")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Supplement 1
Fabrication and characterization of anisotropic nanofiber scaffolds for advanced drug delivery systems
Authors Jalani G, Jung CW, Lee JS, Lim DW
Received 20 July 2013
Accepted for publication 28 August 2013
Published 6 May 2014 Volume 2014:9(Supplement 1) Pages 33—49
DOI https://doi.org/10.2147/IJN.S51842
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Ghulam Jalani,* Chan Woo Jung,* Jae Sang Lee, Dong Woo Lim
Department of Bionano Engineering, College of Engineering Sciences, Hanyang University, Education Research Industry Cluster at Ansan Campus, Ansan, South Korea
*These authors contributed equally to this work
Abstract: Stimuli-responsive, polymer-based nanostructures with anisotropic compartments are of great interest as advanced materials because they are capable of switching their shape via environmentally-triggered conformational changes, while maintaining discrete compartments. In this study, a new class of stimuli-responsive, anisotropic nanofiber scaffolds with physically and chemically distinct compartments was prepared via electrohydrodynamic cojetting with side-by-side needle geometry. These nanofibers have a thermally responsive, physically-crosslinked compartment, and a chemically-crosslinked compartment at the nanoscale. The thermally responsive compartment is composed of physically crosslinkable poly(N-isopropylacrylamide) poly(NIPAM) copolymers, and poly(NIPAM-co-stearyl acrylate) poly(NIPAM-co-SA), while the thermally-unresponsive compartment is composed of polyethylene glycol dimethacrylates. The two distinct compartments were physically crosslinked by the hydrophobic interaction of the stearyl chains of poly(NIPAM-co-SA) or chemically stabilized via ultraviolet irradiation, and were swollen in physiologically relevant buffers due to their hydrophilic polymer networks. Bicompartmental nanofibers with the physically-crosslinked network of the poly(NIPAM-co-SA) compartment showed a thermally-triggered shape change due to thermally-induced aggregation of poly(NIPAM-co-SA). Furthermore, when bovine serum albumin and dexamethasone phosphate were separately loaded into each compartment, the bicompartmental nanofibers with anisotropic actuation exhibited decoupled, controlled release profiles of both drugs in response to a temperature. A new class of multicompartmental nanofibers could be useful for advanced nanofiber scaffolds with two or more drugs released with different kinetics in response to environmental stimuli.
Keywords: stimuli responsiveness, anisotropy, nanofibers, actuation, drug delivery, tissue engineering
Introduction
Three-dimensional polymeric architectures with micro- and nanoscale anisotropy have emerged as a new class of advanced materials, as they are capable of simultaneously presenting different physical and chemical properties to each compartment.1 Recently, multicompartmental micro- or nanoparticles with different optical, electric, or magnetic properties for each compartment have been widely applied in industrial and biomedical applications because of their capability of dynamic switching in response to light, electric, or magnetic fields.2,3 Alternatively, multicompartmental micro- or nanofiber scaffolds with different biological ligands in each compartment have been developed as smart tissue engineering scaffolds because each compartment induces strong adhesion to specific cell lines, resulting in directional cell growth with a compartment, and spatial control of cell proliferation.4,5 However, it is still challenging to introduce completely different physicochemical properties of polymers into each compartment of anisotropic micro- or nanoarchitectures, where each compartment is separately responsive to environmental stimuli, such as temperature, pH, and ionic strength.6
Various original engineering approaches to synthesize anisotropic micro- or nanoarchitectures in the form of various geometries have been explored: electrohydrodynamic (EHD) cojetting,7 microfluidics,8 differential solvent evaporation, flow focusing lithography,9 spinning disks, selective deposition,10 partial modification by masking, Pickering emulsion,11 and self-assembly.12 Of the various methodologies for preparing anisotropic structures, EHD cojetting is specifically interesting as it allows for equilibrated laminar flow of multiple polymer solutions to be ejected from the vertex of a multiphasic Taylor cone. The created thin-jet stream due to charge–charge repulsion results in the formation of multicompartmental architectures.13
Recently, the EHD cojetting platform technology of multiple polymer solutions in miscible solvents with side-by-side needle geometry was established with significant advantages: 1) versatility to introduce dissimilar polymers, different polymer blends, reactive additives, therapeutic biomacromolecules, drugs with low molecular weights (MWs), and a variety of inorganic nanomaterials into each compartment; 2) easy scale-up to prepare micron- or nanosized anisotropic structures composed of different materials in each compartment internally and externally; 3) free control of the composition, geometry, and aspect ratio of anisotropic architectures;10,14 and 4) facile approach to spatio-selectively immobilize multiple biological ligands for three-dimensionally controlled cell adhesion or in situ polymer chain growth in a compartment via atom transfer radical polymerization for chemically-controlled bending of biphasic microcylinders.15 EHD cojetting is a modified form of the EHD jetting process, which is commonly used to fabricate micro- or nanostructures from a variety of polymer solutions.16,17 The original EHD jetting uses the EHD forces to generate micro- or nanometer sized diameters of liquid jets.18 The electrified jet is produced when a high voltage at several kilovolts is applied between the flowing liquid and a conductive substrate separated by a certain distance. The outflowing of polymer solutions with specific viscosity, conductivity, and surface tension through a thin capillary in the presence of an externally applied electric field results in the production of micro- or nanoparticles or fibers of various sizes and shapes.19,20
Multicompartmental characteristics of the anisotropic architectures make them suitable for a number of intriguing applications, including switchable display devices,21 a colloidal stabilizer at an interface of two immiscible solutions,22 self-propelled motors,23 optical sensors,24 and spontaneous assembly for complex structures.25 In addition, multicompartmental nanofibers with core–shell26–29 and side-by-side30,31structures have been explored for drug delivery systems and tissue engineering scaffolds. However, there have been very limited studies regarding anisotropic architectures with actuation at the micro- or nanoscale, which are useful for advanced biomedical applications, specifically for biosensors, shape memory devices, microactuators, tissue engineering, regenerative medicine, and drug delivery. In this respect, environmentally-responsive polymers, such as thermally responsive poly(N-isopropylacrylamide) poly(NIPAM), could be incorporated into anisotropic architectures. Poly(NIPAM) monophasic nanofibers prepared by electrospraying have been developed for pulsatile drug release and cell–sheet formations by thermal aggregation of the poly(NIPAM) chain due to the lower critical solution temperature (LCST) transition behavior.32–34 Stimuli-responsive physically- or chemically-crosslinked poly(NIPAM) copolymer networks have been extensively developed for various biomedical applications in smart drug delivery sensors and actuators, switchable interfaces, and bioengineered surfaces.
With a series of biomedical applications using environmentally-responsive polymers, the composite nanofiber scaffolds produced from two or more bioactive materials carrying various biomolecules or therapeutic agents have been developed. These scaffolds have the potential of tunable drug loading efficiency, drug release profiles, and biodegradability. Antibiotic drug cefoxitin sodium was successfully released from biodegradable poly(lactide-co-glycolide)-based nanofiber scaffolds in a sustained release manner.35 Controlled release of metronidazole benzoate from poly(e-caprolactone) scaffolds was achieved by tuning the polymer-to-solvent ratio. This system was applied to treat the periodontal disease.36 In situ crosslinked chitosan–polyethylene glycol (PEG)–folic acid nanocomplexes produced via electrospinning were loaded with anticancer drugs and explored for potential applications in cancer therapy.37 Nanofiber scaffolds with controlled topographical features have been investigated as potent materials for effective gene delivery with improved human wound healing ability.38 The prolonged release up to 50–60 days of nonviral gene vectors was achieved in the core–sheath structured nanofiber scaffolds composed of PEG and poly(e-caprolactone) in another investigation.39 Furthermore, thermoresponsive nanofiber composites composed of poly(NIPAM), poly(vinyl alcohol), and poly(acrylic acid) showed interesting drug release profiles with burst to sustained release kinetics, depending on the ratios of the constituent polymers.22
In this study, we report a new class of thermally-responsive anisotropic nanofibers with physically and chemically distinct compartments that were first developed as smart drug delivery systems with mechanical actuation on the nanoscale. The bicompartmental nanofiber scaffolds were prepared via EHD cojetting of two different polymer solutions of thermally-responsive poly(NIPAM-co-stearyl acrylate) poly(NIPAM-co-SA) and thermally-unresponsive PEG dimethacrylates (PEGDMA) with a side-by-side geometry. Although totally different polymer solutions were used for the EHD cojetting, a biphasic Taylor cone was maintained due to the balanced viscosity, conductivity, and surface tension of two polymer solutions, resulting in the formation of discrete interfaces between the two compartments. The poly(NIPAM-co-SA) compartment was physically crosslinked by hydrophobic interaction of stearyl chains of poly(NIPAM-co-SA), while the PEGDMA was chemically stabilized via ultraviolet (UV) irradiation. Both compartments of these nanofiber scaffolds were swollen in physiologically relevant buffers due to the hydrophilic polymer network, clearly demonstrating their hydrogel property. The bicompartmental nanofibers showed thermally-triggered shape shifting from an uncoiled to coiled configuration due to thermally-induced aggregation of poly(NIPAM-co-SA) chains and inert PEGDMA in the other compartment. To the best of our knowledge, this is the first study of bicompartmental nanofibers composed of a physically-crosslinked, thermoresponsive compartment, along with a chemically-crosslinked compartment showing a thermally-triggered mechanical actuation and fast response to external stimuli because of high surface area-to-volume ratios, which make them unique choices for the development of smart drug delivery systems for stimuli-triggered, controlled drug release.2,5 Furthermore, when bovine serum albumin (BSA) and dexamethasone 21-phosphate (DMP) were separately loaded into each compartment, the bicompartmental nanofibers with mechanical actuation showed fully decoupled, controlled release profiles in response to temperature. This new class of multicompartmental nanofibers could be useful as advanced nanofiber scaffolds with two or more drugs that can be released under different release kinetics in response to environmental stimuli.
