")
Back to Journals » Drug Design, Development and Therapy » Volume 11
Fabrication, characterization and in vitro evaluation of silibinin nanoparticles: an attempt to enhance its oral bioavailability
Authors Sahibzada MUK , Sadiq A, Khan S , Faidah HS, Naseemullah , Khurram M, Amin MU, Haseeb A
Received 4 February 2017
Accepted for publication 13 April 2017
Published 15 May 2017 Volume 2017:11 Pages 1453—1464
DOI https://doi.org/10.2147/DDDT.S133806
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Qiongyu Guo
Muhammad Umar Khayam Sahibzada,1,2 Abdul Sadiq,1 Shahzeb Khan,1 Hani S Faidah,3 Naseemullah,1 Muhammad Khurram,4 Muhammad Usman Amin,5 Abdul Haseeb6
1Department of Pharmacy, University of Malakand, Chakdara, Lower Dir, 2Department of Pharmacy, Sarhad University of Science and Information Technology, Peshawar, Khyber Pakhtunkhwa, Pakistan; 3Department of Microbiology, Faculty of Medicine, Umm Al Qura University, Makkah, Saudi Arabia; 4Department of Pharmacy, Shaheed Benazir Bhutto University, Sheringal, Upper Dir, 5Department of Pharmacology, KMU Institute of Medical Sciences, Kohat, Khyber Pakhtunkhwa, Pakistan; 6Department of Clinical Pharmacy, College of Pharmacy, Umm Al Qura University, Makkah, Saudi Arabia
Background: Silibinin has gained in importance in the past few decades as a hepatoprotector and is used widely as oral therapy for toxic liver damage, liver cirrhosis, and chronic inflammatory liver diseases, as well as for the treatment of different types of cancers. Unfortunately, it has low aqueous solubility and inadequate dissolution, which results in low oral bioavailability.
Materials and methods: In this study, nanoparticles (NPs) of silibinin, which is a hydrophobic drug, were manufactured using two cost-effective methods. Antisolvent precipitation with a syringe pump (APSP) and evaporative precipitation of nanosuspension (EPN) were used. The prepared NPs were characterized using different analytical techniques such as scanning electron microscopy (SEM), fourier transform infrared spectroscopy (FTIR), differential scanning calorimetry (DSC), and X-ray powder diffractometry (XRD) and were sifted for their bioavailability through in vitro dissolution and solubility studies. Moreover, the prepared NPs were evaluated for antimicrobial activity against a battery of bacteria and yeast.
Results: DSC and XRD studies indicated that the prepared NPs were amorphous in nature, with more solubility and dissolution compared to the crystalline form of this drug. NPs prepared through the EPN method had better results than those prepared using the APSP method. Antimicrobial activities of the NPs were improved compared to the unprocessed drugs, while having comparable activities to standard antimicrobial drugs.
Conclusion: Results indicate that the NPs have significantly increased solubility, dissolution rate, and antimicrobial activities due to the conversion of crystalline structure into amorphous form.
Keywords: silibinin, nanoparticles, antimicrobial activity, solubility, dissolution
Background
One of the most convenient ways of administering drugs is the oral route, but the low aqueous solubility of a number of drugs results in very low oral bioavailability. Formulation scientists who deal with drug development and discovery face major hurdles in formulation design of drugs with low aqueous solubility, thus resulting in a poor dissolution rate. Among the compounds developed in the pharmaceutical industry, 40% either have reduced water solubility or are completely insoluble in the aqueous phase.1–3 For such poorly soluble drugs, major difficulties occur when they are exposed to different formulation development phases, as they are poorly wetted by and dissolved in the solvent. Due to these problems, efforts are undertaken by formulation scientists to improve the solubility and dissolution rates of such drugs in order to achieve maximum drug absorption in the gastrointestinal tract (GIT).4,5
Nanostructured materials form one of the most exciting fields in science today. These materials have great scope in the field of medicines, as nanosized medicines have better pharmacokinetic and pharmacodynamic profiles. They have valuable characteristics that make them of great value in comparison to the conventional macrosized drug molecules. Their smaller size gives them greater surface area and higher surface-to-volume ratios.6–9 According to the Noyes–Whitney equation, particle size reduction to the nano range increases the surface area and surface free energy, which may improve the saturation solubility and dissolution rate of poorly water-soluble drugs.4,5
Nanoparticles (NPs) can be synthesized using top-down and bottom-up approaches using different production methods. If the drug is soluble in an organic solvent, precipitation would be a possible method.10 Fessi et al first developed and got a patent for preparation of nanosuspension (solvent displacement method).11 The major advantages of nanosuspensions are the ease of preparation as well as a fast and straightforward preparation method.11 Antisolvent precipitation with a syringe pump (APSP) and evaporative precipitation of nanosuspension (EPN) are among the ways that address the problems of solubility, dissolution rate, and bioavailability through precipitation.12
Silibinin was first extracted from silymarin, a unique flavonolignan complex also containing silydianine and silychristine. Silymarin is derived from Silybum marianum, commonly known as milk thistle, belonging to the Asteraceae family. Silibinin is also called “silybin” and represents about 50%–60% of the silymarin extract. Silibinin has been widely investigated for its use as a hepatoprotector13,14 and as oral therapy for toxic liver damage,14 liver cirrhosis, and chronic inflammatory liver diseases.13–15 Silibinin has been reported to have other effects, such as antioxidant15 and antimicrobial effects,16 and it can be used in the treatment of different types of cancers.13–15 In addition, silibinin has been reported to effectively treat lung cancer.17 Self-nanoemulsifying drug delivery systems (SNDDS) for silymarin have recently been developed to improve its therapeutic performance.18 Liu et al19 have found silibinin NPs to have strong antiviral effect against hepatitis C virus (HCV) infections. The role of silibinin as a potential candidate to treat breast cancer has been studied by Sun et al.20 Phospholipid carriers have become an attractive tool to address the issue of poorly water-soluble active pharmaceutical ingredients.21 Phytosomes have emerged as a new technology to incorporate phyto constituents into phospholipid complexes, with subsequent improvement in bioavailability and increased absorption of the poorly soluble compounds.22 A substantial increase in the bioavailability and absorption of silibinin and silymarin has been witnessed using phospholipid-based carriers and microspheres.23,24 El-Far et al25 have recently reported that silymarin NPs play an important role in the reduction of blood glucose level using streptozotocin-induced diabetic rats.
Silibinin belongs to the group of Biopharmaceutics Classification System (BCS) class II drugs, which have high permeability and poor solubility (in vitro and in vivo evaluation of silybin nanosuspensions for oral and intravenous delivery). The drugs categorized as BCS II have been proven to be very potential drug candidates; however, their bioavailability is limited by their poor dissolution and solubility.26
Orally administered silibinin is absorbed rapidly, with the time at which maximum concentration of drug is present in the blood (tmax) of 2–4 h and a half-life of 6 h. Following administration, only 20%–50% of silibinin is absorbed from the GIT and is metabolized in the liver; it has low absorption, resulting in poor bioavailability.27–30 Given this profile, silibinin NPs were prepared through the APSP and EPN methods in order to improve the bioavailability of this hydrophobic drug. The prepared NPs were investigated for various parameters, including dissolution, solubility, and antimicrobial properties.
