")
Back to Journals » OncoTargets and Therapy » Volume 8
Extracellular matrix as target for antitumor therapy
Received 16 December 2014
Accepted for publication 14 April 2015
Published 9 June 2015 Volume 2015:8 Pages 1387—1398
DOI https://doi.org/10.2147/OTT.S48883
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Revekka Harisi, Andras Jeney
1st Institute of Pathology and Experimental Cancer Research, Faculty of Medicine, Semmelweis University, Budapest, Hungary
Abstract: The aim of the present review is to survey the accumulated knowledge on the extracellular matrix (ECM) of tumors referring to its putative utility as therapeutic target. Following the traditional observation on the extensive morphological alteration in the tumor-affected tissue, the well-documented aberrant cellular regulation indicated that ECM components have an active role in tumor progression. However, due to the diverse functions and variable expression of proteoglycans, matrix proteins, and integrins, it is rather difficult to identify a comprehensive therapeutic target among ECM components. At present, the elevated level of heparanase and the prominent expression of αvβ5 integrin are considered as promising therapeutic targets. The inhibition of glycosaminoglycan offers another promising approach in the treatment of those tumors which are stimulated by proteoglycans. It can be ascertained that a selective ECM inhibitor would be a great asset to control metastasis driven by ECM-mediated signaling.
Keywords: tumor microenvironment, tumor progression, three-dimensional ECM, heparan sulfate proteoglycan
Introduction
In the multicellular organism, tissue architecture and cellular function are controlled by guidance cues derived from the microenvironment composing both cellular and molecular elements. Recent studies demonstrated the assembly of extracellular matrix (ECM) proteins, proteoglycans (PGs), glycopeptidases, and growth factors to well-defined structures containing approximately 300 molecules. ECM participates in multiple biological events among others in tumor angiogenesis.1,2 ECM, representing a network of biopolymers, offers an adynamic tissue-specific structure that is responsible for the transmission of extracellular signals to the cells.
Both functions require a highly precise attachment of ECM biopolymers to cells executed by the cell surface integrins acting as transmembrane receptors. Several lines of investigation concluded that ECM plays an important role in maintaining cellular phenotype, ie, ECM biopolymers participate in the retrieval of the gene information. Tumor cells – predominantly during metastasis - are exposed to changes of the microenvironment. The metabolic reprogramming ability of ECM can be regarded as an active contributor to the malignant processes.3,4
ECM in tumor progression
ECM has multiple effects on the biological behaviors of tumors.5,6 It was concluded from our previous study that the localization of the tumor in the host organism could modify the malignant phenotype of the same tumor.5
Examination of gene expression of ECM components demonstrated that breast carcinomas could be classified to four subtypes with different clinical outcomes.7 In a xenograft model of human mammary carcinomas, it was shown that tumors with low and high metastatic potential differ in ECM composition. Several matrix proteins such as LTBP3, SNED1, EGLN1, and S100A2 were characteristic for the highly metastatic tumors.8 Subtypes of breast and colon tumors based upon signature of ECM proteins also showed correlation with disease prognosis.9
ECM regulates cell proliferation
In the literature, contradictory findings are published regarding the role of ECM in cell proliferation. There are articles reporting enhanced cell proliferation10–17 and others referring to inhibition of it.18,19
Our data show that the ECM has a magnificent role in the maintenance of tumor cell proliferation. Sarcoma cells, themselves, synthetize ECM components.13,14 The EHS-ECM (ECM derived from Engelbreth-Holm-Swarm [EHS] tumor) and the OSCORT-ECM (ECM produced by the OSCORT osteosarcoma cells) caused a significant increase of tumor cell proliferation and more extensive migration.5 For the mechanism by which ECM augments cell growth and proliferation, we presume that it aids the transition from the G1 to the S phase of the cell cycle. This suspicion is supported both by our flow cytometry and the protein expression results concerning increase of cyclin D1 and low decrease of Rb and p53 expression.5,15 PCNA, a marker for S phase, detected in culture on ECM gel, was complexed with other proteins. The second regulation point where ECM and its components may help the cells to get through the cell cycle control points is the checkpoint at G2/M. In our osteosarcoma cell line, we observed a decrease of the proportion of cells staying in this point, which is also a sign of increased proliferation. This is underscored by the increase of cyclin B1 and increase of topoisomerase II activity.5,15 The latter enzyme is most active in the G2/M phase.16 Another confirmation of this presumption is the stimulation of type IV collagen on Ki-67 production. It is known that the Ki-67 protein is a proliferation marker of G2/M phase.17
Tumor dormancy and resistance to cytotoxic agents are key limiting events in the treatment of malignant diseases. Recent results suggest that the primary rate-limiting step in metastasis is not always the early cell destruction in the circulation or the inability of cells to extravasate, but it could be also due to the failure of extravasated cells in the target organ to initiate growth and failure of micrometastases to continue growing.20 Most cells successfully extravasate, but 95% remain dormant, possibly revealing a substantial role of the new microenvironment in growth regulation. Since cytotoxic agents preferentially act on highly proliferating cells, tumor dormancy and loss of drug susceptibility are inversely related. Dormancy has been attributed by some to avascularity resulting from balanced proliferation and apoptosis, sequestration, absence of growth factors or hormones, or immunologic mechanisms.13,21 Our results suggested that ECM plays an important role in the development of a certain type of tumor dormancy. The inhibition of this process would be a therapeutic target, and our model might prove useful for exploring molecular mechanisms implicated in drug resistance and in testing compounds to circumvent it.13
ECM controls cell migration and metastasis
The critical role of ECM in tumor migration and metastasis has been based partly on the expression of the ECM receptors (integrins) on the surface of the tumor cells and on the ability of the tumor cells to degrade the current ECM by matrix metalloproteinases (MMPs).22–25 Based on our investigations, remarkable tumor cell migration was associated with the elevated expression of β1 integrin as well as MMP-9 and MMP-2.14
Migration and metastasis
It was a further noteworthy observation that migration of osteosarcoma cells into the ECM gel was accompanied by an elevation of topoisomerase II activity, the target molecule for doxorubicin. As topoisomerase II showed higher levels of expression and activity in the migrating cells relative to the proliferating monolayer, it was of interest to study whether the two subpopulations are affected at the same extent by doxorubicin as an inhibitor of topoisomerase II.5 Doxorubicin caused a similar reduction in the activity but a difference in the lowering of protein expression of topoisomerase II in the non-migrating and in the migrating cells. Nevertheless, assuming the critical function of topoisomerase II in tumor invasion, it is conceivable that it could offer a molecular target for the selective antimigratory effect of doxorubicin applied in a low concentration for a long period. This elevation of topoisomerase II in cells migrated to ECM gel may indicate increased transcription, DNA synthesis, and repair functions, since this is one of the enzymes responsible for maintenance of the genome. We assume that the ECM-induced elevation of topoisomerase II may be an underlying event in the reprogramming of the gene expression, which is necessary for the invasive growth. The detected “topoisomerase-switch” in relation to tumor ECM-induced growth means elevation in topoisomerase II and reduction in topoisomerase I activity. It was also induced by heparan sulfate PG (HSPG).5
We found that OSCORT-ECM also supported the invasive growth of a human osteosarcoma cell line. This support was in association with the induction of β1 integrin and MMP-9 enzymatic activity, and was related to the suppression of topoisomerase I and enhancement of topoisomerase II activities.5
Tumor cells may migrate alone (ovarian cancer cells) or in the form of a cluster (osteosarcoma cells). The ECM morphology is different in these two types of migration.26–32 The Boyden chamber and the three-dimensional cell culture (3-DCC) differentiate the antimigratory action of test compounds against tumor.32,33 The Boyden chamber provided a model for single-cell migration and a 3-DCC system for cluster migration.33,34 Investigations with Boyden chamber and 3-DCC models help with finding agents which have the ability to inhibit the activity of PGs, integrins, and MMPs.32
MMPs and their inhibitors
Higher expression of MMPs was associated with severe symptoms, increased tumoral size, and worse grade. MMPs and their inhibitors have been identified as critical modulators of ECM composition and are thus crucial in tumor cell progression, invasion, and metastasis.14 MMPs are members of the metzincin family of proteases, and they are responsible for processing ECM components to respond to physiological regulators or pathological impacts. Nevertheless, uncontrolled activity of MMPs results in alteration in the biological function of ECM biopolymers by degrading the ECM of basement membrane. In addition, the concomitant release of cytokines and growth factors from the degraded ECM accelerates tumor progression.
