")
Back to Journals » International Journal of Nanomedicine » Volume 18
Extracellular Vesicles Expressing CD19 Antigen Improve Expansion and Efficacy of CD19-Targeted CAR-T Cells
Authors Zhang Y, Ge T , Huang M, Qin Y, Liu T, Mu W, Wang G, Jiang L, Li T, Zhao L, Wang J
Received 5 October 2022
Accepted for publication 19 December 2022
Published 5 January 2023 Volume 2023:18 Pages 49—63
DOI https://doi.org/10.2147/IJN.S390720
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Dongwoo Khang
Yuanyuan Zhang,1,* Tong Ge,1,* Meijuan Huang,1 Yun Qin,2 Tianjiao Liu,3 Wei Mu,1 Gaoxiang Wang,1 Lijun Jiang,1 Tongjuan Li,1 Lei Zhao,1 Jue Wang1
1Department of Hematology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, People’s Republic of China; 2Department of Hematology, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, People’s Republic of China; 3Department of Hematology, Harbin Medical University Cancer Hospital, Harbin, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Lei Zhao; Jue Wang, Email [email protected]; [email protected]
Background: CAR-T cell therapy is effective in the treatment of certain hematological malignancies, and the expansion and functional persistence of CAR-T cells in vivo are crucial to clinical efficacy. The aim of this study was to investigate the potential of extracellular vesicles (EVs) modified with the CAR antigen to promote the efficacy of CAR-T cells in vivo.
Methods: We generated HEK293T-derived EVs to present the CD19 antigen as the CAR target. In vitro, EVs expressing CD19 antigen (CD19 EVs) were co-incubated with anti-CD19 CAR-T cells. Then, proliferation, cytokine secretion, CD107a expression, tumor killing, subsets, and immune checkpoint expression were measured to assess CAR-T cell function. After infusion of CD19 EVs pretreated CAR-T cells into a lymphoma xenograft mouse model, flow cytometry and digital PCR were used to measure the expansion of CAR-T cells, and tumor volumes were continuously monitored to assess the anti-tumor efficacy of CAR-T cells in vivo. Another mouse model was created to investigate the effect of in vivo injection of CD19 EVs on the functional persistence of CAR-T cells, and safety was determined by histopathology of the main organs.
Results: CD19 EVs activated CAR-T cells in an antigen-specific and dose-dependent manner and promoted the selective expansion and cytokine secretion of co-cultured CAR-T cells. Specifically, CD19 EVs preferably increased the expansion of the CAR-T subpopulation with a high surface CD19-CAR density and consequently enhanced the anti-tumor activity of CAR-T cells. Futhermore, CD19-EVs-primed CAR-T cells achieved superior proliferation and anti-tumor effects in a mouse model with lymphoma xenograft. In vivo administration of CD19 EVs promoted the functional persistence of CAR-T cells in the xenograft mouse model.
Conclusion: Our findings indicate that antigen-expressing EVs can be utilized as a boost to improve CAR-T cell efficacy in vitro and in vivo.
Keywords: immunotherapy, chimeric antigen receptor T-cell, extracellular vesicles, CD19 antigen
Introduction
Chimeric antigen receptor (CAR)-T cell immunotherapy has been clinically successful in patients with certain types of leukemia and lymphoma, achieving a high percentage of durable complete remissions.1–4 However, poor expansion or persistence of CAR-T cells in some patients may result in disease progression.5,6 It has been reported that robust proliferation of CAR-T cells correlates with better clinical response,7 and the functional persistence of CAR-T cells in vivo is necessary for long-term remission in patients.8,9 Conversely, insufficient expression of antigens is shown to lead to inefficient expansion of CAR-T cells,10 whereas antigen-positive or antigen-dim relapses are associated with rapid loss of functional CAR-T cells.11 To enhance CAR-T cell proliferation and persistence in vivo, several groups have developed different strategies to deliver CAR target antigens to stimulate adoptively transferred CAR-T cells. For example, a RNA vaccine encoding a CAR-specific antigen and a peptide vaccine carrying CAR-ligands was developed to modify dendritic cells (DCs) to display target antigens to activate CAR-T cells, which significantly promoted their expansion and persistence in vivo. In addition, the anti-tumor effect of CAR-T cells was improved in mouse models.12,13 These studies have introduced the concept of activating CAR-T cells with target antigens to promote their efficacy. However, the current approaches require the help of antigen presenting cells (APCs), and whether direct stimulation of CAR-T cells is feasible needs to be explored.
As a fast-developing field in nanomedicine, extracellular vesicles (EVs) have been exploited as cell membrane-based nanovesicle delivery systems.14 EVs are released by almost all cells and are considered as important regulators of intercellular signaling.15 Recent studies confirmed that EVs could affect T-cell function through molecules exposed on the surface of the EV membrane.16,17 Furthermore, surface molecules carried by EVs could interact with receptors on CAR-T cells and thus regulate their functions.18,19 Therefore, stimulating CAR-T cells by EVs armed with target antigen could be a potent strategy for facilitating expansion and functional persistence of CAR-T cells.
In this study, we used CD19, the most widely used target in CAR-T cell therapy, as an example to validate the potential of this strategy. We genetically modified the HEK293T cell line to obtain EVs with high surface CD19 antigen expression (CD19 EVs). CAR-T cells targeting CD19 through a single chain variable fragment (scFv) were tested for function and persistence when stimulated by CD19 EVs both in vitro and in vivo. Our data provide the first proof of concept evidence for the feasibility and safety of the administration of EVs with CAR target antigen for tunable expansion of CAR-T cells.
Materials and Methods
Cell Lines and Cell Culture
The human embryonic kidney cell line HEK293T, the Burkitt lymphoma cell line Raji, and the human chronic myeloid leukemia cell line K562 were obtained from the ATCC. Cells were cultured in Dulbecco’s modified Eagle medium or RPMI 1640 supplemented with 10% fetal bovine serum (Gibco, NY, USA) at 37°C in a 5% CO2 atmosphere.
Animals
All animal studies were approved by the Animal Care and Use committee of Tongji Hospital following National Institutes of Health guidelines (NIH publication 86–23 revised 1985) and in accordance with Institutional Animal Care and Use Committee (IACUC) requirements. Female NCG mice (5–6 weeks old) were purchased from GemPharmatech (China). Mice were kept under specific-pathogen-free (SPF) conditions.
Plasmid
For CD19 expression, the lentiviral vector pCDH-CMV-CD19 was purchased from GeneCopoeia. For CD19 knockdown, short hairpin RNA (shRNA) targeting the CD19 sequence (GGGCATTCTTCATCTTCAAAG) was designed and synthesized by Sangon (Shanghai, China), and then cloned into pLKO.1 lentiviral vector to form recombinant lentiviral shRNA. The anti-CD19 CAR lentiviral plasmid was constructed as previously described.20
Lentivirus Preparation and Transfection
The lentiviral particles were prepared by PEI co-transfection of HEK293T cells with the corresponding lentiviral plasmids and the packaging plasmids. The supernatants were harvested 48 h after transfection and centrifuged for 30 min at 3000 g at 4°C to remove cells and then passed through a 0.45-μm filter (Thermo Scientific) to remove cellular debris. The supernatant was further centrifuged for 2.5 h at 30,000 g at 4°C (Avanti J-26S XPI High-performance Centrifuge, Beckman Coulter). The concentrated lentiviral stocks were frozen at −80°C for future use. HEK-293T and Raji cells were infected with lentivirus and sorted by FACS.
CAR-T Cell Production
Peripheral blood was obtained from healthy donors and patients. Human peripheral blood mononuclear cells (PBMCs) were then isolated using lymphocyte separation solution (Axis-Shield) according to the manufacturer’s instructions. Human T cells isolated from PBMCs using the CD3 MicroBeads kit (Miltenyi, 130-097-043) were activated with CD3/CD28 Dynabeads (Thermo Scientific, 11131D) in medium (Gibco, A3021002) containing 200 IU mL−1 recombinant IL-2 (PeproTech) for 24 h. Activated T cells were transduced with anti-CD19 CAR lentivirus at multiplicity of infection of 3, and cultured for 10–14 days. The medium was renewed every 2–3 days and the cell concentration was adjusted to 0.5–2.5×106 mL−1.