Materials and methods
Materials
All the chemicals used in this study were of analytical grade and had the highest purity. Silibinin (purity <98%) was acquired from Sigma-Aldrich Co, St Louis, MO, USA. Ethanol, n-hexane, potassium phosphate monobasic, sodium hydroxide, HCl (36.5%, w/w), hydroxypropyl methylcellulose (HPMC), and polyvinyl pryrrolidone (PVP) were also from Sigma Aldrich, while the deionized double-distilled water used in this work was obtained using the Milli-Q® system (Merck Millipore, Darmstadt, Germany).
Preparation of silibinin NPs
Preparation of silibinin NPs through the APSP method was carried out using a previously reported method,12 with a slight modification. Briefly, a saturated solution of unprocessed silibinin was prepared in ethanol. A syringe was filled with the prepared drug solution and was quickly injected at a fixed flow rate (2 mL/min) into deionized water (antisolvent) of definite volume under mechanical stirring (3,000 rpm). Different volumes of deionized water were mixed with the same volume of saturated drug solution (1:10, 1:15, and 1:20, v/v), which was evaporated quickly in a vacuum using a rotary evaporator.
For the EPN method, a solution of the unprocessed silibinin was prepared in ethanol followed by quick addition of hexane, which served as an antisolvent, to the prepared saturated drug solution under mechanical stirring (3,000 rpm). Different solvent: antisolvent ratios (1:10, 1:15, and 1:20, v/v) were used. Nanosized drug particles from the nanosuspensions were obtained by quick evaporation of the solvent and antisolvent in a vacuum for both methods (APSP and EPN).
Characterization
Particle size and the associated polydispersity index (PI) of silibinin NPs were characterized by dynamic light scattering (Zetasizer® NanoS; Malvern Instruments, Malvern, UK). All the measurements were carried out in triplicate.
Confirmation of the structure of the unprocessed silibinin and the NPs prepared by the APSP and EPN methods was done by Fourier transform infrared spectroscopy (FTIR; IR Prestige-21 FTIR spectrophotometer; Shimadzu, Tokyo, Japan). FTIR spectra were acquired in the range of 200–4,400 cm−1.
The surface morphology of the prepared NPs was confirmed from the electron photomicrographs obtained by scanning electron microscopy (SEM; Jeol JSM-5910; JEOL Ltd., Tokyo, Japan). The instrument was operated at 20 kV, using a standard procedure for sample preparation.
The X-ray powder diffractometry (XRD) patterns of unprocessed silibinin and the NPs prepared by the two methods were recorded by a PANalytical X-ray diffractometer (X’Pert Pro, USA). The operating voltage was 40 kV, the operating current was 30 mA, the start angle 2θ was 5°, and the finishing angle was 60°.
Differential scanning calorimetry (DSC) studies of unprocessed silibinin and NPs were carried out using Mettler Tolado 822e (Greifensee, Switzerland). The instrument was equipped with a compatible computer program, and the procedure was carried out as per manufacturer specifications at a rate of 10°C/min under even nitrogen flow. Any thermal event that happened in the pans was recorded.
In vitro analysis
Solubility determination
An excess amount of unprocessed silibinin and NPs prepared by APSP and EPN were placed in 25 mL glass volumetric flasks, followed by addition of distilled water to the mark. The flasks were sealed to prevent any loss of solvent and were kept in an orbital shaker (IKA HS501 orbital shaker; IKA®-Werke GmbH, Staufen, Germany) at room temperature for 24 h at 100 rpm. After mixing, samples were kept undisturbed on a flat surface for ~72 h. Thereafter, 5 mL of the supernatant solution from each flask was withdrawn and filtered using Whatman® filter paper number 1. The filtered portion was then analyzed to determine solubility at 286 nm using ultraviolet (UV)–visible spectrophotometer (Pharmaspace 1700 UV–Visible spectrophotometer; Shimadzu). All the samples were analyzed in triplicate.
Dissolution studies
The in vitro dissolution studies were carried out using the US Pharmacopeia (USP) method II, ie, the paddle method. Three different dissolution media, namely, distilled water, 0.1 M HCl, and phosphate buffer solution (pH 6.8), were used in the dissolution studies. The volume of medium used was 900 mL at 37.0°C±0.5°C, at 50 rpm paddle rotation. Subsequently, 70 mg of unprocessed silibinin and the two types of NPs were subjected to the dissolution studies. Thus, 5 mL of the dissolution media was drawn at predetermined intervals (10, 30, 60, and 90 min) and filtered through Whatman filter paper No 1. The same volume of dissolution medium was replaced each time with fresh dissolution medium after withdrawal. Filtered samples were suitably diluted and observed spectrophotometrically using a double-beam spectrophotometer (8453 UV–Visible spectrophotometer; Agilent Technologies, Santa Clara, CA, USA) at a maximum wavelength of 286 nm. Each test was conducted in triplicate.
Dissolution parameters
Dissolution efficiency (DE)
This parameter determines the area under the dissolution curve at time t. The trapezoidal rule was used to determine this value. DE represents the percentage of the area of the rectangle described by the 100% dissolution value at the same time:31
|
|
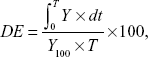
where Y = % drug dissolved at time t.
Mean dissolution time (MDT)
A measurement of the rate of the dissolution process involves estimation of the MDT. It can be calculated from the dissolution data using the following equation:31
|
|
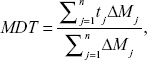
where j = sample number, n = number of dissolution sampling times, tj = time at midpoint between tj and tj–1, ΔMj = additional amount of drug dissolved between tj and tj–1.31
Dissimilarity factor f1 and similarity factor f2
Moore and Flanner proposed a model-independent approach to calculate the dissimilarity factor f1 and similarity factor f2.31 The two factors were used to calculate or to compare the dissolution profiles of various samples. They can be represented by the following equations:
|
|
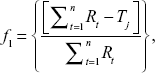
|
|
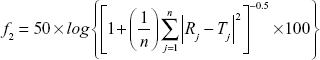
where Rj and Tj = % of reference and test drugs dissolved at each time point j; n = number of withdrawal points.31,32
Antimicrobial assays
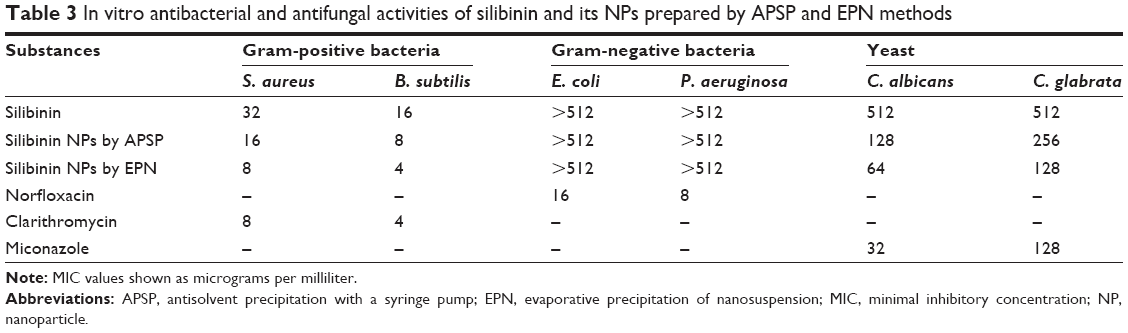
Silibinin and its NPs prepared by the APSP and EPN methods were tested for their in vitro antimicrobial potentials against two Gram-positive bacteria (Staphylococcus aureus, Bacillus subtilis), two Gram-negative bacteria (Escherichia coli, Pseudomonas aeruginosa), and two yeasts (Candida albicans, C. glabrata). The 96-well microtest plate method was used in this study in accordance with the National Committee for Clinical Laboratory Standards guidelines.33,34 The lowest concentration of test NPs that totally inhibited the growth of bacteria and yeast was considered as the minimal inhibitory concentration (MIC, micrograms per milliliter) of the respective tested NPs. Positive controls included norfloxacin (for Gram-negative bacteria), clarithromycin (for Gram-positive bacteria), and miconazole (for yeast), while phosphate-buffered saline was used as a vehicle for the NP solutions from the respective methods.
