")
Back to Journals » OncoTargets and Therapy » Volume 12
FAM111B, a direct target of p53, promotes the malignant process of lung adenocarcinoma
Authors Sun H, Liu K , Huang J, Sun Q, Shao C, Luo J, Xu L, Shen Y, Ren B
Received 15 October 2018
Accepted for publication 14 March 2019
Published 17 April 2019 Volume 2019:12 Pages 2829—2842
DOI https://doi.org/10.2147/OTT.S190934
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Federico Perche
Haijun Sun,1–3,* Kaichao Liu,4,* Jianfeng Huang,1,2,* Qi Sun,5 Chenye Shao,4 Jing Luo,4 Lin Xu,1,2 Yi Shen,4 Binhui Ren1,2
1Department of Thoracic Surgery, Nanjing Medical University Affiliated Cancer Hospital, Nanjing, People’s Republic of China; 2Jiangsu Key Laboratory of Molecular and Translational Cancer Research, Cancer Institute of Jiangsu Province, Nanjing, Jiangsu, People’s Republic of China; 3Department of Thoracic Surgery, the First People’s Hospital of Lianyungang City, Nanjing Medical University Affiliated Lianyungang Clinical College, Lianyungang, Jiangsu, People’s Republic of China; 4Department of Cardiothoracic Surgery, Jinling Hospital, School of Medicine, Nanjing University, Nanjing, Jiangsu, People’s Republic of China; 5Institut für Laboratoriumsmedizin, Klinische Chemie und Pathobiochemie, Charité – Universitätsmedizin Berlin, Campus Virchow-Klinikum, Berlin 13353, Germany
*These authors contributed equally to this work
Purpose: Lung adenocarcinoma (LUAD) is a main subtype of lung cancer, which is the leading cause of cancer-related deaths. The five-year survival rates of lung cancer patients are still comparatively low. Therefore, potential therapeutic targets are urgently needed to improve the survival of lung cancer patients. In this study, we identified FAM111B as an oncogene and potential therapeutic target for LUAD.
Methods: The TCGA database and tissue microarray analysis were used to compare the expression of FAM111B in tumor tissue and normal tissues and evaluate the relationship between FAM111B expression and clinical survival. FAM111B was knocked down and overexpressed to observe whether FAM111B could affect the proliferation, migration, cell cycle, and apoptosis of LUAD cells in vivo and in vitro.
Results: FAM111B was highly expressed in tumor tissues compared with normal tissues (P<0.01). LUAD patients with hyper-expression of FAM111B had a lower recurrence-free survival (P<0.01) and shorter overall survival (P<0.01). Knocking down FAM111B inhibited cell proliferation, migration and invasion in vitro and tumor growth in vivo. Silencing FAM111B could arrest LUAD cells at G2/M phase and increase apoptosis. Overexpression of FAM111B promoted the growth of lung cancer cells. FAM111B was identified as a direct target of p53 in existing researches by chip-seq analysis. Bioinformatics analysis predicted that FAM111B could directly bind to BAG3 (BCL2 associated athanogene 3). When FAM111B was down-regulated, both expression of BAG 3 and BCL2 were significantly reduced, whereas decreasing the expression of BAG3 had no effect on FAM111B.
Conclusions: Our study indicated that FAM111B might be an oncogene and potential therapeutic target in LUAD which could be involved in the regulation of tumor cells by p53 signaling pathway and play an important role in the process of cell cycle and apoptosis by influencing the expression of BAG3 and BCL2.
Keywords: apoptosis, p53, BAG3, FAM111B, lung adenocarcinoma, oncogene
Background
Lung cancer is the most common type of malignant tumors worldwide and the incidence of lung cancer is still on the rise.1 Non-small cell lung cancer (NSCLC) occupies 85% of lung cancer and includes two pathological types of lung cancer, namely lung adenocarcinoma (LUAD) and lung squamous carcinoma. Although most patients received suitable treatment, the five-year survival rates of NSCLC patients are still <15%.2,3 Therefore, therapy targets are still urgently needed to improve the survival of patients with NSCLC.
With the development of microarray and high-throughput sequencing technologies that provide a large number of open data resources [such as the Cancer Genome Atlas (Tumor Genome Atlas; TCGA] database and GEO datasets), novel cancer-related genes can be identified efficiently by mining big data sets.4–6 In this study, we carefully analyzed the TCGA database and found FAM111B was highly expressed in LUAD patients and related with the prognosis of LUAD patients.
FAM111B, also known as CANP or POIKTMP, is located at human chromosome 11q12.1 and encodes a protein with a trypsin-like cysteine/serine peptidase domain in the C-terminus (provided by PubMed RefSeq, Apr 2014). By reading previous literature, we found that FAM111B is a direct target of p53.7 However, the function of the FAM111B gene is still unclear and little is known about its role in human cancers. In this study, we would like to investigate the relationship of FAM111B expression with LUAD malignant phenotype both in vitro and in vivo.
Methods
Data sources and bioinformatics
To evaluate the difference of the FAM111B expression between LUAD tissue and normal tissues, we first used the TCGA LUAD database (TCGA_LUAD_exp_HiSeqV2-2015–02-24) including 511 LUAD tissues with 57 paired normal tissues. The expression data were extracted from the “genomicMatrix” file. The age, gender, survival days, TNM stage, and other clinical data were obtained from the “clinical_data” file. The BioGRID database (
Cell lines, cell culture, and siRNA transfection
The human LUAD cell lines A549, H1650, PC9, H1975, SPC-A-1, and the normal human bronchial epithelial cell line HBE were purchased from the Shanghai Institute of Life Sciences (Shanghai, China). Cells were cultured in DMEM (KeyGEN) supplemented with 10% FBS and penicillin/streptomycin (KeyGEN). Cells were grown in incubator at 37°C with 5% CO2. The overexpression plasmid of FAM111B was purchased from Nanjing Kanghao Biological Co., Ltd.
A total of 8 × 104 cells were seeded per well in six-well plates 24 hrs before transfection. When confluency reached 60–70%, the transfection with FAM111B siRNA using Lipofectamine RNAiMAX Reagent (Invitrogen, Waltham, MA, USA) was performed, the procedure is as follows: the amount of liquid added to each well should be: (OptiMEM 190 µL + 10 µL siRNA) + (OptiMEM 190 µL + 10 µL RNAiMAX). Nonsense siRNA was used as the negative control. The transfection efficiency was evaluated by quantitative real-time RT-PCR (qRT-PCR) and western blot analysis.
Primer sequences used were as follows: siRNA-1 for FAM111B 5ʹ-GCAUGCCCUUAUUGAAUUUTT-3ʹ (sense), and 5ʹ-AAAUUCAAUAAGGGCAUGCTT-3ʹ (anti-sense), siRNA-2 for FAM111B 5ʹ-GUAGUGUGUUUACAGCAUAUG-3ʹ (sense), and 5ʹ-UAUGCUGUAAACACACUACGG-3ʹ (anti-sense), siRNA for BAG3 5ʹ- GGCUGUAGACAACUUUGAAGG-3ʹ (sense), 5ʹ-UUCAAAGUUGUCUACAGCCUG-3ʹ (anti-sense). Nonsense siRNA 5ʹ-UUCUCCGAACGUGUCACGUTT-3ʹ sense), and 5ʹ-ACGUGACACGUUCGGAGAATT-3ʹ (anti-sense).