Flow Cytometry
The following antibodies were used for flow cytometry analysis: anti-human CD19-APC (BioLegend), anti-human CD3-BV510 (BD Biosciences), anti-human CD4-PerCP–Cy5.5 (BioLegend), anti-human CD8-PE–Cy7 (BioLegend), anti-human CD45-FITC (BD Biosciences), anti-human CD45RO-PE (BioLegend), anti-human CD62L-BV605 (BD Biosciences), anti-human CD62L-APC-Cy7 (BioLegend), anti-human PD-1-BV785 (BioLegend), anti-human TIGIT-BV421 (BioLegend), anti-human LAG3-BV650 (BioLegend), anti-human PD-L1-PE (BioLegend), PE-conjugated CD19 protein (ACROBiosystems). For the CFSE dilution assay, T cells were labeled with 5 μM CFSE (Thermo Fisher Scientific) before culture. The degranulation of CAR-T cells was detected by staining with APC-conjugated CD107a antibody (BioLegend) for 4 h. For flow cytometry analysis of tumor from mice, tumors were excised from mice on day 4 after adoptive T cell transfer (ACT) and dissociated into a single-cell suspension by using tumor dissociation kit (Miltenyi, 130-095-929).
Flow cytometry staining was carried out in a 1:100 antibody dilution with 1:100 of Fc block (Biolegend) and analyzed on a NovoCyte Flow Cytometer (ACEA Biosciences) or a MACSQuant Analyzer 10 Flow Cytometer (Miltenyi Biotec). Flow cytometry analysis of the EVs was performed on a CytoFLEX flow cytometer (Beckman Coulter), as previously described.21 The data analysis was performed using the FlowJo software (Tree Star).
EVs Isolation and Quantification
HEK293T and HEK293T-CD19 were transferred into serum-free medium for 24 hours. Then, control EVs and CD19 EVs were isolated from the supernatants of HEK293T and HEK293T-CD19 cells, respectively, as previously described.21 Briefly, the supernatants were centrifuged at 500 g for 10 min and then passed through 0.45-μm sterile syringe filters. The supernatant was then centrifuged at 14,000 g at 4°C for 1 h (Beckman Coulter) to pellet EVs.
For quantification, isolated EVs were lysed with RIPA lysis buffer (Beyotime, China) containing a protease inhibitor cocktail (Roche, Switzerland), and the protein content was quantified by using a BCA assay kit (Beyotime, China).
Nanoparticle Tracking Analysis
After EVs were purified and diluted in PBS, nanoparticle tracking analysis (NTA) was performed on the ZetaView Multiple Parameter Particle Tracking Analyzer (Particle Metrix) to observe the Brownian motion of EVs and determine the particle sizes.
Western Blotting
Following the lysis and quantification of EVs, samples were loaded with loading buffer and heated at 100°C for 10 min. Equal amounts of protein (40 μg) from each sample were separated on SDS-PAGE gels and transferred to PVDF membranes (Millipore, USA). The membrane was blocked for 1 h at 25°C using 5% non-fat milk and then incubated overnight at 4°C with gentle rocking in the presence of anti-Annexin A1 antibody (Abcam, ab214486, 1:2000), and anti-CD19 antibody (Abcam, ab245235, 1:1000). The membrane was incubated with HRP-conjugated secondary antibodies for 1 h at room temperature. Immunoblots were visualized with chemiluminescence using Pierce ECL reagent (Thermo Fisher, USA).
Cytokine Detection
For CAR-T cell cytokine release assay in vitro, CAR-T cells were resuspended at a concentration of 1×106 mL−1. Then, 300 μL of suspension was transferred to a 48-well plate and incubated with PBS or EVs for 24 h. The supernatant was separated from cells by centrifugation and stored at −80°C until testing. IFN-γ, TNF-α and IL-2 secretion by CAR-T cells was analyzed using cytokine-specific ELISA kits (Neobioscience Technology, China).
Cytotoxicity Assay
Target cells, including Raji WT cells, Raji Dim cells, and K562 cells, were labeled with 1 μM CellTraceTM CFSE or Violet (ThermoFisher) and subsequently co-cultured with CAR-T cells for 24 h with an effector to target (E:T) ratio of 1:1. Dead target cells were determined by using propidium iodide (PI) (BD Pharmingen) staining. The percentage of deaths of CellTrace-labeled target cells was used to evaluate the cytotoxicity of CAR-T cells.
Engraftment of Tumor Cells
Raji cells were injected subcutaneously into the right flank of mice at 5×106 cells in 200 μL of PBS. Every 3 or 4 days, tumor sizes were calculated according to the formula (width2 x length)/2. Mice were euthanized when they showed signs of illness or tumor ulceration with volumes exceeding 1500 mm3.
Adoptive Cell Transfer and EVs Treatment in vivo
A total of 1×106 CAR-T cells were infused through tail vein injection and 200 μg of CD19 EVs was administered by tail vein injection on day 1 and 5 after ACT. Mice treated with control EVs or PBS of the same volume served as controls.
CD19 EVs Toxicity Analysis
Serum was extracted 72 h after CAR-T cell infusion. Serum cytokine levels were quantified using human Th1/Th2 cytokine cytometric beads array kit (BD Biosciences) following the manufacturer’s instructions. CAR-T cell therapy and CD19 EVs-induced body weight fluctuations were measured every 3 or 4 days.
To assess liver and kidney toxicity induced by EVs, liver and kidney tissue were collected at designated time points after therapy and grinded with 9 times the volume of saline to make 10% tissue homogenate. Alanine aminotransferase (ALT), aspartate aminotransferase (AST) and creatinine (CRE) levels were analyzed according to the manufacturer’s protocol (BIOBASE, 70911, 70910, 70927).
Droplet Digital PCR
The expansion and persistence of CAR-T cells in vivo were measured by ddPCR, as previously reported.22 Peripheral blood was collected from mice at designated time points. The sequence of the CAR primers is as follows: forward primer, 5’-CAGCAAAAATACGACCTCCTCACT-3’; reverse primer, 5’-TGGTGCTGCCTTTGATCTCA-3’; and probe, 5’-FAM-TTGGCGGAGGGACC-3’.
Histopathological Analysis and Immunohistochemistry
Tumor tissues and organs were removed from mice, fixed in formalin, embedded in paraffin, and dissected into 3 mm-thick sections. Organ slides were stained with H&E. Fixed tumor slides were incubated with anti-human CD3 antibody (Abcam, Ab1669, 1:100) overnight at 4°C to detect the presence of human T cells in tumors. The slices were then treated with a secondary antibody and visualized using 3,3′-diaminobenzidine. The cell nuclei were stained with hematoxylin.
Statistical Analysis
All experiments were performed at least three times and all measurement data were presented as the mean ± SEM. All data were analyzed using GraphPad Prism 7.0. Statistical significance of differences between two groups was assessed using a two-tailed paired or unpaired t-test, comparisons between more than two groups were carried out by one-way ANOVA and two-way ANOVA. Animal survival was analyzed using Log-rank (Mantel-Cox) test. p < 0.05 was considered statistically significant and significance is designated with asterisks (ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001).
Study Approval
All experiments in human subjects were approved by the Ethics Committee of Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China (decision on 30.12.2020) and carried out in accordance with the approved protocols.