Results
Characterization of the NPs prepared by APSP and EPN methods
Particle size measurement
The mean particle sizes of silibinin NPs were found to be in the range of 35±5 to 180±7 nm. The mean particle size of NPs prepared by APSP method (Figure 1A) was 104.52 nm, while those prepared by the EPN method (Figure 1B) had a size of 60.33 nm. The average values are as 104.52±3.2 nm and 60.33±2.5 nm. Additionally, the PI values found 0.3±0.02 and 0.2±0.01 for the NPs produced by APSP and EPN methods respectively.
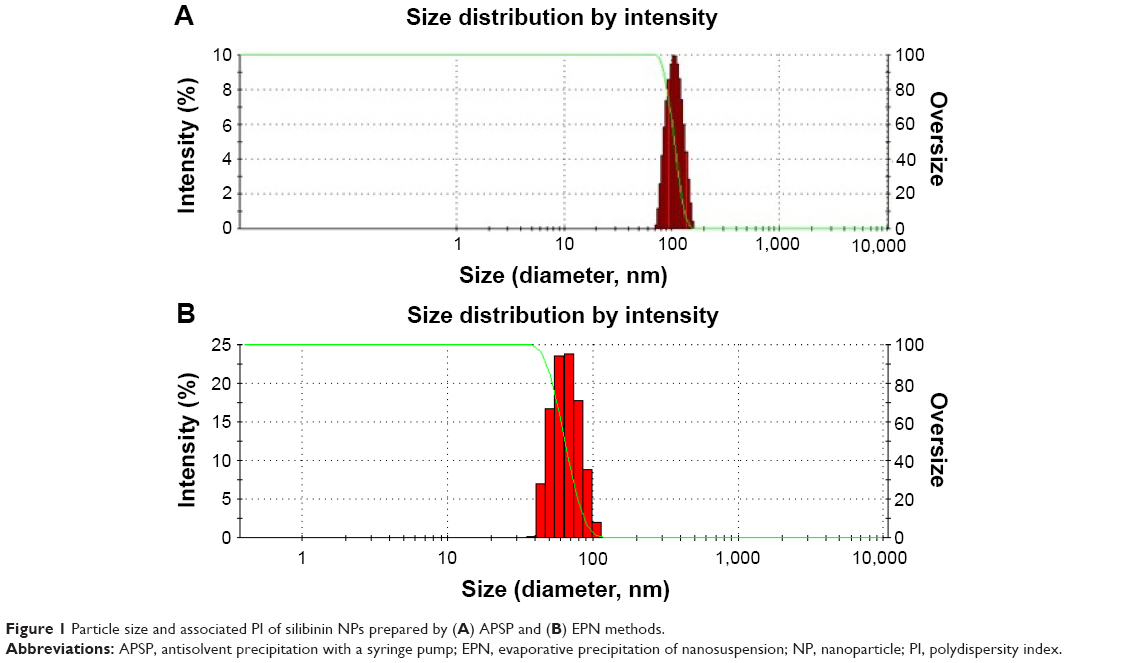 | Figure 1 Particle size and associated PI of silibinin NPs prepared by (A) APSP and (B) EPN methods. |
FTIR studies
FTIR images of unprocessed silibinin (Figure 2A) and NPs prepared by APSP (Figure 2B) and EPN (Figure 2C) show characteristic peaks with different intensities, as different functional groups vibrate at different frequencies.
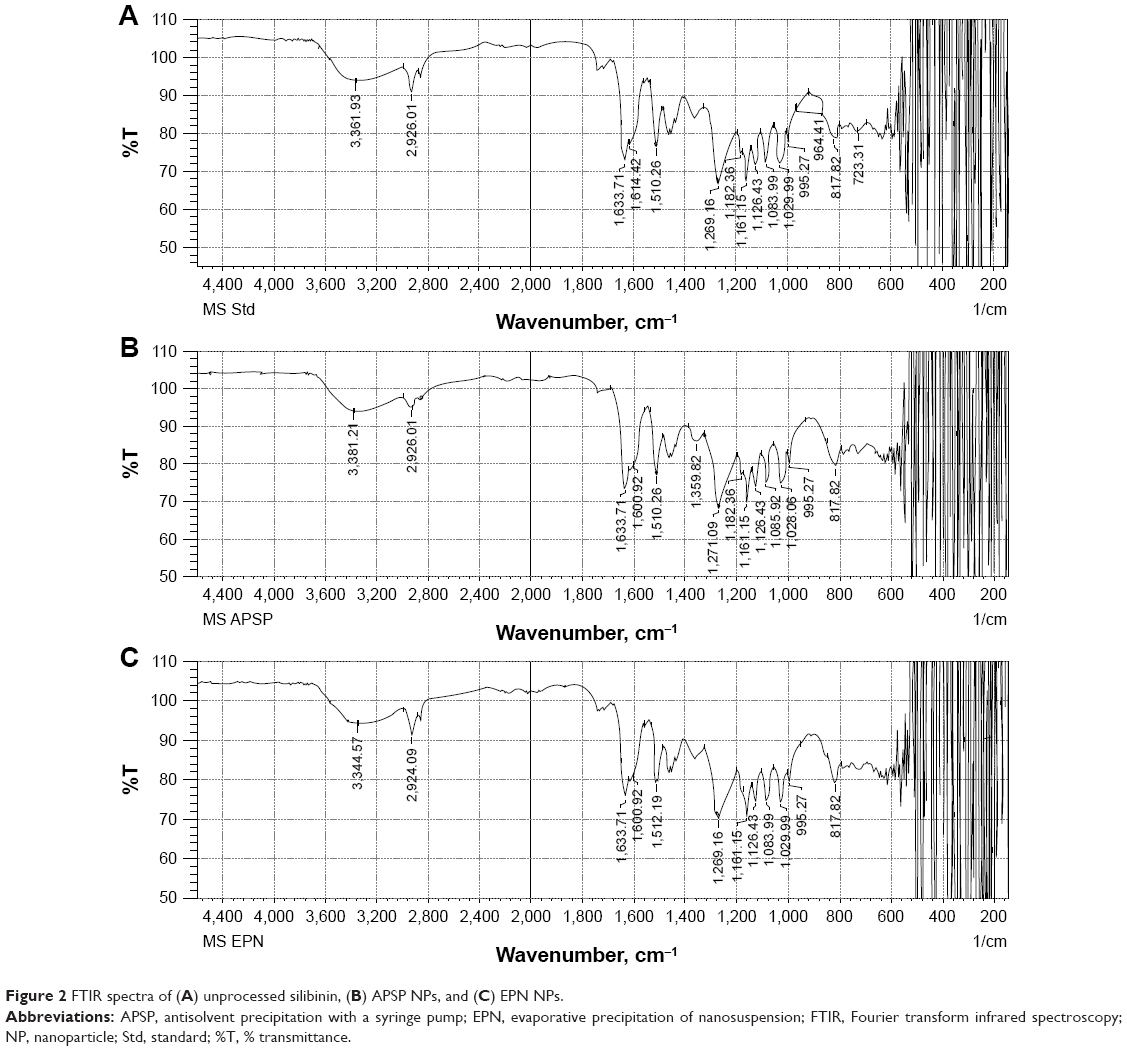 | Figure 2 FTIR spectra of (A) unprocessed silibinin, (B) APSP NPs, and (C) EPN NPs. |
Scanning electron microscopy
NPs prepared by the APSP method (Figure 3) and EPN method (Figure 4) were subjected to SEM studies. Images were obtained at different magnifications. The white dots in the images represent silibinin NPs prepared by APSP and EPN methods, respectively. The size of NPs was reduced to a greater extent. As shown in Figures 3 and 4, NPs ~60.33 nm in size were obtained.