RNA extraction, reverse transcription, and real-time quantitative PCR
TRIzol reagent (Invitrogen) was used to extract total RNA of tissue samples and cell lines. And cDNA was synthesized using Primer Script RT reagent DRR036A (TAKARA). The reaction was performed according to the protocol. For qRT-PCR, SYBR Select Master Mix (KeyGEN) was used, and the reaction was performed in a QuantStudio 6 Flex Real-Time PCR System as follows: initial denaturation step at 95°C for 10 mins, followed by 40 cycles at 92°C for 15 s and 60°C for 1 min. Primers are shown in Table 1, and the housekeeping gene ACTIN was used as a control. Changes in gene expression were calculated using the 2−ΔΔCT method.
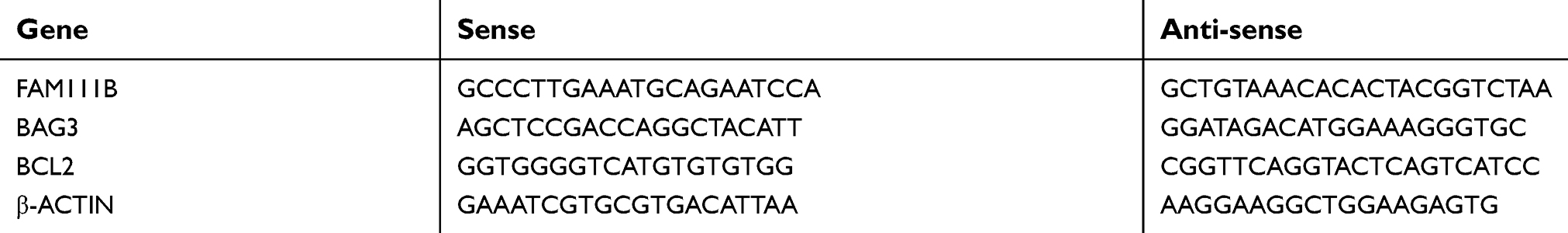 | Table 1 Primer sequences |
Protein extraction and western blot analysis
For protein extraction, cells were lysed on ice by RIPA lysis buffer (KeyGEN), and the BCA Kit (KeyGEN) was used to determine the protein concentration. Similar amounts of protein were loaded on SDS–PAGE gels and subjected to electrophoresis, then transferred onto a PVDF membrane and blocked with Tris-buffered saline with 0.1% (vol/vol) Tween 20 (TBST) containing 5% (wt/vol) BSA for 2 hrs. Then, membranes were washed by TBST 3 times and incubated overnight at 4°C with primary antibodies against FAM111B (PA5-58474; Invitrogen), BAG3 (AB47124; Abcam), BCL2 (TA806639S; Origene), β-actin (8H10D10; Cell Signaling Technology) at a 1:1,000 dilution in 5% BSA. After washing by TBST 3 times, PVDF membranes were incubated with the appropriate fluorescent secondary antibody (1:10,000; Odyssey) for 2 hrs at room temperature. Proteins were visualized by Odyssey two-color infrared fluorescence imaging system (LI-COR Company, USA), cropped with Photoshop CS3 (Adobe Systems Incorporated, San Jose, CA, USA). All experiments were independently repeated at least three times.
Colony formation assay
A549 and PC9 cells were harvested 24 hrs after transfection. A total of 200 cells were seeded per well in six-well plates in medium containing 10% FBS which is not a soft agarose. Cells were cultured in a 37°C incubator and medium was replaced every 4 days. After 14 days, the cell culture medium was removed and cells were fixed with 4% paraformaldehyde for 30 mins, then stained with 0.1% crystal violet for 15 mins. The number of colonies was counted and images were taken after washing the cells with PBS. All experiments were independently repeated at least three times.
Cell proliferation assay
At 24 hrs after transfection, cells were seeded in a 96-well plate at a density of 4 × 104 cells per well in 100 μL. Next, 20 μL of CCK-8 reagent was added to the wells and cells were incubated at 37°C for 2 hrs on the first, second, third, fourth, and fifth day. The OD value was measured at 450 nm. The OD value measured after seeding cells on the first day for 6 hrs was taken as the baseline value.
We also used the Edu Proliferation Assay Kit of Guangzhou Ruibo Co., Ltd. to resuspend and count the cells. After 48 hrs of cell transfection, the cells were resuspended and counted and seeded in 96-well plates. Then, according to the experimental description procedure, the percentage of the EdU signal in the FAM111B-overexpression group and the control group in the well plate was measured. All experiments were independently repeated at least three times.
Cell migration, invasion and wound healing assay
For the migration assay, transfected cells were diluted to 4 × 105/mL in serum-free medium. A total of 100 µL cell suspension was added to the upper chamber of transwell assay inserts (8-mm pores; Millipore, Billerica, MA, USA) containing 200 μL of serum-free DMEM. A total of 800 µL DMEM containing 10% FBS was added to the lower chamber. After incubating at 37°C for 24 hrs, the chamber was removed, and the medium was discarded. Cells on the filter surface were fixed with methanol for 30 mins, stained with crystal violet for 15 mins and imaged using confocal microscopy. The migration ability was assessed by counting the number of stained cells in five random fields per filter in each group.
For the invasion assay, transfected cells were diluted to 4 × 105/mL in serum-free medium. Add 100 µL cell suspension to the upper chamber with a matrigel-coated membrane (BD Biosciences, San Jose, CA, USA) in 300 µL of serum-free DMEM. The bottom chambers were filled with DMEM containing 10% FBS. The invasion ability was determined after 48 hrs of incubation.
For the wound healing assay, cells were plated into a six-well plate and transfected when reaching 60% confluence. After culturing for 24 hrs, the monolayer was wounded by a pipette tip, and floating cells were removed by two washes with PBS. The cells were cultured for another 24 hrs, and the distance between the lines of the wounded area was measured to assess the wound-healing ability using a confocal microscope.
Each experiment was independently repeated at least three times.
Flow-cytometry analysis
Cells were collected after transfection with si-FAM111B or control siRNA after 48 hrs, were fixed with 70% alcohol and stored overnight at −20°C. The cells were then stained with propidium oxide by the Cycle TEST PLUS DNA kit (BD Biosciences) and analyzed by FACScan according to the manufacturer’s guidelines. The percentages of cells in the G1, S and G2-M phases were determined and compared.
Apoptosis was measured by an Annexin-V kit according to the manufacturer’s guidelines. In brief, cells were digested with trypsin, pelleted and washed once with cold PBS. Then, cells were re-suspended in 300 µL of binding buffer (1–5 × 106 cells per condition), then 5 µL of Annexin-V-FITC solution was added to the binding buffer. After incubating in the dark for 15 mins, nuclei were stained with 5 µL of PI for 5 mins at room temperature. Finally, 200 µL of binding buffer was added, and apoptosis was evaluated by flow cytometry.
Xenograft studies
All animal studies were conducted in accordance with NIH animal user guidelines and the research was approved by the Nanjing Medical University Animal Care Committee. In brief, 12 female athymic nude mice (4–6 weeks old) were purchased from the Medical School of Nanjing Medical University. All animals were tested according to the protocol approved by the Animal Health Committee of Nanjing Medical University. A549 and PC9 cells were transfected with FAM111B-siRNA or NC-siRNA using Lipofectamine 3000 as mentioned above. At 24 hrs after transfection, a total of 1.0 × 106/100 µL exponentially growing cells were subcutaneously injected into mice, and tumor volume was calculated weekly ([length x width2] × 0.5). Six weeks after injection of tumor cells, animals were sacrificed, and tumors were collected and weighed. Finally, tumors were cut into sections and stained immunohistochemically for Ki67 and FAM111B expression.