Materials
NIPAM (97%) and stearyl acrylate (SA) obtained from Sigma-Aldrich (St Louis, MO, USA) were separately purified by recrystallization from n-hexane and ethanol, respectively. In addition, 2,2’-azobis(2-methylpropionitrile) known as azobisisobutyronitrile (98%) obtained from Acros (Thermo Fisher Scientific, Waltham, MA, USA) was purified by recrystallization from methanol. PEG (MW, 20,000 g/mol), methacrylic anhydride (MA), 2-hydroxy-2-methyl-propiophenone (97%) as a photoinitiator, fluorescein diacetate, Nile red, ethanol, 2,2,2-trifluoroethanol (TFE), DMP disodium salt, BSA, and phosphate buffered saline (PBS) were obtained from Sigma-Aldrich. Deionized water purified by Milli-Q (Millipore Water Purification Systems; EMD Millipore, Bedford, MA, USA) was used throughout all of the experiments.
Synthesis of poly(NIPAM-co-SA)
The physically crosslinkable polymer, poly(NIPAM-co-SA), was synthesized as previously reported.6 NIPAM was dissolved in dimethylformamide at 30°C and recrystallized below 10°C to remove any impurities. In a typical batch, 20 g of NIPAM were dissolved in 200 mL of dimethylformamide in a conical flask at room temperature to produce a final concentration of 10.0 weight/volume (w/v)%. This solution was then transferred to a 4°C refrigerator for recrystallization. The solution was filtered through filter paper (Whatman® qualitative filter paper, grade 1; Whatman Ltd, Kent, UK) via an aspirator assembly (A-1000S, EYELA, USA; Tokyo Rikakikai Co, Ltd, Tokyo, Japan). Shiny, crystalline NIPAM was obtained, which was then dried to remove any residual organic solvent. SA was also recrystallized from ethanol before use. Two grams of SA was dissolved in 50 mL of ethanol at room temperature to produce a final concentration of 4.0 w/v%, followed by incubation at 4°C for recrystallization. Monomers of NIPAM and SA were dissolved in pure ethanol with a feed molar ratio of 97:3 for copolymerization. Azobisisobutyronitrile was used as a free radical initiator up to 0.005 w/w% of the total monomer weight. The mixture was heated to boiling with magnetic stirring at 260 rpm under reflux for at least 5 hours in a nitrogen atmosphere to complete the reaction. When the resultant transparent solution was poured into deionized water to purify poly(NIPAM-co-SA), the copolymers immediately turned into aggregates, which were then collected and dried for further use. Unreacted NIPAM was dissolved in the water and unreacted; water-insoluble SA was precipitated and removed. The copolymers were removed and freeze-dried by a lyophilizer (MCFD8508; Ilshin Lab Co, Ltd, Gyeonggi-do, Korea) under vacuum to completely remove any residual solvent. The resulting white, spongy solid was used for further characterization and experiments.
Synthesis of PEGDMA
As previously reported, PEGDMA was synthesized from the reaction of MA and PEG (MW, 20,000 g/mol) at room temperature.40 PEG, MA, and triethylamine were dissolved in 15 mL of DCM at a feed molar ratio of 0.025:0.22:0.01, respectively, and stirred at room temperature for 4 days. The product was precipitated in diethyl ether, filtered, and freeze dried, while unreacted monomer was removed.
Polymer characterization
Poly(NIPAM-co-SA) was analyzed by gel permeation chromatography (GPC) to determine the average MW and MW distribution. GPC analysis was performed with a Waters-515 high-performance liquid chromatography pump system (Waters Corporation, Milford, MA, USA) connected with a refractive index detector. Tetrahydrofuran was used as the mobile phase at a flow rate of 1.0 mL/minute. Polystyrenes in the MW range of 820 to 1,070,000 g/mol were used as a standard, while the column temperature was maintained at 40°C. Poly(NIPAM-co-SA) was confirmed from the 1H nuclear magnetic resonance (NMR) analysis (AVANCE III 400; Bruker BioSpin AG, Fällanden, Switzerland) and the apparent molar ratio of each monomer was determined through the relative area under the curves of the corresponding peaks in the 1H NMR spectrum. When poly(NIPAM-co-SA) was dissolved in deionized water at a concentration of 1.0 w/v%, 1.5 w/v%, and 2.0 w/v%, the thermal property was confirmed by measuring the hydrodynamic size of the self-assembled micelle nanostructures as a function of the temperature using dynamic light scattering (DLS) (Zetasizer Nano ZS90; Malvern Instruments, Malvern, UK). The micelle polymer solution was prepared in ethanol at a final concentration of 10 w/v%. The polymer solution was then transferred to deionized water while maintaining the final concentrations of 1.0 w/v%, 1.5 w/v%, and 2.0 w/v%, followed by ultrasonication using an ultrasonicator (VC 505; Vibra-Cell, Sonics and Materials, Inc, Newton, CT, USA) for 40 seconds with 3/2 seconds of on/off cycles to prepare the polymeric micelles. A UV–visible spectrometer (Cary-100 Bio; Varian Biotech, USA) with Peltier thermostated temperature control was used to measure the absorbance of the polymer solution at 350 nm as a function of the temperature at a heating rate of 1°C/minute.6
Preparation of anisotropic nanofibers by EHD cojetting
Two polymer solutions were separately prepared for the EHD cojetting of biphasic nanofibers. Fluorescein diacetate and Nile red, as hydrophobic fluorescence dyes, were separately used to characterize the biphasic Taylor cone and the degree of anisotropy using different emission spectra. Prior to the EHD cojetting for the preparation of anisotropic nanofibers, EHD jetting of each solution for the monophasic nanofibers was carried out to determine the critical concentrations to form a stable cone–jet mode for each solution. One polymer solution comprising of 0.36 g of poly(NIPAM-co-SA) and 1.0 mg of fluorescein dissolved in 1.0 mL of pure ethanol resulted in a final concentration of 36.0 w/v% and 0.01 w/v%, respectively. The other polymer solution contained 0.24 g of PEGDMA and 0.5 mg of Nile red dissolved in a solvent mixture of ethanol and TFE at a 3:1 volume ratio to produce a final concentration of 24.0 w/v% and 0.05 w/v%, respectively. Each polymer solution was loaded into a 1.0 mL syringe (BD, Franklin Lakes, NJ, USA) connected to a dual-channel needle (FibriJet® SA-3610; Micromedics, Inc, St Paul, MN, USA) with side-by-side geometry. The two syringes were fixed with an applicator (Micromedics, Inc) to identically control both flow rates of the polymer solutions. The precisely controlled flow rate of the two polymer solutions was achieved through a micro syringe pump (KD Scientific, Inc, Holliston, MA, USA). The positive terminal of a high-voltage power supply (Nano NC, Seoul, Korea) was connected to dual needles, and a ground was applied to aluminum foil with a thickness of 0.018 mm (Fisherbrand; Thermo Fisher Scientific, Waltham, MA, USA) that served as a collecting substrate. The distance between two electrodes was vertically maintained in the range of 10–12 cm. High direct current electrical potential was applied in the range of 9.0–10.0 kV, and a flow rate of both solutions was kept at 0.40–0.50 mL/hour. A high-resolution digital camera (D-90; Nikon Corporation, Tokyo, Japan) was focused at the tip of the dual needles to visualize the biphasic Taylor cone, jet stream, and jet breakup during the EHD cojetting.
Physical and chemical stabilization of the biphasic nanofibers
For chemical crosslinking, 0.005 w/w% 2-hydroxymethylpropiophenone was added to the PEGDMA polymer solution as a photoinitiator. The PEGDMA compartment of the bicompartmental nanofibers in the solid state was chemically crosslinked by UV irradiation for 10 minutes using a UV lamp (Omnicure-1500A; Lumen Dynamics Group Inc, Mississauga, ON, Canada) operating at a power of 60 mW/cm2 for 2 minutes. Alternatively, the other compartment composed of poly(NIPAM-co-SA) was stabilized through physical crosslinking because of hydrophobic interactions among the long alkyl side chains.6 The resulting biphasic nanofibers were stabilized in an aqueous environment, which was confirmed by imaging analysis with fluorescence and confocal laser scanning microscopy (CLSM).