Results
Establishment and Characterization of CD19 EVs
To obtain CD19 EVs, we established a HEK293T monoclonal cell line to express CD19 on the cell surface by transfection of lentivirus. Control EVs were produced by HEK293T cells transfected with a control vector. Control EVs and CD19 EVs were examined by NTA, and their Brownian motions and size distribution were monitored (Figure S1A and S1B). The particle sizes of the control EVs and CD19 EVs were both ranged between 50 and 450 nm, which was consistent with the typical particle size of EVs.23 The data indicated that CD19 expressed on the EV membrane surface did not significantly influence its physical properties, such as size and motion state. Based on the centrifugation speed used in this study, the EVs obtained were mainly microvesicles (MVs), a type of EVs directly shed from the cell membrane, and their specific marker was Annexin A1.23 Western blotting confirmed that Annexin A1 was expressed by both types of EVs, and CD19 was expressed significantly only in CD19 EVs (Figure S1C). Flow cytometry was used to identify CD19 expression on the membrane surface of CD19 EVs (Figure S1D). PD-L1 was hardly expressed on the surface of both control and CD19 EVs (Figure S1E).
CD19 EVs Activate CAR-T Cells and Promote Their Expansion and Functional Maturation
First, we characterized CD19 CAR-T cells that were derived from healthy donors in combination with EVs in vitro. CD19 EVs promoted stimulation, cytokine secretion and proportion of co-cultured CAR-T cells in a dose-dependent manner, reaching a peak at a concentration of 700 μg/mL of CD19 EVs (Figure 1A and B). At a concentration of 1350 μg/mL of CD19 EVs, the levels of cytokine secretion and percentage of CAR-T cells plateaued after reaching high levels, suggesting a saturation threshold presumably for CAR-T cells being fully activated by a sufficient amount of target antigens presented by 700 μg/mL of CD19 EVs. Therefore, we adopted 700 μg/mL as the concentration for co-culturing of EVs for the subsequent experiments.
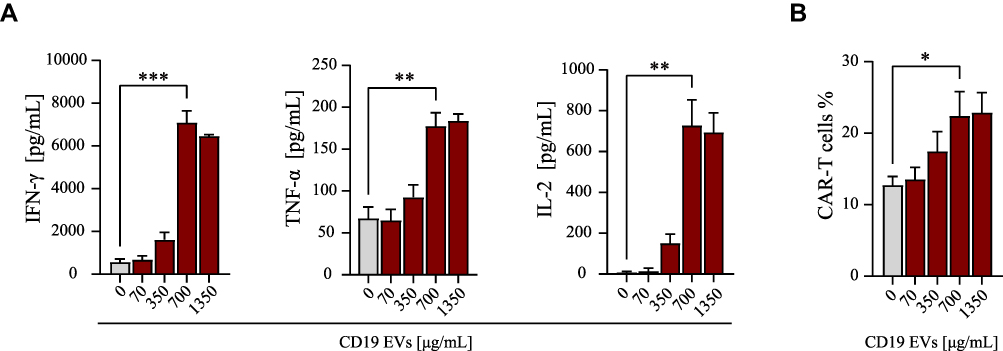 |
Figure 1 Activation of CAR-T cells by CD19 EVs is dose-dependent. (A) Cytokine secretion of CAR-T cells analyzed by ELISA after 24 h of co-culture with escalating amounts of CD19 EVs. (B) Proportion of CAR-T cells analyzed by flow cytometry after 72 h of co-culture with escalating amounts of CD19 EVs. *P < 0.05, **P < 0.01, ***P < 0.001. |
After co-incubation of CAR-T cells with PBS, control EVs, or CD19 EVs for 72 h, we found that the proportion of CAR-T cells was significantly higher in the CD19 EVs group compared to the PBS group, and further analysis revealed that the proportion of CAR-T cells was higher in both CD4+ T cells and CD8+ T cells, while the proportion of CAR-T cells in the control EVs group did not differ from that in the PBS group (Figures 2A and S2A). The CAR-T cell proliferation in the presence of CD19 EVs was boosted, while control EVs lacking antigen were unable to induce proliferation (Figure 2B). The above results indicated that CD19 EVs promoted antigen-specific expansion of CAR-T cells, leading to an increase in the CAR-T cell ratio.
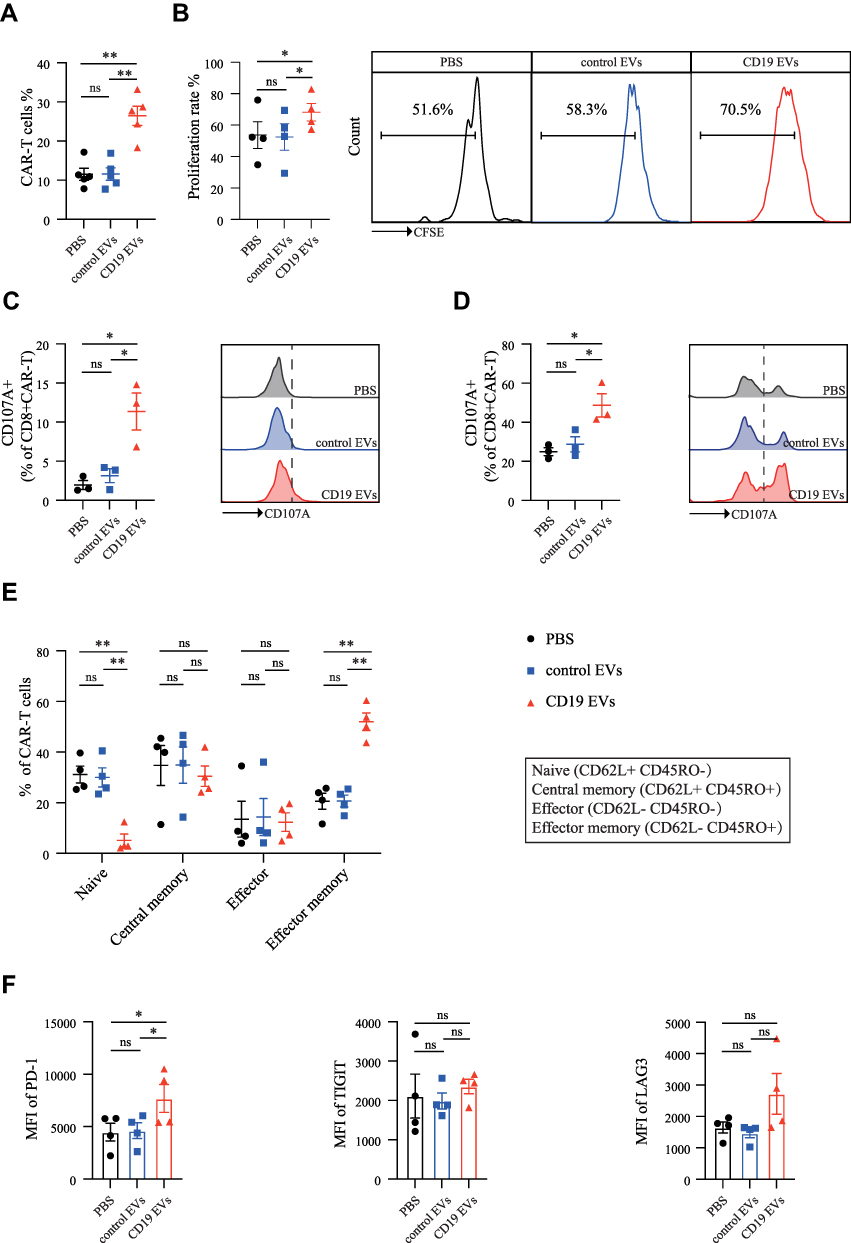 |
Figure 2 CD19 EVs promote the expansion and functional maturation of CAR-T cells. (A) Proportion of CAR-T cells analyzed by flow cytometry after 72 h of co-culture with PBS, control EVs, and CD19 EVs. (B) CFSE-labeled CAR-T cells were treated with PBS, control EVs or CD19 EVs respectively, for 72 h, then proliferation of CAR-T cells was analyzed by flow cytometry. (C) CD107a expression in CAR-T cells stimulated by PBS, control EVs and CD19 EVs respectively. (D) CD107a expression in CAR-T cells after a 72 h-treatment with PBS, control EVs and CD19 EVs mixed with Raji cells at effector to target ratio of 1:1. (E and F) Subsets and immune checkpoint expression was detected using flow cytometry in CAR-T cells after a 7-day treatment with PBS, control EVs, and CD19 EVs respectively. *P < 0.05, **P < 0.01. Abbreviation: ns, not significant. |
To verify whether stimulation of CAR-T cells by CD19 EVs promotes functional maturation, we analyzed the degranulation levels, which can be indicated by surface expression of CD107a (LAMP-1).24 In the absence of tumor cells, PBS or control EVs had minimal effect on degranulation of CAR-T cells, while CD19 EVs considerably increased degranulation, with an average of 11.36% of CAR-T cells expressing CD107a (Figure 2C). In the presence of tumor cells, CD19 EVs pretreated CAR-T cells had more pronounced degranulation compared to the other groups (Figure 2D). We also analyzed the effects of CD19 EVs on CAR-T cell subsets and immune checkpoint expression. The flow cytometry analysis revealed that CD19 EVs induced differentiation of CAR-T cells towards the effector-memory phenotype (Figure 2E), with relatively higher levels of PD-1 expression than the other groups, no significant difference in TIGIT expression, and some elevated expression of LAG3 without statistical difference (Figure 2F). The increased PD-1 expression may be associated with CAR-T cell activation.25 Taken together, these data demonstrated that CD19 EVs promoted antigen-specific expansion of CAR-T cells and induced their functional maturation.