Tissue collection and immunohistochemistry
The LUAD tissue microarray (Xinchao Company, Shanghai, China) used contained a paired sample of 92 LUAD patients and adjacent normal tissues from patients who underwent surgical resection and contained all the required clinical prognostic information. Subcutaneous tumor tissue samples were dewaxed and dehydrated by the following steps: xylene I 10 mins; xylene II 10 mins; 100% alcohol 3–5 mins; 100% alcohol 3–5 mins; 95% alcohol 1–3 mins; 80% alcohol 1–3 mins; 70% alcohol 1–3 mins; distilled water for 5 mins. Rinse 3 times for 5 mins to inactivate endogenous peroxidase. Immerse in a 0.01 M citrate buffer (pH 6.0) autoclave for high-temperature cooking and rinse 3 times with PBS for 5 mins each time; 1% BSA was blocked for 1 hr and the corresponding antibody (1:100) was added at 4°C overnight. A negative control group (PBS buffer group) was also established. The next day, the sections were taken out, the anti-incubation solution was discarded, and washed 3 times with PBS. After 5 mins, EnVision reagent (horseradish peroxidase) was added, and the mixture was washed three times for 30 mins at room temperature, and DAB/H2O2 reaction was added. Then, the samples were desiccated with hematoxylin, gradient alcohol, transparent xylene, neutral gel seal, observed under a microscope and photographed. The immunohistochemical staining scores of tissue microarrays were observed by two independent pathologists and scoring was dependent on the intensity and percentage of positive cells. The staining intensity was assessed at 0 (no staining), 1 (weak staining), 2 (moderate staining) or 3 (strong staining). The use of the human tissue microarrays was approved by the ethics committee of Jiangsu Cancer Hospital.
Statistics analysis
For data analysis, SPSS version 20.0 statistical software was used (SPSS Inc., Chicago, IL, USA). Data were expressed as the mean ± SD. Kaplan–Meier survival analysis and multivariate Cox regression analysis was used for clinical data. Paired sample t-test was used for the comparison of paired cancer tissues, Student’s t-test and one-way ANOVA were used for statistical analysis. P<0.05 was considered statistically significant. Graphs were prepared using the GraphPad Prism 6.0 software package (La Jolla, CA, USA).
Results
Bioinformatics analysis suggested that FAM111B may be a potential oncogene in LUAD
By analyzing the TCGA_LUAD_exp_HiSeqV2-2015–02-24 LUAD dataset, we found that the expressions of FAM111B were 7.71±0.51 in tumor tissues and 5.02±0.10 in normal tissues (P<0.0001, Figure 1A). And the PCR analysis of tumor and para-tumor tissues obtained from our hospital showed that FAM111B was upregulated expression in 45 of 52 patients (Figure 1B).
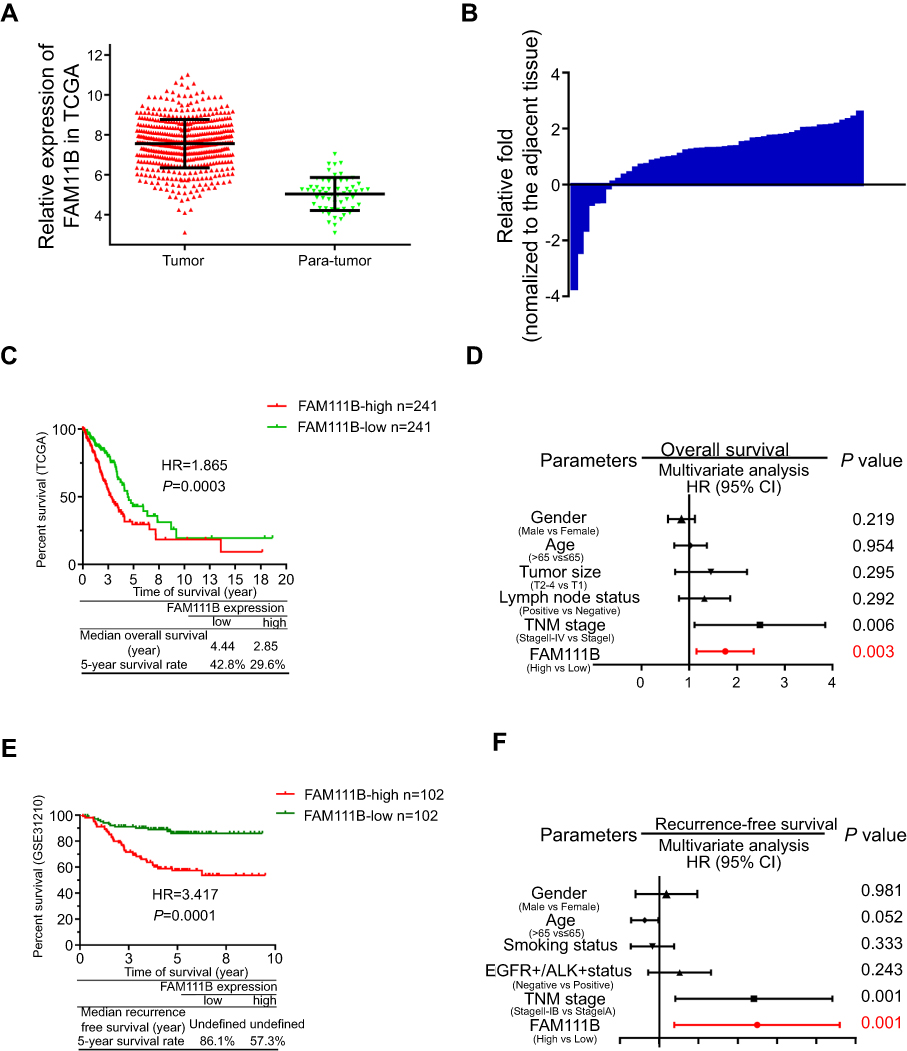 | Figure 1 mRNA of FAM111B was overexpressed in LUAD and correlated with poor clinical survival in TCGA and GSE database. (A) FAM111B was overexpressed in LUAD tissues in the TCGA LUAD database. (B) FAM111B was upregulated in 45 of 52 paired LUAD patients obtained from the Jiangsu Cancer Hospital (Jiangsu, China). (C and E) Kaplan–Meier survival analysis showed that the OS of patients with hyper-expression of FAM111B in tumor tissues were significantly shorter when compared to those with lower expression of FAM111B, while the progression-free survival of a total of 204 patients from GSE31210 showed a larger HR value. (D and F) Multivariate Cox regression analysis suggested that high expression of FAM111B (P=0.0001) and more advanced TNM stage (P=0.0001) were two independent risk factors affecting OS of LUAD.Abbreviations: LUAD, lung adenocarcinoma; TCGA, Tumor Genome Atlas; OS, overall survival. |
After removing cases with incomplete clinical information or lost follow-up, we collected 484 LUAD patients had available overall survival (OS) and 348 patients had available OS and tumor progression. Kaplan–Meier survival analysis showed that the OS of patients with hyper-expression of FAM111B in tumor tissues were significantly shorter when compared to those with lower expression of FAM111B, while the progression-free survival (PFS) of a total of 204 patients from GSE31210 showed a larger HR value (Figure 1C and E). Meanwhile, multivariate Cox regression analysis from comprehensive clinical parameters such as FAM111B expression level, gender, age, T stage, N stage, and TNM stage suggested that high expression of FAM111B (P=0.0001) and more advanced TNM stage (P=0.0001) were two independent risk factors affecting OS and PFS of LUAD (Figure 1D and F).