The elevation of MMP-9, which is responsible target for the cluster-type migration, correlates with the higher invasive capacity of neoplastic cells.30 Reports on the elevated MMP activity in human tumors have stimulated attempts to identify MMP inhibitors for many years.14 Although MMPs were considered an attractive target for therapeutic intervention, recent clinical trials did not fulfill the expectations. Data from clinical trials with the MMP inhibitor batimastat (BB-94) in the 1990s emphasized the need for better understanding of the complexity of MMP activity and the consequences of its suppression in tumor growth.35 Later results have provided data for the multiple roles of MMPs, thereby tumor progression and metastasis could be not only inhibited but also promoted after treatment with an MMP inhibitor depending on the function of the affected ECM protein. Since certain ECM components have tumor growth-controlling potency, their degradation must be avoided. This situation has opened avenues for a strategy involving the normalization of the tumor microenvironment as a novel therapeutic approach to cancer treatment.36 Endostar, a human recombinant endostatin decreases MMP expression. This inhibitory effect could be correlated with the reduced migration and invasive growth of MDA-MB-435 cells through reconstituted ECM.37 At present, the concept recognizes MMP inhibitors acting against a signal mechanism implicated in the induction of MMPs. MMP-9 expression is reduced by oroxylin A (a bioactive flavonoid) through the inhibition of extracellular signal-regulated kinase (ERK)1/2, which resulted in the control of metastasis in animal models.38
ECM biopolymers and their inhibitors
The biopolymers of ECM (ECM biopolymers or ECM components) appearing in tumors were reported to have a priming function in microenvironment for cancer development and metastasis. The interaction of tumor cells and ECM components is profoundly altered at all stages of tumor progression. The microscopical injuries of basement membrane are signs of invasive growth of tumor.6,39
The ECM biopolymers are classified as polysaccharides and fibrous proteins and are localized either in the interstitial space or in the well-defined morphological entry of the basement membrane surrounding epithelial cells. Basement membrane is built from laminin and collagen IV networks stabilized by nidogen/perlecan bridges.6 In tumor biological studies, the most well documented ECM polysaccharides are the glycosaminoglycans (GAGs), which usually link to proteins forming PGs such as perlecan, decorin, agrin, and syndecan HSPGs. These PGs have two important characteristics: the hydrophilic property and the ability to store and transmit growth factors. The other main groups of ECM biopolymers, the family of the fibrous proteins, include collagen, laminin, and fibronectin.
The ECM proteins have been classified as a family of nonstructural matrix proteins, and they are a crucial component of tumor microenvironment because they not only represent a structural element in tissue organization, but also have an active role in the maintenance of malignant phenotype. This implies that transformed malignant cells are, to a certain extent, under the surveillance of ECM proteins as well. These proteins induced by either tumor cells or tumor stromal components initiate downstream signaling events that lead to proliferation, invasion, matrix remodeling, and dissemination (Figure 1). As the architecture of the tumors show remarkable diversity, the composition and the amount of ECM proteins differ from normal tissue and allow novel interactions to influence tumor cell function.40 The accumulated data from numerous relevant studies suggest that basic behavior of tumor cells, including altered intercellular contacts, invasive growth, and metastasis, can be associated with the dysregulated ECM proteins. Modification of these events of tumor microenvironment could lead to potential new methods of tumor therapy.5
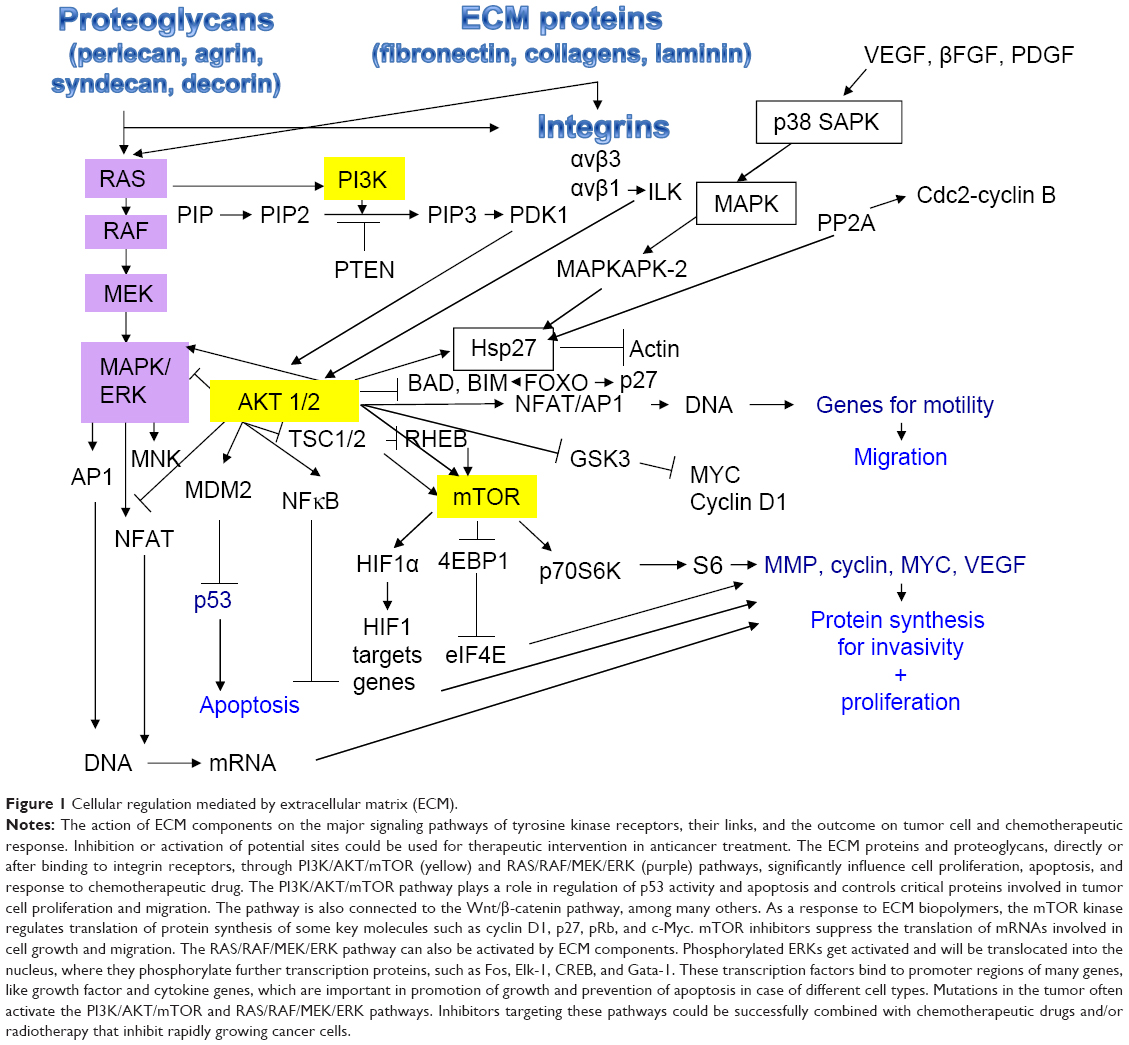 | Figure 1 Cellular regulation mediated by extracellular matrix (ECM). |
A brief introduction to the expression and cellular functions and also the enzymatic degradation of ECM proteins intends to offer an insight into the relevance of ECM proteins as a promising target for antitumor agents.
Collagen