CD19 EVs Enrich High CD19-CAR-Expressing T Cells and Promote Anti-Tumor Activity
Interestingly, CD19 EVs not only induced CAR-T cell proliferation but also promoted CAR expression of CAR-T cells (Figure 3A). The median fluorescent intensity (MFI) of surface anti-CD19 CAR in the CD19 EVs group was more than two times higher than that of the control EVs group, and the MFI of CAR was significantly higher in both CD4+ and CD8+ CAR-T cells (Figure S2B). To clarify the underlying mechanism, we evaluated the proliferation rate of CAR-T cells with different levels of CAR expression following stimulation with EVs. We found that CAR-T cells with higher CAR MFI showed greater expansion ability when activated by CD19 EVs than CAR-T cells with low CAR MFI, which generated more CAR-T cells with high CAR MFI in the newly expanded CAR-T cells, while CAR MFI had no influence on CAR-T cell proliferation in the control EVs group (Figure 3B). This finding suggested that CD19 EVs enriched high CAR-expressing T cells.
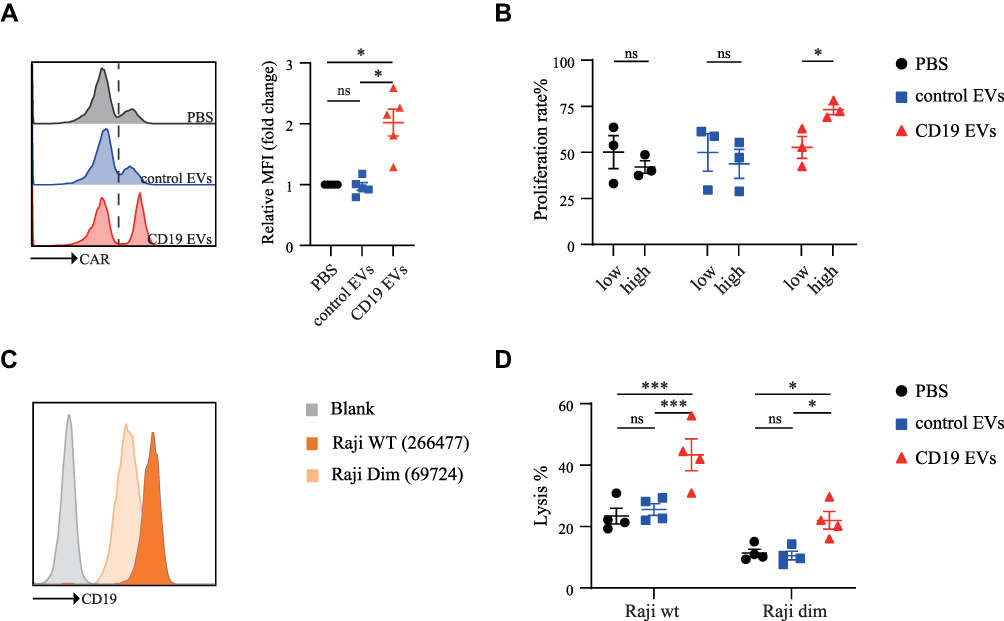 |
Figure 3 CD19 EVs promote CAR expression of CAR-T cells and enhance their anti-tumor activity. (A) CAR MFI of CAR-T cells analyzed by flow cytometry after 72 h of co-culture with PBS, control EVs and CD19 EVs. (B) PBS, control EVs and CD19 EVs were added to CFSE-labeled CAR-T cells for 72 h, 50% of which with low CAR MFI were classified as “low” and the 50% of which with high CAR MFI were classified as “high”, and then their proliferation rate was evaluated by flow cytometry. (C) Expression of cell surface CD19 protein was evaluated and quantified by flow cytometry. (D) CAR-T cells were treated with PBS, control EVs and CD19 EVs respectively for 72 h and then mixed with Raji cells, after 24 h incubation the death of Raji cells was determined by using PI (BD Pharmingen) and analyzed using flow cytometry. *P < 0.05, ***P < 0.001. Abbreviation: ns, not significant. |
Many studies have demonstrated that CAR surface expression and target antigen density regulated the cytotoxic activity of CAR-T cells.26–30 To further confirm this, we sorted high, medium, and low CAR-expressing T cells by fluorescence-activated cell sorting (FACS), and used Raji cells with normal (Raji WT) or low CD19 expression (Raji Dim) as target cells to determine the levels of degranulation of CAR-T cells with varying CAR expression (Figure 3C). The results showed that the higher the CAR expression and antigen density, the higher the level of CAR-T cell degranulation (Figure S3). To determine whether high CAR-expressing T cells enriched by CD19 EVs have enhanced cytotoxic activity, we treated CAR-T cells with PBS, control EVs, and CD19 EVs respectively, then co-incubated them with Raji cells. The CD19 EVs significantly enhanced the anti-tumor activity of CAR-T cells against Raji WT or Raji Dim, defined by the death ratio of Raji cells (Figure 3D). The anti-tumor activity of CAR-T cells against Raji Dim was lower than against Raji WT, which is consistent with previous studies.26,29 The above results suggested that CD19 EVs preferentially promoted the expansion of CAR-T cells with high CAR expression and thereby enhanced anti-tumor activity, even against lower antigen expression target.
Effect of CD19 EVs on CAR-T Cells Prepared from Patient-Derived T Cells
The CAR-T cells used in the above experiments were derived from healthy donors. To further evaluate the translational potential of our strategy, we applied CD19 EVs to CAR-T cells prepared from T cells derived from patients with B-cell malignancies. CD19 EVs promoted CAR-T cell proliferation and CAR expression in most patient-derived CAR-T cells (Figure 4A, B and Figure S4A), although CAR-T cells from approximately 20% of tested patients did not respond to CD19 EVs (Figure S4B). The CD19 EVs expanded CAR-T cells were fully functional, with higher level of cytokine secretion and increased degranulation (Figure 4C–E). Furthermore, we examined subsets of CAR-T cells prepared from three patient-derived T cells after co-culture with CD19 EVs. CD19 EVs induced significant increases in the proportion of cells with an effector-memory or central-memory phenotype (Figure S4C).