Imaging of bicompartmental nanofibers using fluorescence microscopy and SEM
The bicompartmental nanofibers were characterized in the dry state using an IX81 inverted phase contrast fluorescence microscope (Olympus Corporation, Tokyo, Japan) in the fluorescence and bright-field mode. Physically- and chemically-crosslinked bicompartmental nanofibers were dispersed in PBS at pH 7.4, sonicated using an ultrasonicator (VC 505; Vibra-Cell, Sonics and Materials, Inc), and imaged to characterize both the stability and swelling property under aqueous conditions. The anisotropic property of the bicompartmental nanofibers was confirmed using the TCS SL CLSM equipped with He/Ne– and argon lasers (Leica Microsystems, Wetzlar, Germany). The corresponding laser excited each dye in each compartment, and the emission spectral range was controlled to avoid overlap of the emission signals of the two dyes. The wavelength ranges of the fluorescein and Nile red for the spectral emission were adjusted to 543–579 nm and 592–647 nm, respectively. A temperature-controlled stage (Tempcontrol 37-2 digital; Leica Microsystems) was used to observe the thermally-triggered mechanical actuation within each compartment when these nanofibers were immersed in PBS at pH 7.4. Any structural changes of the biphasic nanofibers were measured as the temperature gradually increased. The temperature was controlled over approximately 4°C–50°C, and any shape changes were observed in real time and were recorded as CLSM images in monophasic nanofibers and bicompartmental nanofibers composed of poly(NIPAM-co-SA) and PEGDMA. Nanofibers homogeneously suspended in PBS were applied on a cover glass and placed on the temperature-controlled stage. A stage temperature was controlled in four different ranges: 1) 5°C to 15°C; 2) 15°C to 25°C; 3) 25°C to 35°C; and 4) 35°C to 45°C. The identical CLSM imaging conditions were also used for the monophasic fibers composed of poly(NIPAM-co-SA) or PEGDMA as controls. In addition, scanning electron microscopy (SEM) was used to characterize the diameter, diameter distribution, and surface morphology of both the monophasic nanofibers and bicompartmental nanofibers in the dry state. These nanofibers were coated with platinum using a K575X Turbo Sputter Coater (Emitech Ltd, Ashford, UK) to introduce a conductive layer, and imaged using an SEM (VEGA-SB3; TESCAN-USA Inc., Cranberry, PA, USA) operated at an accelerating voltage of 0.5 kV to 30 kV.
Preparation of dual drug-loaded bicompartmental nanofibers
The model drugs, DMP and BSA, were used as small molecular and biomacromolecular drugs, respectively. In a representative experiment, two polymer solutions were separately prepared. Solution “A” was comprised of 0.36 g of poly(NIPAM-co-SA) and 2.0 mg of 1) DMP or 2) BSA dissolved in 1.0 mL of a solvent mixture of ethanol and TFE at a 3:1 volume ratio, while solution “B” contained 0.24 g of PEGDMA and 2.0 mg of 1) BSA or 2) DMP dissolved in 1.0 mL of the solvent mixture of ethanol and TFE at a 3:1 volume ratio. The dual drugs of DMP and BSA were alternately loaded into each of solution “A” and solution “B”, such as the DMP-loaded poly(NIPAM-co-SA) compartment and BSA-loaded PEGDMA compartment, respectively. Two polymer solutions with dual drugs were loaded into a 1.0 mL syringe connected to a dual-channel needle (FibriJet SA-3610; Micromedics, Inc) with side-by-side geometry. A high-voltage power was applied for the EHD cojetting, as previously mentioned.
In vitro release of DMP and BSA from the drug-loaded bicompartmental nanofibers
Ten milligrams of dual drug-loaded bicompartmental nanofibers were suspended in 1 mL of PBS at pH 7.4 to study the drug release kinetics decoupled from each compartment as a function of the temperature. These nanofiber solutions were maintained in the suspension state in a shaker operating at 100 rpm, and the temperature was maintained at 4°C or 37°C to observe temperature-controlled drug release. Five different batches were prepared to study the batch-to-batch variability of the drug release from each compartment. The aliquots were periodically taken from the nanofiber solutions after fixed time intervals, and the fresh PBS buffer was replenished to maintain an identical solution volume. The concentration of DMP and BSA in the aliquot was determined by measurement of the optical absorbance at 242 nm for DMP and 278 nm for BSA, and then by the deconvolution of overlapping absorbance peaks. In addition, the nanofiber solutions were incubated in a shaker operating at 100 rpm at 4°C or 37°C every 10 minutes to study the pulsatile drug release. The temperature was periodically changed from 4°C to 37°C and from 37°C to 4°C every 10 minutes. The concentration of the released model drugs in the aliquot was analyzed with a UV–visible spectrophotometer, as previously described.
Results and discussion
Figure 1A shows a schematic of the instrumental setup of EHD cojetting of two separate polymer solutions with side-by-side needle geometry to synthesize a new class of stimuli-responsive, bicompartmental nanofibers with distinct physically- and chemically-crosslinked compartments. As represented in the insets of Figure 1A, totally dissimilar polymer solutions, including: 1) thermally responsive and physically crosslinkable poly(NIPAM-co-SA); and 2) nonthermally responsive and chemically crosslinkable PEGDMA at different concentrations were loaded into individual syringes connected to a dual-channel needle with a side-by-side geometry, which were fixed at a dual-channel applicator assembly to propel equal amounts of polymer solution during EHD cojetting.8,11 A microsyringe pump maintained a continuous flow of the two polymer solutions through the channels. High voltage at 9–10 kV applied between the needle and collecting substrate made jet streams that were simultaneously ejected from the vertex and rapidly evaporated, forming polymeric bicompartmental nanofibers. A digital image of the biphasic Taylor cone in Figure 1A shows that there was no severe interdiffusion between the two polymer solutions during EHD cojetting, aiding the production of more bicompartmental nanofibers. As reported elsewhere, it is important to maintain a discrete interface between two outflowing polymer solutions within the biphasic Taylor cone, which is critical for producing anisotropic nanoarchitectures with distinct multicompartments.12,13 Figure 1B represents a diagram of stimuli-responsive actuation of bicompartmental nanofibers with distinct compartments of: 1) thermoresponsive and physically-crosslinked poly(NIPAM-co-SA) on one side; and 2) nonthermoresponsive and chemically-crosslinked PEGDMA via UV irradiation on the other side, in a swollen state under aqueous conditions as a function of temperature. We hypothesized that conformational changes of poly(NIPAM-co-SA) chains above its LCST could thermally induce mechanical actuation when immersed in aqueous solutions, which makes these nanofibers ideal for multimodal drug delivery systems where one drug could be released via external stimuli and the other drug could be delivered through diffusion.
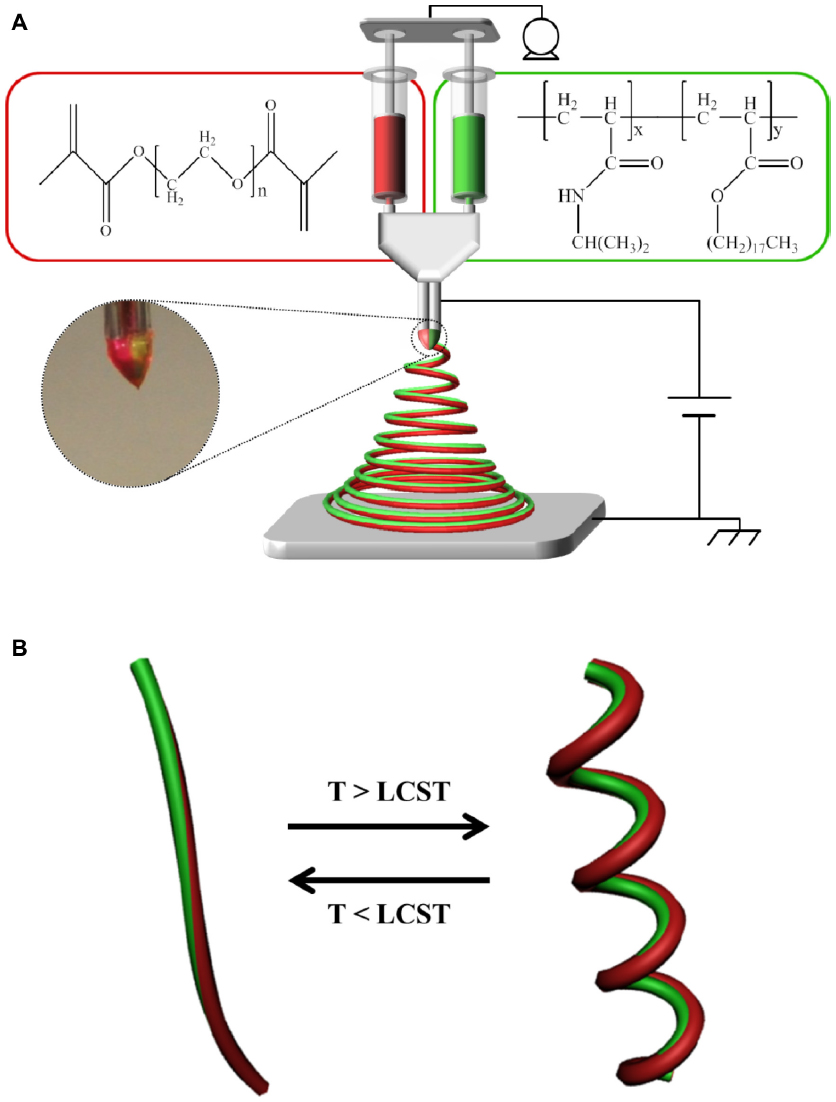 | Figure 1 Schematic of the setup of EHD cojetting and diagram of the thermally-triggered actuation of biocompartmental nanofibers. |
As shown in Figure 1A, poly(NIPAM-co-SA) dissolved in ethanol served as the thermo-sensitive compartment, while photocrosslinkable and biocompatible PEGDMA in the ethanol–TFE solvent mixture was used to generate the nonthermoresponsive compartment. Organic solvents, such as ethanol and TFE, were used to clearly observe the thermally responsive actuation of the bicompartmental nanofibers because the organic solvents prevented thermal aggregation of poly(NIPAM-co-SA) during EHD cojetting. It is notable that the evaporation of the aqueous solutions of poly(NIPAM) copolymers during EHD jetting caused the polymer chains to aggregate due to the LCST, resulting in bead-on-strings or fibrous structures with different diameters.23,24 Additionally, different polymer concentrations in organic solvents were optimized so that their viscosities were balanced. When the bicompartmental nanofibers were prepared by EHD cojetting, the photocrosslinkable PEGDMA compartment was then irradiated under UV light to chemically crosslink the PEGDMA chains.
