")
Back to Journals » International Medical Case Reports Journal » Volume 12
Familial glioblastoma clustering in adult patients: a case report of two non-twin siblings and review of the literature
Authors Sander C, Reuschel V, Eisenlöffel C, Nestler U , Meixensberger J
Received 14 January 2019
Accepted for publication 3 April 2019
Published 5 July 2019 Volume 2019:12 Pages 205—211
DOI https://doi.org/10.2147/IMCRJ.S201488
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ronald Prineas
Caroline Sander,1 Vera Reuschel,2 Christian Eisenlöffel,3 Ulf Nestler,1 Jürgen Meixensberger1
1Department of Neurosurgery, University Hospital Leipzig, Leipzig 04103, Germany; 2Department of Neuroradiology, University Hospital Leipzig, Leipzig 04103, Germany; 3Institute of Neuropathology, University Hospital Leipzig, Leipzig 04103, Germany
Purpose: Familial glioblastoma multiforme (gbm) has been described in children with hereditary tumor syndromes. The occurrence of gbm in adult members of the same family and in the absence of tumor syndromes is extremely rare. We describe the cases of a brother and a sister with multifocal gbm diagnosed at the age of 63 years. We discuss three further paired gbm in adult patients from the literature.
Patients and results: The sister was diagnosed with multifocal primary gbm in 2014 at the age of 63 years and 6 months. In 2018, her younger brother had to be operated on for a multifocal primary gbm at the age of 63 years and 9 months. Extended neuropathological examination revealed most markers to be similar, except for the percentage of O6,-methylguanine-DNA methyltransferase promotor methylation, the presence of intratumoral immune cells and the immunohistochemical expression of C12ORF75. Comparison with further published cases of familial adult GBM reveals that most of these patients are male, about 65 years old and the tumor is localized predominantly in the left temporal lobe.
Conclusion: Paired adult familial gbm occurs mainly in the elderly male patient with an integrative diagnosis of primary gbm. Whereas a statistical coincidence seems to be most likely in these rare cases, supplementary and improved genetic studies may identify pathogenetic causes of gbm.
Keywords: multifocal glioblastoma, familial tumor clustering, IDH-1 wildtype, tumor syndrome
Introduction
Glioblastoma multiforme (gbm) is the most malignant tumor of the central nervous system with an extremely poor prognosis.1 The average overall survival is 12–15 months after first diagnosis. Two molecular subtypes of gbm are described, divided into primary and secondary gbm. Primary gbm is characterized by de novo development and usually occurs in elderly patients over 50 years of age. Molecular analysis reveals an overexpression of EGFR, loss of heterozygosity of chromosome 10q and phosphatase and tensin homolog mutations. Secondary GBM occurs mainly in younger patients and its genetic profile is characterized by tumor protein p53 (TP53) mutations and isocitrate dehydrogenase 1 (IDH-1) mutations.2
There is faint evidence for the involvement of environmental factors in the genesis of glioblastoma, such as nonionizing electromagnetic fields and ionizing radiations.3 Furthermore, several hereditary tumor syndromes are associated with an increased occurrence of gbm in childhood and adolescence, for example, neurofibromatosis type-1, familial melanoma-astrocytoma syndrome, Li-Fraumeni syndrome, and Turcot syndrome.4–7
Familial gbm in the adult is extremely rare. A review of the literature revealed many well-documented descriptions of familial gbm in children, but there were only three case reports dealing with familial gbm in adult patients.8–10
This report describes familial gbm in two adult siblings. Interestingly, both patients suffered from multifocal gbm and were diagnosed at the same age of 63 years.