Tissue microarray analysis of LUAD showed that elevated expression of FAM111B was associated with poor patient prognosis
A total of 86 patients with LUAD were followed up for 5 years. The median OS time of FAM111B low expression group was 4.7 years, whereas which of the FAM111B high expression group was 2.2 years. The staining score in LUAD tumor tissues was higher compared with adjacent normal tissues (Figure 2A and B, P<0.01). As shown in Figure 2C and D, poorly differentiated LUAD samples showed higher FAM111B protein expression, compared with those well-differentiated LUAD samples. As shown in Tables 2 and 3, in the subsequent validation of tissue samples, we used immunohistochemistry to evaluate the associations between the FAM111B expression and clinical characteristics as well as survival time of LUAD. The Kaplan–Meier analysis indicated OS rate of the FAM111B high expression group was decreased when compared with that of the low expression group (P<0.01) (Figure 2E).
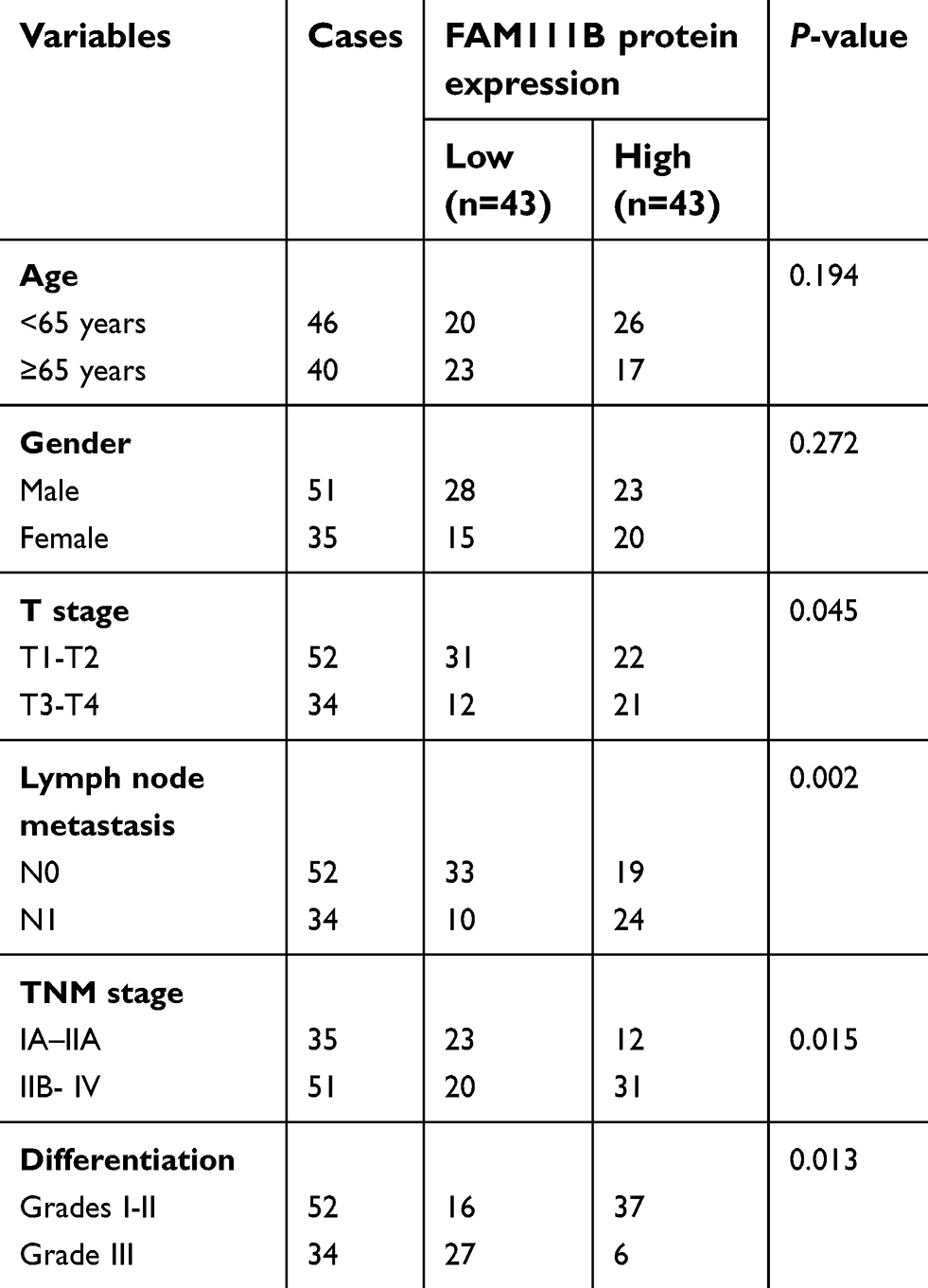 | Table 2 Correlation of FAM111B protein expression with clinicopathological characteristics of 86 LUAD cases |
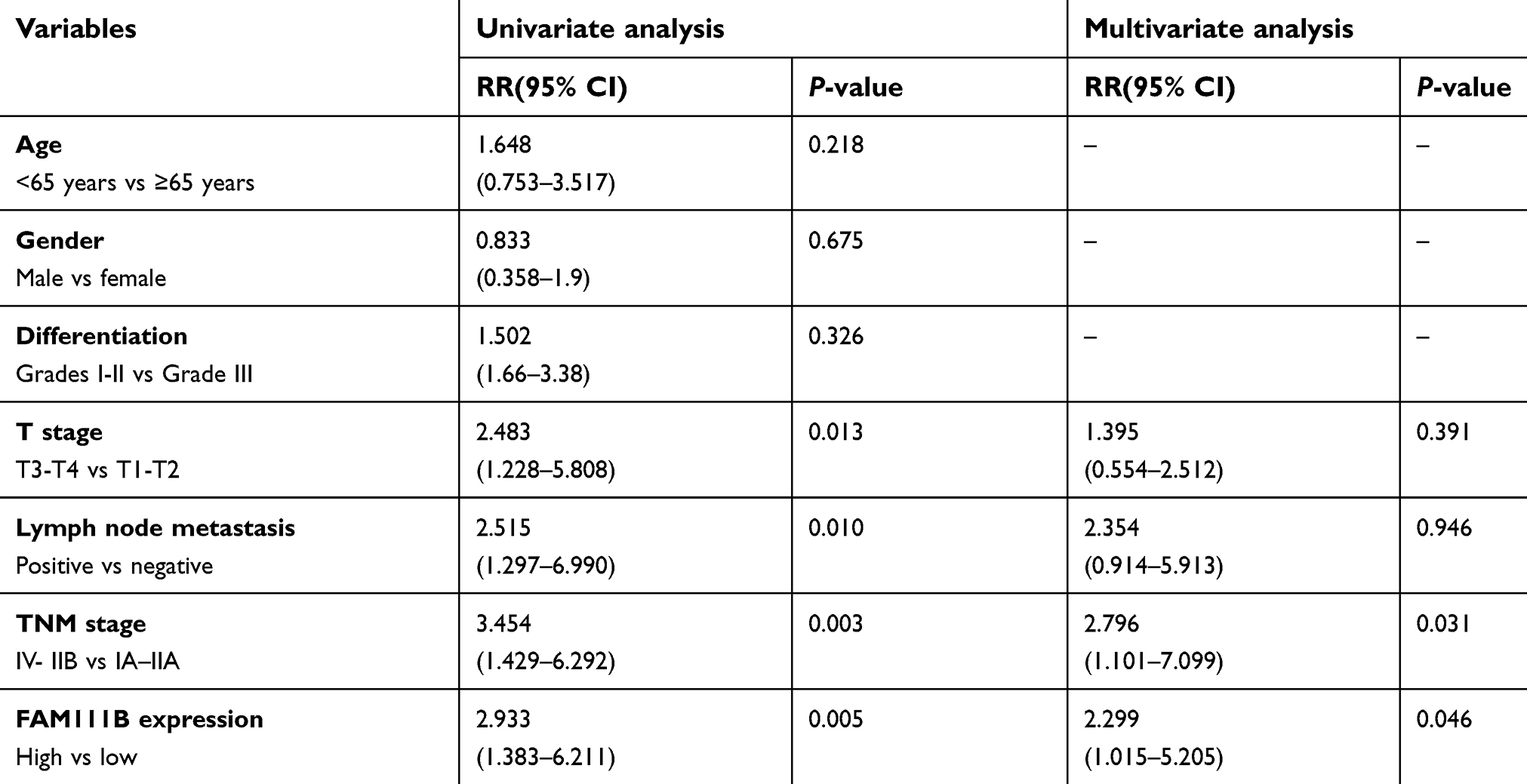 | Table 3 Univariate and multivariate analyses of prognostic factors for overall survival in 86 LUAD cases |
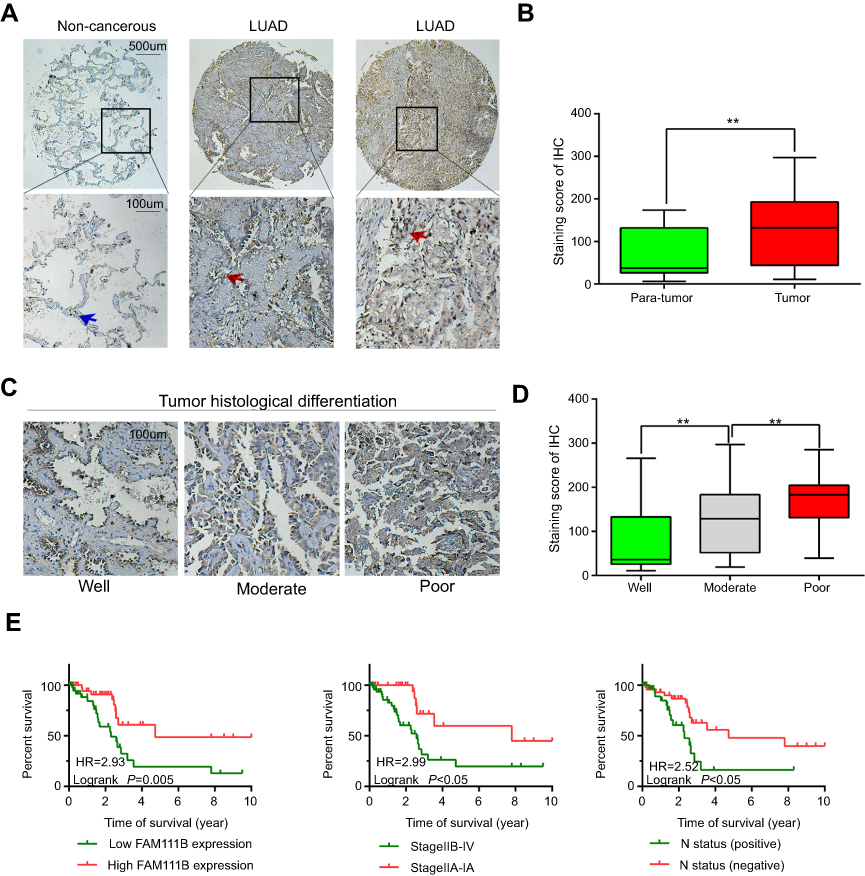 | Figure 2 The protein level of FAM111B was upregulated in LUAD tissues with a worse OS. Abbreviations: LUAD, lung adenocarcinoma; OS, overall survival. |
Knockdown of FAM111B inhibited proliferation, migration and invasion of LUAD cell lines and increased apoptosis, whereas overexpression of FAM111B increased proliferation in vitro
Figure 3A showed the expression of FAM111B in different LUAD cell lines. As a result, FAM111B were up-regulated in most LUAD cell lines when compared to normal human bronchial epithelial (HBE) cells. We choose A549 and PC9 cell lines to conduct our following experiments because A549 is a common LUAD cell line and PC9 showed the highest FAM111B levels in these cell lines.
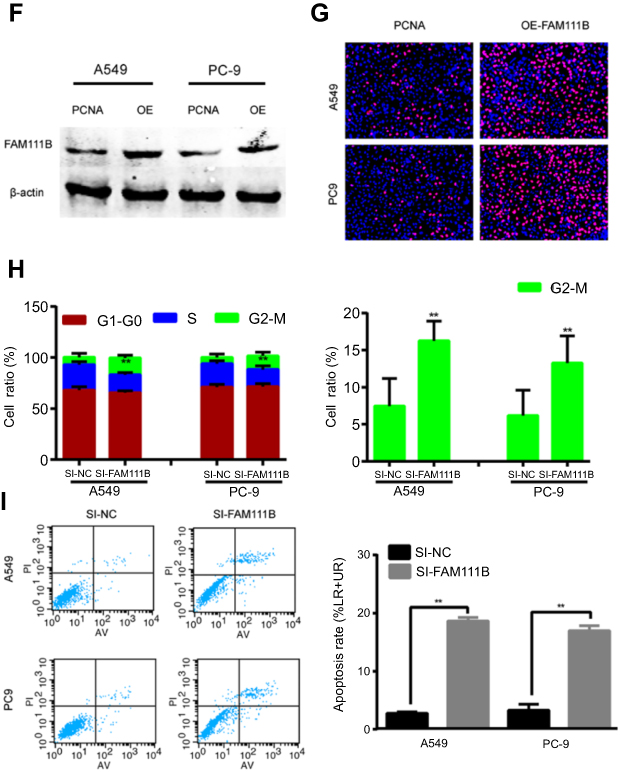