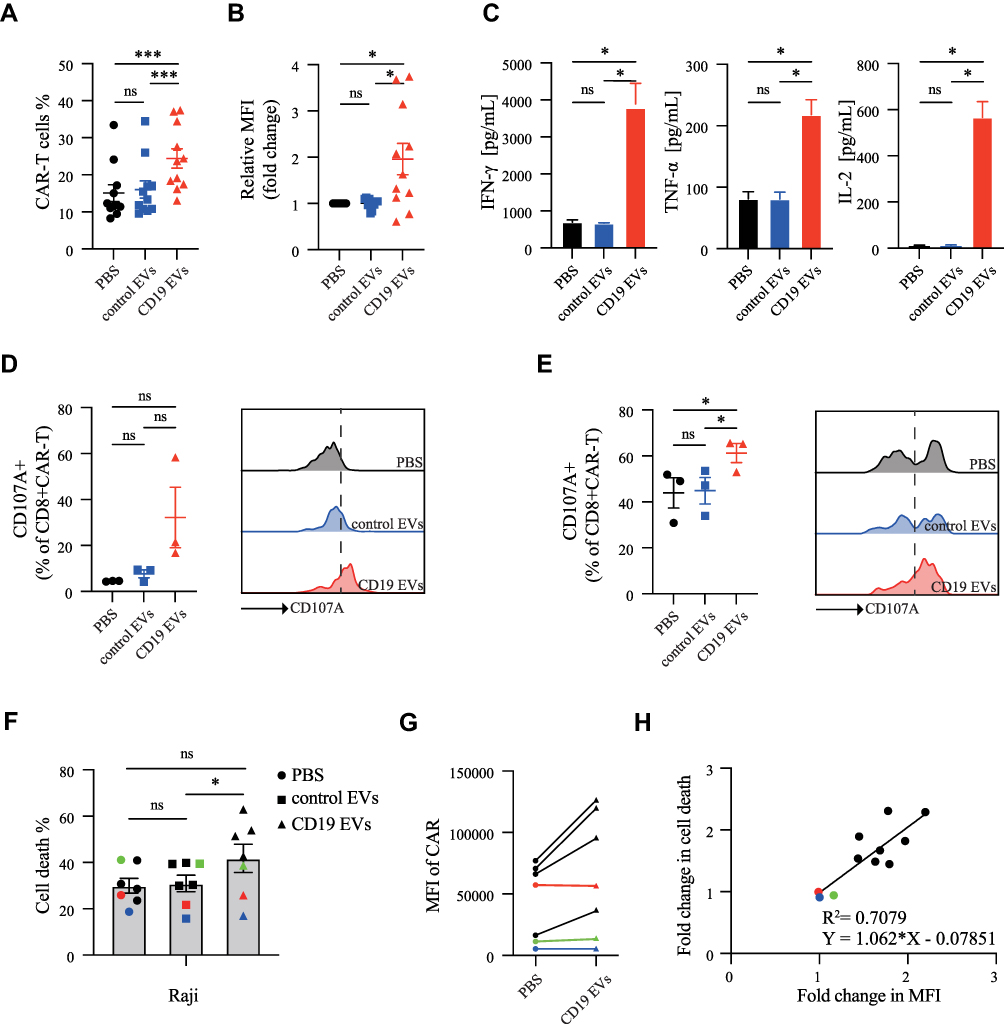 |
Figure 4 CD19 EVs promote the efficacy of CAR-T cells prepared from patient-derived T cells. (A) Proportion and (B) CAR MFI of CAR-T cells analyzed by flow cytometry after 72 h of co-culture with PBS, control EVs and CD19 EVs. (C) Cytokine secretion of CAR-T cells analyzed by ELISA after 24 h of co-culture with PBS, control EVs and CD19 EVs. (D) CD107a expression in CAR-T cells stimulated by PBS, control EVs and CD19 EVs respectively. (E) CD107a expression in CAR-T cells after 72 h treatment with PBS, control EVs and CD19 EVs mixed with Raji cells at effector to target ratio of 1:1. (F) CAR-T cells derived from seven patients were treated with PBS, control EVs, or CD19 EVs respectively for 72 h and then mixed with Raji cells. After a 24 h incubation, the death rate of Raji cells was analyzed by flow cytometry. (G) CAR MFI variation of CAR-T cells in (F) induced by CD19 EVs. The colored lines represent three patients whose CAR-T cells’ CAR expression and anti-tumor activity could not be enhanced by CD19 EVs. (H) Based on cytotoxicity data from the 7 patients and the previous 4 healthy donors, simple linear regression was performed on the fold change in CAR MFI of CD19 EVs treated CAR-T cells compared to the PBS treated CAR-T cells and the fold change in cytotoxic activity. *P < 0.05, ***P < 0.001. Abbreviation: ns, not significant. |
Next, we confirmed that CD19 EVs improved the cytotoxic activity of CAR-T cells (Figure 4F). However, no enhancement was seen in three cases. Additional analysis indicated that CAR expression in CAR-T cells from these three patients was not increased by CD19 EVs (Figure 4G), implying that the effective enhancement of anti-tumor activity by CD19 EVs was closely related to increased CAR expression. Furthermore, linear regression analysis revealed that the fold change in CAR MFI and the fold change in CAR-T cell cytotoxic activity after treatment with CD19 EVs were proportional with R2=0.7079 (P=0.0012), indicating a strong relationship between them (Figure 4H). This suggested that the more CAR-T cells with increased CAR expression induced by CD19 EVs, the higher anti-tumor activity could be achieved. Overall, these results demonstrated that CD19 EVs could enhance proliferation, cytokine secretion, and degranulation of patient-derived CAR-T cells, as well as enhance CAR expression and anti-tumor ability.
CD19 EVs-Primed CAR-T Cells Have Better Expansion and Anti-Tumor Activity in vivo
To explore the functional role of CD19 EVs in CAR-T cells in vivo, tumor-bearing mice were injected intravenously with a sub-therapeutic dose of CAR-T cells primed with PBS, control EVs, CD19 EVs or Mock-T cells (Figure S5A). Tumors were incompletely inhibited by PBS- or control EVs-pretreated CAR-T cells. In contrast, CAR-T cells primed with CD19 EVs completely eradicated tumors on day 21 post infusion in mice, with a significantly higher median survival rate (Figure 5A and B). Effective tumor control in the CD19 EVs group (Figure S5B) correlated with a higher percentage of CAR-T cells in the peripheral blood, which was confirmed by flow cytometry and droplet digital PCR (Figure 5C and D). The CD19 EVs induced a slight increase in the proportion of cells with effector phenotype or effector memory phenotype in the CD19 EVs treatment group, while a significant decrease in the proportion of cells with central memory phenotype. The expression of exhaustion markers did not differ between groups (Figure S5C and S5D).
 |
Figure 5 CD19 EVs primed CAR-T cells exhibit enhanced proliferation and anti-tumor activity in vivo. (A) Tumor growth and (B) survival of Raji tumor-bearing mice (n = 5 animals per group) treated with 1×106 CAR-T cells primed with PBS, control EVs or CD19 EVs for 72 h. (C) Proportion of CAR-T cells and (D) CAR transgene copy number in peripheral blood on day 9 after ACT. (E) Cytokine serum levels on day 3 after adoptive T cell transfer (ACT). (F–H) on day 4 after ACT, (F) Proportion and (G) CAR MFI of CAR-T cells in the tumors analyzed by flow cytometry. (H) CD3+T cell infiltration into the tumors analyzed by IHC (n = 3 animals per group). Scale bars, 50 μm. *P < 0.05, **P < 0.01, ****P < 0.0001. Abbreviation: ns, not significant. |
T cell infiltration in tumors from treated mice was confirmed by both flow cytometry (Figure 5F and G) and IHC (Figure 5H). Pretreatment of CAR-T cells with CD19 EVs improved infiltration of CAR-T cells, as well as CAR expression in tumors, which could explain the accelerated tumor regression in the CD19 EVs group. Thus, CD19 EVs favored the expansion of transferred CAR-T cells, and enabled effective tumor control at a sub-therapeutic dose of CAR-T cells.
