelphia, PA, 19104, USA, Tel +1 215 615 0044, Fax +1 215 615 1263, Email [email protected]
Abstract: Hypertrophic cardiomyopathy (HCM) is widely recognized as one of the most common inheritable cardiac disorders. Since its initial description over 60 years ago, advances in multimodality imaging and translational genetics have revolutionized our understanding of the disorder. The diagnosis and management of patients with HCM are optimized with a multidisciplinary approach. This, along with increased safety and efficacy of medical, percutaneous, and surgical therapies for HCM, has afforded more personalized care and improved outcomes for this patient population. In this review, we will discuss our modern understanding of the molecular pathophysiology that underlies HCM. We will describe the range of clinical presentations and discuss the role of genetic testing in diagnosis. Finally, we will summarize management strategies for the hemodynamic subtypes of HCM with specific emphasis on the rationale and evidence for the use of implantable cardioverter defibrillators, septal reduction therapy, and cardiac myosin inhibitors.
Keywords: hypertrophic cardiomyopathy, genetics, heart failure, myosin, mavacamten
Introduction
Hypertrophic cardiomyopathy (HCM) affects more than 750,000 individuals in the United States.1–3 While a substantial proportion of individuals with HCM can expect a normal life expectancy without the need for HCM-specific therapies, data from referral-based cohorts demonstrate that 30–40% of the patients with HCM experience adverse cardiovascular events.4,5 Advances in contemporary cardiovascular diagnostics and therapeutics have decreased the burden of malignant cardiovascular outcomes and have helped shift focus to the early identification of patients and at-risk family members.6 Herein, we review the diagnosis, mechanisms, and management of HCM with a focus on emerging therapies.
Epidemiology
Screening studies of asymptomatic adults in the general population indicate that the prevalence of HCM ranges from 0.17% to 0.23%.1,3,7–9 The frequency of HCM is not expected to differ by ancestry or sex as the rate of spontaneous disease-causing rare variants is not thought to be impacted by selection pressures.3 Data from the Sarcomeric Human Cardiomyopathy Registry (SHaRe), the largest HCM registry to date, indicate that the median age of HCM diagnosis is 45.8 years.5 In this registry, patients diagnosed before the age of 40 years had higher rates of adverse cardiac outcomes including atrial fibrillation, ventricular arrhythmias, and heart failure. Young patients with HCM aged 20–29 years had a nearly fourfold increase in the risk of death as compared to the general population.
Sex also plays an important role in diagnosis. Women are often diagnosed later in life than men10 and at more advanced stages of the disease with more symptoms, a higher risk of heart failure, and a greater risk of mortality.11 Women are also more likely to have pathogenic sarcomeric variants.12 The exact reason for this disparity remains unclear. One increasingly recognized explanation is the role of bias in disease recognition and treatment.13 Indeed, there may be a lower clinical suspicion of HCM in younger women which could thereby delay the time to diagnosis. Furthermore, women are less likely to be referred for implantable cardiac defibrillators when compared to men.11,13 A potential physiological explanation for the sex disparity in diagnosis is that estrogen may be able to suppress hypertrophic signaling in females.14–16 However, this hypothesis remains challenging to confirm clinically and may be difficult to address pharmacologically.17 Regardless of the underlying cause, women continue to face many disparities in diagnosis and treatment as compared to their male counterparts.
Diagnosis and Classification
In adults, the diagnosis of HCM is made in the presence of myocardial hypertrophy ≥1.5 cm in the absence of abnormal loading conditions.6 The threshold for diagnosis is reduced to ≥1.3 cm in adults with familial HCM to improve the diagnostic yield in this at-risk population.6 In children, a BSA normalized Z score of myocardial thickness greater than 2 is used as the diagnostic threshold. Disorders that mimic HCM, such as Fabry’s disease, hypertensive heart disease, mitochondrial cardiomyopathies, and cardiac amyloidosis, must also be ruled out before diagnosis.18–20 In children, additional care must be taken to rule out additional syndromic diseases such as glycogen storage disease (eg, Danon disease, Pompe disease, gamma-2 regulatory subunit of adenosine monophosphate-activated protein kinase-2 disease), mitochondrial cytopathies, and inborn errors of metabolism.19
While hypertrophy is most commonly identified in the interventricular septum, it can be present globally or in any myocardial segment.2 The location of hypertrophy can be described using a variety of different classification frameworks. The initial schema was devised using echocardiography and differentiated hypertrophy based on the anatomic location of hypertrophy.21 This schema denotes four main subtypes: basilar anterior hypertrophy (type I), hypertrophy of the entire septum (type II), hypertrophy of the entire septum and additional hypertrophy of the posterior lateral wall (type III), and hypertrophy involving the apex, posterior or lateral wall (type IV).21 A more recent schema that utilized cardiac MRI uses phenotypic rather than anatomic classifications (Figure 1).22 These phenotypes include reverse curvature HCM (type A – mid-septal convexity), sigmoid septum HCM (type B – concave septum towards to LV cavity), neutral HCM (type C – neither a concavity nor convexity), apical HCM (type D – predominant apically localized HCM), and mid-cavitary HCM (type E). Mid-cavitary and apical HCM are further differentiated by the presence or absence of an apical aneurysm.