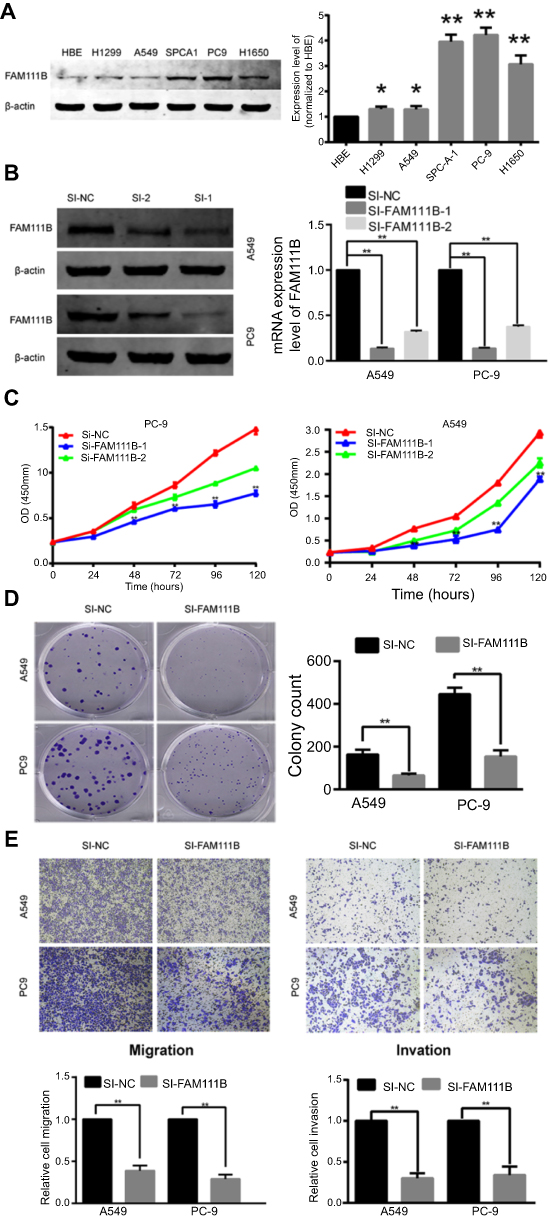 | Figure 3 In vitro experiments showed that FAM111B could improve proliferation and migration of LUAD cells and affect cell cycle and apoptosis. (A) FAM111B was up-regulated in all cancer cell lines (H1975, A549, SPC-A-1, PC9 and H1650) compared with HBE. (B) The two designed siRNAs presented satisfied inhibition efficiency in both A549 and PC9 cell lines. (C) Knocking down FAM111B decreased the proliferations in A549 and PC9 cells. (D) Cells transfected with si-FAM111B had fewer colonies than cells transfected with control siRNA. (E) The migrations and invasions of A549 and PC9 cells were inhibited by silencing FAM111B. (F) The protein expression level of FAM111B was greatly increased after using the overexpression plasmid. (G) The Edu signal of the FAM111B overexpression group was significantly enhanced compared to the control group. (H) Knockdown of FAM111B increased the percentage of A549 and PC9 cells in the G2 phase and decreased the cells in the S phase. (I) Silencing FAM111B increased cell apoptosis. *P<0.05, **P<0.01.Abbreviation: LUAD, lungadenocarcinoma; OE, overexpression. |
 | Figure 3 Continued. |
To study the biological function of FAM111B in vitro, two small interfacial RNAs targeting FAM111B (siRNA-1 and siRNA-2) were designed and transfected into A549 and PC9 cells to knock down the expression of FAM111B. As shown in Figure 3B, both siRNA constructs effectively reduced the expression of FAM111B mRNA and protein.
As shown in Figure 3C, CCK-8 analysis showed that knocking down FAM111B reduced the proliferation of both A549 and PC9 cells. In addition, we found that cells transfected with si-FAM111B showed fewer colonies when compared to cells transfected with control siRNA (Figure 3D).
Since SI-1 can achieve higher inhibition efficiency, we use SI-1 in the following experiments. Trans-well analysis showed that the migration and invasion ability of A549 and PC9 cells were significantly inhibited by silencing FAM111B (Figure 3E).
As is shown in Figure 3F, the expression of FAM111B was dramatically increased after the application of plasmid according to the result of western blot. And the result of Edu experiment showed that while FAM111B was overexpressed, the proliferation ability of cancer cells was largely increased (Figure 3G).