Collagen represents the main ECM protein in tumors as well as in normal tissues. The elevation of collagen is a risk factor in the development of breast carcinoma. In the metastatic process, collagen facilitates cluster formation for the spreading tumor cell. In animal models, it was also confirmed that increased stromal collagen significantly increased tumor formation and results in a more invasive phenotype.32,41,42 In our investigations, type IV collagen decreased proliferation5,15 – interestingly, not because of decreased adhesivity of the cells, since our experiments with type IV collagen-coated culture dishes showed greater adhesivity of the cells to type IV collagen than to poly-L-lysine. Inhibition of melanoma cell proliferation was previously reported for a distinct domain of type IV collagen: a fragment of alpha3 chain (C-terminal amino acids: 185–203).43 The background of cell growth decrease upon addition of type IV collagen to the culture medium, on the other hand, is likely due to cell cycle arrest in the G2/M phase.44,45 Collagen type IV inhibited the invasive growth of osteosarcoma. The elevation of topoisomerase II activity was related to the accumulation of osteosarcoma cells in the G2/M phase.5 Diphosphorylated active ERK2 phosphorylated topoisomerase IIα and increased its specific activity sevenfold, as recorded by DNA relaxation assays.5,46 ERK2 is activated by mitogen-activated protein kinase 1/2 (MAPK 1/2), which is the target of collagen IV-mediated signaling.5,47 Collagen environment controls the Wnt pathway.5,48
Laminin
The laminin family of glycoproteins is an important constituent of basal lamina, which forms networks to stabilize adhesion of cells. Laminins bind other ECM components such as collagen, nidogen, and entactin. The laminins influence cell differentiation, migration, and viability of tissues.49 Serving as an early and useful model of tumor cell–ECM interactions is the human fibrosarcoma cell line (HT1080).50 Due to the in vivo infiltrative growth of a fibrosarcoma, it inevitably interacts with ECM components at the site of intra- and extravasation. HT1080 cells adhere strongly to laminin-1 and laminin-5 utilizing both α3β1 and α6β1 integrins and can attach to collagen I and fibronectin using α2β1 and α5β1 integrins, respectively.51 Under appropriate conditions, rapid HT1080 cell aggregation leads to the formation of extensive cellular networks.13 Rather than being evidence of a malignant phenotype, network formation may more precisely be related to basement membrane-induced cell motility and can be observed in normal primary cell culture.50,52 On the contrary, our results concerning lack of laminin effect on osteosarcoma cell proliferation, while it enhanced β1 integrin production, might be explained by Kawaguchi and Uede: the decreased expression of laminin receptors in human osteosarcomas.53
Fibronectin
Fibronectin, a glycoprotein, binds to ECM and enhances its binding to integrins. Fibronectin expression is elevated in poorly differentiated tumors, where it has a major role in tumor growth and migration, which could either be due to increased fibronectin production by the more anaplastic tumor cells or internalization of exogenous fibronectin bound to its receptor.54 Several mechanisms of cell growth control were clarified during the last few years, in particular the negative regulation of integrin activation by RAS/RAF and the TGF-β inhibition of p33cdk2 phosphorylation and cyclin A- and E-associated histone H1 kinase activities.55,56 Another model of growth inhibition resulting in dormancy of human carcinoma Hep3 cells is the reduced urokinase plasminogen activator receptor (uPAR) level leading to a reduced avidity of the α5β1 integrin and a lower adhesion of cells to fibronectin.57 In our results, it seems that from ECM biopolymers, in case of osteosarcoma, HSPG and fibronectin provide a stimulatory signal for the tumor, for increased proliferation observed on ECM gel.5 These two components are not more than 10% of ECM gel but may trigger signal routes resulting in increased cell reproduction.58,59 Kawaguchi and Uede examined cell surface receptors of human osteosarcomas.53 They found the integrins that bind to fibronectin: α4β1 in 100% of the cases, and α5β1 in 75% of the cases. β1 integrin complexes are known to interact with fibronectin, which explains the increased growth of OSCORT cells in fibronectin-containing medium.