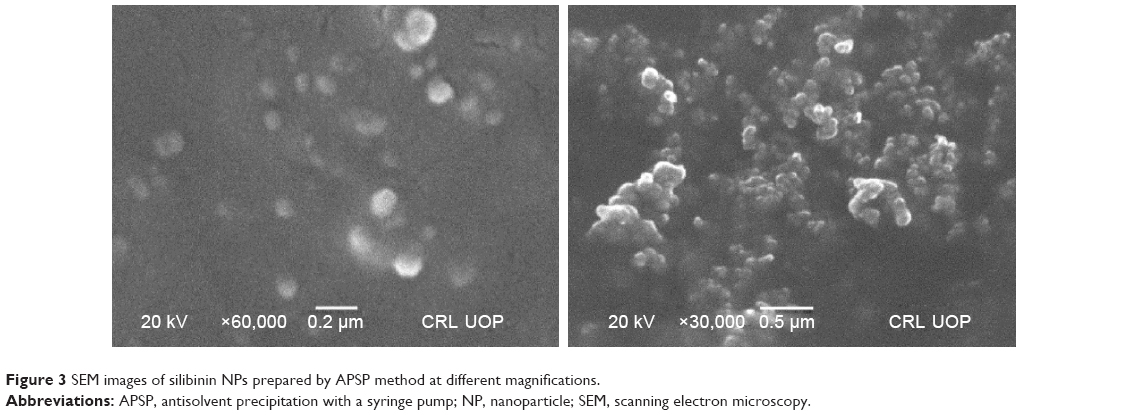 | Figure 3 SEM images of silibinin NPs prepared by APSP method at different magnifications. |
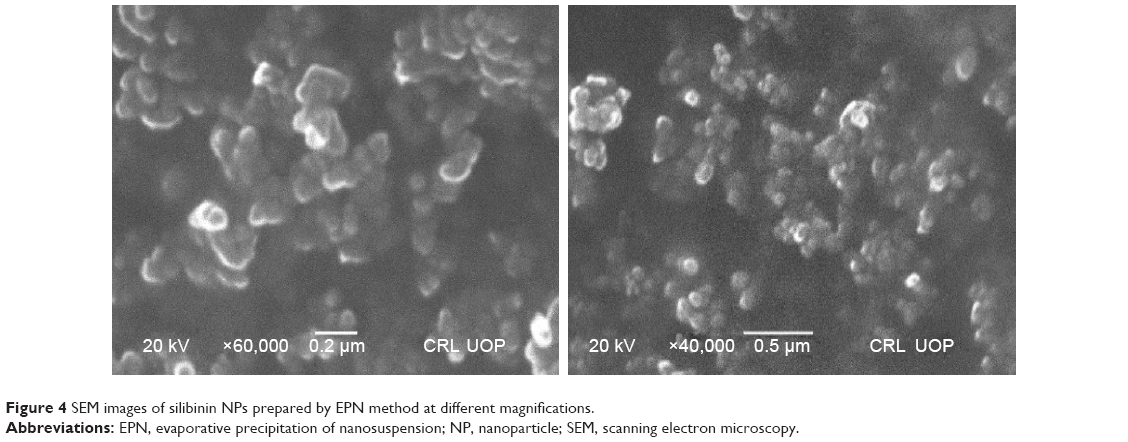 | Figure 4 SEM images of silibinin NPs prepared by EPN method at different magnifications. |
XRD studies
From the XRD studies, it is clear that unprocessed silibinin (Figure 5A) shows diffraction peaks at 2θ of 11.75, 23.8°, 26.25°, 26.85°, and 28.29°. Some of the peaks are sharp and intense, which shows that unprocessed silibinin is crystalline in nature (Figure 5A). The NPs prepared by APSP (Figure 5B) and EPN (Figure 5C) show diffraction peaks in the same range with less intensity, which represents a reduction of the crystalline nature to the amorphous form.
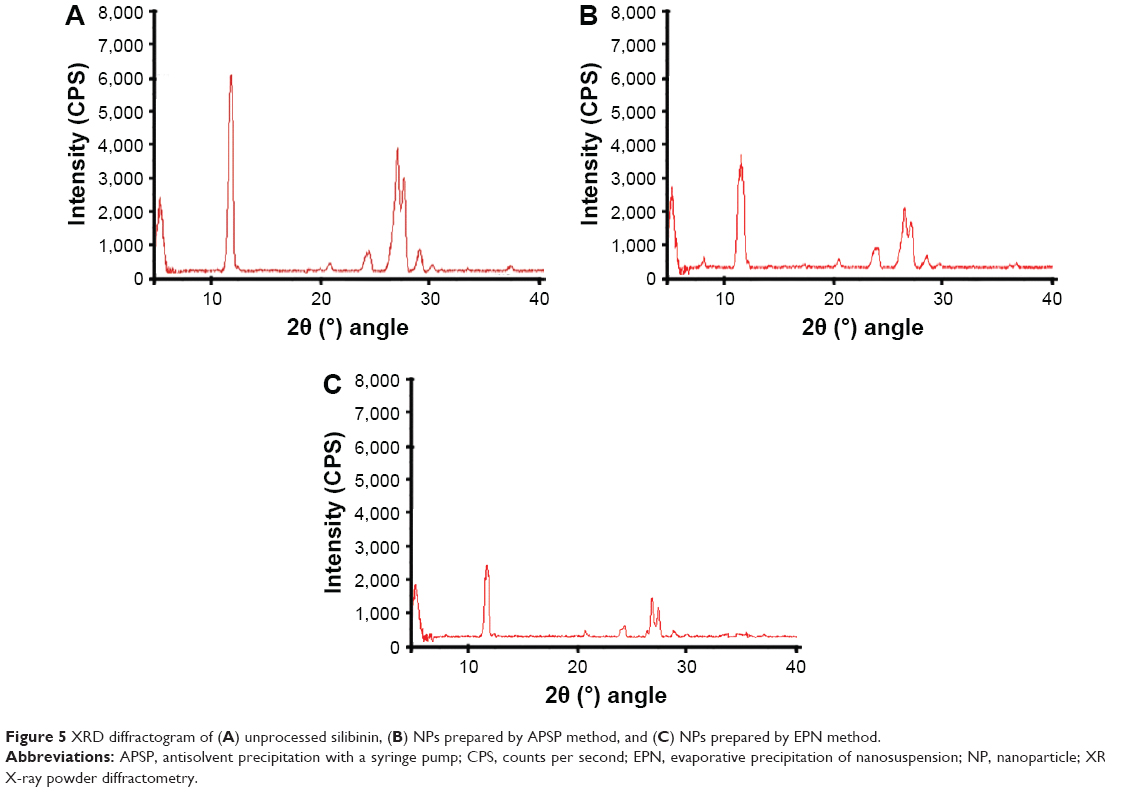 | Figure 5 XRD diffractogram of (A) unprocessed silibinin, (B) NPs prepared by APSP method, and (C) NPs prepared by EPN method. |
DSC analysis
DSC studies of unprocessed silibinin and NPs prepared by the APSP and EPN methods (Figure 6) were carried out to evaluate the thermal effects at melting temperature (Tm). The particle size of the material or the surface area of the material has profound effects on its melting. Little broadening and decrease in the melting point peaks of the processed silibinin was observed, which is due to the low packing density of the NPs.26
 | Figure 6 DSC analysis of unprocessed silibinin and NPs prepared by APSP and EPN methods. |
In vitro characterization
Solubility studies
Table 1 shows the solubility of unprocessed silibinin and the NPs of silibinin prepared by the APSP and EPN methods in distilled water. The acquired data clearly show that solubility of silibinin increases when it is converted to NPs (Figure 7).