Case description
Case 1
Sibling 1 was the older sister, a 63-year-old female without malignant disease in her history (Table 1). She suffered from sudden paresthesia, ataxia, and vertigo. Brain magnetic resonance imaging (mri) disclosed mass lesions with gadolinium enhancement in the left parieto-temporal and the left frontal lobe infiltrating the splenium to the contralateral hemisphere with a total volume of 54.52 cm3 (Figure 1A-C). Craniotomy and partial resection of the tumor were performed 1 week after the first occurrence of clinical symptoms. Histopathologic examination of the tissue revealed a primary gbm, IDH-1 wild type (Table 2). The patient postoperatively developed focal epilepsia in the left upper limb, which was successfully treated with levetiracetam. She was discharged 14 days after surgery.
 |
Table 1 Clinical data and oncologic history of both cases |
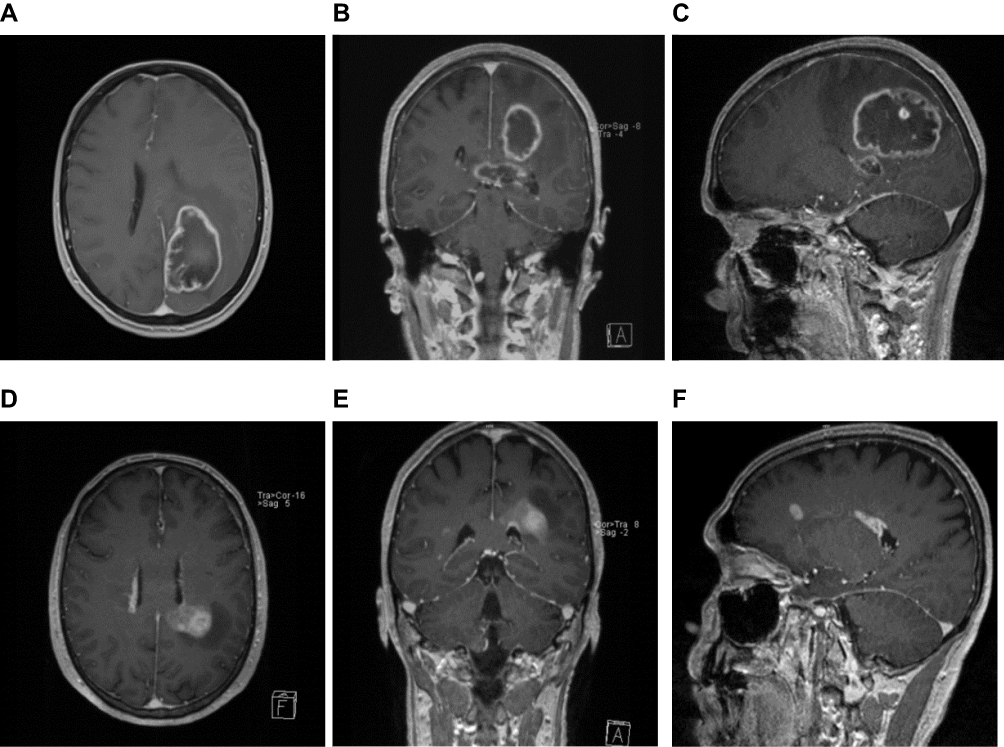 |