Periostin
Periostin, a multifunctional matricellular protein, has been detected at elevated levels in tumors and demonstrated its active contribution to tumor progression.60
PGs, heparanases, and inhibitors
PGs – synthetized in both parenchymal and stromal cells – consist of a “core protein” in which serine residues are covalently linked to GAG through a tetrasaccharide bridge. GAGs, mainly heparan sulfate or chondroitin sulfate, are long linear polymers, which are negatively charged due to the sulfate groups. PGs have an important role in cell–cell and cell–ECM interactions and signaling. The complex structure of PGs allows their participation in a variety of cellular functions (motility, adhesion, growth).5 However, the individual PGs (perlecan, syndecan, decorin) have distinct mechanisms and, in certain cases, opposite cellular action.
The GAG chains recognize “binding” motifs of the ECM components, including integrins and receptors. The participation of heparan sulfate as a receptor in endocytosis is notable. The storage and transport of growth factors is a rather important function of PGs, because it may be viewed as a rate-limiting process in signaling mechanisms. Recent studies provided evidence for the translocation of syndecan-1 (Sdc1), glypican, and perlecan into nucleus. In another study, the reduced topoisomerase I activity and inhibition of gene transcription were documented.61,62
PG structures are substrates of heparanase, which cleaves the β(1,4)-glycosidic bond between glucuronic acid and glucosamine residue, releasing heparan sulfate polymer with a shorter chain length. Besides the remodeling of the ECM, uncontrolled heparanase activity also results in an extended release of many HS-linked molecules similarly to growth factors, cytokines, and enzymes, which contribute to various pathological conditions. In fact, heparanase regulates HSPG Sdc1 shedding from the cell surface, which promotes tumor growth and metastasis.
In tumors, changes in the expression and degradation of the PGs contribute to the altered composition of ECM and result in abnormal PG function. PGs after the structural changes may have altered biological effects. On the basis of the above data, it has been proposed that PGs may be potentially useful as molecular markers for cancer personalized diagnosis and therapy.63
Syndecan and glycosylphosphatidylinositol-anchored glypican are the major, and betaglycan and V-3 isoform of CD44 the minor, compounds of membrane HSPGs. In the ECM, and especially in basement membrane, the multidomain perlecan, agrin, and collagen XVIII core proteins are the main heparan sulfate-bearing species. HSPG contributes to development of malignant phenotype and tumor progression.5,15 Alterations of HSPG represent the most tumor-specific changes in the ECM structure. It depends on the variability of the heparan-sulfate synthesis.5,24
Perlecan and agrin
Perlecan is a multidomain HSPG PG that binds to and crosslinks many ECM components and cell-surface receptors. Perlecan as a constituent of basement membrane is affected by growth factors when tumor cells cross this barrier.64 HSPGs were proved to have regulatory roles.65–68 Perlecan’s expression reduces in vitro the tumor cells formed by larger colonies. Enhanced perlecan release causes faster growth in nude mouse cells compared to wild-type tumor cells. A low perlecan level substantially inhibits the growth of nude mouse tumor cells.65–68 The HSPGs, including perlecan, act as promoters or suppressors for proliferation, migration, and invasion of the tumors.5,69 It is well known that the Wnt/β-catenin pathway is involved in the regulation of cell migration, and HSPGs are important regulators of this signaling pathway.5,67 HSPGs induce the expression and activity of topoisomerase II through the stimulation of ERK2 and via integrin-mediated signaling to the Wnt signaling pathway.5,46,48,70–72
HSPG is the most active ECM component for increased proliferation and migration of human OSCORT osteosarcoma cells. That is why its inhibition could be a target in the treatment of metastatic osteosarcoma.5,14 These results are in line with findings referring to HSPG’s multiple role in tumor invasion.5
Agrin, another HSPG, is involved in neuromuscular differentiation and synaptogenesis, as well as being a constituent of renal glomerular basement membrane.5 We have found that agrin is also present in the osteosarcoma tumorous ECM.5 This was the first time that the presence of agrin in a tumor was verified.5 Agrin was the only HSPG present in ECM of OSCORT osteosarcoma cells.5 Tumorous ECM is important in supporting the metastatic ability. The stimulatory effects of HSPG might be attributed to heparan sulfate chains belonging to agrin.5,14 A potential of the Wnt/β-catenin pathway to crosstalk with the agrin signaling cascade has also been reported.5,70
Syndecan
Transmembrane HSPGs have the capacity to interact with a large number of ligands and receptors from the Sdc1.
Docking sites in the syndecan extracellular domains capture αvβ3 integrin and insulin-like growth factor 1 receptor (IGF1R), forming a ternary receptor complex which initiates signaling downstream of IGF1R. It is noteworthy that a peptide inhibitor called synstatin derived from a mutant syndecan can competitively displace the αvβ3 or αvβ5 integrin and IGF1R kinase from the syndecan.73,74
Decorin
Decorin is a small, leucine-rich cellular or pericellular matrix PG and binds to type I collagen fibrils. In contrast to syndecan, decorin acts as a tumor repressor by interfering with the PDGF signaling pathway.75 Upregulation of decorin A549 tumor cells resulted in decreased TGF-β1, cyclin D1 expression, and phosphorylation of EGFR, but higher levels of p53 and of p21 (Waf1/Cip1). The changes of these signaling molecules could be correlated with the decreased invasive growth and cell cycle blockage at G1.76,77
The expression of CCN1, a secreted matrix-associated molecule, has been reported to correlate inversely with the aggressiveness of non-small-cell lung carcinoma; recombinant CCN1 protein led to a permanent cell cycle arrest in G1 phase.
The phenotype characteristic of senescent cells is influenced by abundance of hypo-phosphorylated Rb, accumulation of p53 and p21 (Waf1/Cip1) and senescence-associated β-galactosidase.78
Heparanase
Heparanase cleaves heparan sulfate and participates in degrading and remodeling the ECM. Since a high level of heparanase in tumor was shown to correlate with distant metastasis and reduced survival of cancer patients, heparanase and its inhibitors have been listed among the targets of antitumor agents.79
The following heparanase inhibitors are documented in detail:80,81
- PI-88 contains a mixture of highly sulfated, monophosphorylated mannose oligosaccharides.
- Oligomannurarate sulfate (JG3), a novel marine-derived oligosaccharide, inhibited tumor angiogenesis and metastasis by combating heparanase activity.
- RK-682 was found to display inhibitory activity against heparanase.
- The selective inhibition of heparanase with 4-benzyl-(R)-3-hexadecanoyl-5-hydroxymethyltetronic acid has been demonstrated to display anti-invasive activity.
- PG545, a tetrasaccharide heparan sulfate, shows affinity for heparanase a clinical anticancer candidate.