 |
Figure 1 Phenotypic subtypes of hypertrophic cardiomyopathy, differentiated by region of hypertrophy. (A) Representation of a normal human heart. (B) Type A/type I hypertrophy results in a sigmoid septum with discrete hypertrophy of the basilar septum with less severe hypertrophy of the basolateral wall. (C) Type B/Type II hypertrophy involves the majority of the basilar septum and in three dimensions spirals anteriorly as it tracts down to the apex. (D) Type C hypertrophy (a subclass of Type III hypertrophy) denotes primarily uniform hypertrophy of both the septum and the posterior wall. (E) Type D (a subclass of Type IV hypertrophy) primarily involves the apex with minimal hypertrophy in the septum or posterior wall. (F) Type E (a subclass of Type III hypertrophy) involves both the septum and the posterior wall with minimal apical involvement and can lead to mid-cavitary gradients. |
Ninety percent of the HCM cases present with hypertrophy of the interventricular septum (type A and type B).8,23 However, diagnosis of type A HCM is challenging to clinically differential from the more benign discrete upper septal thickening.24–26 Key features that differentiate between the two include systolic anterior motion of the mitral valve (SAM), left ventricular outflow tract (LVOT) obstruction, mitral valve abnormalities, age of onset, symptoms, and/or identification of a pathogenic sarcomere variant.27 Patients with a sigmoid septal hypertrophy phenotype are generally younger than patients with discrete upper septal thickening and present with symptomatic LVOT obstruction and/or SAM. However, clinical scenarios consistent with but potentially unrelated to HCM, such as syncope, make diagnosis often challenging.28 Additional information, such as diastolic dysfunction, increased left ventricular posterior wall thickness, or the presence of myocardial fibrosis on cardiac magnetic resonance imaging (CMR) can help provide clarity in these situations.29
While left ventricular septal thickness is routinely quantified by echocardiography, other regions are not routinely sampled for wall thickness. For this reason, subtle hypertrophy in other regions can be easily missed or misconstrued as artifact with this modality.30 CMR is more sensitive to detect myocardial hypertrophy in regions outside of the septum. However, it is costlier and less readily available than echocardiography. CMR does have the additional advantage of being able to quantify myocardial fibrosis which can be incorporated into sudden death risk stratification. Therefore, a CMR is strongly recommended for confirmatory testing in patients with newly diagnosed HCM. Prescreening should then be repeated every 3–5 years to monitor for progression.6
Genetics
Approximately 40–60% of the HCM is caused by autosomal dominant genetic variants in genes that encode myocardial sarcomere proteins.8 Within this subset, the majority (~70%) of patients have disease-causing variants in either myosin-binding protein C3 (MYBPC3) or myosin heavy chain 7 (MYH7) (Table 1).31–33 The remaining ~30% harbor variants in other regions of the sarcomere including myosin light chain 2 (MYL2), myosin light chain 3 (MYL3), troponin T2 (TNNT2), troponin I3 (TNNI3), tropomyosin 1 (TPM1), and actin alpha cardiac muscle 1 (ACTC1).5,34 Alterations in these proteins interfere with the mechanical properties of the sarcomere and lead to myocyte hypertrophy and disarray, diastolic dysfunction, and myocardial fibrosis. Over time, these changes impair global cardiac function and can cause hyper-contractility, atrial and ventricular arrhythmias, and heart failure.
 |
Table 1 Genes Responsible for the Development of Hypertrophic Cardiomyopathy |
The remainder of the genetic variants that are known to be associated with HCM lie in genes outside the sarcomere.8 These include proteins integral to the Z-disk (eg, ACTN2, ANKRD1, MYOZ2, TCAP, VCL), calcium handling (eg, PLN, RYR2, CASQ2, TNNC1, JPH2), and the proteasome (TRIM63).35 However, there remains a large population of patients with HCM who do not carry disease-causing genetic variants. This suggests that additional host factors – genetic, epigenetic, and environmental – that influence penetrance and expressivity. In monozygotic twin studies, there appears to be a strong epigenetic or environmental role in the expression of the HCM phenotype.36,37 For example, one possible environmental exposure that is thought to potentiate HCM is obesity. Multiple studies have shown that a higher body mass index is strongly associated with higher LVOT gradients and worse clinical outcomes.38–41 The mechanism of this effect remains unclear, and it is unclear if obesity is causal or simply a confounding variable due to its associated cardiometabolic and functional impacts. However, the possibility of treating or preventing HCM with weight management strategies such as exercise, caloric restriction, or weight loss treatments is an area of active investigation.42–45
Major Hypertrophic Cardiomyopathy Subtypes
Obstructive Hypertrophic Cardiomyopathy
HCM phenotype plays a major role in the development of obstruction, which is a key physiologic differentiator in HCM. Approximately two-thirds of patients with HCM develop obstruction, which is strongly associated with symptoms and heart failure progression.6 Patients with LVOT obstruction typically have asymmetric septal hypertrophy (ie types A and B). Obstructive HCM is characterized by a peak LVOT gradient of ≥30 mmHg. In patients with LVOT gradients of ≤30 mmHg, provocative maneuvers such as Valsalva or exercise should be performed to evaluate for latent obstruction.6 Obstruction can also occur at the mid-cavitary or apical levels. The cause of LVOT obstruction is variable and dependent on several factors, including the degree of septal hypertrophy, mitral valve apparatus structure, hemodynamic loading conditions, and flow dynamics. An additional factor that remains under investigation is the left ventricle to aortic root angle (Figure 2).46,47
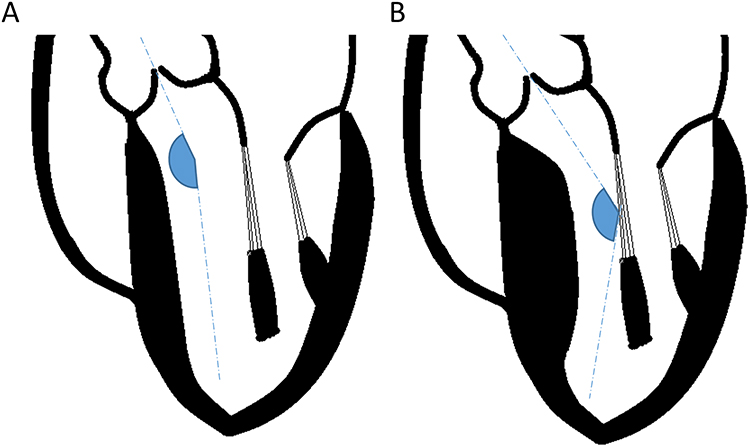 |
Figure 2 Left ventricle to aortic root angulation and its impact on left ventricular outflow tract gradient. (A) In structurally normal hearts, the left ventricular to aortic root angle (LVARA) is greater than 140°. (B) With septal hypertrophy, the LVARA decreases, and more blood is directed toward the anterior leaflet of the mitral valve. This is thought to promote a Venturi effect and drag forces that increase the left ventricular outflow tract gradient and promote systolic anterior motion of the mitral valve. |
Another key factor that influences obstruction is the presence of SAM.2 SAM is thought to result from both the presence of disproportionate hypertrophy in the interventricular septum as well as structural abnormalities of the mitral valve apparatus. Abnormalities include anterior displacement of the papillary insertion, bifid papillary muscles, papillary muscle elongation, abnormal leaflet coaptation, and chordae elongation.48 The abnormalities in any combination can cause laxity in the mitral apparatus which can move into the LVOT during isovolumic contraction. This further narrows the LVOT and causes worsening obstruction.