Next, flow cytometry was performed to assess the effect of FAM111B on cell cycle changes and apoptosis. As shown in Figure 3H, knockdown of FAM111B significantly increased the percentage of A549 and PC9 cells in the G2 phase and decreased the cells in the S phase when compared to the control group, and the level of apoptosis in the si-FAM111B treatment group increased when compared to the control group (Figure 3I).
Knocking-down of FAM111B suppressed tumor growth in vivo
To verify the effect of FAM111B on cell proliferation in vivo, we established a xenograft tumor model in nude mice using A549 cells and PC9 cells transfected with SI-FAM111B or SI-NC. All mice developed xenograft tumors at the subcutaneous injection sites, and tumor growth was inhibited after silencing the expression of FAM111B. The xenograft tumors were harvested 6 weeks after injection, and tumors of SI-FAM111B group were smaller in volume (Figure 4A and B). Xenograft tumors were immunohistochemically stained with anti-Ki-67 and anti-FAM111B antibodies. As shown in Figure 4C, the expression of FAM111B in the SI-FAM111B group was impaired and the proliferation maker Ki-67 was also reduced when compared to the control group.
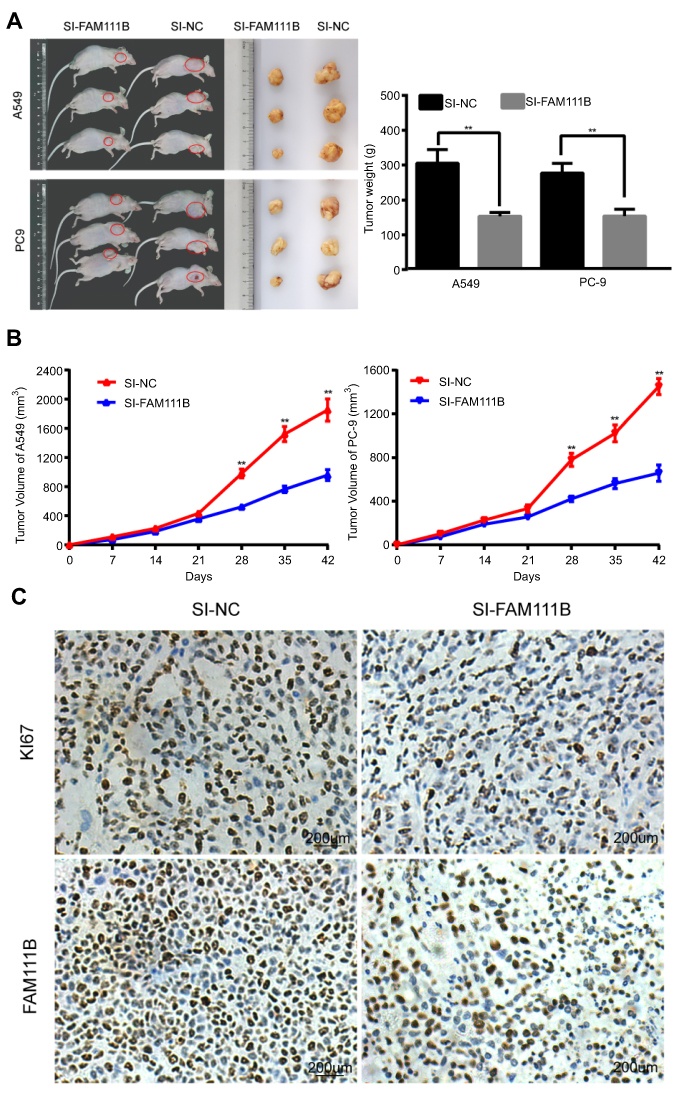 | Figure 4 FAM111B could promote tumor growth in vivo. (A) Tumors taken from Xenograft mice infected with SI-FAM111B were smaller when compared to those from control groups. (B) The tumor growth rate of the si-NC group was significantly faster than that of the si-FAM111B group. (C) Immunohistochemical results revealed that the expression of FAM111B in the SI-FAM111B group was impaired and the proliferation maker Ki-67 was reduced when compared to the control group. |
FAM111B promoted the development of LUAD by inhibiting the expression of BAG3 and BCL2
After KEGG bioinformatics analysis, we found that genes similar to FAM111B expression were enriched in the cell cycle and p53 signaling pathway, which is also confirmed by our involvement in the cell cycle of FAM111B involved in the signaling pathway of p53 (Figure 5A). Subsequently, we used correlation analysis to find that the expression of FAM111B is highly correlated with the mRNA expression of CCNB1 (r=0.7505) and CDC25C (r=0.7975, Figure 5B). Both of these genes play an important role in the G2M phase cell cycle regulated by the p53 signaling pathway.