Figure 1 Magnetic resonance imaging (mri) scan of the neurocranium, gadolinium-enhanced T1-weighted sequences. (A-C) Sibling 1. mri axial (A), coronal (B) and sagittal view (C) large mass located in the parieto-temporal lobe, multifocal localization, extension into the corpus callosum and infiltration to the right hemisphere. (D-F) Sibling 2. mri axial (D), coronal (E) and sagittal view (F). Tumor mass located in the parieto-temporal lobe with periventricular, ependymal spread. Multifocal localization, up to the right frontal and temporal regions. |
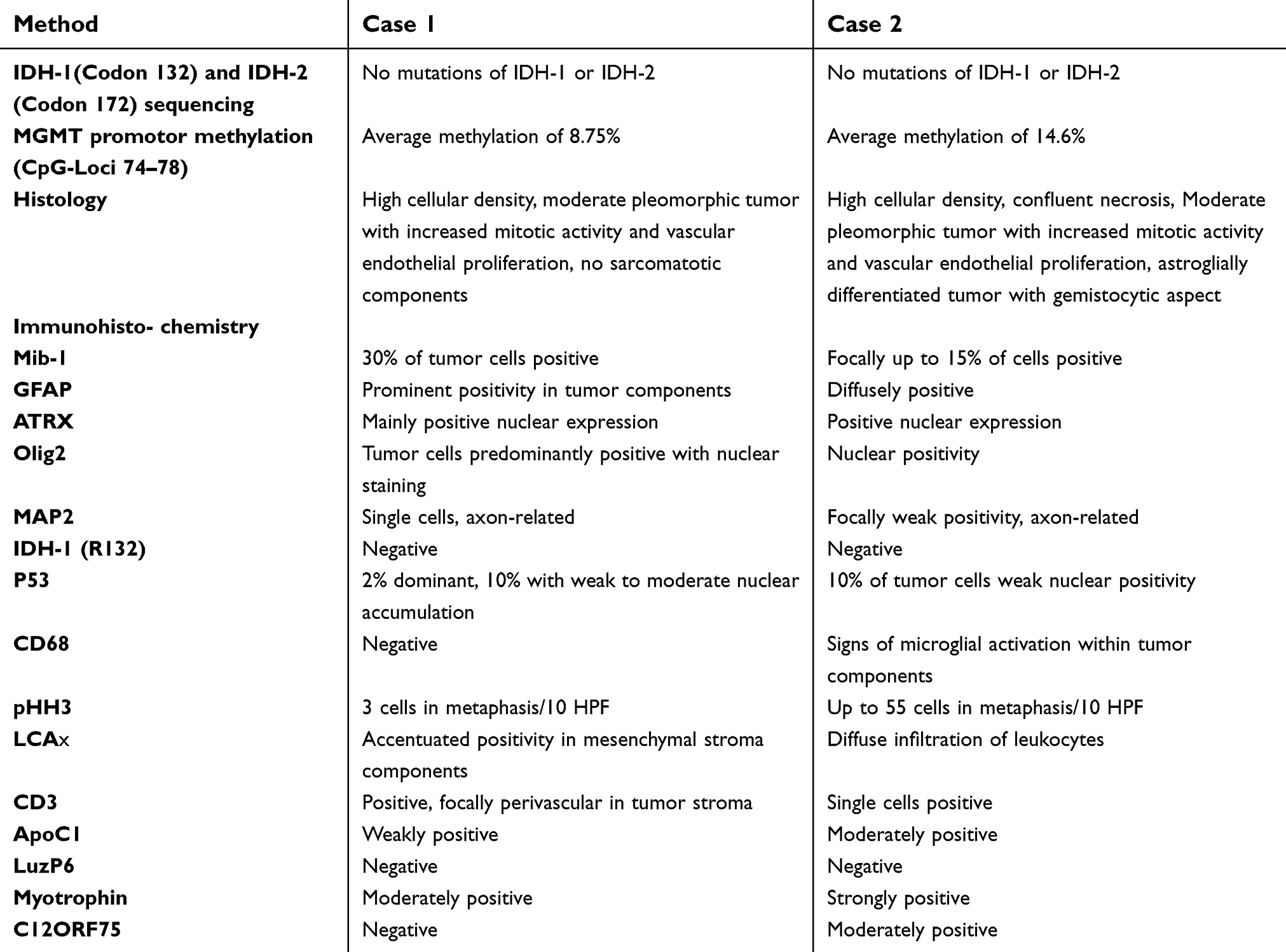 |
Table 2 Neuropathologic results of tumor samples |
Concomitant radiation with temozolomide (tmz) was performed, followed by chemotherapy with tmz alone. The first recurrence occurred in the dorsal corpus callosum, left occipital lobe and left temporal lobe 1 year after the first diagnosis of gbm. A second palliative radiation of the recurrence was undertaken. The patient died 63 weeks after the first diagnosis of multifocal gbm.