Nonobstructive Hypertrophic Cardiomyopathy
The remaining one-third of patients with HCM have nonobstructive physiology.2 When symptomatic, patients with nonobstructive HCM can experience similar symptoms to those with obstructive HCM including dyspnea, palpitations, exercise intolerance, syncope, chest pain, and reduced functional capacity. Patients with nonobstructive HCM can have a variety of LV hypertrophy morphologies. Patients with nonobstructive HCM are thought to have a more favorable prognosis on average.49 They are 2–4 times less likely to develop heart failure than those with obstructive HCM and typically have preserved systolic function.2 However, a significant proportion of patients do become symptomatic. Even when asymptomatic, patients with nonobstructive HCM warrant routine longitudinal screening to monitor for progression.
Management
Medical and Interventional Therapies for Obstructive Hypertrophic Cardiomyopathy
Given the diversity of HCM phenotypes, patient-centered treatment plans and therapies are necessary to reduce the overall morbidity and mortality of this disease. One fundamental dividing line in therapeutic decisions is the presence or absence of obstruction. In patients with obstruction, those without symptoms may be monitored closely without medical management.4,6,15,46–49 Once symptoms develop, pharmacotherapy should be initiated with beta‐blockers or non‐dihydropyridine calcium channel blockers.6 Beta-blockers are particularly effective at reducing provocable gradients caused by exercise and may also improve resting gradients.2 Non-dihydropyridine calcium channel blockers can be an alternative for patients that have contraindications or are intolerant to beta-blockade. If patients have symptoms refractory to maximum medical management with beta-blockers and calcium channel blockers, disopyramide, a sodium channel blocker with potent negative inotropic effects can be utilized.50 There is little evidence that any of these therapies alter the natural history of obstructive HCM, likely because they fail to address the underlying molecular pathophysiology of the disease.51
For patients with LVOT gradients exceeding 50mmHg at rest or with physiologic provocation despite maximally tolerated medical management and persistent New York Heart Association (NYHA) III/IV moderate-to-severe symptoms, the 2020 American College of Cardiology/American Heart Association guidelines recommend septal reduction therapy. Options include surgical septal myectomy and alcohol septal ablation, both of which aim to reduce overall septal mass, improve obstruction and thereby, improve the functional capacity of patients.52,53 Surgical myectomy is the preferred invasive option and is typically performed in patients at acceptable surgical risk and in patients with indications for other surgical procedures (ie, concomitant obstructive coronary artery disease, mitral valve abnormalities, valvular aortic stenosis). Myectomy alleviates symptoms of heart failure and improves quality of life in over 90% of the patients, with 70% becoming asymptomatic after the procedure.2,54 In older patients with comorbidities and prohibitive surgical risk, percutaneous alcohol septal ablation may be a more suitable option as it is less invasive and is without the attendant risks of cardiothoracic surgery.4 This procedure causes alcohol-induced transmural infarct, which mimics the effects of myectomy by resulting in septal thinning and improved subaortic gradients. Heart block is the most common complication of the procedure, with 10% of patients requiring permanent pacemaker placement.4 Approximately 10% eventually require repeat ablation but is an overall well-tolerated procedure causing significant improvement in symptoms for patients.2,51–54
Cardiac Myosin Inhibitors
HCM is now widely recognized as a disease of enhanced ensemble force generation within the myocyte.55,56 This fundamental discovery led to the hypothesis that targeted pharmacologic inhibition of sarcomeric force generation could abrogate the disease process.51 Mavacamten (Bristol Myers Squibb, Inc.) was approved by the United States Food and Drug Administration in April 2022 as a first-in-class negative allosteric modulator of human cardiac beta myosin. Mavacamten inhibits the ATPase rate of beta myosin by shifting the equilibrium of beta myosin away from its activated state toward a super relaxed state.57,58 This reduction in beta myosin activity in turn inhibits contractility and reduces excitotoxic calcium handling. Beneficial effects of mavacamten in rodent models include reducing myocardial contractility, preventing left ventricular (LV) hypertrophy, reducing myocardial fibrosis, and suppressing pro-fibrotic signaling pathways.51 Together, these effects were found to improve functional capacity and prevent hypertrophic remodeling in animal models.