However, we were concerned that increased CAR-T cell expansion could result in excessive cytokine secretion, an undesired effect known as the cytokine release syndrome (CRS).11 To evaluate systemic cytokine release in conjunction with exposure to CD19 EVs, we analyzed IFN-γ, IL-6 and TNF-α serum concentrations. Except for an obvious increase of IFN-γ, no significant changes in IL-6 or TNF-α levels were observed (Figure 5E). Mice treated with CD19 EVs-primed CAR-T cells were of normal appearance and showed regular weight gain over time (Figure S5E). Taken together, CAR-T cells stimulated by CD19 EVs showed better proliferation and anti-tumor activity in vivo, without causing additional adverse events.
In vivo Application of CD19 EVs Promotes the Functional Persistence of CAR-T Cells
According to a previous study, poor persistence of CAR-T cells can lead to tumor relapse.8 To assess the impact of repeated administrations of CD19 EVs in vivo on CAR-T cells, mice engrafted with CAR-T cells received two doses of EVs followed by tumor inoculation (Figure 6A). None of the mice showed behavioral abnormalities or significant changes in body weight gain (Figure S6A). Furthermore, no significant differences were observed in the indicators of liver and kidney function among the groups (Figure S6B and S6C). The physiological structure of the vital organs of the mice was normal (Figure S6D). This suggested that the in vivo administration of CD19 EVs was generally tolerable. In mice that received PBS or control EVs, the engraftment of CAR-T cells gradually decreased over time without antigen stimulation (Figure 6B). Notably, in mice treated with CD19 EVs, CAR-T cells were rapidly amplified after being exposed to the first dose of CD19 EVs, and were robustly re-expanded after the second treatment with CD19 EVs and were maintained at a higher level. CAR transgene copy number tested on day 6 after CAR-T cell transfer was highest in the CD19 EVs treatment group, which was approximately twice as high as that observed in the control groups, and was consistent with flow cytometry analysis (Figure 6C).
 |
Figure 6 In vivo administration of CD19 EVs improves CAR-T cell functional persistence. (A) Flow chart of administration of every group treatment (n = 5 mice per group). (B) Cellular kinetics of CAR-T cells transferred into mice analyzed by flow cytometry. (C) CAR transgene copy number in peripheral blood on day 6 after ACT. (D)Tumor growth and (E) survival of mice with tumors. C represents CAR-T cells injection, and E represents EVs administration, and T represents tumor cells inoculation. The arrows indicate the injection time. **P < 0.01. Abbreviation: ns, not significant. |
To test whether the application of CD19 EVs could promote the functional persistence of CAR-T cells, we performed tumor inoculation in EV-treated mice. After tumor inoculation, mice receiving repeated CD19 EVs enhancing doses had significantly delayed tumor growth and extended survival (Figure 6D and E). The above results indicated that CD19 EVs promote the persistence of CAR-T cells in vivo and CAR-T cells maintained by CD19 EVs effectively exerted anti-tumor activity.
Discussion and Conclusions
In this study, we generated HEK293T-derived EVs to present CD19 antigen as CAR target. CAR-T cells were activated upon scFv recognition of antigens presented by CD19 EVs, which lead to secretion of cytokines, cell proliferation with increased CAR expression that favored better cytotoxicity (Figure 7). This approach also exhibited strong cytotoxic ability in tumor-bearing mice and improved the functional persistence of CAR-T cells. Our study provides a novel intervention strategy that leverages the antigen delivery function of EVs to enhance the proliferation and persistence of CAR-T cells, possibly promoting clinical efficacy.
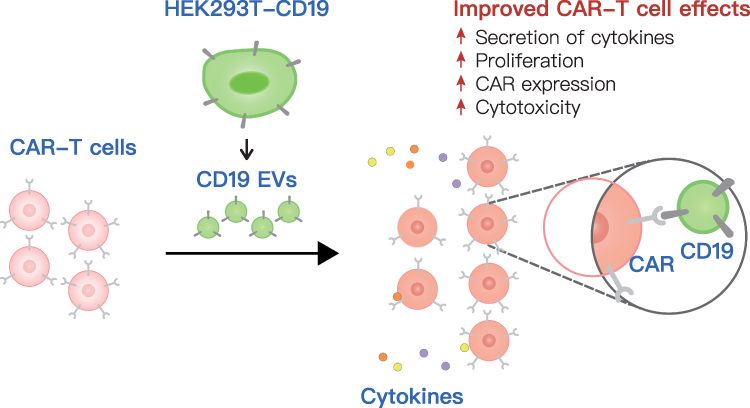 |
Figure 7 A schematic model of the effect of CD19 EVs on CAR-T cell function. EVs secreted by engineered HEK293T cells stably expressed CD19 (CD19 EVs) and were administered to CAR-T cells, causing a marked enhancement in CAR-T cell expansion and cytokine secretion, with elevated CAR expression, which was attributed to the higher cytotoxicity ex vivo and in vivo. |
There have been several attempts to activate CAR-T cells by displaying CAR target antigens. In a clinical trial, episodic antigen exposure using truncated human CD19 expressing T cells triggered CD19 CAR-T cell proliferation in vivo, resulting in more durable CAR-T cell persistence and diminished risk of CD19+ relapse (NCT03186118). In addition, some studies have investigated into modifying DCs with proteins or RNA vaccines, followed by DCs presenting target antigens to activate CAR-T cells.12,13 These studies have demonstrated the concept of activating CAR-T cells with target antigens to obtain better therapeutic effects. However, they were confronted with technical difficulties and high preparation costs. Moreover, they relied heavily on the function of antigen-presenting cells or T cells, which may not be feasible for some immunocompromised patients, and the use of synthetic substances introduces nonhuman-antigens that may trigger an immune response in the host.
Unlike previous approaches, we employed EVs as a vehicle for delivering target antigens to CAR-T cells, with the following considerations. Firstly, EVs are membrane-enclosed cell-derived nanoparticles, a natural carrier with lower cytotoxicity and immunogenicity,31–33 thus EVs are relatively safe as antigen delivery vehicles. In ongoing clinical trials, the EV method is being used to generate CD8+ T cell responses against a spectrum of different tumor antigens with early clinical data supporting the execution of the intended mode-of-action in humans.34,35 Second, the EVs used in this study are microvesicles, which bud directly from the plasma membrane, so their surface molecules are very similar to those of their derived cell line.36,37 Based on this, EVs can be designed and obtained by modifying the parent cell line for any protein-based antigen. Third, EVs can be produced quickly and inexpensively on a large scale. In addition, studies on the purification and quality control of clinical-grade EVs are ongoing.38 In summary, EVs equipped with the target antigen may offer a feasible and simple solution to boost CAR-T cells with any potential antigen specificity. Their time- and cost-effective synthesis and tolerability are also favorable for clinical translation.
Tumor-derived extracellular vesicles (TEVs) are natural carriers of tumor-associated antigens, which can be used as neoantigen vaccines to induce active anti-tumor immune responses39 or as target cell surrogates to accelerate the proliferation to optimize the in vitro culture of CAR-T cells.18 However, many studies have shown that TEVs promote tumor growth or metastasis,40,41 and inhibit immune cell functions to help tumor escape immune surveillance and clearance.42 It is worth noting that TEVs may induce exhaustion of CAR-T cells, impair their proliferation and anti-tumor activity.19,43–45 Specifically, the immunological checkpoints (IC) including PD-L1 found in TEVs could be responsible for the negative effect of TEVs in CAR-T cells.19,45 Therefore, in our study, we adopted EVs derived from HEK293T cells to deliver target antigen. The HEK293T cell line is derived from human embryonic kidney cells and is frequently used in various experiments and studies associated with EVs.46 It has been reported that HEK293T cells derived EVs have little effect of T cells.17 Our findings also suggest that HEK293T cells derived EVs hardly express PD-L1 on their surface and do not functionally impact on CAR-T cells. In addition, our findings demonstrate that in vivo administration of EVs derived from HEK293T cells is safe and tolerable, which is consistent with previous studies.16,47 This indicates that HEK293T cells could be used as parent cells to produce EVs with target antigen. Other non-tumor cells, such as mesenchymal stem cells or T cells, may potentially be viable EV parent cells and need to be further explored.