Case 2
Sibling 2 was the younger brother of the patient described as case 1, a 63-year-old male without malignant diseases (Table 1). He presented to our institution with a 1-year history of progressive gait disturbance and motor aphasia. The mri scan revealed multifocal lesions in the left parieto-temporal and frontal lobe combined with ependymal spread and a total tumor volume of 8.97 cm3 (Figure 1D-F). Craniotomy and partial resection of the tumor in the left parietal lobe were performed. Histopathology disclosed primary gbm, IDH-1 wild type (Table 2). The patient was discharged 5 days after surgery with motor dysphasia, but the absence of further neurologic sequelae. Combined radiation and chemotherapy had been advised, but, in view of his sister’s fate, the patient refused any further therapy or clinical follow-up examinations and died 3 months after the first diagnosis of gbm.
Discussion
In this study, we describe a sister and her younger brother who both suffered from gbm at an identical age with nearly the same tumor localization and multifocal appearance. Furthermore, the histological, immunohistochemical, and molecular findings can be considered to be nearly identical.
Familial gbm has been associated in pediatric patients with specific tumor syndromes (Table 3).
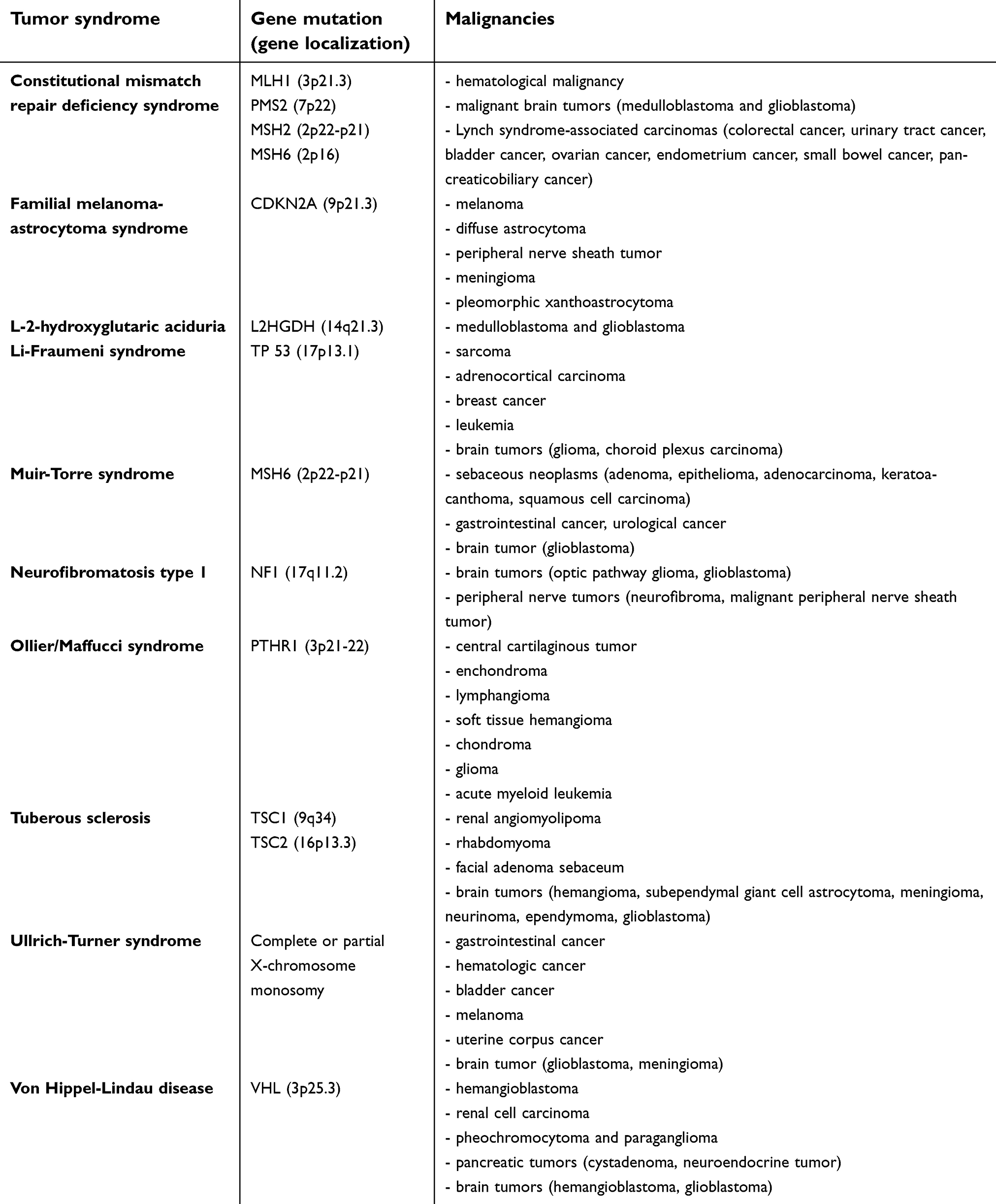 |
Table 3 Pediatric tumor syndromes with reported familial clustering of glioblastoma multiforme |
In contrast to this, the familial occurrence of gbm in adult patients is an extremely rare event, with only a few case reports found in the recent literature (Table 4).