Mavacamten was then evaluated in Phase II trials in patients with obstructive and nonobstructive HCM. Following positive data from the phase II PIONEER-HCM trial,59 the Phase III EXPLORER-HCM trial was designed to test if mavacamten was efficacious and safe at reducing LVOT obstruction and improving functional status in patients with obstructive HCM.60 This multicenter, randomized, placebo-controlled study randomized 251 patients with symptomatic obstructive HCM to an echocardiography-guided dose up-titration regimen of mavacamten or placebo. Participants were allowed to continue previously stabilized beta blockers and calcium channel blockers but had to discontinue disopyramide. The study met its primary composite endpoint of ≥ 1 point improvement in NYHA class with a ≥ 1.5 mL/kg/min increase in peak oxygen consumption (peak VO2) or a ≥ 3.0 mL/kg/min increase in peak VO2 with no worsening of NYHA class. Thirty-seven percent of patients assigned to mavacamten reached the primary endpoint as compared with 17% of patients assigned to placebo (p = 0.0005). Twenty-seven percent of the patients had a complete therapeutic response, which was defined as LVOT obstruction <30mmHg and NYHA class I at final evaluation, versus 1% of placebo. Side effects were limited with the primary adverse event being transient and reversible episodes of LV systolic dysfunction. A subsequent echocardiographic substudy of EXPLORER-HCM demonstrated that treatment with mavacamten was associated with improvement in absolute intracellular myocardial mass index, LV mass index, maximum LV wall thickness, and left atrial volume index, which suggests the disease-modifying potential of the drug.61
A follow-up study to EXPLORER-HCM, the phase III multicenter randomized trial VALOR-HCM, attempted to address whether mavacamten provided similar benefits in a sicker population of patients who were eligible for septal reduction therapy.62 Patients underwent a similar echocardiography-guided dose up-titration protocol and were evaluated for septal reduction therapy candidacy following a 16-week treatment course. Following this period, only 18% of the patients remained septal reduction therapy candidates as compared to 77% for placebo (p < 0.001). The trial also met multiple secondary endpoints including significant decreases in LVOT gradients and NYHA class with only two patients (3.6%) requiring temporary drug cessation for drops in LV ejection fraction. Open-label extension studies of mavacamten are underway, and preliminary results indicate that long-term use of mavacamten can reduce LV wall thickness and myocardial fibrosis.61
Additional cardiac myosin inhibitors are under development. Aficamten (formerly CK-3773274, Cytokinetics, Inc.) is a next-in-class selective cardiac myosin inhibitor that has a similar mechanism of action but acts to a distinct allosteric binding site on cardiac myosin. In contrast with mavacamten, it has a shorter half-life and fewer drug–drug interactions and has shown promise in preliminary results from its initial phase II clinical trial program.63,64 The REDWOOD-HCM trial evaluated the safety and tolerability of aficamten in patients with symptomatic obstructive HCM and demonstrated that compared with placebo, treatment with aficamten resulted in rapid and sustained reduction in LVOT gradients, improvement in NYHA class, and decrease in cardiac biomarkers.65 Importantly, there were no serious adverse events related to aficamten and no treatment interruptions or discontinuations due to adverse events. These data have set the stage for the phase III randomized controlled trial testing the clinical efficacy and safety of aficamten in symptomatic obstructive HCM, SEQUOIA-HCM (Safety, Efficacy, and Quantitative Understanding of Obstruction Impact of Aficamten in HCM), which is planned to be completed in September 2023.66 In animal studies, a newer mavacamten surrogate, MYK-581, has been shown to alleviate hypercontractility and LVOT obstruction and improve intracardiac hemodynamics.67,68
Medical Therapy for Nonobstructive Hypertrophic Cardiomyopathy
Pharmacologic therapy for symptomatic nonobstructive HCM includes the use of beta-blockers and calcium and channel blockers as primary agents to improve diastolic filling. Patients with symptomatic nonobstructive HCM are not eligible for septal reduction therapy. Mavacamten was evaluated in the phase II randomized MAVERICK-HCM trial and resulted in reduced NT-proBNP and troponin I levels in patients with nonobstructive HCM with NYHA class II/III symptoms.69 The phase III ODYSSEY-HCM trial (NCT05582395), a randomized trial of mavacamten versus placebo in symptomatic nonobstructive hypertrophic cardiomyopathy is expected to be completed in March 2025. For patients with early HCM but without symptoms, valsartan has shown promise in a phase II clinical trial as a possible disease-modifying therapy.70 Compared with placebo, valsartan demonstrated a statistically significant improvement in a composite endpoint of serum NTproBNP, serum high-sensitivity cardiac troponin, LV mass, left atrial volume, LV end-diastolic volume, LV end-systolic volume, maximal LV wall thickness, and echocardiographic markers of diastolic function.
Arrhythmia Management in Hypertrophic Cardiomyopathy
All patients with HCM should undergo risk stratification for sudden death.71 Major risk factors for sudden death include a family history of HCM with sudden cardiac death, unexplained syncope, prolonged nonsustained ventricular tachycardia, LV apical aneurysm, massive LV hypertrophy (> 30mm), extensive late gadolinium enhancement and LV ejection fraction <50%. Patients at elevated risk should be referred for primary prevention implantable cardioverter defibrillator placement.6 Patients with a history of cardiac arrest or sustained ventricular tachycardia should be referred for secondary prevention implantable cardioverter defibrillator placement.71 Risk stratification is an ongoing process and should be repeated annually.
Patients with HCM are at risk of increased morbidity from atrial fibrillation and are often symptomatic with this tachyarrhythmia due to the combination of myocardial structural abnormalities and diastolic dysfunction.5,71 Frequent episodes of paroxysmal atrial fibrillation may interfere with quality of life and may require acute intervention with cardioversion. Episodes may be suppressed with the use of antiarrhythmic drugs or catheter ablation.72 Anticoagulation with either warfarin or a direct oral-acting anticoagulant for the prevention of thromboembolism should be strongly considered in anyone with HCM and atrial fibrillation independent of their CHA2DS2-VASc score.6
Gene Therapy for Hypertrophic Cardiomyopathy
Gene therapy is an emerging strategy that seeks to directly address the underlying genetic causes of HCM.72,73 The various strategies being taken have been extensively reviewed elsewhere.74 Strategies currently being explored include gene replacement with adeno-associated viral vectors, gene editing, allele-specific silencing, trans-splicing, and exon skipping. These gene replacement tools have shown some promise in studies of induced pluripotent stem cells deficient of MYBPC3.75,76 Recently, base editing of a common disease-causing variant in HCM, MYH7 p.R403Q, has been shown to rescue the HCM phenotype in induced pluripotent stem cell cardiomyocytes and in a mouse model.77 While each approach has its unique potential benefits and risks, the ethical limitations of gene therapy trial design must remain paramount.78 Additional early challenges in early phase, first-in-human studies of gene therapy for inherited cardiomyopathies will include patient selection, identification of outcomes, and recognition and management of off-target effects.