GAG biosynthesis and FAF inhibitors
Taking into account the importance of GAG chains in the biological action of PGs, small molecular compounds related to carbohydrate metabolism were investigated as putative GAG inhibitors. In tumor cell cultures, beta-D-xyloside, 2-deoxy-D-glucose, ethane-l-hydroxy-l,l-diphosphonate, and 5-hexyl-2-deoxyuridine (HUdR) induced reduced incorporation of [14C]-glucosamine into GAG, but to varying degrees. The most effective HUdR at 15 μg/mL concentration selectively inhibited GAG relative to other glycoconjugates.82,83
The first studies on the antimetastatic efficacy of HUdR aimed to compare the highly metastatic variant of LLT3 tumor containing elevated PG concentration with the parent tumor. The significant reduction in the metastatic nodules in the lung indicated an inverse correlation with GAG concentration and metastatic potency. However, in the subsequent experiments, the metastatic pattern of the mice transplanted intracardially with the HHLLT3 tumor showed an organ-specific action of HUdR as the number of metastatic nodules decreased only in the lung and kidney but not in the liver.84 Table 1 describes the antimetastatic action of HUdR.
 | Table 1 The effect of HUdR on the metastasis formation of 3LL-HH tumor cells injected into the left ventricle |
Integrins as ECM receptor
Integrin-mediated signaling
The multiple attachments of ECM biopolymers to the cell surface are executed mainly from a family of transmembrane adhesion receptors composed of non-covalently linked α and β subunits. The 18 α and eight β subunits form around 24 unique integrins, which have a ligand-binding region at the cell surface for the ECM biopolymers. Integrins on the cell surface in a distinct activation state represent different affinities for ECM biopolymers. They are responsible for the formation of the dynamic contacts among the cellular and macromolecular elements of a tissue. In addition, the extracellular signal transduction, as an essential process for the cells, is mediated by the integrins. Several cellular signaling pathways are initiated from the integrins toward cytoskeletons for migration and also to the RAS/MAPK and PI3 kinase/mTOR pathways for survival. The outcome of the signal mechanisms are influenced by the correct linkage between ECM biopolymer and integrin. Survival pathways may be activated providing the appropriate ligand–integrin complex has been formed and stimulated by the relevant growth factor.5,14,32 Conversely, faulty, deficient, or blocked integrin ligand complexes can promote apoptosis under otherwise permissive growth conditions.
Integrin-linked kinase and focal adhesion kinase are key components of the signal transduction pathway triggered by integrin receptors to control cell growth, invasion, and survival.
Integrins in tumor progression
The invasion of cancer cells in tumor progression is mainly regulated by their proteolytic remodeling of their surrounding ECM and by their integrin expression. The expression of the actual member of the integrin family in healthy conditions is mostly tissue/cell-specific, whereas, in pathological lesions, remarkable changes could be seen, including cancer. Integrin expression exhibits considerable heterogeneity according to tumor origin. Proliferation and invasion of many tumors have been associated with the overexpression of integrins.
The receptors for the extracellular signals providing increased proliferation of the cells are presumed to be integrin molecules, mainly β1 integrin-containing complexes.85 As previously described, osteosarcomas produce β1-integrin complexes (α2β1, α3β1, α4β1, α5β1), and β-1 integrin could be induced by ECM biopolymers in human osteosarcoma cell culture.5,86 There was a linear correlation between β1 integrin production and cell proliferation.85,87
The αv subunit was generally moderately to highly expressed in most tumors; nevertheless, the integrin profile may be modulated by the microenvironment. It was reported that αvβ8 binding to type I collagen activated the MEK/ERK signaling pathway.88
The high expression of integrins αvβ3 and αvβ5 was proposed as a prognostic and diagnostic marker and therapeutic target for gastric cancer.89 High expression and activation of αvβ3 integrin was recognized in growing tumor vasculature and has correlated with the most metastatic and invasive breast tumors. Since tumor cells are exposed by a variety of ECM biopolymers during metastasis, for survival, their integrin profile must adapt to the new environment. Therefore, it is important to collect data about the expressed integrin profiles of tumor metastases. At the first step of peritoneal dissemination, ovarian carcinoma cells enhanced expression of α2β1 integrin that was related to spheroid disaggregation and proteolysis.90
In the permanent migratory capacity of tumor cells, the fibronectin-binding integrins, such as αvβ3, have been suggested to have an important function in metastatic renal cell carcinomas and melanomas. αvβ3 and cytoplasmic β3 were weakly to moderately detectable. In addition, αvβ5 was prominently expressed in metastatic renal and colorectal carcinomas. Interestingly, in metastatic lung adenocarcinomas, αvβ6 was the most abundantly detectable integrin, but was absent in melanomas. Thus, the dominant integrins in metastatic tumors show certain heterogeneity.
Integrin inhibitors
Some integrins (αvβ3, αvβ5, αvβ1, αIIbvβ3) interact with ligands of ECM containing the Arg-Gly-Asp sequence (RGD sequence).
Cilengitide is a cyclic RGD peptide (cyclo arg-gly-asp-D-Phe-1-amino cyclohexane carboxylic acid) and was reported to reduce invasiveness of melanoma cells through the inhibition of αvβ5 integrin.91
Another integrin inhibitor, S247 is a small peptidomimetic antagonist of αvβ3 and αvβ5 integrins, and it is able to inhibit lung metastasis in mice.