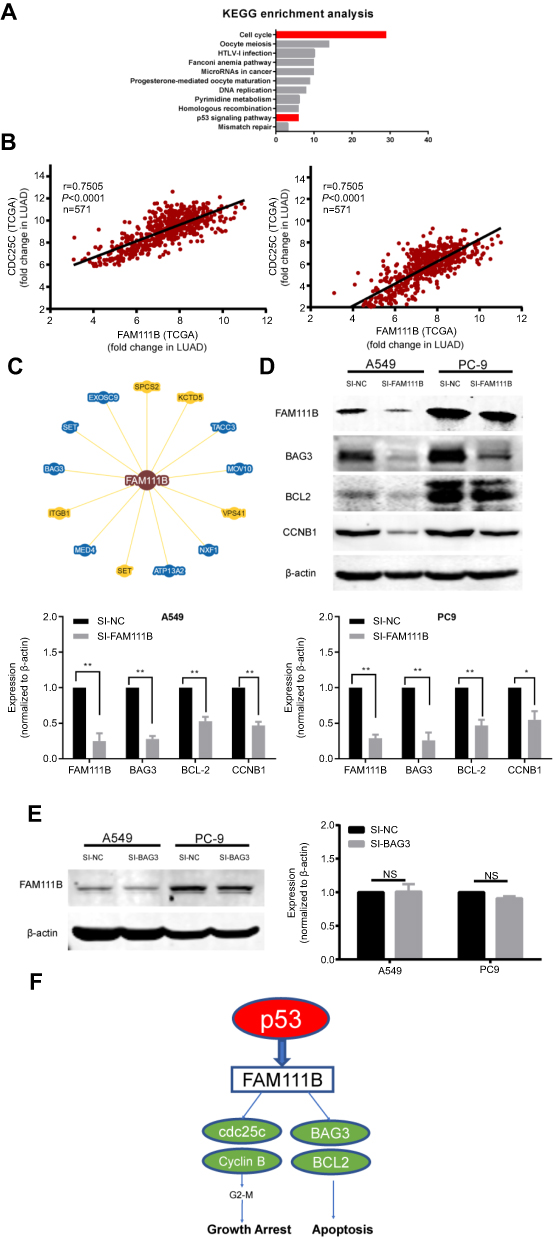 | Figure 5 FAM111B might affect the expression of p53 related protein BAG3, BCL2 and CCNB1. (A) The result of KEGG bioinformatics analysis showed that genes similar to FAM111B expression were enriched in the cell cycle and p53 signaling pathway. (B) The mRNA expression of G2M regulate proteins CCNB1 and CDC25C have a close correlation with that of FAM111B. (C) Bioinformatics analysis indicated that the expression of FAM111B correlated with BAG3. (D) The expression of G2M regulated protein CCNB1 and apoptosis-related proteins BAG3 and BCL-2 was significantly reduced when FAM111B was knocked down. (E) Knockdown of BAG3 had no significant effect on the expression of FAM111B. (F) FAM111B might participate in the regulation of tumor by p53 signaling pathway while promoting the malignancy of LUAD through cell cycle and apoptosis. Abbreviation: LUAD, lungadenocarcinoma. |
Our results also showed that knockdown of FAM111B could promote apoptosis of LUAD cells, which is also regulated by p53 signaling pathway. Therefore, we attempted to identify a pathway in which FAM111B might regulate the apoptotic process by analyzing the biogrid database (
Discussion
FAM111B is a member of the FAM111 family and was named for its similar sequences. In 2012 Akamatsu and his colleagues9 discovered three novel susceptibility loci of SNP’s in prostate cancer and the chromosome region of FAM111B contained one of the three susceptibility loci. Next, Mercier and his colleagues10 demonstrated by whole-exome sequencing that FAM111B played a role as causative mutations in HFP in which three FAM111B missense mutations were identified. Subsequently, most studies on FAM111B focused on its relationship with HFP11–13 and only a very few studies14,15 were focused on the impact of FAM111B on human tumors. Moreover, the function of FAM111B in the progression of human tumors has not yet been reported.
In this study, we found that FAM111B was over-expressed in LUAD tissues when compared to adjacent tissues by analyzing the TCGA database and clinical samples of LUAD patients in our institute. Furthermore, the immunohistochemistry data of patients in the tissue microarray showed that hyper-expression of FAM111B positively correlated with the clinical prognosis of patients with LUAD, and Cox regression analysis showed that the FAM111B expression was an independent risk factor of LUAD. These results suggested which we were the first time to present evidence that up-regulation of FAM111B was common among LUAD patients and positively correlates with more advanced clinic-pathological characteristics.
In our in vitro studies, we used siRNA to reduce the expression of FAM111B in different LUAD cell lines. Our findings showed that knocking-down of FAM111B, decreased cell proliferation and migration whereas increased apoptosis. Moreover, we showed that down-regulated FAM111B inhibited the growth of LUAD tumors in nude mice. Meantime, overexpression of FAM111B could increase cell proliferation in vitro. These findings suggested that FAM111B might play an important role in the development of LUAD.
As we know, HFP displays mottled pigmentation, telangiectasia, tendon contractures, and progressive pulmonary fibrosis, among which progressive pulmonary fibrosis is the main cause of death for patients.16 Cell apoptosis is tightly associated with the pulmonary fibrosis process17 and our results showed that that knockdown of FAM111B increased apoptosis of LUAD cells. Studies have shown that apoptosis and inflammation could also result in pulmonary fibrosis in mice.18 Therefore, we speculated that increase anti-apoptotic ability might be the way for FAM111B to promote LUAD. However, the mechanism of pulmonary fibrosis may be different from that in cancer.
Thus, it is difficult to say whether FAM111B knockdown-induced apoptosis may trigger or result in pulmonary fibrosis. In our study, we analyzed the online protein interaction database BioGRID (
BAG3 is a member of BAG family, which is having a tight relationship with BCL2.19,20 Overexpression of BAG3 can synergistically act with BCL-2 to induce anti-apoptotic effects and play an important role in the progression of tumors.21 The expression of BCL2 proved to inhibit the process of apoptosis and lots of studies have shown that BCL2 was an important oncogene in the development of lung cancer.22,23 In this study, western blot analysis showed that BAG3 and BCL2 were significantly decreased after silencing FAM111B. Whereas, qRT-PCR analysis showed that knockdown FAM111B did not significantly influence mRNA levels of BAG3 and BCL2. These results indicated that FAM111B regulated BAG3 and BCL2 post-transcriptionally, although the underlying mechanisms involved remained unclear.
The tumor suppressor p53 plays a key regulatory role in many aspects such as cell cycle, apoptosis and genomic stability.24,25 Previous chip-seq experiments performed by other researchers have found that p53 can directly target the transcriptional initiation region of FAM111B and reduce the expression of FAM111B.7,26 However, during the development of lung cancer, p53 may have a weaker inhibitory effect on FAM111B due to deletion or mutation, thereby making it highly expressed in lung cancer. Since in vitro experiments showed that FAM111B silencing can arrest the cell cycle at the G2/M phase and lead to more apoptosis, which is also the same effect of p53 signaling pathway on tumor cells. Thus, we hypothesized that FAM111B might participate in the regulation of tumor by p53 signaling pathway.