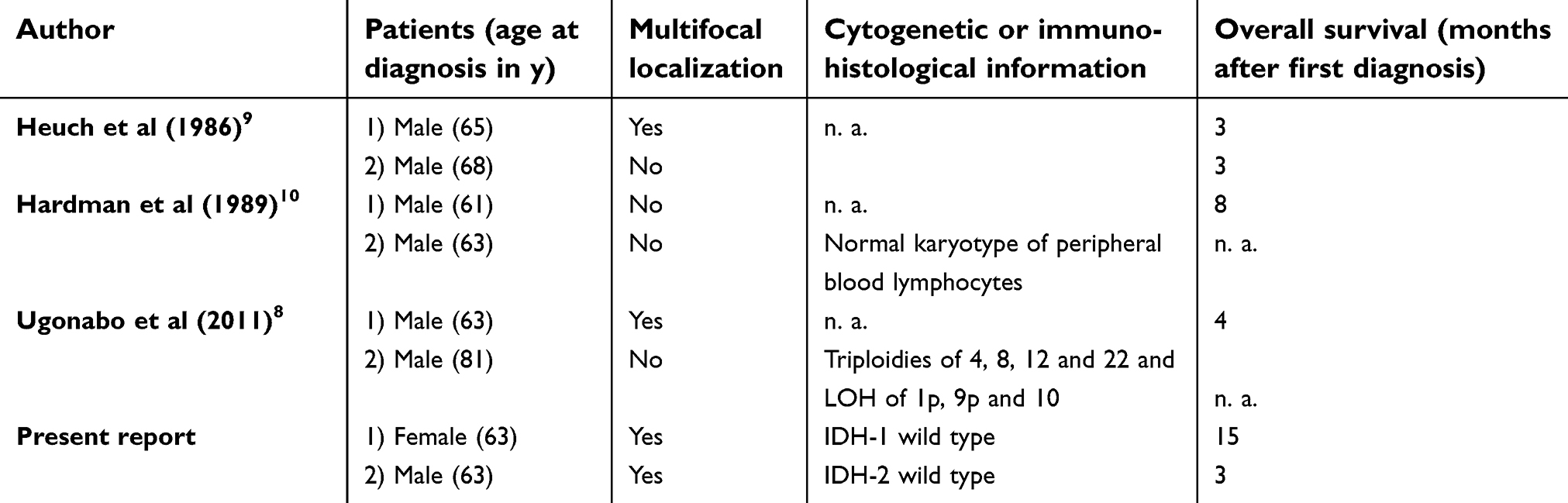 |
Table 4 Reports on familial gbm in the adult |
Heuch et al described two brothers aged 65 and 68, one case only diagnosed at autopsy.9 The second report of adult familiar gbm was illustrated by Hardman: one pair of identical twins suffered from gbm, the first at the age of 61 and the second at 63. Normal karyotypes of blood leukocytes and similar environmental exposure are described.10 Thirdly, Ugonabo published a report of two brothers with the diagnosis of gbm at the age of 63 and 81 years.8 There was no clinical information given about any environmental risk factors or genetic alterations to depict pathophysiologic mechanisms.