Recently, humanized monoclonal antibodies have been developed to inactivate integrin αv and α4β7. Abituzumab targeting integrin αv heterodimers has demonstrated preclinical activity. Vedolizumab binds to the α4β7 integrin complex and inhibits adhesion of certain molecules.92
Salvicine, a modified diterpenoid quinone, inhibited the adhesion of human breast cancer MDA-MB-435 cells to fibronectin and collagen. Salvicine downregulated β1 integrin ligand affinity, clustering and signaling via dephosphorylation of focal adhesion kinase.93
Secreted threonyl-tRNA synthetase stimulates cell migration. This enzyme could be inhibited by borrelidin, showing selective antimigratory and antimetastatic effects in several animal models. In vitro antimigratory efficacy of borrelidin was attenuated in the presence of ECM proteins, preferentially by fibronectin. This observation demonstrated the selective reduction of expression of integrin αvβ5, because α2, α3 and β1 subunit were not affected in borrelidin-treated tumor cells.5,94,95
ECM as modulator of chemotherapeutic drug efficiency and as target in antitumor treatment
It is well known that ECM remodeling is crucial for tumor malignancy and metastatic progression.13,14,87,96 However, the further question arises whether ECM could provide protection against chemotherapeutic drugs and contribute to tumor cell repopulation after the treatment. A large number of experiments have provided evidence that apoptosis induced by chemotherapeutic agents is suppressed in the presence of ECM.14,69,97,98 Failure in antitumor therapy is mostly due to repopulation of the surviving tumor cells. In this respect, the repopulation is viewed as a continuing proliferation of cells with the capacity to regenerate the tumor shrinkage after anticancer treatment.69 ECM promotes both a “dormant-like” state and increases resistance of certain tumor cells to cytoreductive treatment, thereby modeling two key processes that limit tumor medication.13,69
Studying ECM and ECM biopolymers is an important requirement of successful anticancer therapy. Nevertheless, ECM-mediating pathways are not well modeled in classical cell culture systems.13,14 The ECM gel used model provides an extracellular matrix-based culture system (3-DCC) as an alternative option to the conventional plastic cell culture. Using this system, the protective effect of ECM against cytotoxic damage is recognizable.13,14
In the presence of ECM (ECM gel), a reduction of chemotherapeutic drug-induced apoptosis was detected in glioma, myeloma, and small-cell lung cancer cells.99 Stromal ECM provides protection against cytotoxicity of topoisomerase poisons in colon tumor cells and small-cell lung cancer cells.100 This is achieved by stimulation of protein tyrosine kinase (PTK) and/or inhibition of caspase-3 activation.100
In the case of osteosarcoma, clinical trials did not show any survival benefit following intensified therapy in poor responders.101 It was found that dynamic ECM remodeling is involved in the chemotherapy resistance of osteosarcoma.5 It includes bone degradation and, simultaneously, synthesis of novel, modified, tumor-specific ECM.5 In the presence of tumorous ECM, the apoptotic index, the parameters of the comet assay and the expression of p53 were lower.69 The detected reduction of apoptosis has been associated with a decrease of bax expression. This lowering is due to the invalidation of p53 cell nuclear localization.69 This event was induced by the tumorous ECM.15 Decreased p53 protein synthesis correlated with increased survival and decreased apoptosis of tumor cells following doxorubicin treatment.15 ECM can increase the repair capacity of cells and may contribute to increased repair after cytostatic damage.69,102 Since DNA damage occurs subsequent to radiochemotherapy, the above data provide further evidence for ECM-enhanced repair.13
Up to now, anticancer drug research has focused on inhibition of a tumor growth, and a little attention was paid to controlling tumor cell migration.32,33 The antimigratory effects of drugs can be well investigated by newly developed assays.14,32 The Boyden chamber in combination with the 3-DCC ECM-based model makes it possible to differentiate between single-cell or cluster-type migration.14,32 Our data provide evidence that there are cytotoxic/cytostatic agents which, depending on concentration, preferentially inhibit migration compared to proliferation.32,33 Taxol, a widely used anticancer drug, selectively inhibits the single-cell migration when it is used at a very low, subtoxic concentration.32,33 Depending on microenvironment or antitumor treatment, tumor cells switch their mechanism of migration and therefore escape the effect of applied drug.14 Since doxorubicin inhibits cluster-type migration at subtoxic concentrations, the combination of Taxol with doxorubicin offers a possibility of acting against both single-cell and cluster-type tumor cell migration.32,33 Therefore, successful antimetastatic therapy will be achieved when agents acting against single-cell and cluster-type migration become weapons in the arsenal of clinical oncology.32,33
Concluding remarks
The drastic structural alteration in tumor microenvironment was recognized more than 100 years ago. Its relevance in tumor development and progression has been understood only in the last two decades. A large body of evidence has been accumulated that indicates matrix proteins are basic constituents of the extracellular space and that their cell surface receptors, mainly integrins, have an active role at all steps of tumor progression. This participation of ECM includes the uncontrolled cell proliferation, disruption of cellular interaction, migration, and settlement in remote organs. ECM is not only a major component of tumors, but also a contributor to the transformed phenotype by reprogramming cellular phenotype. ECM provides cues that promote proliferation and migration. Considering all the complex impact of matrix proteins and PGs surrounding the malignant cells, it has been suggested that ECM acts as a driving force in tumor progression and represents a novel perspective for examining the prognosis and developing therapeutic agents. Composition of ECM in tumors not only differs from in normal tissue, but also shows great heterogeneity among various tumors, which allows the classification of tumors according to their ECM profile. As a matter of fact, the structural and functional changes in tumorous ECM may not be viewed as a reactive response exclusively by the host because gene expression of ECM components was modified both in neoplastic and stromal cells.8
The clinical studies indicate that high expression of integrins and metalloproteinases predicted poorer prognosis, whereas low expression of laminin chains represented a rather favorable outcome. Since preclinical and clinical studies demonstrated the multiple mechanism by which the various ECM components constantly exert a driving force on the progression of tumors, the question has been raised whether the ample knowledge may be utilized in antitumor therapy. Certainly, to design drugs selectively targeting ECM represents an attractive actual and novel approach, but raises several unresolved problems. The ECM components, such as decorin and syndecan, exert opposite actions on proliferation; similarly, PGs, after loss of their carbohydrate chain, show different biological activity. Perhaps the most severe limiting aspect is the duality in the function of some ECM-associated molecules, as was revealed when loss of β1 integrin attenuated breast tumor growth but markedly enhanced tumor cell dissemination.103
These circumstances explain the modest steps in the transition of the results from preclinical studies on ECM-targeted anticancer therapy into clinical practice. Presently, it is worth focusing on agents that act on heparanase and proteases, which restructure ECM. In addition, there are noteworthy clinical experiences with cilengitide inactivating αvβ3 and αvβ5 integrins. Furthermore, the inhibition of GAG offers another promising approach in the treatment of those tumors which are stimulated by PGs. In spite of all the difficulties, ECM must be considered as a highly rewarding target in tumor therapy, because nearly all steps in the metastatic process – ie, disruption and adhesion in intercellular contacts, migration, and settlement – are controlled by the aberrantly operating tumor ECM.
Disclosure
The authors report no conflicts of interest in this work.