For the thermoresponsive and physically crosslinkable compartment, poly(NIPAM-co-SA) was synthesized by free radical polymerization of NIPAM monomers with hydrophobic SA, while PEGDMA was synthesized for the nonthermoresponsive and chemically crosslinkable compartment by the reaction of MA and PEG with a MW of 20,000 g/mol. Figure 2A and B show the 1H NMR spectra of poly(NIPAM-co-SA) and PEGDMA, respectively. First, triplets near 0.84 ppm indicate the presence of stearyl groups in the polymer backbone, demonstrating that SA monomers were copolymerized with NIPAM, as previously reported.34 The apparent molar ratio of NIPAM and SA was 96.6:3.4, which compares to its feed molar ratio of 97:3, indicating enhanced incorporation of the hydrophobic SA monomers into the polymer chains, potentially due to their good compatibility in ethanol and preferential reactivity. Second, the chemical shifts of methylene protons on PEGDMA were 5.76 ppm and 6.18 ppm, and the protons next to the terminal MA groups shifted to 4.37 ppm in Figure 2B, as previously reported.40 In addition, the MW and MW distribution of poly(NIPAM-co-SA) were determined by GPC. Poly(NIPAM-co-SA) has a weight average MW of 22,000 g/mol and a number average MW of 9,700 g/mol, leading to a polydispersity index of 2.3. More importantly, poly(NIPAM-co-SA) and PEGDMA have similar MWs, which helped to maintain the stable biphasic Taylor cone during EHD cojetting, potentially due to balanced viscosities in ethanol and TFE.
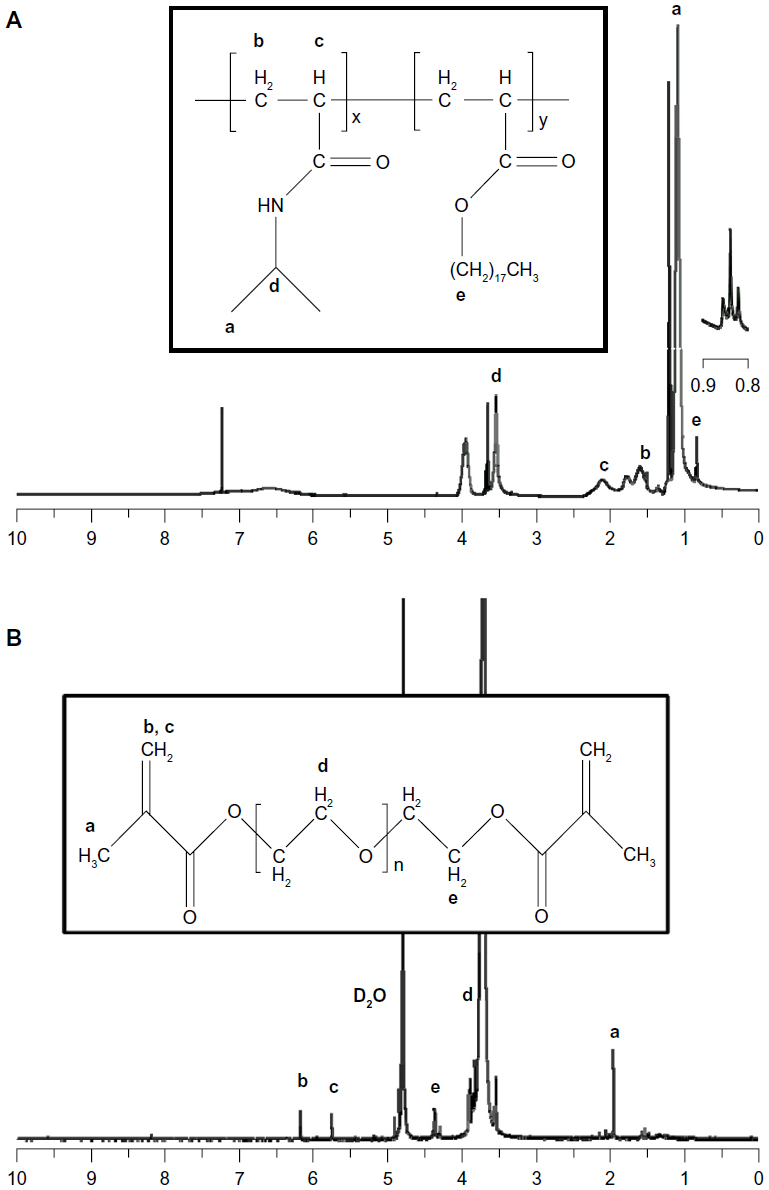 | Figure 2 1H nuclear magnetic resonance spectra of poly(NIPAM-co-SA) and PEGDMA. |
Figure 3A and B represent the intrinsic physicochemical properties of poly(NIPAM-co-SA) characterized by DLS and UV absorbance. As previously reported,34 poly(NIPAM-co-SA) has limited solubility in water because of long hydrophobic alkyl chains, while its backbone has hydrophilic properties because of the amide functional groups. Along with its amphipathic characteristics, poly(NIPAM-co-SA) molecules were spontaneously self-assembled into nanoscaled micellar structures in dilute conditions upon dissolution of poly(NIPAM-co-SA) in ethanol, mixing with water and subsequent ultrasonication. These micelles have the hydrophilic poly(NIPAM) backbone chain facing outward and the hydrophobic SA side chains merging inward, forming the self-assembled core–shell nanostructures. As shown from the DLS measurement in Figure 3A, a sharp single transition occurred at a critical temperature (the LCST), which is characteristic of thermally responsive poly(NIPAM)-based copolymers. The micelle size collapsed from approximately 170–190 nm to 40–50 nm for the 1.0–2.0 w/v% polymer solution, as the temperature increased above its LCST, which is in good agreement with a previous report.34 The LCST varied as a function of the poly(NIPAM-co-SA) concentration, potentially due to different degrees of entanglement and mobility of the polymer chains.34 As the polymer concentration increased, more interweaving between the polymer chains occurred, resulting in the reduction of the LCST. Figure 3B represents the 350 nm UV absorbance that clearly shows the LCST behavior of poly(NIPAM-co-SA). The polymer chains were aggregated above the LCST at 30.2°C–34.7°C, causing an increase in turbidity, as was previously reported.6,34 As the polymer concentration increased from 1.0 w/v% to 2.0 w/v%, the LCST changed from 34.7°C to 30.2°C. The LCSTs determined by UV absorbance were similar to those analyzed by DLS as a function of the polymer concentration. Furthermore, the increased polymer concentration caused an increase of the initial micelle size, likely due to more entanglement among the polymer chains and less mobility, as observed in the UV absorption spectra below the LCST. These results clearly show a sharp thermal transition of environmentally-responsive poly(NIPAM-co-SA).