In our study, CD19 EVs activate CAR-T cells generated from healthy donors as well as patients-derived T cells, and unleash effector responses including proliferation, release of cytokines and degranulation in CAR-T cells, which are similar to the findings of a recently published study.18 Our data also suggest that following CD19 EVs activation, CAR-T cells undergo effector-memory programming. The effector-memory T cell subset was reported to secrete a higher level of IFN-γ and Granzyme B.48,49 Notably, chronic CD19 stimulation is considered as a major cause of T-cell dysfunction,44 which raises concerns that the long-term exposure to CD19-EVs may lead to an exhaustion of the CART-cells. However, in the present study, the doses of CD19 EV administration were limited, and no exhaustion or dysfunction was observed after the transient stimulation of CD19 EVs both in vitro and in vivo.
In addition, while CAR-T cells expand faster after CD19 EVs treatment, the CAR MFI was also elevated possibly because CAR-T cells with high CAR expression are more sensitive to CD19 EVs. Previous studies suggest that antigen and CAR molecule density regulate efficacy of CAR-T cells, as low expression of either the antigen or CAR contributes to a limited anti-tumor response.26,27 The ability of CD19 EVs to sensitize CAR-T cells to kill Raji cells with dim CD19 expression by increasing the percentage of high CAR-expression T cells implied that such approach may help eliminate tumor cells with low target antigen density which likely leads to insufficient activation of CAR-T cells10 or resistance to CAR-T cell therapy.
Rapid manufacturing of CAR-T cells is crucial in patients with progressive disease, and CAR-T cells cultured in less time also retain better anti-tumor activities.50,51 Our study suggests that CD19 EVs primed CAR-T cells had a better expansion profile which may reduce the ex vivo culture time needed. Furthermore, repetitive administration of CD19 EVs in vivo maintained a relatively high level of circulating CAR-T cells. This artificial setting of animal model was made for two reasons: 1. To assess the dynamic proliferation of CAR-T cells in vivo stimulated only by CD19 EVs but without tumor-derived first signal; 2. To test whether the repeated dosing of CD19 EVs in vivo lead to any functional impairment of CAR-T cells, such as T-cell exhaustion. The results show that when limited doses of CD19 EV were used, the CAR-T cell function was maintained and enhanced by CD19 EVs in vivo. This strategy can increase the therapeutic window of CAR-T cells and may be beneficial in preventing disease recurrence, as immune surveillance.8,52 Therefore, engineered target-presenting EVs may serve as a highly versatile platform for tunable functional behavior of the CAR-T cells both in vitro and in vivo, thereby providing potential tools for individualized cellular therapy with current available CAR-T products.
In summary, we present the strategy to generate EVs equipped with a CAR target antigen to drive the expansion and function of CAR-T cells. The efficacy and safety of this strategy need to be further investigated in clinical trials. We speculate that genetically engineered EVs will be developed as adjuvants for CAR-T cell immunotherapy in the near future.
Acknowledgments
The work was supported by grants from the National Natural Science Foundation of China (NSFC) (Nos: 82100247, 81630006 and 82000176).
Disclosure
Drs Yuanyuan Zhang, Dr Lei Zhao, and Prof. Dr. Jue Wang report a patent CD19 EVs pending to wipo. The authors report no conflicts of interest in this work.
References
1. Locke FL, Ghobadi A, Jacobson CA, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, Phase 1–2 trial. Lancet Oncol. 2019;20(1):31–42. doi:10.1016/S1470-2045(18)30864-7
2. Park JH, Rivière I, Gonen M, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. 2018;378(5):449–459. doi:10.1056/NEJMoa1709919
3. Schuster SJ, Bishop MR, Tam CS, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019;380(1):45–56. doi:10.1056/NEJMoa1804980
4. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–448. doi:10.1056/NEJMoa1709866
5. Fraietta JA, Lacey SF, Orlando EJ, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. 2018;24(5):563–571. doi:10.1038/s41591-018-0010-1
6. Porter DL, Hwang WT, Frey NV, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7(303):303ra139. doi:10.1126/scitranslmed.aac5415
7. Mueller KT, Maude SL, Porter DL, et al. Cellular kinetics of CTL019 in relapsed/refractory B-cell acute lymphoblastic leukemia and chronic lymphocytic leukemia. Blood. 2017;130(21):2317–2325. doi:10.1182/blood-2017-06-786129
8. Ghorashian S, Kramer AM, Onuoha S, et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat Med. 2019;25(9):1408–1414. doi:10.1038/s41591-019-0549-5
9. Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. doi:10.1056/NEJMoa1407222
10. Rust BJ, Kean LS, Colonna L, et al. Robust expansion of HIV CAR T cells following antigen boosting in ART-suppressed nonhuman primates. Blood. 2020;136(15):1722–1734. doi:10.1182/blood.2020006372
11. Mueller KT, Waldron E, Grupp SA, et al. Clinical pharmacology of tisagenlecleucel in B-cell acute lymphoblastic leukemia. Clin Cancer Res. 2018;24(24):6175–6184. doi:10.1158/1078-0432.CCR-18-0758
12. Reinhard K, Rengstl B, Oehm P, et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science. 2020;367(6476):446–453. doi:10.1126/science.aay5967
13. Ma L, Dichwalkar T, Chang JYH, et al. Enhanced CAR–T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science. 2019;365(6449):162–168. doi:10.1126/science.aav8692
14. Wiklander OPB, Brennan MÁ, Lötvall J, Breakefield XO, El Andaloussi S. Advances in therapeutic applications of extracellular vesicles. Sci Transl Med. 2019;11(492):eaav8521. doi:10.1126/scitranslmed.aav8521
15. Lener T, Gimona M, Aigner L, et al. Applying extracellular vesicles based therapeutics in clinical trials – an ISEV position paper. J Extracell Vesicles. 2015;4(1):30087. doi:10.3402/jev.v4.30087
16. Casella G, Rasouli J, Boehm A, et al. Oligodendrocyte-derived extracellular vesicles as antigen-specific therapy for autoimmune neuroinflammation in mice. Sci Transl Med. 2020;12(568):eaba0599. doi:10.1126/scitranslmed.aba0599
17. Xu Z, Tsai H-I, Xiao Y, et al. Engineering programmed death Ligand-1/Cytotoxic T-lymphocyte-associated antigen-4 dual-targeting nanovesicles for immunosuppressive therapy in transplantation. ACS Nano. 2020;14(7):7959–7969. doi:10.1021/acsnano.9b09065
18. Ukrainskaya V, Rubtsov Y, Pershin D, et al. Antigen-specific stimulation and expansion of CAR-T cells using membrane vesicles as target cell surrogates. Small. 2021;17(45):2102643. doi:10.1002/smll.202102643
19. Cox MJ, Lucien F, Sakemura R, et al. Leukemic extracellular vesicles induce chimeric antigen receptor T cell dysfunction in chronic lymphocytic leukemia. Mol Ther. 2021;29(4):1529–1540. doi:10.1016/j.ymthe.2020.12.033