References
Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol. 2012;196(4):395–406. | ||
Langlois B, Saupe F, Rupp T, et al. AngioMatrix, a signature of the tumor angiogenic switch-specific matrisome, correlates with poor prognosis for glioma and colorectal cancer patients. Oncotarget. 2014;5(21):10529–10545. | ||
Iozzo RV. Matrix proteoglycans: from molecular design to cellular function. Annu Rev Biochem. 1998;67:609–652. | ||
Ali MY, Chuang CY, Saif MT. Reprogramming cellular phenotype by soft collagen gels. Soft Matter. 2014;10(44):8829–8837. | ||
Harisi R, Dudás J, Timár F, et al. Invasive growth and topoisomerase-switch induced by tumorous extracellular matrix in osteosarcoma cell culture. Cell Biol Int. 2005;29(11):959–967. | ||
Glentis A, Gurchenkov V, Matic Vignjevic D. Assembly, heterogeneity, and breaching of the basement membranes. Cell Adh Migr. 2014;8(3): 236–245. | ||
Triulzi T, Casalini P, Sandri M, et al. Neoplastic and stromal cells contribute to an extracellular matrix gene expression profile defining a breast cancer subtype likely to progress. PLoS One. 2013;8(2): e56761. | ||
Naba A, Clauser KR, Whittaker CA, Carr SA, Tanabe KK, Hynes RO. Extracellular matrix signatures of human primary metastatic colon cancers and their metastases to liver. BMC Cancer. 2014;14:518. | ||
Bergamaschi A, Tagliabue E, Sørlie T, et al. Extracellular matrix signature identifies breast cancer subgroups with different clinical outcome. J Pathol. 2008;214(3):357–367. | ||
Adamson IY, Young L, Bakowska J. Enhanced alveolar type II cell growth on a pulmonary extracellular matrix over fibroblasts. Am J Physiol. 1997;272(3 Pt 1):L413–L417. | ||
Vlodavsky I, Lui GM, Gospodarowicz D. Morphological appearance, growth behavior and migratory activity of human tumor cells maintained on extracellular matrix versus plastic. Cell. 1980;19(3):607–616. | ||
Niedbala MJ, Crickard K, Bernacki RJ. Adhesion, growth and morphology of human mesothelial cells on extracellular matrix. J Cell Sci. 1986;85:133–147. | ||
Pogány G, Timár F, Oláh J, et al. Role of the basement membrane in tumor cell dormancy and cytotoxic resistance. Oncology. 2001;60(3):274–281. | ||
Harisi R, Dudás J, Tímár F, et al. Antiproliferative and antimigratory effects of doxorubicin in human osteosarcoma cells exposed to extracellular matrix. Anticancer Res. 2005;25(2A):805–813. | ||
Harisi R, Dudas J, Nagy-Olah J, Timar F, Szendroi M, Jeney A. Extracellular matrix induces doxorubicin-resistance in human osteosarcoma cells by suppression of p53 function. Cancer Biol Ther. 2007;6(8):1240–1246. | ||
Larsen AK, Skladanowski A, Bojanowski K. The roles of DNA topoisomerase II during the cell cycle. Prog Cell Cycle Res. 1996;2:229–239. | ||
Gerdes J, Lemke H, Baisch H, Wacker HH, Schwab U, Stein H. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol. 1984;133(4):1710–1715. | ||
He CJ, Striker LJ, Tsokos M, Yang CW, Peten EP, Striker GE. Relationships between mesangial cell proliferation and types I and IV collagen mRNA levels in vitro. Am J Physiol. 1995;269(3 Pt 1):C554–C562. | ||
Williams DF, Burke JM. Modulation of growth in retina-derived cells by extracellular matrices. Invest Ophthalmol Vis Sci. 1990;31(9):1717–1723. | ||
Rutka JT. The K.G. McKenzie award lecture – 1986. Effects of extracellular matrix proteins on the growth and differentiation of an anaplastic glioma cell line. Can J Neurol Sci. 1986;13(4):301–306. | ||
Holmgren L, O’Reilly MS, Folkman J. Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat Med. 1995;1(2):149–153. | ||
Liotta LA, Kohn EC. The microenvironment of the tumor-host interface. Nature. 2001;411(6835):375–379. | ||
Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer. 2001;1(1):46–54. | ||
Sasisekharan R, Shriver Z, Venkataraman G, Narayanasami U. Roles of heparan-sulphate glycosaminoglycans in cancer. Nat Rev Cancer. 2002;2(7):521–528. | ||
Thorns V, Walter GF, Thorns C. Expression of MMP-2, MMP-7, MMP-9, MMP-10 and MMP-11 in human astrocytic and oligodendroglial gliomas. Anticancer Res. 2003;23(5A):3937–3944. | ||
Friedl P, Noble PB, Walton PA, et al. Migration of coordinated cell clusters in mesenchymal and epithelial cancer explants in vitro. Cancer Res. 1995;55(20):4557–4560. | ||
Hegerfeldt Y, Tusch M, Bröcker EB, Friedl P. Collective cell movement in primary melanoma explants: plasticity of cell-cell interaction, beta1-integrin function, and migration strategies. Cancer Res. 2002;62(7):2125–2130. | ||
Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer. 2003;3(5)362–374. | ||
Wolf K, Wu YI, Liu Y, et al. Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat Cell Biol. 2007;9(8):893–904. | ||
Friedl P, Wolf K. Tube travel: the role of proteases in individual and collective cancer cell invasion. Cancer Res. 2008;68(18):7247–7249. | ||
Friedl P, Wolf K. Proteolytic interstitial cell migration: a five-step process. Cancer Metastasis Rev. 2009;28(1–2):129–135. | ||
Harisi R, Kenessey I, Olah JN, et al. Differential inhibition of single and cluster type tumor cell migration. Anticancer Res. 2009;29(8):2981–2985. | ||
Jeney A, Kenessey I, Timár F, et al. [Study of drugs against neoplastic metastasis]. Magy Onkol. 2006;50(2):93–100. Hungarian. | ||
Arboleda MJ, Lyons JF, Kabbinavar FF, et al. Overexpression of AKT2/protein kinase Bbeta leads to up-regulation of beta1 integrins, increased invasion, and metastasis of human breast and ovarian cancer cells. Cancer Res. 2003;63(1):196–206. | ||
Tauro M, McGuire J, Lynch CC. New approaches to selectively target cancer-associated matrix metalloproteinase activity. Cancer Metastasis Rev. 2014;33(4):1043–1057. | ||
Stetler-Stevenson WG, Gavil NV. Normalization of the tumor microenvironment: evidence for tissue inhibitor of metalloproteinase-2 as a cancer therapeutic. Connect Tissue Res. 2014;55(1):13–19. | ||
Lu N, Ling Y, Gao Y, et al. Endostar suppresses invasion through downregulating the expression of matrix metalloproteinase-2/9 in MDA-MB-435 human breast cancer cells. Exp Biol Med (Maywood). 2008;233(8):1013–1020. | ||