End-Stage Disease
As HCM progresses, patients can develop symptoms that are refractory to drug therapy. This includes worsening systolic progressive exercise intolerance, systolic dysfunction, diastolic heart failure, and progressive VT burden. In the SHaRe registry, patients diagnosed before the age of 40 years had a higher cumulative risk of the development of adverse cardiovascular events, including heart failure, when compared with patients diagnosed after 40 years old.5 In other studies, up to 50% of patients with NYHA functional classes III/IV go on to develop an LV ejection fraction of less than 50%.2,4 Once heart failure with reduced ejection fraction, defined as <50% in HCM, is discovered, patients are managed with typical guideline-directed therapies such as beta-blockers, renin-angiotensin-aldosterone system inhibitors, and diuretic therapy; however, these therapies have little evidence for efficacy in this population. Cardiac resynchronization therapy may be indicated for patients with intraventricular conduction delay and may improve overall symptoms and ejection fraction.6 Patients with advanced NYHA functional class III/IV symptoms and LV systolic dysfunction with LV ejection fraction <50% should be referred for an advanced therapies evaluation.6 Withdrawal of negative inotropic therapy, evaluation for other causes of systolic dysfunction, and referral for cardiac resynchronization therapy are indicated. Survival after cardiac transplant for HCM is 85%, 75%, and 61% at 1, 5, and 10 years with a gradually improved survival rate over the last decade.79,80 Patients with end-stage HCM and dilated LV cavities can also be eligible for ventricular assist devices if technically feasible.
Conclusion
Despite many advances, the care of patients with HCM presents many ongoing challenges. Younger patients and women represent an important subset of patients that require special consideration. The incomplete penetrance and variable expressivity of HCM pathogenic variants highlight the possible role of environmental or epigenetic factors in disease presentation. The arrival of cardiac myosin inhibitors provides patients and physicians with an ever-expanding treatment armamentarium. However, the long-term risk-benefit of this intervention as compared to more established surgical interventions remains unclear. Risk stratification remains an inexact science with primary prevention cardioverter defibrillator implantation requiring shared decision-making. Treatment of end-stage HCM also requires additional investigation as our current therapies for systolic dysfunction are inadequate for this unique population. Fortunately, the treatment of HCM remains a field of active inquiry and ingenuity.
Disclosure
N.R. reports support from the National Center for Advancing Translational Sciences of the National Institutes of Health under award number KL2TR001879 and honoraria from Zoll. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The remaining authors report no conflicts of interest in this work.
References
1. Maron BJ, Gardin JM, Flack JM, et al. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary artery risk development in (young) adults. Circulation. 1995;92:785–789. doi:10.1161/01.cir.92.4.785
2. Maron BJ. Clinical Course and management of hypertrophic cardiomyopathy. N Engl J Med. 2018;379:1977. doi:10.1056/NEJMc1812159
3. Zou Y, Song L, Wang Z, et al. Prevalence of idiopathic hypertrophic cardiomyopathy in China: a population-based echocardiographic analysis of 8080 adults. Am J Med. 2004;116:14–18. doi:10.1016/j.amjmed.2003.05.009
4. Maron BJ, Desai MY, Nishimura RA, et al. Management of hypertrophic cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol. 2022;79:390–414. doi:10.1016/j.jacc.2021.11.021
5. Ho CY, Day SM, Ashley EA, et al. Genotype and lifetime burden of disease in hypertrophic cardiomyopathy: insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation. 2018;138:1387–1398. doi:10.1161/CIRCULATIONAHA.117.033200
6. Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: executive summary: a report of the American college of cardiology/American heart association joint committee on clinical practice guidelines. Circulation. 2020;142:e533–e557. doi:10.1161/CIR.0000000000000938
7. Hada Y, Sakamoto T, Amano K, et al. Prevalence of hypertrophic cardiomyopathy in a population of adult Japanese workers as detected by echocardiographic screening. Am J Cardiol. 1987;59:183–184. doi:10.1016/s0002-9149(87)80107-8
8. Marian AJ, Braunwald E. Hypertrophic cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res. 2017;121:749–770. doi:10.1161/CIRCRESAHA.117.311059
9. Maron BJ, Mathenge R, Casey SA, Poliac LC, Longe TF. Clinical profile of hypertrophic cardiomyopathy identified de novo in rural communities. J Am Coll Cardiol. 1999;33:1590–1595. doi:10.1016/s0735-1097(99)00039-x
10. Siontis KC, Ommen SR, Geske JB. Sex, survival, and cardiomyopathy: differences between men and women with hypertrophic cardiomyopathy. J Am Heart Assoc. 2019;8:e014448. doi:10.1161/JAHA.119.014448