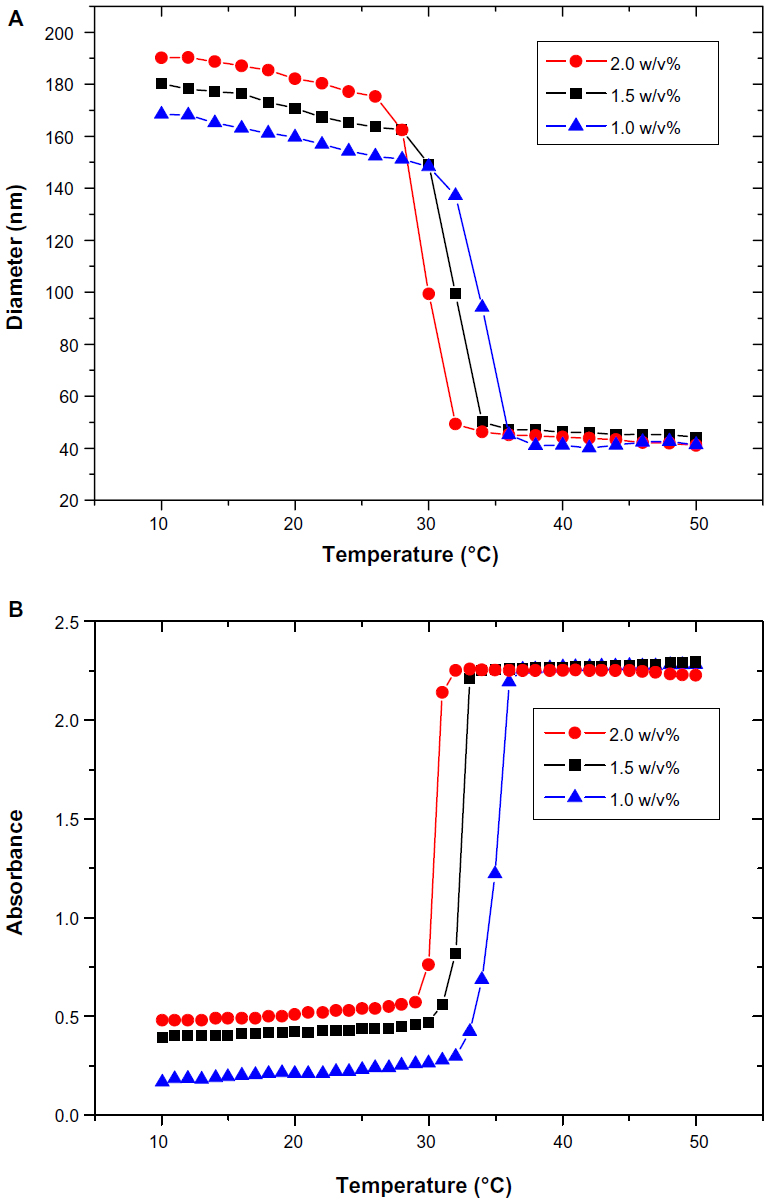 | Figure 3 Hydrodynamic diameters and UV absorbance of amphipathic poly(NIPAM-co-SA). |
Previously, EHD jetting of a poly(NIPAM-co-SA) solution in methanol homogeneously produced thermally responsive poly(NIPAM-co-SA) nanofiber matrices with a rapid swelling–deswelling property at the single nanofiber level, as the organic solvent-based method prevented aggregation during evaporation of the jet stream.24 Alternatively, EHD jetting of a poly(NIPAM)-based copolymer solution in water induced aggregation of the polymer chains due to the water evaporation, resulting in the formation of beads-on-strings and nanofibrous structures.34 Similarly, hydrogen bonding between water and polymer chains also plays an important role, as it is the most critical factor with respect to the thermo-responsive property when a poly(NIPAM)-based copolymer solution in water was electrospun.14 In addition, solubility of polymer chains in a solvent is another important parameter, which causes aggregation instead of interacting with the solvent. The relative solubility of poly(NIPAM-co-SA) in water was lower than that of the other organic solvents such as methanol, ethanol, TFE, and acetone. Therefore, ethanol and an ethanol–TFE mixture were selected as organic solvents for poly(NIPAM-co-SA) and PEGDMA, respectively.
Figure 4 shows the SEM and CLSM images of poly(NIPAM-co-SA) and PEGDMA nanofibers in the dry and swollen states at a low temperature of 10°C and a higher temperature of 40°C, respectively. As determined by the SEM images of five different batches (N=5) per nanofiber, poly(NIPAM-co-SA) nanofibers in Figure 4A have an average diameter of 185 ± 7 nm, while PEGDMA nanofibers in Figure 4B have an average diameter of 266 ± 11 nm, which is in good agreement with the diameters of their CLSM images in Figure 4C and D. These images show uniform nanofibrous structures without any beads-on-a-string structures. When poly(NIPAM-co-SA) and PEGDMA nanofibers were immersed in PBS at pH 7.4, they were swollen at similar degrees due to stabilization via physical and chemical crosslinking. As previously reported,6 poly(NIPAM-co-SA) nanofibers stabilized by physical crosslinks of hydrophobic long alkyl side chains clearly showed a thermally responsive volume change as a function of temperature. Figure 4E and G indicate that the average diameter of poly(NIPAM-co-SA) nanofibers in the swollen state significantly changed from 1.48 μm to 0.94 μm as the temperature increased from 10°C below its LCST to 40°C above its LCST because the poly(NIPAM-co-SA) chains instantaneously collapsed above its LCST. The nanoscaled thermal transition of poly(NIPAM-co-SA) nanofibers was consistently observed in a series of experiments conducted with various sizes of poly(NIPAM-co-SA) nanofibers. Alternatively, Figure 4F and H shows that PEGDMA nanofibers with the average diameter of 1.84 μm in the swollen state showed no volume transition at either temperature due to the nonthermoresponsive PEGDMA chains and stability in PBS due to UV-initiated chemical crosslinking. It is important to physically or chemically crosslink water-soluble polymers in various nanostructures to make them resistant to water dissolution.3 Intermolecular crosslinking of the polymer chains help to retain the original geometry under aqueous conditions and act as a nanoscaled, soft hydrogel embedded with water. Crosslinking of poly(NIPAM)- or PEGDMA-based hydrogels has been achieved through various mechanisms including physical-,3 chemical-,25 or photo-initiated crosslinking.34
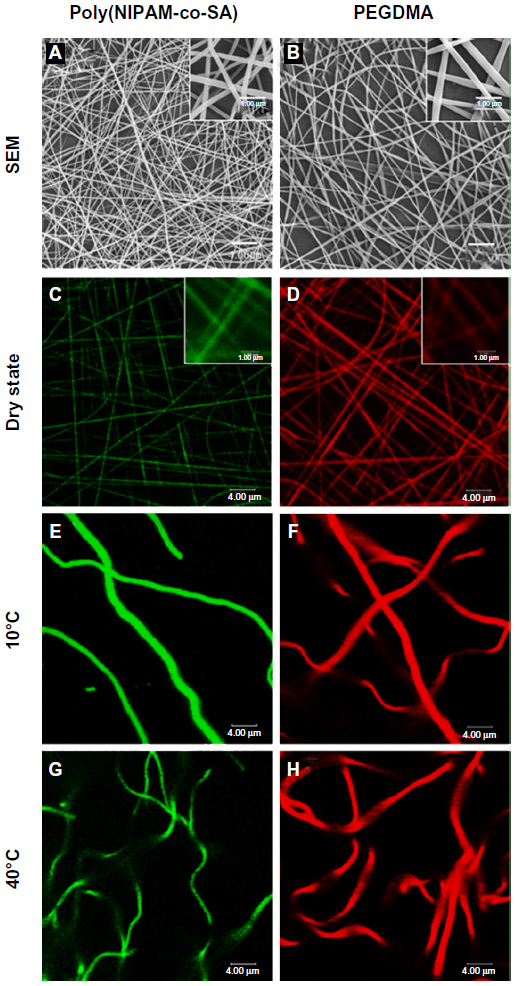 | Figure 4 SEM and CLSM images of nanofibers of poly(NIPAM-co-SA) and PEGDMA. |
With advances in the preparation of single compartmental poly(NIPAM-co-SA) and PEGDMA nanofibers, multicompartmental polymeric nanofibers with physically and chemically distinct regions produced via EHD cojetting act as novel materials for multiple functions.4 Separate compartments of biphasic nanofibers were loaded with different nanomaterials or drugs to provide multiple functionalities.11 Generally, two compartments are originally generated and then chemically modified to introduce different physical and chemical properties.34 It was reported that various shape reconfigurations at the microscale, such as bending hydrogels,34,41 coiled structures,14,41 and chemomechanical actuators,42 occurred when one compartment was stimuli-responsive, while the counter compartment was stimuli-unresponsive. Preparation of multicompartmental nanostructures is largely affected by rheological properties and fluid dynamics of different polymer solutions under an externally applied electric field during the EHD cojetting.34 When various methodologies to stabilize multicompartmental nanostructures are introduced, chemical crosslinking requires extra crosslinkers in the polymer jetting solution, possibly changing its rheological properties during EHD jetting.34 However, physical crosslinking based on electrostatic and hydrophobic interactions makes the rheological properties constant throughout the EHD jetting process without any crosslinking agents.6 Different polymers were modified to introduce hydrophobic interactions for physical and chemical stability, resulting in the formation of original nanostructures that contained cyclodextrins with pendant-hydrophobic groups for reversible molecular interaction of polymer backbone chains.43 In addition, thermoresponsive poly(NIPAM) core–shell microstructures, where the shell was highly modified with hydrophobic long alkyl chains, were stabilized when exposed to water.44 In this respect, totally dissimilar jetting solutions separately containing 1) thermoresponsive and physically crosslinkable poly(NIPAM-co-SA), and 2) nonthermoresponsive and UV-initiated, chemically crosslinkable PEGDMA, were EHD cojetted by maintaining balanced rheological properties and fluid dynamics of both solutions, resulting in the formation of bicompartmental nanofibers with distinct compartments for stimuli-responsive actuation.