 | Table 1 Aqueous solubility of pure silibinin and prepared nanoformulations of silibinin |
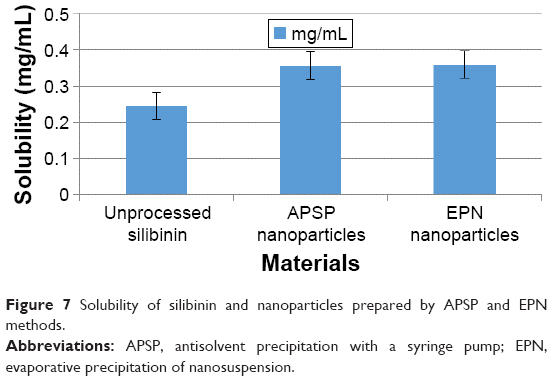 | Figure 7 Solubility of silibinin and nanoparticles prepared by APSP and EPN methods. |
Dissolution studies
Dissolution studies of unprocessed silibinin and the two prepared NPs (Figure 8A–C) were conducted in distilled water, 0.1 M HCl, and phosphate buffer, pH 6.8, over a time period of 90 min at different intervals: 10, 30, 60, and 90. The dissolution analysis clearly shows that dissolution of unprocessed silibinin was very low in distilled water, 0.1 M HCl, and phosphate buffer of pH 6.8. The NPs prepared by the two methods had better dissolution than unprocessed silibinin over the same time period (90 min). The improved dissolution of the NPs can be seen in Figure 8B and C, demonstrating >70% dissolution within 30 min in all three dissolution media, thus clearly showing improvement in the dissolution rate. Among the two prepared NPs, by comparison, the NPs prepared by the EPN method had a better dissolution rate than those prepared by the APSP method.
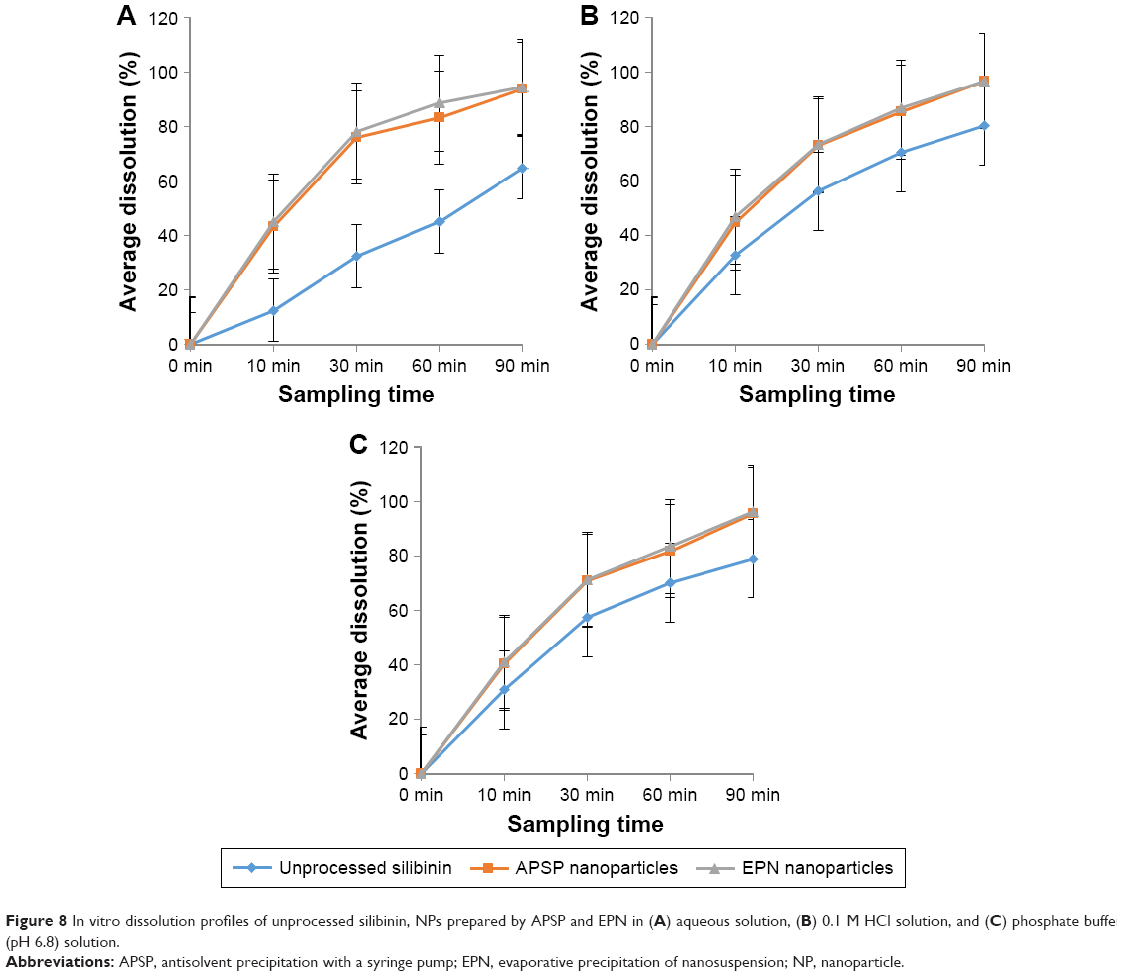 | Figure 8 In vitro dissolution profiles of unprocessed silibinin, NPs prepared by APSP and EPN in (A) aqueous solution, (B) 0.1 M HCl solution, and (C) phosphate buffer (pH 6.8) solution. |
Data on dissimilarity and similarity factors (Table 2) indicates that the release profile of silibinin is quite different for the NPs prepared by the two different methods. Among the two methods, NPs prepared by the EPN method show more dissimilarity than those prepared by APSP. Therefore, we can conclude that EPN is the best method to improve the dissolution rate of silibinin.