In summary, all four case reports of adult familiar gbm describe elderly patients with an average age of 65.9 years at first diagnosis and seven of eight patients are male. Frequent tumor localizations are the temporal lobe (54%, 6 of 11 tumors), the parietal lobe (36%, 4 of 11), occipital lobe (27%, 3 of 11), and the frontal lobe (18%, 2 of 11). A multifocal localization was detected in half of the patients. The left hemisphere was more often implicated than the right hemisphere (63% left vs 36% right). The average overall survival was 6 months. These findings are in good accordance with the natural epidemiology of primary gbm, pointing to a statistical coincidence as an explanation for the familial occurrence, rather than to a causal connection.11
Integrated diagnosis was only available in the two current cases due to the retrospective nature of case reports. In the present cases, the integrated diagnosis and the detailed immunohistochemical markers can be considered to be almost identical, revealing the diagnosis of primary gbm, IDH-1 wild type.
However, we detected discrete differences concerning MGMT promoter methylation and the number of immune cells within the tumor stained by cluster of differentiation 68. In case 1 MGMT promoter methylation was classified as absent, whereas in case 2 the MGMT promoter was strongly methylated. These findings would have suggested a better overall survival and chemotherapeutical response for case 2 compared to case 1, as has been detailed by the MGMT promoter methylation score,12 but unfortunately patient 2 declined further therapy.
Differing results were also shown for the immunohistochemical staining of C12orf75, whereas the staining for ApoC1, leucine zipper protein 6 (LuzP6), and myotrophin were rather similar, all of them proteins that have been associated with cystic appearance of gbm (Figure 1, Table 2).13
The intriguing finding of tumor occurrence at nearly the same age with a large amount of corresponding radiological and neuropathological results inspires the search for a potential common pathogenetic mechanism. Further detailed genetic analysis would help to describe the precise driver genes to distinguish between familial hereditary occurrence and mere coincidence.
Ugonabo et al performed cytogenetic analysis in one sibling and illustrated multiple chromosomal abnormalities, such as triploidies 4, 8, 12 and 22, and loss of heterozygosity of 1 p, 9 p and 10.8 All of these chromosomal aberrations have already been well described in gbm.14–19 However, the frequent hallmarks of gbm, gain at chromosome 7 and loss of chromosome 10, were absent, thus, the genetic imbalances illustrated did not allow to define a “familial clustering subtype”.
Conclusion
In addition to the two gbm siblings presented in this report, a review of the literature revealed only a very limited number of reports on familial gbm clustering in adults. Different from the pediatric population and although supplementary and improved genetic studies may disclose further common aberrations in the future, our results support the hypothesis of statistical coincidence without an underlying genetic cause.
Informed Consent
Both patients gave their written informed consent to the operation and the use of their data in pseudonymized form for research purposes and publication of the case details prior to the neurosurgical intervention.
Ethics approval
Ethics approval had been obtained from the ethics committee of the University of Leipzig (277/15-ff). The study was conducted in accordance with the principles of the Declaration of Helsinki (as revised in Tokyo 2004).
Abbreviation list
ApoC1, apolipoprotein C1; C12ORF75, chromosome 12 open reading frame 75; CD68, cluster of differentiation 68; gbm, glioblastoma multiforme; IDH-1, isocitrate dehydrogenase 1; IDH-2, isocitrate dehydrogenase 2; LOH, loss of heterozygosity; LuzP6, leucine zipper protein 6; MGMT, O6-methylguanine-DNA methyltransferase; mri, magnetic resonance imaging; PTEN, phosphatase and tensin homologue; tmz, temozolomide; TP53, tumor protein p53.
Acknowledgment
This study had no funding source.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Veliz I, Loo Y, Castillo O, Karachaliou N, Nigro O, Rosell R. Advances and challenges in the molecular biology and treatment of glioblastoma-is there any hope for the future? Ann Transl Med. 2015;3(1):7. doi:10.3978/j.issn.2305-5839.2014.10.06
2. Ohgaki H, Dessen P, Jourde B, et al. Genetic pathways to glioblastoma: a population-based study. Cancer Res. 2004;64(19):6892–6899. doi:10.1158/0008-5472.CAN-04-1337
3. Daniels LB, Shaya M, Nordberg ML, Shorter CD, Fowler M, Nanda A. Glioblastoma multiforme in two non-nuclear family members. J La State Med Soc. 2007;159(4):215–222.