In general, the viscosity of a polymeric solution is directly proportional to its concentration, which has a pronounced effect on the stability of the cone–jet mode and on the morphology of resulting nanofibers.15,18 The liquid jet tends to break into droplets at low polymer concentrations, while keeping other parameters constant. On the other hand, the jet stream starts extending continuously and then forms continuous nanofibers at a critical polymer concentration. A further increase of the polymer concentration produces fibrous structures with the larger diameter. At very high polymer concentrations, the fluid is unable to flow, and clogs in the spinneret. In addition, the surface tension of a polymer solution largely depends on the solvent, which is critical to produce a stable Taylor cone.17 In order to avoid detachment of the two polymer solutions during EHD cojetting, a miscible solvent set of ethanol for poly(NIPAM-co-SA), an ethanol–TFE mixture for poly(NIPAM-co-SA), and PEGDMA was employed. The distance between two electrodes also affects the stability of the cone–jet mode.11 An increased distance between the electrodes produces nanofibers with decreased diameters and noncontinuous structures. The voltage applied between the electrodes is one of the most important parameters used to generate a stable and continuous cone–jet stream.17,18,31 Dripping of the polymer solutions was observed at 4.0 kV, and as the voltage increased to 6.5 kV, the Taylor cone started thinning and elongating at the vertex. A further increase in the voltage up to 9.0–10.0 kV resulted in the generation of a stable and continuous jet stream. As the voltage was increased above this threshold, the Taylor cone started wobbling and lost swirl-free meridian flow, while multiple jet streams were produced. A combination of solvents, polymer concentrations, applied voltage, and the distance between electrodes were optimized to prepare continuous anisotropic nanofibers.
Figure 5 shows the SEM and fluorescence images of the bicompartmental nanofibers composed of poly(NIPAM-co-SA) and PEGDMA compartments in the dry state, with separate fluorescence signals of the fluorescein and Nile red channels. The bicompartmental nanofibers in Figure 5A and B have an average diameter of 327 ± 8 nm, as determined by SEM images of five different batches (N=5). Although elliptical morphology of the nanofibers was observed at high magnification, their uniform nanofibrous structures without beads-on-a-string were also observed for the poly(NIPAM-co-SA) and PEGDMA nanofibers, which is in good agreement with the previous report on multicompartmental microcylinders.45 Similarly, Figure 5C and D clearly illustrate that the discrete nanofibrous structures in the dry state were retained while having two fluorescence signals of both the fluorescein and Nile red channels without any phase separation during the EHD cojetting.
 | Figure 5 SEM and fluorescence images of the bicompartmental nanofibers composed of poly(NIPAM-co-SA) and PEGDMA compartments. |
Figure 6 shows CLSM images of the bicompartmental nanofibers in the dry and swollen states at a low temperature of 10°C and at a higher temperature of 40°C with fluorescein, Nile red, and combined fluorescence signals. The CLSM images indicate that both compartments maintained the nanofibrous structures and distinct interfaces in both the dry and swollen state at 10°C and 40°C were observed. Therefore, the hydrophobic stearyl side chains in poly(NIPAM-co-SA) were physically crosslinked via inter- and intramolecular hydrophobic interactions, while PEGDMA chains were chemically crosslinked through a facile and mild reaction mechanism of photo-initiated crosslinking.46 Both crosslinking strategies applied to each compartment helped to produce homogeneous biphasic nanofibers because the rheological properties (including complex viscosity and surface tensions of both polymer solutions) were balanced throughout the EHD cojetting process. As previously mentioned,34 it is difficult to control the rheological properties in the other chemical crosslinking mechanisms, which could produce beads-on-a-string structure or a mixture of nanofibers and nanoparticles. When the bicompartmental nanofibers were immersed in PBS at pH 7.4, their average diameter changed from 0.32 μm to 2.05 μm because both compartments were swollen at similar degrees, as observed in Figure 4E and F. Furthermore, CLSM images of the bicompartmental nanofibers in Figure 6F and I showed that their average diameter in the swollen state changed from 2.05 μm to 1.79 μm. Dramatic structural changes at the nanoscale occurred from the uncoiled to coiled state due to significant conformational changes of the polymer chains within the poly(NIPAM-co-SA) compartment as the temperature increased above its LCST from 10°C to 40°C, while the PEGDMA compartment showed no volume transition due to the nonthermoresponsive PEGDMA chains. As observed in real-time monitoring of their physical changes through thermally-induced actuation within the poly(NIPAM-co-SA) compartment, the reversible structural transition from an uncoiled form in the extended, relaxed structure at 10°C in Figure 6D–F to a coiled form in the collapsed, tense structure at 40°C in Figure 6G–I is potentially due to completely different physical and chemical properties of two distinct compartments. As the temperature increased from 10°C to 40°C, the uncoiled nanofibers turned into coiled structures while maintaining distinct interfaces between the two compartments. It was also confirmed that the thermoresponsive and physically-crosslinked poly(NIPAM-co-SA) compartment was always on the inside of the coiled structure, while nonthermoresponsive and chemically-crosslinked PEGDMA was located on the outside of the coils, as clearly shown in the inset of Figure 6I. This similar behavior was observed elsewhere, potentially due to different degrees of hydration of each compartment.15 Additionally, bending of the poly(NIPAM)-based hydrogel structures was previously reported,47 which is similar to the bimetal strip used in temperature-controlled electrical switches, where one metal has a greater thermal coefficient of expansion.
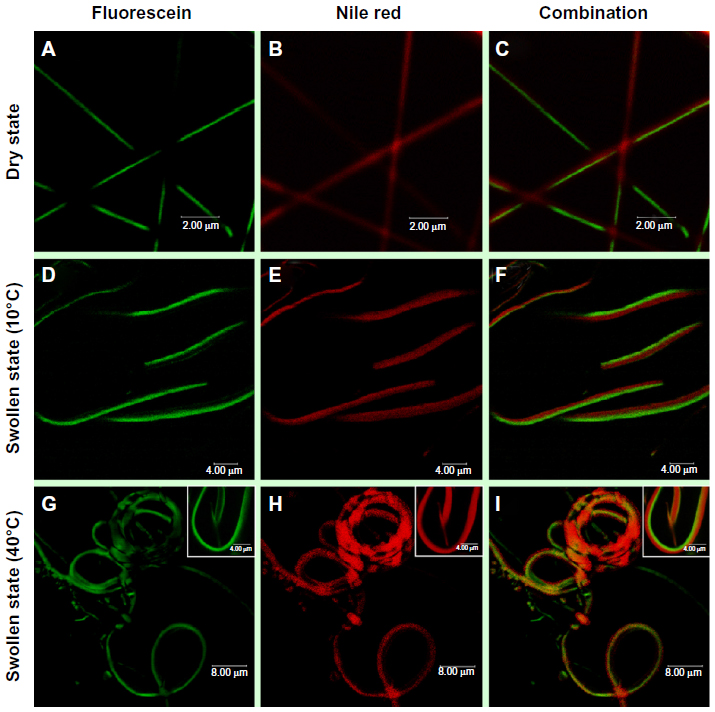 | Figure 6 CLSM images of the bicompartmental nanofibers in the dry and swollen state. |
Figure 7 shows decoupled in vitro drug release kinetics from the dual drug-loaded, bicompartmental nanofibers composed of poly(NIPAM-co-SA) and PEGDMA at various temperatures. Two different model drugs, DMP and BSA, were alternatively loaded into each compartment as small molecular and biomacromolecular drugs, respectively. Two different sets of the dual drug-loaded bicompartmental nanofibers were prepared: 1) a BSA-loaded poly(NIPAM-co-SA) compartment and a DMP-loaded PEGDMA compartment; and 2) a DMP-loaded poly(NIPAM-co-SA) compartment and a BSA-loaded PEGDMA compartment. The different sets of the dual drug-loaded nanofiber solutions were maintained in a suspension state at 4°C or 37°C to observe the temperature-controlled drug release. Five different batches were prepared to study batch-to-batch variability of the in vitro drug release from each compartment. As shown in Figure 7A, BSA from the BSA-loaded poly(NIPAM-co-SA) compartment was released at a slower rate at 37°C, which is above the LCST, rather than at 4°C, which is below the LCST, because aggregation of the poly(NIPAM-co-SA) chains at 37°C caused the hydrated pore size of the poly(NIPAM-co-SA) compartment to decrease, trapping BSA within the physically-crosslinked poly(NIPAM-co-SA) compartment. While the hydrated pore size of the chemically-crosslinked PEGDMA compartment was maintained at 4°C and 37°C, the DMP from the DMP-loaded PEGDMA compartment was released faster at 37°C than at 4°C, potentially due to enhanced mobility of DMP as the small molecular drug through the hydrated pores of PEGDMA network under dynamic suspension condition. In addition, Figure 7B shows the effect of the MWs of the drugs loaded into each compartment on the controlled release kinetics at 4°C and 37°C. Although the hydrated pore size of the physically-crosslinked poly(NIPAM-co-SA) compartment decreased at 37°C due to the collapsed poly(NIPAM-co-SA) chains, DMP was released at faster rates at 37°C rather than 4°C because DMP is smaller than the collapsed hydrated pore size, resulting in enhanced molecular mobility; this is in good agreement with the DMP release profiles, as shown in Figure 7A. Alternatively, BSA was released at similar controlled release rates at 4°C and 37°C, potentially because the BSA (as a biomacromolecule) was mostly entrapped within the hydrated, chemically-crosslinked PEGDMA compartment. Furthermore, Figure 8 shows the temperature-dependent drug release profiles of the different sets of dual drug-loaded nanofiber solutions when incubated in suspension at 4°C or 37°C for every 10 minutes. The incubation temperature was periodically changed from 4°C to 37°C and from 37°C to 4°C every 10 minutes. As shown in Figure 8A, the BSA from the BSA-loaded poly(NIPAM-co-SA) compartment was released at different rates at 4°C and 37°C, which was due to the reversible conformational change of the poly(NIPAM-co-SA) chains as a function of temperature, indicating temperature-triggered pulsatile drug release profiles. On the other hand, the others showed diffusion-controlled drug release, as shown in Figure 8B. This systematic study suggests that thermally-responsive bicompartmental nanofibers composed of poly(NIPAM-co-SA) and PEGDMA compartments could be useful for advanced drug delivery systems with decoupled in vitro drug release kinetics of dual drugs.