11. Geske JB, Ong KC, Siontis KC, et al. Women with hypertrophic cardiomyopathy have worse survival. Eur Heart J. 2017;38:3434–3440. doi:10.1093/eurheartj/ehx527
12. Butters A, Lakdawala NK, Ingles J. Sex differences in hypertrophic cardiomyopathy: interaction with genetics and environment. Curr Heart Fail Rep. 2021;18:264–273. doi:10.1007/s11897-021-00526-x
13. Patlolla SH, Schaff HV, Nishimura RA, et al. Sex and race disparities in hypertrophic cardiomyopathy: unequal implantable cardioverter-defibrillator use during hospitalization. Mayo Clin Proc. 2022;97:507–518. doi:10.1016/j.mayocp.2021.07.022
14. Pedram A, Razandi M, Lubahn D, et al. Estrogen inhibits cardiac hypertrophy: role of estrogen receptor-beta to inhibit calcineurin. Endocrinology. 2008;149:3361–3369. doi:10.1210/en.2008-0133
15. Haines CD, Harvey PA, Luczak ED, et al. Estrogenic compounds are not always cardioprotective and can be lethal in males with genetic heart disease. Endocrinology. 2012;153:4470–4479. doi:10.1210/en.2012-1391
16. Trongtorsak A, Polpichai N, Thangjui S, et al. Gender-related differences in hypertrophic cardiomyopathy: a systematic review and meta-analysis. Pulse. 2021;9:38–46. doi:10.1159/000517618
17. Dubey RK, Imthurn B, Zacharia LC, Jackson EK. Hormone replacement therapy and cardiovascular disease: what went wrong and where do we go from here? Hypertension. 2004;44:789–795. doi:10.1161/01.HYP.0000145988.95551.28
18. Sankaranarayanan R, Fleming EJ, Garratt CJ. Mimics of hypertrophic cardiomyopathy - diagnostic clues to aid early identification of phenocopies. Arrhythm Electrophysiol Rev. 2013;2:36–40. doi:10.15420/aer.2013.2.1.36
19. Hoss S, Habib M, Silver J, et al. Genetic testing for diagnosis of hypertrophic cardiomyopathy mimics: yield and clinical significance. Circ Genom Precis Med. 2020;13:e002748. doi:10.1161/CIRCGEN.119.002748
20. Wilcox NS, Prenner SB, Cevasco M, et al. End stage mitochondrial cardiomyopathy and heart transplantation due to biallelic pathogenic C1QBP variants. Circulation. 2022;15:e003559. doi:10.1161/CIRCGEN.121.003559
21. Maron BJ, Gottdiener JS, Epstein SE. Patterns and significance of distribution of left ventricular hypertrophy in hypertrophic cardiomyopathy. A wide angle, two dimensional echocardiographic study of 125 patients. Am J Cardiol. 1981;48:418–428. doi:10.1016/0002-9149(81)90068-0
22. Syed IS, Ommen SR, Breen JF, Tajik AJ. Hypertrophic cardiomyopathy: identification of morphological subtypes by echocardiography and cardiac magnetic resonance imaging. JACC Cardiovasc Imaging. 2008;1:377–379. doi:10.1016/j.jcmg.2008.02.008
23. Turer AT, Samad Z, Valente AM, et al. Anatomic and clinical correlates of septal morphology in hypertrophic cardiomyopathy. Eur J Echocardiogr. 2011;12:131–139. doi:10.1093/ejechocard/jeq163
24. Azechi N, Morita Y, Inoue M, Kuzuyama R, Imataka K. Age-associated morphological change in interventricular septum. Rinsho Byori. 1993;41:285–288. Japanese.
25. Krasnow N. Subaortic septal bulge simulates hypertrophic cardiomyopathy by angulation of the septum with age, independent of focal hypertrophy. An echocardiographic study. J Am Soc Echocardiogr. 1997;10:545–555. doi:10.1016/s0894-7317(97)70009-9
26. Toth AB, Engel JA, McManus AM, McManus BM. Sigmoidity of the ventricular septum revisited: progression in early adulthood, predominance in men, and Independence from cardiac mass. Am J Cardiovasc Pathol. 1988;2:211–223.
27. Canepa M, Pozios I, Vianello PF, et al. Distinguishing ventricular septal bulge versus hypertrophic cardiomyopathy in the elderly. Heart. 2016;102:1087–1094. doi:10.1136/heartjnl-2015-308764
28. Diaz T, Pencina MJ, Benjamin EJ, et al. Prevalence, clinical correlates, and prognosis of discrete upper septal thickening on echocardiography: the Framingham Heart Study. Echocardiography. 2009;26:247–253. doi:10.1111/j.1540-8175.2008.00806.x
29. Mendez C, Soler R, Rodríguez E, et al. Differential diagnosis of thickened myocardium: an illustrative MRI review. Insights Imaging. 2018;9:695–707. doi:10.1007/s13244-018-0655-9
30. Valente AM, Lakdawala NK, Powell AJ, et al. Comparison of echocardiographic and cardiac magnetic resonance imaging in hypertrophic cardiomyopathy sarcomere mutation carriers without left ventricular hypertrophy. Circ Cardiovasc Genet. 2013;6:230–237. doi:10.1161/CIRCGENETICS.113.000037
31. Niimura H, Bachinski LL, Sangwatanaroj S, et al. Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. N Engl J Med. 1998;338:1248–1257. doi:10.1056/NEJM199804303381802
32. Millat G, Bouvagnet P, Chevalier P, et al. Prevalence and spectrum of mutations in a cohort of 192 unrelated patients with hypertrophic cardiomyopathy. Eur J Med Genet. 2010;53:261–267. doi:10.1016/j.ejmg.2010.07.007
33. Kaski JP, Syrris P, Esteban MTT, et al. Prevalence of sarcomere protein gene mutations in preadolescent children with hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2009;2:436–441. doi:10.1161/CIRCGENETICS.108.821314
34. Bick AG, Flannick J, Ito K, et al. Burden of rare sarcomere gene variants in the Framingham and Jackson heart study cohorts. Am J Hum Genet. 2012;91:513–519. doi:10.1016/j.ajhg.2012.07.017