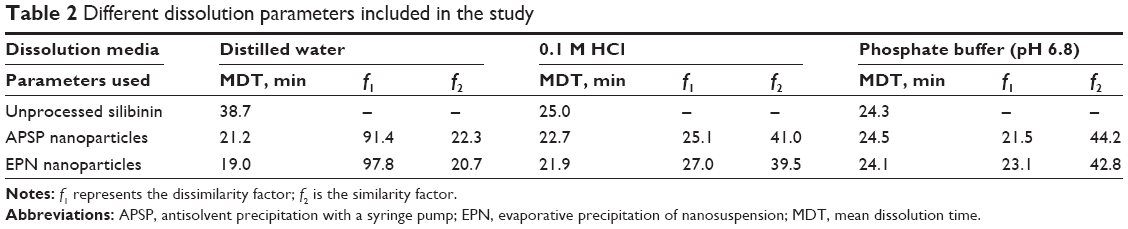 | Table 2 Different dissolution parameters included in the study |
Antimicrobial assay
The results stated in Table 3 indicate that silibinin NPs prepared by the APSP and EPN methods exhibit greater antibacterial and antifungal potential than silibinin (unprocessed) and the controls including norfloxacin, clarithromycin, and miconazole. The antibacterial activity of silibinin NPs prepared by the EPN method was increased greatly against Gram-positive bacteria and was similar to that of the control drug clarithromycin. The APSP-prepared NPs showed an increase in activity against Gram-positive bacteria, but this was lower than that of EPN-prepared NPs. However, the results indicate a significant increase in antibacterial activity of silibinin NPs compared to that of unprocessed silibinin. Silibinin NPs prepared by the EPN method were found to have equivalent activity to that of miconazole.
 | Table 3 In vitro antibacterial and antifungal activities of silibinin and its NPs prepared by APSP and EPN methods |
Discussion
Particle size and the associated PI of silibinin NPs show that both NPs have nanosizes and narrow size distributions.35 A reduction of size results in greater surface area and surface free energy, which helps to improve solubility and dissolution rate, which in turn enhances bioavailability.
FTIR images of unprocessed silibinin and the NPs prepared by APSP and EPN show characteristic peaks at 2,962.01 cm−1 (O–H stretching), 1,126.43 cm−1 (C=C stretching), 1,161.15 cm−1, 1,182.36 cm−1 (aldehyde C=O stretching), 1,510.26 cm−1, 1,614.42 cm−1, 1,633.71 cm−1 (skeletal vibration due to aromatic C=C ring stretching and ketone C=O stretching), 1,269.16 cm−1 (C–O–C stretching), 995.27 cm−1 (O–H out-of-plane bending), 1,029.99116 cm–1, 1,510.26 cm−1 (aromatic alkene stretching), and 3,361.93 cm−1 (–OH, =C–H).36 The results indicate that the prepared NPs and the unprocessed sample have the same chemical structure. This analysis confirmed that there is no formation of a new complex among the formulation components and that silibinin’s structure is maintained.
XRD analysis confirmed the change of crystalline nature to amorphous form, which helps to increase the solubility and dissolution rate, as well as ultimately improving the bioavailability.35 Less-crystalline and amorphous materials have greater free energy compared to the corresponding crystalline forms. Therefore, less-crystalline or amorphous forms of drugs can get solubilized more easily and show enhanced dissolution rates compared to their respective crystalline forms.37–40 Thus, modification in the crystalline nature through nanosizing may be an ideal approach for enhancement of the solubility and the dissolution rates of drug molecules, which will further improve their bioavailability.40
A sharp peak at the melting temperature (Tm) was recorded for the crystalline structure, representing an endothermic process due to the breakage of the crystalline lattice. In the case of amorphous or less-crystalline structures, the peaks at the melting temperature (Tm) were less intense. The enthalpy of fusion (ΔH) value, which is directly proportional to the crystallinity of a sample, is low for the amorphous sample compared to that for the crystalline sample. A sharp endothermic peak was observed at the melting temperature (168°C) in the case of unprocessed silibinin, and although endothermic peaks for the prepared NPs were observed at almost the same temperature, the peak intensities were lower compared to that of the unprocessed drug. Particle size reduction due to conversion of the unprocessed drug to the NPs caused a reduction in crystallinity and thus attainment of the amorphous form, which has a lower enthalpy value than the parent unprocessed drug.11,32,41,42
Data from solubility studies (Table 1) indicate that enhancement of the solubility may be due to 1) the conversion of silibinin to the amorphous form, which is more soluble than its crystalline form, and 2) reduction in the particle size to the nano range, which increases the interfacial surface area and thus increases the wettability and surface free energy. All these factors collectively increase the solubility and dissolution rate of poorly water-soluble drugs.4,5
Improvement in the dissolution of silibinin NPs can be attributed to certain factors, such as increased surface area, the conversion of drug to the amorphous form, good dispersibility, better wettability, and decrease in agglomeration and aggregation between the hydrophobic drug particles. Additionally, the dissolution study showed that NPs of silibinin maintained the particle size and no agglomeration occurred during the process. Increase in dissolution rate is the key attribute of NPs, which is due to large surface area and high dissolution pressure.43 Moreover, the NPs, due to small particle sizes, cause increase in adhesiveness to the cell membrane, which subsequently enhances the bioavailability of the poorly water-soluble drugs.44 For BCS class II drugs, dissolution is the rate-limiting step for improved bioavailability. Therefore, it is imperative to address the issue of poor water solubility of BCSII drugs; herein, for silibinin, bioavailability is subsequently substantially enhanced. The dissolution studies showed effectively enhanced dissolution rate of the poorly water-soluble silibinin, which is indicative of improved bioavailability.45 Additionally, Prandtl and Noyes–Whitney developed an equation, wherein the relationship among the particle size, hydrodynamic boundary layer, velocity of the surrounding liquid, surface area, and dissolution rate has been expressed, showing that greater surface area and smaller thickness of the diffusion layer can increase the dissolution rate of the particles.46 In this study, the silibinin NPs produced by APSP and EPN interestingly showed enhanced dissolution rate compared to raw silibinin, which circumvents the barrier related to its bioavailability.4,5,22
Currently, it has been found that pathogens such as P. aeruginosa, S. aureus, E. coli, and C. albicans are responsible for the infections that have a high global prevalence and are responsible for the increased worldwide morbimortality due to infections. Insufficient supplies and indiscriminate use of antimicrobials, especially in underdeveloped and poor countries, has led to an increase in resistance. Thus, there is a need to develop new treatment modalities for such infections. Hence, in the past 10–15 years, use of plants and their derivatives has gained popularity as a cure for infections caused by microorganisms, especially in developing countries.47
Silibinin has been established to reduce the infections in both animals and humans caused by different microorganisms such as bacteria, fungi, andprotozoa.47–49 Previous literature has shown that silibinin has negligible activity against Gram-negative bacteria.16 The same trend was observed for the NPs against Gram-negative bacteria. In this study, silibinin effectively inhibited S. aureus (32 μg/mL) and B. subtilis (16 μg/mL). However, silibinin NPs prepared by the EPN method were more effective in the control of S. aureus and B. subtilis, with MIC values of 8 μg/mL and 4 μg/mL, respectively. In comparison, NPs prepared through APSP inhibited S. aureus and B. subtilis at 16 μg/mL and 8 μg/mL, respectively, indicating the superiority of the EPN method. This also indicates a substantial increase in the activity of this drug with a two- to threefold enhancement of inhibition of the Gram-positive bacteria tested. The antifungal activity of silibinin NPs also was improved compared to that of unprocessed silibinin, which was 512 μg/mL against the test yeasts. With the EPN method, inhibitory concentrations were reduced to 64 μg/mL and 128 μg/mL against C. albicans and C. glabrata, which are almost the same as that of the standard drug miconazole, indicating the impact of nanosizing this drug.