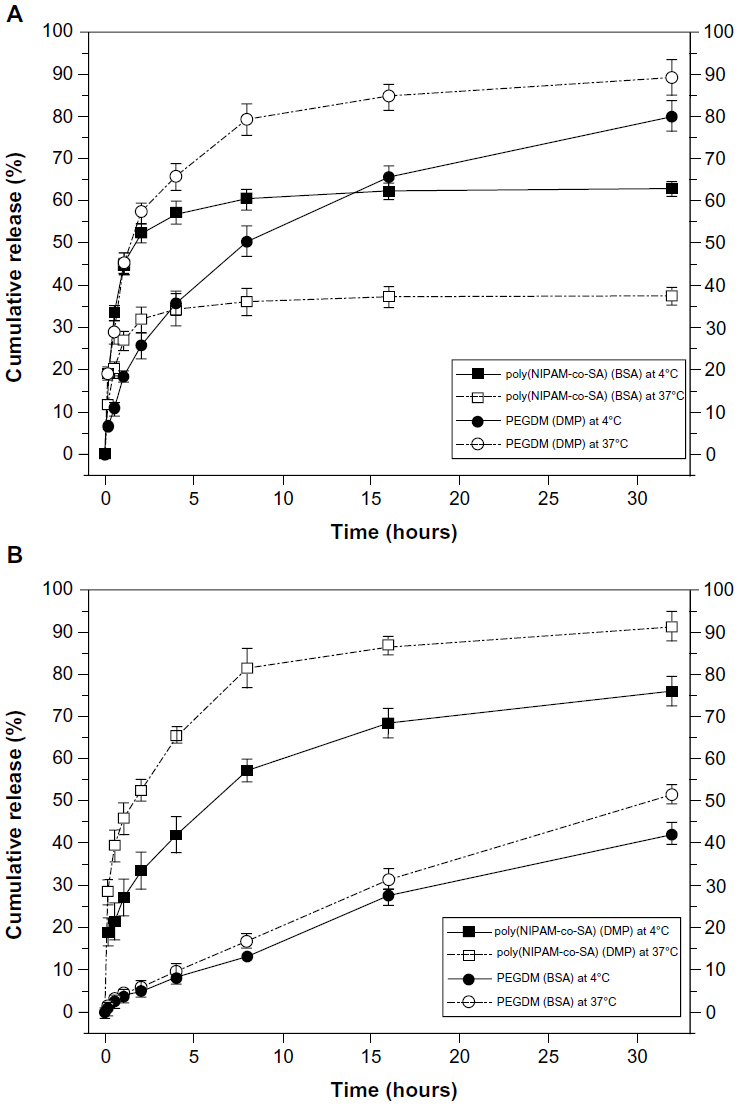 | Figure 7 Decoupled in vitro drug release kinetics from the dual drug-loaded bicompartmental nanofibers composed of poly(NIPAM-co-SA) and PEGDMA compartments at various temperatures. |
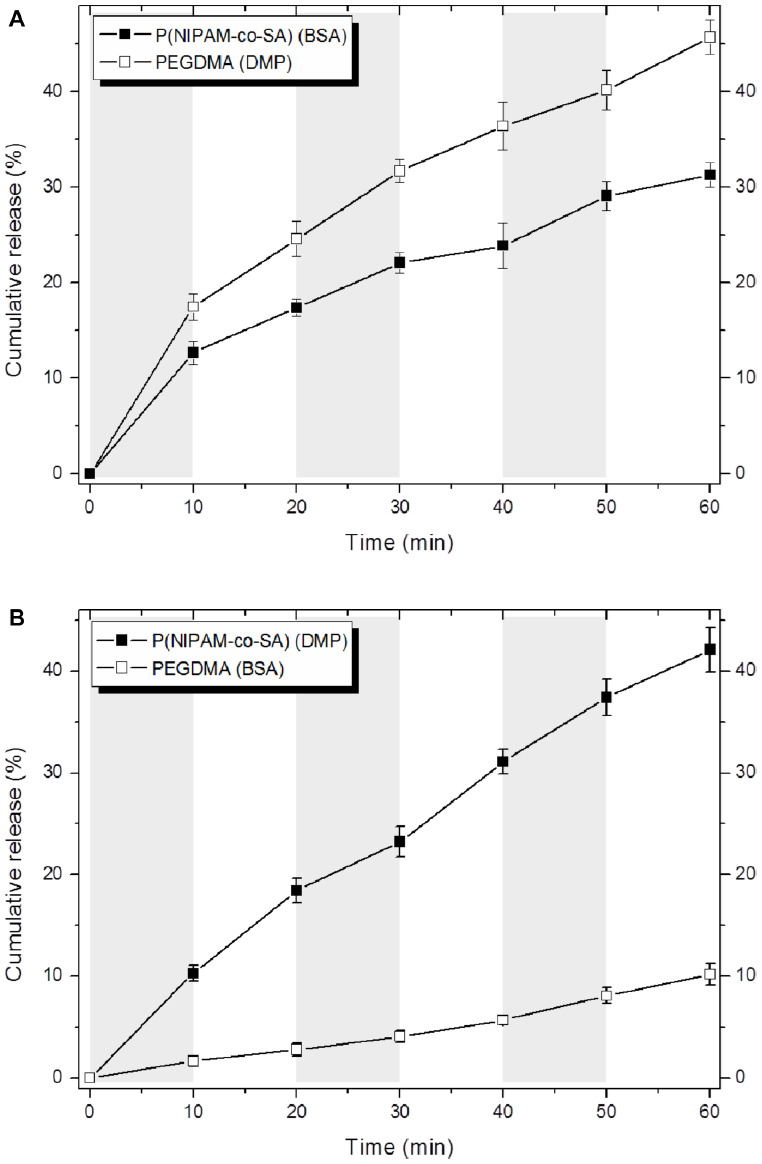 | Figure 8 Temperature-dependent drug release profiles of the different sets of dual drug-loaded nanofiber solutions when incubated in suspension at 4°C or 37°C for every 10 minutes. |
Conclusion
In summary, a new class of thermally-responsive bicompartmental nanofibers with physically- and chemically-distinct compartments was developed for a variety of multifunctional biomedical applications. The bicompartmental nanofibers were prepared via the EHD cojetting of totally different polymer solutions of thermoresponsive poly(NIPAM-co-SA) and nonthermoresponsive PEGDMA. Rational experimental design including stimuli-responsive polymers, their MWs, solvent composition, and crosslinking mechanisms produced uniform bicompartmental nanofibers with a discrete interface between two compartments on a large scale, by maintaining balanced rheological and fluid dynamic properties of both polymer solutions during EHD cojetting. Thermally-triggered shape shifting of the bicompartmental nanofibers from an uncoiled to coiled configuration at the nanoscale was due to thermally-induced aggregation of poly(NIPAM-co-SA) chains and the PEGDMA of the counter compartment. Future work will focus on multicompartmental nanofibers or nanoparticles with more than two compartments having distinct stimuli-responsive properties for therapeutic applications. Therefore, the thermally-triggered actuation of the bicompartmental nanofibers makes them useful for advanced drug delivery systems with the stimuli-triggered, decoupled drug release of multiple drugs.
Acknowledgments
This work was supported by the National Research Foundation of Korea, and was funded by the Ministry of Education, Science and Technology, Korea (2010-0013284, 2012R1A1A2043792, 2012-0009838, and R11-2008-044-01002-0), the Ministry of Science, Information and Communication Technology (ICT) and Future Planning, Korea (2013R1A2A2A04016796), and by the Agency for Defense Development through the Chemical and Biological Research Center.
Disclosure
The authors report no conflicts of interest in this work.