35. Teekakirikul P, Zhu W, Huang HC, Fung E. Hypertrophic cardiomyopathy: an overview of genetics and management. Biomolecules. 2019;9:878. doi:10.3390/biom9120878
36. Littler WA. Twin studies in hypertrophic cardiomyopathy. Br Heart J. 1972;34:1147–1151. doi:10.1136/hrt.34.11.1147
37. Repetti GG, Kim Y, Pereira AC, et al. Discordant clinical features of identical hypertrophic cardiomyopathy twins. Proc Natl Acad Sci USA. 2021;118. doi:10.1073/pnas.2021717118
38. Olivotto I, Maron BJ, Tomberli B, et al. Obesity and its association to phenotype and clinical course in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2013;62:449–457. doi:10.1016/j.jacc.2013.03.062
39. Fumagalli C, Maurizi N, Day SM, et al. Association of obesity with adverse long-term outcomes in hypertrophic cardiomyopathy. JAMA Cardiol. 2020;5:65–72. doi:10.1001/jamacardio.2019.4268
40. Nollet EE, Westenbrink BD, de Boer RA, Kuster DWD, van der Velden J. Unraveling the genotype-phenotype relationship in hypertrophic cardiomyopathy: obesity-related cardiac defects as a major disease modifier. J Am Heart Assoc. 2020;9:e018641. doi:10.1161/JAHA.120.018641
41. Chen Z, Xu B. Clinical significance of overweight in patients with hypertrophic cardiomyopathy: a retrospective cohort study. Medicine. 2021;100:e27445. doi:10.1097/MD.0000000000027445
42. Klempfner R, Kamerman T, Schwammenthal E, et al. Efficacy of exercise training in symptomatic patients with hypertrophic cardiomyopathy: results of a structured exercise training program in a cardiac rehabilitation center. Eur J Prev Cardiol. 2015;22:13–19. doi:10.1177/2047487313501277
43. Limongelli G, Monda E, D’Aponte A, et al. Combined effect of Mediterranean diet and aerobic exercise on weight loss and clinical status in obese symptomatic patients with hypertrophic cardiomyopathy. Heart Fail Clin. 2021;17:303–313. doi:10.1016/j.hfc.2021.01.003
44. Petto J, DE Oliveira EC, DE Almeida RVA, et al. Reverse myocardial remodeling in hypertrophic cardiomyopathy: little explored benefit of exercise. Int J Exerc Sci. 2021;14:1018–1026.
45. Saberi S, Wheeler M, Day SM. Exercise for patients with hypertrophic cardiomyopathy-reply. JAMA. 2017;318:481–482. doi:10.1001/jama.2017.8201
46. Kwon DH, Smedira NG, Popovic ZB, et al. Steep left ventricle to aortic root angle and hypertrophic obstructive cardiomyopathy: study of a novel association using three-dimensional multimodality imaging. Heart. 2009;95:1784–1791. doi:10.1136/hrt.2009.166777
47. Critoph CH, Pantazis A, Tome Esteban MT, et al. The influence of aortoseptal angulation on provocable left ventricular outflow tract obstruction in hypertrophic cardiomyopathy. Open Heart. 2014;1:e000176. doi:10.1136/openhrt-2014-000176
48. Maron MS, Olivotto I, Harrigan C, et al. Mitral valve abnormalities identified by cardiovascular magnetic resonance represent a primary phenotypic expression of hypertrophic cardiomyopathy. Circulation. 2011;124:40–47. doi:10.1161/CIRCULATIONAHA.110.985812
49. Makavos G, Κairis C, Tselegkidi M-E, et al. Hypertrophic cardiomyopathy: an updated review on diagnosis, prognosis, and treatment. Heart Fail Rev. 2019;24:439–459. doi:10.1007/s10741-019-09775-4
50. Sherrid MV, Barac I, McKenna WJ, et al. Multicenter study of the efficacy and safety of disopyramide in obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2005;45:1251–1258. doi:10.1016/j.jacc.2005.01.012
51. Green EM, Wakimoto H, Anderson RL, et al. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science. 2016;351:617–621. doi:10.1126/science.aad3456
52. Maron BJ, Dearani JA, Ommen SR, et al. Low operative mortality achieved with surgical septal myectomy: role of dedicated hypertrophic cardiomyopathy centers in the management of dynamic subaortic obstruction. J Am Coll Cardiol. 2015;66:1307–1308. doi:10.1016/j.jacc.2015.06.1333
53. Levine GN, Bates ER, Blankenship JC, et al. 2011 ACCF/AHA/SCAI guideline for percutaneous coronary intervention. A report of the American College of Cardiology Foundation/American Heart Association task force on practice guidelines and the society for cardiovascular angiography and interventions. J Am Coll Cardiol. 2011;58:e44–122. doi:10.1016/j.jacc.2011.08.007
54. Ommen SR, Maron BJ, Olivotto I, et al. Long-term effects of surgical septal myectomy on survival in patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2005;46:470–476. doi:10.1016/j.jacc.2005.02.090
55. Day SM, Tardiff JC, Ostap EM. Myosin modulators: emerging approaches for the treatment of cardiomyopathies and heart failure. J Clin Invest. 2022;132. doi:10.1172/JCI148557
56. Nag S, Trivedi DV, Sarkar SS, et al. The myosin Mesa and the basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Nat Struct Mol Biol. 2017;24:525–533. doi:10.1038/nsmb.3408
57. Scellini B, Piroddi N, Dente M, et al. Mavacamten has a differential impact on force generation in myofibrils from rabbit psoas and human cardiac muscle. J Gen Physiol. 2021;153. doi:10.1085/jgp.202012789
58. Anderson RL, Trivedi DV, Sarkar SS, et al. Deciphering the super relaxed state of human beta-cardiac myosin and the mode of action of mavacamten from myosin molecules to muscle fibers. Proc Natl Acad Sci USA. 2018;115:E8143–E8152. doi:10.1073/pnas.1809540115