S. aureus has been routinely isolated in systemic infections such as bacteremia, necrotizing pneumonia, osteomyelitis, and septic arthritis. It also has a proven common association with infections of soft and cutaneous tissues.50 Mycoses by yeasts of genus Candida occur on cutaneous and mucosal surfaces, and these species also may find their way to the systemic circulation, with grave consequences, especially for immunocompromised patients.51 The situation becomes grimmer as therapeutics for mycotic infections are limited, particularly resistant ones.52 Moreover, irrational and indiscriminate use of broad-spectrum antibiotics, especially in developing and poor countries, has contributed to a surge of microbial resistance. It has been reported that silibinin possesses activity against the Gram-positive bacteria S. epidermidis and B. subtilis.16 It has also shown synergism when combined with antibacterial drugs.47 In this present study, silibinin NPs produced by both APSP and EPN methods have shown to be promising therapeutic options in the case of Gram-positive bacteria, especially S. aureus, which is developing resistance against last-resort drugs such as vancomycin,53 as well as against the yeasts C. albicans and C. glabrata causing important mycotic infections. The results are very encouraging and require further research and standardization in the form of suitable dosage forms that may be utilized after titration through in vivo studies.
Conclusion
From the study, it has been concluded that the NPs of silibinin prepared by the APSP and EPN methods are highly soluble, with a higher dissolution rate compared to that of unprocessed silibinin. Owing to the small particle size in the nano range, the poor water solubility issue of silibinin has successfully been addressed, which in turn would exhibit improved bioavailability as well. However, it has been noted that the NPs prepared by the EPN method were more efficient after assessing different physical parameters and passing through analytical tests. Furthermore, the produced silibinin NPs demonstrated strong antibacterial potential compared to the raw counterpart. In future works, the produced NPs of silibinin could be subjected to development of a suitable dosage form and subsequently assessed for in vivo performance.
Acknowledgments
The authors thank Dr Mohammad Sadiq Qadir and M/S Ferring Controlled Therapeutics, East Kilbride, Glasgow, G74 5PB, UK, for their generous donation of silibinin for this project. Funds for covering the costs to publish in open access were arranged by Hani S Faidah and Abdul Haseeb.
Author contributions
MUKS and AS conceived and designed the experiments; MUKS performed the experiments; MUKS, SK, MUA, HSF, and MK analyzed the data; N, AS, HSF, and AH contributed reagents and materials. All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Radtke M. Pure drug nanoparticles for the formulation of poorly soluble drugs. New Drugs. 2001;3:62–68. | ||
Lipinski C. Poor aqueous solubility-an industry wide problem in drug discovery. Am Pharm Rev. 2002;5(3):82–85. | ||
Sailor G, Seth AK, Parmar G, Chauhan S, Javia A. Formulation and in-vitro evaluation of berberine containing liposome optimized by 32 full factorial designs. J Appl Pharm Sci. 2015;5(7):023–028. | ||
Dressmann JB, Reppas C. In vitro, in-vivo correlations for lipophilic, poorly water soluble drugs. Eur J Pharm Sci. 2000;11:73–80. | ||
Kesisoglou F, Panmai S, Wu Y. Nanosizing-oral formulation development and biopharmaceutical evaluation. Adv Drug Deliv Rev. 2007;59(7):631–644. | ||
Salah N, Habib SS, Khan ZH, et al. High-energy ball milling technique for ZnO nanoparticles as antibacterial material. Int J Nanomedicine. 2011;6:863–886. | ||
Puckett SD, Taylor E, Raimondo T, Webster TJ. The relationship between the nanostructure of titanium surfaces and bacterial attachment. Biomaterials. 2010;31(4):706–713. | ||
Seo WS, Lee JH, Sun X. FeCo/graphitic shell nanocrystals as advanced magnetic-resonance-imaging and near infrared agents. Nat Mater. 2006;5(12):971–976. | ||
Dilnawaz F, Singh A, Mohanty C, Sahoo SK. Dual drug loaded super-paramagnetic iron oxide nanoparticles for targeted cancer therapy. Biomaterials. 2010;31(13):3694–3706. | ||
Jacobs C, Müller RH. Production and characterization of a budesonide nanosuspension for pulmonary administration. Pharm Res. 2002;19(2):189–194. | ||
Bilati U, Allémann E, Doelker E. Development of a nanoprecipitation method intended for the entrapment of hydrophilic drugs into nanoparticles. Eur J Pharm Sci. 2005;24(1):67–75. | ||
Kakran M, Sahoo NG, Tan IL, Li L. Preparation of nanoparticles of poorly water-soluble antioxidant curcumin by antisolvent precipitation methods. J Nanopart Res. 2012;14(3):1–11. | ||
Hussain I, Khan FA, Ullah R, Khan N, Muhammad Z. Analysis of silymarin in the seeds of white and blue capitulum of Silybum marianum plants. Afr J Pharm Pharmacol. 2011;5(6):742–745. | ||
Dixit N, Baboota S, Kohli K, Ahmad S, Ali J. Silymarin: a review of pharmacological aspects and bioavailability enhancement approaches. Ind J Pharmacol. 2007;39(4):172–179. | ||
Loguercio C, Festi D. Silybin and the liver: from basic research to clinical practice. World J Gastroenterol. 2011;17(18):2288–2301. | ||
Lee DG, Kim HK, Park Y, et al. Gram-positive bacteria specific properties of silybin derived from Silybum marianum. Arch Pharm Res. 2003;26(8):597–600. | ||
Amirsaadat S, Pilehvar-Soltanahmadi Y, Zarghami F, Alipour S, Ebrahimnezhad Z, Zarghami N. Silibinin-loaded magnetic nanoparticles inhibit hTERT gene expression and proliferation of lung cancer cells. Artif Cells Nanomed Biotechnol. 2017;12:1–8. | ||
Chen CH, Chang CC, Shih TH, Aljuffali IA, Yeh TS, Fang JY. Self-nanoemulsifying drug delivery systems ameliorate the oral delivery of silymarin in rats with Roux-en-Y gastric bypass surgery. Int J Nanomedicine. 2015;10:2403–2416. | ||
Liu CH, Lin CC, Hsu WC, et al. Highly bioavailable silibinin nanoparticles inhibit HCV infection. Gut. 2016:doi:10.1136/gutjnl-2016-312019 Epub 2016 Jul 19. | ||