20. Xu Q, Zhang Z, Zhao L, et al. Tropism-facilitated delivery of CRISPR/Cas9 system with chimeric antigen receptor-extracellular vesicles against B-cell malignancies. J Control Release. 2020;326:455–467. doi:10.1016/j.jconrel.2020.07.033
21. Kim SM, Yang Y, Oh SJ, Hong Y, Seo M, Jang M. Cancer-derived exosomes as a delivery platform of CRISPR/Cas9 confer cancer cell tropism-dependent targeting. J Control Release. 2017;266:8–16. doi:10.1016/j.jconrel.2017.09.013
22. Lou Y, Chen C, Long X, et al. Detection and quantification of chimeric antigen receptor transgene copy number by droplet digital PCR versus real-time PCR. J Mol Diagnos. 2020;22(5):699–707. doi:10.1016/j.jmoldx.2020.02.007
23. Jeppesen DK, Fenix AM, Franklin JL, et al. Reassessment of exosome composition. Cell. 2019;177(2):428–445.e18. doi:10.1016/j.cell.2019.02.029
24. Aktas E, Kucuksezer UC, Bilgic S, Erten G, Deniz G. Relationship between CD107a expression and cytotoxic activity. Cell Immunol. 2009;254(2):149–154. doi:10.1016/j.cellimm.2008.08.007
25. Ai L, Xu A, Xu J. Roles of PD-1/PD-L1 pathway: signaling, cancer, and beyond. Adv Exp Med Biol. 2020;1248:33–59. doi:10.1007/978-981-15-3266-5_3
26. Watanabe K, Terakura S, Martens AC, et al. Target antigen density governs the efficacy of anti-CD20-CD28-CD3 ζ chimeric antigen receptor-modified effector CD8+ T cells. J Immunol. 2015;194(3):911–920. doi:10.4049/jimmunol.1402346
27. Arcangeli S, Rotiroti MC, Bardelli M, et al. Balance of anti-CD123 chimeric antigen receptor binding affinity and density for the targeting of acute myeloid leukemia. Mol Ther. 2017;25(8):1933–1945. doi:10.1016/j.ymthe.2017.04.017
28. Drent E, Themeli M, Poels R, et al. A rational strategy for reducing on-target off-tumor effects of CD38-chimeric antigen receptors by affinity optimization. Mol Ther. 2017;25(8):1946–1958. doi:10.1016/j.ymthe.2017.04.024
29. Walker AJ, Majzner RG, Zhang L, et al. Tumor antigen and receptor densities regulate efficacy of a chimeric antigen receptor targeting anaplastic lymphoma kinase. Mol Ther. 2017;25(9):2189–2201. doi:10.1016/j.ymthe.2017.06.008
30. Juillerat A, Tkach D, Busser BW, et al. Modulation of chimeric antigen receptor surface expression by a small molecule switch. BMC Biotechnol. 2019;19(1):44. doi:10.1186/s12896-019-0537-3
31. Zlotogorski-Hurvitz A, Dayan D, Chaushu G, et al. Human saliva-derived exosomes: comparing methods of isolation. J Histochem Cytochem. 2015;63(3):181–189. doi:10.1369/0022155414564219
32. Yang C, Robbins PD. The roles of tumor-derived exosomes in cancer pathogenesis. Clin Dev Immunol. 2011;2011:1–11. doi:10.1155/2011/842849
33. Clayton A, Harris CL, Court J, Mason MD, Morgan BP. Antigen-presenting cell exosomes are protected from complement-mediated lysis by expression of CD55 and CD59. Eur J Immunol. 2003;33(2):522–531. doi:10.1002/immu.200310028
34. Escudier B, Dorval T, Chaput N, et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: results of the first Phase I clinical trial. J Transl Med. 2005;3(1):10. doi:10.1186/1479-5876-3-10
35. Besse B, Charrier M, Lapierre V, et al. Dendritic cell-derived exosomes as maintenance immunotherapy after first line chemotherapy in NSCLC. Oncoimmunology. 2016;5(4):e1071008. doi:10.1080/2162402X.2015.1071008
36. van Dommelen SM, Vader P, Lakhal S, et al. Microvesicles and exosomes: opportunities for cell-derived membrane vesicles in drug delivery. J Control Release. 2012;161(2):635–644. doi:10.1016/j.jconrel.2011.11.021
37. Cocucci E, Racchetti G, Meldolesi J. Shedding microvesicles: artefacts no more. Trends Cell Biol. 2009;19(2):43–51. doi:10.1016/j.tcb.2008.11.003
38. Pirisinu M, Pham TC, Zhang DX, Hong TN, Nguyen LT, Le MT. Extracellular vesicles as natural therapeutic agents and innate drug delivery systems for cancer treatment: recent advances, current obstacles, and challenges for clinical translation. Semin Cancer Biol. 2020;S1044579X20301796. doi:10.1016/j.semcancer.2020.08.007
39. Yong T, Li X, Wei Z, Gan L, Yang X. Extracellular vesicles-based drug delivery systems for cancer immunotherapy. J Control Release. 2020;328:562–574. doi:10.1016/j.jconrel.2020.09.028
40. Hu W, Liu C, Bi ZY, et al. Comprehensive landscape of extracellular vesicle-derived RNAs in cancer initiation, progression, metastasis and cancer immunology. Mol Cancer. 2020;19(1):102. doi:10.1186/s12943-020-01199-1
41. Wortzel I, Dror S, Kenific CM, Lyden D. Exosome-mediated metastasis: communication from a distance. Dev Cell. 2019;49(3):347–360. doi:10.1016/j.devcel.2019.04.011
42. Li Q, Cai S, Li M, et al. Tumor-derived extracellular vesicles: their role in immune cells and immunotherapy. Int J Nanomedicine. 2021;16:5395–5409. doi:10.2147/IJN.S313912
43. Ali S, Toews K, Schwiebert S, et al. Tumor-derived extracellular vesicles impair CD171-specific CD4+ CAR T cell efficacy. Front Immunol. 2020;11:513.
44. Zhu X, Hu H, Xiao Y, et al. Tumor-derived extracellular vesicles induce invalid cytokine release and exhaustion of CD19 CAR-T cells. Cancer Lett. 2022;536:215668. doi:10.1016/j.canlet.2022.215668
45. Böttcher M, Böttcher-Loschinski R, Kahlfuss S, et al. CLL-derived extracellular vesicles impair T-cell activation and foster T-cell exhaustion via multiple immunological checkpoints. Cells. 2022;11(14):2176. doi:10.3390/cells11142176
46. Zaborowski MP, Balaj L, Breakefield XO, Lai CP. Extracellular vesicles: composition, biological relevance, and methods of study. Bioscience. 2015;65(8):783–797. doi:10.1093/biosci/biv084
47. Zhu X, Badawi M, Pomeroy S, et al. Comprehensive toxicity and immunogenicity studies reveal minimal effects in mice following sustained dosing of extracellular vesicles derived from HEK293T cells. J Extracell Vesicles. 2017;6(1):1324730. doi:10.1080/20013078.2017.1324730
48. Neal LR, Bailey SR, Wyatt MM, et al. The basics of artificial antigen presenting cells in T cell-based cancer immunotherapies. J Immunol Res Ther. 2017;2(1):68–79.
49. Liu L, Bi E, Ma X, et al. Enhanced CAR-T activity against established tumors by polarizing human T cells to secrete interleukin-9. Nat Commun. 2020;11(1):5902. doi:10.1038/s41467-020-19672-2
50. Zhang DKY, Cheung AS, Mooney DJ. Activation and expansion of human T cells using artificial antigen-presenting cell scaffolds. Nat Protoc. 2020;15(3):773–798. doi:10.1038/s41596-019-0249-0
51. Ghassemi S, Nunez-Cruz S, O’Connor RS, et al. Reducing ex vivo culture improves the antileukemic activity of chimeric antigen receptor (CAR) T cells. Cancer Immunol Res. 2018;6(9):1100–1109. doi:10.1158/2326-6066.CIR-17-0405
52. Stein AM, Grupp SA, Levine JE, et al. Tisagenlecleucel model-based cellular kinetic analysis of chimeric antigen receptor-T cells. CPT Pharmacomet Syst Pharmacol. 2019;8(5):285–295. doi:10.1002/psp4.12388
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.