59. Heitner SB, Jacoby D, Lester SJ, et al. Mavacamten treatment for obstructive hypertrophic cardiomyopathy: a clinical trial. Ann Intern Med. 2019;170:741–748. doi:10.7326/M18-3016
60. Olivotto I, Oreziak A, Barriales-Villa R, et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, Phase 3 trial. Lancet. 2020;396:759–769. doi:10.1016/S0140-6736(20)31792-X
61. Saberi S, Cardim N, Yamani M, et al. Mavacamten favorably impacts cardiac structure in obstructive hypertrophic cardiomyopathy: EXPLORER-HCM cardiac magnetic resonance substudy analysis. Circulation. 2021;143:606–608. doi:10.1161/CIRCULATIONAHA.120.052359
62. Desai MY, Owens A, Geske JB, et al. Myosin inhibition in patients with obstructive hypertrophic cardiomyopathy referred for septal reduction therapy. J Am Coll Cardiol. 2022;80:95–108. doi:10.1016/j.jacc.2022.04.048
63. Chuang C, Collibee S, Ashcraft L, et al. Discovery of aficamten (CK-274), a next-generation cardiac myosin inhibitor for the treatment of hypertrophic cardiomyopathy. J Med Chem. 2021;64:14142–14152. doi:10.1021/acs.jmedchem.1c01290
64. Malik FI, Robertson LA, Armas DR, et al. A phase 1 dose-escalation study of the cardiac myosin inhibitor aficamten in healthy participants. JACC Basic Transl Sci. 2022;7:763–775. doi:10.1016/j.jacbts.2022.04.008
65. Maron MS, Masri A, Choudhury L, et al. Phase 2 study of aficamten in patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2023;81:34–45. doi:10.1016/j.jacc.2022.10.020
66. ClinicalTrials.gov. CY 6031 study will evaluate the effects of treatment with aficamten (CK-3773274) over a 24-week period on cardiopulmonary exercise capacity and health status in patients with symptomatic oHCM (SEQUOIA-HCM). Available from: https://clinicaltrials.gov/ct2/show/NCT05186818.
67. Del Rio CL, Yadav A, Ferguson BS, et al. Chronic treatment with a mavacamten-like myosin-modulator (MYK-581) blunts disease progression in a mini-pig genetic model of non-obstructed hypertrophic cardiomyopathy: in vivo evidence for improved relaxation and functional reserve. Circulation. 2019;140:A14585–A14585.
68. Ferguson B, Stern JA, Oldach MS, et al. Acute effects of a mavacamten-like myosin-inhibitor (MYK-581) in a feline model of obstructed hypertrophic cardiomyopathy: evidence of improved ventricular filling (beyond obstruction reprieve). Eur Heart J. 2020;41:ehaa946. doi:10.1093/ehjci/ehaa946.3713
69. Ho CY, Mealiffe ME, Bach RG, et al. Evaluation of mavacamten in symptomatic patients with nonobstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2020;75:2649–2660. doi:10.1016/j.jacc.2020.03.064
70. Ho CY, Day SM, Axelsson A, et al. Valsartan in early-stage hypertrophic cardiomyopathy: a randomized phase 2 trial. Nat Med. 2021;27:1818–1824. doi:10.1038/s41591-021-01505-4
71. Maron BJ, Rowin EJ, Maron MS. Evolution of risk stratification and sudden death prevention in hypertrophic cardiomyopathy: twenty years with the implantable cardioverter-defibrillator. Heart Rhythm. 2021;18:1012–1023. doi:10.1016/j.hrthm.2021.01.019
72. Iavarone M, Monda E, Vritz O, et al. Medical treatment of patients with hypertrophic cardiomyopathy: an overview of current and emerging therapy. Arch Cardiovasc Dis. 2022;115:529–537. doi:10.1016/j.acvd.2022.06.003
73. Packard E, de Feria A, Peshin S, Reza N, Owens AT. Contemporary therapies and future directions in the management of hypertrophic cardiomyopathy. Cardiol Ther. 2022;11:491–507. doi:10.1007/s40119-022-00283-5
74. Repetti GG, Toepfer CN, Seidman JG, Seidman CE. Novel therapies for prevention and early treatment of cardiomyopathies. Circ Res. 2019;124:1536–1550. doi:10.1161/CIRCRESAHA.119.313569
75. Prondzynski M, Krämer E, Laufer SD, et al. Evaluation of MYBPC3 trans-splicing and gene replacement as therapeutic options in human iPSC-derived cardiomyocytes. Mol Ther Nucleic Acids. 2017;7:475–486. doi:10.1016/j.omtn.2017.05.008
76. da Rocha AM, Guerrero-Serna G, Helms A, et al. Deficient cMyBP-C protein expression during cardiomyocyte differentiation underlies human hypertrophic cardiomyopathy cellular phenotypes in disease specific human ES cell derived cardiomyocytes. J Mol Cell Cardiol. 2016;99:197–206. doi:10.1016/j.yjmcc.2016.09.004
77. Chai AC, Cui M, Chemello F, et al. Base editing correction of hypertrophic cardiomyopathy in human cardiomyocytes and humanized mice. Nat Med. 2023;11:1.
78. Iyer AA, Saade D, Bharucha-Goebel D, et al. Ethical challenges for a new generation of early-phase pediatric gene therapy trials. Genet Med. 2021;23:2057–2066. doi:10.1038/s41436-021-01245-3
79. Maron MS, Kalsmith BM, Udelson JE, Li W, DeNofrio D. Survival after cardiac transplantation in patients with hypertrophic cardiomyopathy. Circ Heart Fail. 2010;3:574–579. doi:10.1161/CIRCHEARTFAILURE.109.922872
80. Fowler CC, Helmers MR, Smood B, et al. The modified US heart allocation system improves transplant rates and decreases status upgrade utilization for patients with hypertrophic cardiomyopathy. J Heart Lung Transplant. 2021;40:1181–1190. doi:10.1016/j.healun.2021.06.018
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.