4. Gutmann DH, Ferner RE, Listernick RH, Korf BR, Wolters PL, Johnson KJ. Neurofibromatosis type 1. Nat Rev Dis Primers. 2017;3:17004. doi:10.1038/nrdp.2017.4
5. Wimmer K, Kratz CP, Vasen HFA, et al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium ‘care for CMMRD’ (C4CMMRD). J Med Genet. 2014;51(6):355–365. doi:10.1136/jmedgenet-2014-102284
6. Schiffman JD, Chun N, Fisher PG, Dahl GV, Ford JM, Eggerding FA. Identification of a novel p53 in-frame deletion in a Li-Fraumeni-like family. Pediatr Blood Cancer. 2008;50(4):914–916. doi:10.1002/pbc.21247
7. Chan AK, Han SJ, Choy W, et al. Familial melanoma-astrocytoma syndrome: synchronous diffuse astrocytoma and pleomorphic xanthoastrocytoma in a patient with germline CDKN2A/B deletion and a significant family history. Clin Neuropathol. 2017;36(5):213–221. doi:10.5414/NP301022
8. Ugonabo I, Bassily N, Beier A, et al. Familial glioblastoma: A case report of glioblastoma in two brothers and review of literature. Surg Neurol Int. 2011;2:153. doi:10.4103/2152-7806.86833.
9. Heuch I, Blom GP. Glioblastoma multiforme in three family members, including a case of true multicentricity. J Neurol. 1986;233(3):142–144.
10. Hardman PD, Bell J, Whittle IR, Gregor A. Familial glioma: a report of glioblastoma in identical twins and oligo-astrocytoma in siblings. Br J Neurosurg. 1989;3(6):709–715.
11. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131(6):803–820. doi:10.1007/s00401-016-1545-1
12. De Carlo E, Gerratana L, De Maglio G, et al. Defining a prognostic score based on O6-methylguanine-DNA methyltransferase cut-off methylation level determined by pyrosequencing in patients with glioblastoma multiforme. J Neurooncol. 2018;140(3):559–568. doi:10.1007/s11060-018-2981-7
13. Groll M, Frenzel J, Krause M, et al. MALDI-TOF mass spectrometric analysis of brain tumor cyst fluid reveals a protein peak corresponding to ApoC1 and LuzP6. Ojmn. 2018;08(03):251–263. doi:10.4236/ojmn.2018.83021
14. Zuber MA, Krupp W, Holland H, Froster UG. Characterization of chromosomal aberrations in a case of glioblastoma multiforme combining cytogenetic and molecular cytogenetic techniques. Cancer Genet Cytogenet. 2002;138(2):111–115.
15. Holland H, Koschny T, Ahnert P, Meixensberger J, Koschny R. WHO grade-specific comparative genomic hybridization pattern of astrocytoma - a meta-analysis. Pathol Res Pract. 2010;206(10):663–668. doi:10.1016/j.prp.2010.04.002
16. Jesionek-Kupnicka D, Szybka M, Potemski P, et al. Association of loss of heterozygosity with shorter survival in primary glioblastoma patients. Pol J Pathol. 2013;64(4):268–275.
17. McLendon R, Friedman A, Bigner D, et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–1068. doi:10.1038/nature07385
18. Bigner SH, Friedman HS, Biegel JA, et al. Specific chromosomal abnormalities characterize four established cell lines derived from malignant human gliomas. Acta Neuropathol. 1986;72(1):86–97.
19. Dahlback H-S-S, Brandal P, Meling TR, Gorunova L, Scheie D, Heim S. Genomic aberrations in 80 cases of primary glioblastoma multiforme: pathogenetic heterogeneity and putative cytogenetic pathways. Genes Chromosomes Cancer. 2009;48(10):908–924. doi:10.1002/gcc.20690
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.