References
Zapotoczny S. Stimuli responsive polymers for nanoengineering of biointerfaces. Methods Mol Biol. 2012;811:51–78. | |
Cole MA, Voelcker NH, Thissen H, Griesser HJ. Stimuli-responsive interfaces and systems for the control of protein-surface and cell-surface interactions. Biomaterials. 2009;30(9):1827–1850. | |
Wang N, Zhao Y, Jiang L. Low-cost, thermoresponsive wettability of surfaces: poly(N-isopropylacylamide)/polystyrene composite films prepared by electrospinning. Macromol Rapid Commun. 2008;29(6):485–489. | |
Chen M, Dong M, Havelund R, et al. Thermo-responsive core-sheath electrospun nanofibers from poly (N-isopropylacrylamide)/polycaprolactone blends. Chem Mater. 2010;22(14):4214–4221. | |
Valiaev A, Abu-Lail NI, Lim DW, Chilkoti A, Zauscher S. Microcantilever sensing and actuation with end-grafted stimulus-responsive elastin-like polypeptides. Langmuir. 2007;23(1):339–344. | |
Okuzaki H, Kobayashi K, Hishiki F, Su SJ, Yan H. Thermo-responsive nanofiber mats fabricated by electrospinning of poly(N-isopropylacrylamide-co-stearyl acrylate). J Nanosci Nanotechnol. 2011;11(6):5193–5198. | |
Loh XJ, Peh P, Liao S, Sng C, Li J. Controlled drug release from biodegradable thermoresponsive physical hydrogel nanofibers. J Control Release. 2010;143(2):175–182. | |
Bhaskar S, Hitt J, Chang SW, Lahann J. Multicompartmental microcylinders. Angew Chem Int Ed Engl. 2009;48(25):4589–4593. | |
Abu-Lail NI, Kaholek M, LaMattina B, Clark RL, Zauscher S. Micro-cantilevers with end-graft stimulus-responsive polymer brushes for actuation and sensing. Sens Actuators B Chem. 2006;114(1):371–378. | |
Song M, Guo D, Pan C, et al. The application of poly(N-isopropylacrylamide)-co-polystyrene nanofibers as an additive agent to facilitate the cellular uptake of an anticancer drug. Nanotechnology. 2008;19(16):165102. | |
Bhaskar S, Lahann J. Microstructured materials based on multicompartmental fibers. J Am Chem Soc. 2009;131(19):6650–6651. | |
Lim DW, Hwang S, Uzun O, Stellacci F, Lahann J. Compartmentalization of gold nanocrystals in polymer microparticles using electrohydrodynamic co-jetting. Macromol Rapid Commun. 2010;31(2):176–182. | |
Roh KH, Yoshida M, Lahann J. Compartmentalized, multiphasic nanocolloids with potential applications in drug delivery and biomedical imaging. Materwiss Werksttech. 2007;38(12):1008–1011. | |
Tzeng P, Kuo CC, Lin ST, Chiu YC, Chen WC. New thermoresponsive luminescent electrospun nanofibers prepared from poly[2,7-(9,9dihexyfluorene)]-block-poly(N-isopropylacrylamide)/PPMA blends. Macromol Chem Phys. 2010;211(13):1408–1416. | |
Saha S, Copic D, Bhaskar S, et al. Chemically controlled bending of compositionally anisotropic microcylinders. Angew Chem Int Ed Engl. 2012;51(3):660–665. | |
Roh KH, Martin DC, Lahann J. Biphasic Janus particles with nanoscale anisotropy. Nat Mater. 2005;4(10):759–763. | |
Roh KH, Martin DC, Lahann J. Triphasic nanocolloids. J Am Chem Soc. 2006;128(21):6796–6797. | |
Hayati I, Bailey AI, Tadros THF. Investigations into the mechanisms of electrohydrodynamic spraying of liquids. I. Effect of electric field and environment on pendant drops and factors affecting the formation of stable jets and atomization. J Colloid Interface Sci. 1987;117(1):205–221. | |
Li D, Xia Y. Electrospinning of nanofibers: reinventing the wheel? Adv Mater. 2004;16(14):1151–1170. | |
Thompson CJ, Chase GG, Yarin AL, Reneker DH. Effects of parameters on nanofiber diameter determined from electrospinning model. Polymer. 2007;48(23):6913–6922. | |
Okuzaki H, Kobayashi K, Yan H. Non-woven fabric of poly(N-isopropylacrylamide) nanofibers fabricated by electrospinning. Synth Met. 2009;169(21–22):2273–2276. | |
Sinha-Ray S, Zhang Y, Placke D, Megaridis CM, Yarin AL. Resins with “nano-raisins”. Langmuir. 2010;26(12):10243–10249. | |
Gu SY, Wang ZM, Li JB, Ren J. Switchable wettability of thermo-responsive biocompatible nanofibrous films created by electrospinning. Macromol Mater Eng. 2010;295(1):32–36. | |
Okeyoshi K, Abe T, Noguchi Y, Furukawa H, Yoshida R. Shrinking behavior of surfactant-grafted thermosensitive gels and the mechanism of rapid shrinking. Macromol Rapid Commun. 2008;29(11):897–903. | |
Rockwood DN, Chase DB, Akins RE, Rabolt JF. Characterization of electrospun poly(N-isopropyl acrylamide) fibers. Polymer. 2008;49(18):4025–4032. | |
Yu JH, Fridrikh SV, Rutledge GC. Production of submicrometer diameter fibers by two-fluid electrospinning. Adv Mater. 2004;16(17):1562–1566. | |
Yu DG, Zhou J, Chatterton NP, Li Y, Huang J, Wang X. Polyacrylonitrile nanofibers coated with silver nanoparticles using a modified coaxial electrospinning process. Int J Nanomedicine. 2012;7:5725–5732. | |
Yu DG, Zhu LM, Branford-White CJ, et al. Solid dispersions in the form of electrospun core-sheath nanofibers. Int J Nanomedicine. 2011;6:3271–3280. | |
Gulfam M, Lee JM, Kim JE, Lim DW, Lee EK, Chung BG. Highly porous core-shell polymeric fiber network. Langmuir. 2011;27(17):10993–10999. | |
Liu Z, Sun DD, Guo P, Leckie JO. An efficient bicomponent TiO2/SnO2 nanofiber photocatalyst fabricated by electrospinning with a side-by-side dual spinneret method. Nano Lett. 2007;7(4):1081–1085. | |
Gupta P, Wilkes GL. Simultaneous electrospinning of two polymer solutions in a side-by-side approach to produce bicomponent fibers. In: Reneker DH, Fong H, editors. Polymeric Nanofibers: ACS Symposium Series. Washington, DC: American Chemical Society; 2006. | |
Gao J, Frisken BJ. Influence of secondary components of the synthesis of self-cross-linked N-isopropylacrylamide microgels. Langmuir. 2005;21(2):545–551. | |
Kurisawa M, Chung JE, Yang YY, Gao SJ, Uyama H. Injectable biodegradable hydrogels composed of hyaluronic acid-tyramine conjugates for drug delivery and tissue engineering. Chem Commun (Camb). 2005;34:4312–4314. | |
Tang C, Saquing CD, Harding JR, Khan SA. In situ cross-linking of electrospun poly(vinyl alcohol) nanofibers. Macromolecules. 2009;43(3):630–637. | |
Kim K, Luu YK, Chang C, et al. Incorporation and controlled release of a hydrophilic antibiotic using poly(lactide-co-glycolide)-based electrospun nanofibrous scaffolds. J Control Release. 2004;98(1):47–56. | |
Zamani M, Morshed M, Varshosaz J, Jannesari M. Controlled release of metronidazole benzoate from poly epsilon-caprolactone electrospun nanofibers for periodontal diseases. Eur J Pharm Biopharm. 2010;75(2):179–185. | |
Park Y, Kang E, Kwon OJ, et al. Ionically crosslinked Ad/chitosan nanocomplexes processed by electrospinning for targeted cancer gene therapy. J Control Release. 2010;148(1):75–82. | |
Galassi G, Brun P, Radice M, et al. In vitro reconstructed dermis implanted in human wounds: degradation studies of the HA-based supporting scaffold. Biomaterials. 2000;21(21):2183–2191. | |
Saraf A, Baggett LS, Raphael RM, Kasper FK, Mikos AG. Regulated non-viral gene delivery from coaxial electrospun fiber mesh scaffolds. J Control Release. 2010;143(1):95–103. | |
Lin-Gibson S, Bencherif S, Cooper JA, et al. Synthesis and characterization of PEG dimethacrylates and their hydrogels. Biomacromolecules. 2004;5(4):1280–1287. | |
Beines PW, Klosterkamp I, Menges B, Jonas U, Knoll W. Responsive thin hydrogel layers from photo-cross-linkable poly(N-isopropylacrylamide) terpolymers. Langmuir. 2007;23(4):2231–2238. | |
Stuart MA, Huck WT, Genzer J, et al. Emerging applications of stimuli-responsive polymer materials. Nat Mater. 2010;9(2):101–113. | |
Mahammad S, Abdala A, Roberts GW, Khan SA. Manipulation of hydrophobic interactions in associative polymers using cyclodextrin and enzyme. Soft Matter. 2010;6:4237–4245. | |
MacKinnon N, Guérin G, Liu B, Gradinaru CC, Rubenstein JL, Macdonald PM. Triggered instability of liposomes bound to hydrophobically modified core-shell PNIPAM hydrogel beads. Langmuir. 2010;26(2):1081–1089. | |
Lee KJ, Yoon J, Rahmani S, et al. Spontaneous shape reconfigurations in multicompartmental microcylinders. Proc Natl Acad Sci U S A. 2012;109(40):16057–16062. | |
Anderson KD, Lu D, McConney E, Han T, Reneker DH, Tsukruk VV. Hydrogel microstructures combined with electrospun fibers and photopatterning for shape and modulus control. Polymer. 2008;49(24):5284–5293. | |
Pester CW, Konradi A, Varnholt B, van Rijn P, Boker A. Responsive macroscopic materials from self-assembled cross-linked SiO2-PNIPAAm core/shell structures. Adv Funct Mater. 2012;22(8):1724–1731. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.