LUAD is the predominant pathological type of lung cancer, which has a poor prognosis because the etiology remains poorly understood.27 Investigating molecules that play important roles in the initiation and progression of LUAD not only helps to elucidate its pathogenic mechanisms involved but also might provide novel molecular markers and therapeutic targets for LUAD.27 In this study, we demonstrated that FAM111B might be an oncogene of LUAD and play an important role in the progression of LUAD.
In conclusion, our study showed that FAM111B expression was elevated in LUAD and correlated with worse clinical characteristics and poorer prognosis. FAM111B promoted proliferation, migration, and invasion, and inhibited apoptosis of LUAD cells in vitro and in vivo. Moreover, FAM111B might participate in the regulation of tumor by p53 signaling pathway while promoting the malignancy of LUAD through cell cycle and apoptosis.
Ethical approval
All applicable international, national, and institutional guidelines for the care and use of animals were followed.
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Acknowledgments
This study was funded by the National Natural Science Foundation of China (81672869), Jiangsu Provincial Special Program of Medical Science Funding (BL2012030), Jiangsu Provincial Science Foundation (BK20161596), Jiangsu Provincial Medical Outstanding Talent (Lin Xu) and Jiangsu Provincial Medical Youth Talent (Binhui Ren, QNRC2016657).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Forde PM, Chaft JE, Smith KN, et al. Neoadjuvant PD-1 blockade in resectable lung cancer. N Engl J Med. 2018;378(21):1976–1986. doi:10.1056/NEJMoa1716078
2. Song Z, Ge Y, Wang C, et al. Challenges and perspectives on the development of small-molecule EGFR inhibitors against T790M-mediated resistance in non-small-cell lung cancer. J Med Chem. 2016;59(14):6580–6594. doi:10.1021/acs.jmedchem.5b00840
3. Lee CK, Man J, Lord S, et al. Checkpoint inhibitors in metastatic EGFR-mutated non-small cell lung cancer-a meta-analysis. J Thorac Oncol. 2017;12(2):403–407. doi:10.1016/j.jtho.2016.10.007
4. Zhan C, Yan L, Wang L, et al. Identification of reference miRNAs in human tumors by TCGA miRNA-seq data. Biochem Biophys Res Commun. 2014;453(3):375–378. doi:10.1016/j.bbrc.2014.09.086
5. Barrett T, Troup DB, Wilhite SE, et al. NCBI GEO: archive for functional genomics data sets–10 years on. Nucleic Acids Res. 2011;39(Databaseissue):D1005–D1010. doi:10.1093/nar/gkq1184
6. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. doi:10.1126/scisignal.2004088
7. Xiong W, Wu X, Starnes S, et al. An analysis of the clinical and biologic significance of TP53 loss and the identification of potential novel transcriptional targets of TP53 in multiple myeloma. Blood. 2008;112(10):4235–4246. doi:10.1182/blood-2007-10-119123
8. Hein MY, Hubner NC, Poser I, et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell. 2015;163(3):712–723. doi:10.1016/j.cell.2015.09.053
9. Akamatsu S, Takata R, Haiman CA, et al. Common variants at 11q12, 10q26 and 3p11.2 are associated with prostate cancer susceptibility in Japanese. Nat Genet. 2012;44(4):426–429, S421. doi:10.1038/ng.1104
10. Mercier S, Kury S, Shaboodien G, et al. Mutations in FAM111B cause hereditary fibrosing poikiloderma with tendon contracture, myopathy, and pulmonary fibrosis. Am J Hum Genet. 2013;93(6):1100–1107. doi:10.1016/j.ajhg.2013.10.013
11. Kury S, Mercier S, Shaboodien G, et al. CUGC for hereditary fibrosing poikiloderma with tendon contractures, myopathy, and pulmonary fibrosis (POIKTMP). Eur J Hum Genet. 2016;24(5). doi:10.1038/ejhg.2015.205.
12. Mercier S, Kury S, Salort-Campana E, et al. Expanding the clinical spectrum of hereditary fibrosing poikiloderma with tendon contractures, myopathy and pulmonary fibrosis due to FAM111B mutations. Orphanet J Rare Dis. 2015;10:135. doi:10.1186/s13023-015-0352-4
13. Takeichi T, Nanda A, Yang HS, et al. Syndromic inherited poikiloderma due to a de novo mutation in FAM111B. Br J Dermatol. 2017;176(2):534–536. doi:10.1111/bjd.14845
14. Seo A, Walsh T, Lee MK, et al. FAM111B mutation is associated with inherited exocrine pancreatic dysfunction. Pancreas. 2016;45(6):858–862. doi:10.1097/MPA.0000000000000529
15. Fernandez-Retana J, Zamudio-Meza H, Rodriguez-Morales M, et al. Gene signature based on degradome-related genes can predict distal metastasis in cervical cancer patients. Tumour Biol. 2017;39(6). 1010428317711895. doi:10.1177/1010428317711895
16. Khumalo NP, Pillay K, Beighton P, et al. Poikiloderma, tendon contracture and pulmonary fibrosis: a new autosomal dominant syndrome? Br J Dermatol. 2006;155(5):1057–1061. doi:10.1111/j.1365-2133.2006.07473.x
17. Drakopanagiotakis F, Xifteri A, Polychronopoulos V, Bouros D. Apoptosis in lung injury and fibrosis. Eur Respir J. 2008;32(6):1631–1638. doi:10.1183/09031936.00176807
18. Hagimoto N, Kuwano K, Miyazaki H, et al. Induction of apoptosis and pulmonary fibrosis in mice in response to ligation of fas antigen. American Journal of Respiratory Cell and Molecular Biology. 1997;17(3):272–278. doi:10.1165/ajrcmb.17.3.2893
19. Doong H, Vrailas A, Kohn EC. What’s in the ‘BAG’? – A functional domain analysis of the BAG-family proteins. Cancer Lett. 2002;188(1–2):25–32.
20. Rosati A, Graziano V, De Laurenzi V, Pascale M, Turco MC. BAG3: a multifaceted protein that regulates major cell pathways. Cell Death Dis. 2011;2:e141. doi:10.1038/cddis.2011.82
21. Zhang Y, Wang JH, Lu Q, Wang YJ. Bag3 promotes resistance to apoptosis through Bcl-2 family members in non-small cell lung cancer. Oncol Rep. 2012;27(1):109–113. doi:10.3892/or.2011.1486
22. Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2(9):647–656. doi:10.1038/nrc883
23. Sun PL, Sasano H, Gao H. Bcl-2 family in non-small cell lung cancer: its prognostic and therapeutic implications. Pathol Int. 2017;67(3):121–130. doi:10.1111/pin.12507
24. Tang Y, Wang J, Lian Y, et al. Linking long non-coding RNAs and SWI/SNF complexes to chromatin remodeling in cancer. Molecular Cancer. 2017;16(1):42. doi:10.1186/s12943-017-0612-0
25. Shakya R, Tarulli GA, Sheng L, et al. Mutant p53 upregulates alpha-1 antitrypsin expression and promotes invasion in lung cancer. Oncogene. 2017;36(31):4469. doi:10.1038/onc.2017.66
26. Hünten S. Integrated Analysis of p53 Targets [Dissertation, LMU München]2016.
27. Hirsch FR, Scagliotti GV, Mulshine JL, et al. Lung cancer: current therapies and new targeted treatments. Lancet. 2017;389(10066):299–311. doi:10.1016/S0140-6736(16)30958-8
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.