Sun HP, Su JH, Meng QS, et al. Silibinin and indocyanine green-loaded nanoparticles inhibit the growth and metastasis of mammalian breast cancer cells in vitro. Acta Pharmacol Sin. 2016;37(7):941–949. | ||
Bhushan SP, Vladimir VC, Vladimir PT. New developments in liposomal drug delivery. Chem Rev. 2015;115(19):10938–10966. | ||
Bhattacharya S, Ghosh AK. Phytosomes: the emerging technology for enhancement of bioavailability of botanicals and nutraceuticals. Int J Aesthetic Anti Ageing Med. 2009;2:87–91. | ||
Kurkin VA, Ryzhov VM, Biryukova OV, Mel’nikova NB, Selekhov VV. Interaction of milk-thistle-fruit flavanonols with Langmuir monolayers of lecithin and bilayers of liposomes. Pharm Chem J. 2009;43(2):101–109. | ||
Garg R, Gupta GD. Gastroretentive floating microspheres of silymarin: preparation and in vitro evaluation. Trop J Pharm Res. 2010;9:59–66. | ||
El-Far YM, Zakaria MM, Gabr MM, El Gayar AM, El-Sherbiny IM, Eissa LA. A newly developed silymarin nanoformulation as a potential antidiabetic agent in experimental diabetes. Nanomedicine (Lond). 2016;11(19):2581–2602. | ||
Khan S, de Matas M, Zhang J, Anwar J. Nanocrystal preparation: low-energy precipitation method revisited. Cryst Growth Des. 2013;13(7):2766–2777. | ||
Wu JW, Lin LC, Tsai TH. Drug–drug interactions of silymarin on the perspective of pharmacokinetics. J Ethnopharmacol. 2009;121(2):185–193. | ||
Wu JW, Lin LC, Hung SC, Chi CW, Tsai TH. Analysis of silibinin in rat plasma and bile for hepatobiliary excretion and oral bioavailability application. J Pharm Biomed Anal. 2007;45(4):635–641. | ||
Arunprasad K, Narayanan N, Rajalakshmi G. Preparation and evaluation of solid dispersion of terbinafine hydrochloride. Int J Pharm Sci Rev Res. 2010;3:130–134. | ||
Khan GM, Zhu JB. Ibuprofen release kinetics from controlled-release tablets granulated with aqueous polymeric dispersion of ethylcellulose II: influence of several parameters and coexcipients. J Control Release. 1998;56(1):127–134. | ||
Costa P, Lobo JM. Modeling and comparison of dissolution profiles. Eur J Pharm Sci. 2001;13(2):123–133. | ||
Patel RP, Patel MM. Solid-state characterization and dissolution properties of lovastatin hydroxypropyl-beta-cyclodextrin inclusion complex. Pharm Tech. 2007;31(2):72–81. | ||
National Committee for Clinical Laboratory Standards Approved Standard. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts M 27-A2. Wayne PA: National Committee for Clinical Laboratory Standards Approved Standard; 2002. | ||
Zhang SL, Chang JJ, Damu GL, et al. Novel berberine triazoles: synthesis, antimicrobial evaluation and competitive interactions with metal ions to human serum albumin. Bioorg Med Chem Lett. 2013;23(4):1008–1012. | ||
Dang L, Yang H, Black S, Wei H. The effect of temperature and solvent composition on transformation of β-to α-glycine as monitored in situ by FBRM and PVM. Org Proces Res Dev. 2009;13(6):1301–1306. | ||
Devaraj P, Kumari P, Aarti C, Renganathan A. Synthesis and characterization of silver nanoparticles using cannonball leaves and their cytotoxic activity against MCF-7 cell line. J Nanotechnol. 2013:1–5. | ||
Murdande SB, Pikal MJ, Shankar RM, Bogner RH. Solubility advantage of amorphous pharmaceuticals: I. A thermodynamic analysis. J Pharm Sci. 2010;99(3):1254–1264. | ||
Murdande SB, Pikal MJ, Shankar RM, Bogner RH. Solubility advantage of amorphous pharmaceuticals: II. Application of quantitative thermodynamic relationships for prediction of solubility enhancement in structurally diverse insoluble pharmaceuticals. Pharm Res. 2010;27(12):2704–2714. | ||
Müller RH, Junghanns J-UAH. “Drug nanocrystals/nanosuspensions for the delivery of poorly soluble drugs. In: Torchilin VP, editor. Nanoparticulates as Drug Carriers”. London: Imperial College Press; 2006:236. | ||
Kakran M, Sahoo NG, Li L, et al. Fabrication of drug nanoparticles by evaporative precipitation of nanosuspension. Int J Pharm. 2010;383(1–2):285–292. | ||
Shid RL, Dhole SN, Kulkarni N, Shid SL. Formulation and evaluation of nanosuspension delivery system for simvastatin. Int J Pharm Sci Nanotechnol. 2014;7:2459–2476. | ||
Jiang T, Han N, Zhao B, Xie Y, Wang S. Enhanced dissolution rate and oral bioavailability of simvastatin nanocrystal prepared by sonoprecipitation. Drug Dev Ind Pharm. 2012;38(10):1230–1239. | ||
Mauludin R, Müller RH, Keck CM. Development of an oral rutin nanocrystal formulation. Int J Pharm. 2009;370(1–2):202–209. | ||
Pardeike J, Müller RH. Nanosuspensions: a promising formulation for the new phospholipase A2 inhibitor PX-18. Int J Pharm. 2010;391(1–2):322–329. | ||
Shah SMH, Ullah F, Khan S, et al. Smart nanocrystals of artemether: fabrication, characterization, and comparative in vitro and in vivo antimalarial evaluation. Drug Des Devel Ther. 2016;10:3837–3850. | ||
Kayaert P, Van den Mooter G. Is the amorphous fraction of a dried nanosuspension caused by milling or by drying? A case study with naproxen and cinnarizine. Eur J Pharm Biopharm. 2012;81(3):650–656. | ||
Rakelly de Oliveira D, Relison Tintino S, Morais Braga MF, et al. In vitro antimicrobial and modulatory activity of the natural products silymarin and silibinin. Biomed Res Int. 2015;2015:292797. | ||
Park KS, Kang KC, Kim JH, Adams DJ, Johng TN, Paik YK. Differential inhibitory effects of protoberberines on sterol and chitin biosyntheses in Candida albicans. J Antimicrob Chemother. 1999;43(5):667–674. | ||
Quan H, Cao YY, Xu Z, et al. Potent in vitro synergism of fluconazole and berberine chloride against clinical isolates of Candida albicans resistant to fluconazole. Antimicrob Agents Chemother. 2006;50(3):1096–1099. | ||
Otto M. Basis of virulence in community-associated methicillin-resistant Staphylococcus aureus. Annu Rev Microbiol. 2010;64:143–162. | ||
Fidel PL, Vazquez JA, Sobel JD. Candida glabrata: review of epidemiology, pathogenesis, and clinical disease with comparison to C. albicans. Clin Microbiol Rev. 1999;12(1):80–96. | ||
Kanafani ZA, Perfect JR. Resistance to antifungal agents: mechanisms and clinical impact. Clin Infect Dis. 2008;46(1):120–128. | ||
Gardete S, Tomasz A. Mechanisms of vancomycin resistance in Staphylococcus aureus. J Clin Invest. 2014;124(7):2836–2840. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.