Lu Z, Lu N, Li C, et al. Oroxylin A inhibits matrix metalloproteinase-2/9 expression and activation by up-regulating tissue inhibitor of metalloproteinase-2 and suppressing the ERK1/2 signaling pathway. Toxicol Lett. 2012;209(3):211–220. | ||
Saha S, Lo PK, Duan X, Chen H, Wang Q. Breast tumour initiating cell fate is regulated by microenvironmental cues from an extracellular matrix. Integr Biol (Camb). 2012;4(8):897–904. | ||
Steelman LS, Chappell WH, Abrams SL, et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging (Albany NY). 2011;3(3):192–222. | ||
Provenzano PP, Inman DR, Eliceiri KW, et al. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008;6:11. | ||
Liang X, Osman TA, Sapkota D, et al. Rapid adherence to collagen IV enriches for tumour initiating cells in oral cancer. Eur J Cancer. 2014;50(18):3262–3270. | ||
Han J, Ohno N, Pasco S, Monboisse JC, Borel JP, Kefalides NA. A cell binding domain from the alpha3 chain of type IV collagen inhibits proliferation of melanoma cells. J Biol Chem. 1997;272(33):20395–20401. | ||
Sudan S, Rupasinghe HP. Flavonoid-enriched apple fraction AF4 induces cell cycle arrest, DNA topoisomerase II inhibition, and apoptosis in human liver cancer HepG2 cells. Nutr Cancer. 2014;66(7):1237–1246. | ||
De Nictolis M, Garbisa S, Lucarini G, et al. 72-kilodalton type IV collagenase, type IV collagen, and Ki 67 antigen in serous tumors or the ovary: a clinicopathologic, immunohistochemical, and Serological study. Int J Gynecol Pathol. 1996;15(2):102–109. | ||
Shapiro PS, Whalen AM, Tolwinski NS, et al. Extracellular signal-regulated kinase activates topoisomerase I alpha through a mechanism independent of phosphorylation. Mol Cell Biol. 1999;19(5):3551–3560. | ||
Anbalagan M, Rao AJ. Collagen IV-mediated signalling is involved in progenitor Leydig cell proliferation. Reprod Biomed Online. 2004;9(4):391–403. | ||
Schambony A, Kunz M, Gradl D. Cross-regulation of Wnt signaling and cell adhesion. Differentiation. 2004;72(7):307–318. | ||
Pal S, Moulik S, Dutta A, Chatterjee A. Extracellular matrix protein laminin induces matrix metalloproteinase-9 in human breast cancer cell line mcf-7. Cancer Microenviron. 2014;7(1–2):71–78. | ||
Kramer RH, Bensch KG, Wong J. Invasion of reconstituted basement membrane matrix by metastatic human tumor cells. Cancer Res. 1986;46(4 Pt 2):1980–1989. | ||
Schifferli KP, Henrich CJ. Analysis of integrin expression and function in HT1080 cells using inhibitory anti-integrin antibodies. Focus. 1996;18:13–15. | ||
Noël AC, Callé A, Emonard HP, et al. Invasion of reconstituted basement membrane matrix is not correlated to the malignant metastatic cell phenotype. Cancer Res. 1991;51(1):405–414. | ||
Kawaguchi S, Uede T. Distribution of integrins and their matrix ligands in osteogenic sarcomas. J Orthop Res. 1993;11(3):386–395. | ||
Christensen L. The distribution of fibronectin, laminin and tetranectin in human breast cancer with special attention to the extracellular matrix. APMIS Suppl. 1992;26:1–39. | ||
Assoian RK, Marcantonio EE. The extracellular matrix as a cell cycle control element in atherosclerosis and restenosis. J Clin Invest. 1997;100(11 Suppl):S15–S18. | ||
Hughes PE, Renshaw MW, Pfaf M, et al. Suppression of integrin activation: a novel function of a Ras/Raf-initiated MAP kinase pathway. Cell. 1997;88(4):521–530. | ||
Aguirre Ghiso JA, Kovalski K, Ossowski L. Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. J Cell Biol. 1999;147(1):89–104. | ||
Kleinman HK, McGarvey MJ, Liotta LA, Robey PG, Tryggvason K, Martin GR. Isolation and characterization of type IV procollagen, laminin, and heparan sulfate proteoglycan from the EHS sarcoma. Biochemistry. 1982;21(24):6188–6193. | ||
Timpl R, Fujiwara S, Dziadek M, et al. Laminin, proteoglycan, nidogen, and collagen IV: structural models and molecular interactions. In: Porter R, Whelan J, editors. Basement Membranes and Movement. London: Pitman; 1984:25–74. | ||
Liu AY, Zheng H, Ouyang G. Periostin, a multifunctional matricellular protein in inflammatory and tumor microenvironments. Matrix Biol. 2014;37:150–156. | ||
Kovalszky I, Hjerpe A, Dobra K. Nuclear translocation of heparan sulfate proteoglycans and their functional significance. Biochim Biophys Acta. 2014;1840(8):2491–2497. | ||
Kovalszky I, Dudás J, Oláh-Nagy J, et al. Inhibition of DNA topoisomerase I activity by heparan sulfate and modulation by basic fibroblast growth factor. Mol Cell Biochem. 1998;183(1–2):11–23. | ||
Suhovskih AV, Mostovich LA, Kunin IS, et al. Proteoglycan expression in normal human prostate tissue and prostate cancer. ISRN Oncol. 2013;2013:680136. | ||
Mishra M, Chandavarkar V, Naik VV, Kale AD. An immunohistochemical study of basement membrane heparan sulfate proteoglycan (perlecan) in oral epithelial dysplasia and squamous cell carcinoma. J Oral Maxillofac Pathol. 2013;17(1):31–35. | ||
Mathiak M, Yenisey C, Grant DS, Sharma B, Iozzo RV. A role for perlecan in the suppression of growth and invasion in fibrosarcoma cells. Cancer Res. 1997;57(11):2130–2136. | ||
Sharma B, Handler M, Eichstetter I, Whitelock JM, Nugent MA, Iozzo RV. Antisense targeting of perlecan blocks tumor growth and angiogenesis in vivo. J Clin Invest. 1998;102(8):1599–1608. | ||
Merz DC, Alves G, Kawano T, Zheng H, Culotti JG. UNC-52/perlecan affects gonadal leader cell migrations in C. Elegans hermaphrodites through alterations in growth factor signaling. Dev Biol. 2003;256(1):173–186. | ||
Fuster MM, Esko JD. The sweet and sour of cancer: glycans as novel therapeutic targets. Nat Rev Cancer. 2005;5(7):526–542. | ||
Harisi R, Dudás J, Pogány G, et al. Repopulation of osteosarcoma cells after treatment with doxorubicin in the presence of extracellular matrix biopolymers. Cancer Chemother Pharmacol. 2006;58(3):334–342. | ||
Sancéau J, Truchet S, Bauvois B. Matrix metalloproteinase-9 silencing by RNA interference triggers the migratory-adhesive switch in Ewing’s sarcoma cells. J Biol Chem. 2003;278(38):36537–36546. | ||
Hsiang YH, Wu Hy, Liu LF. Proliferation-dependent regulation of DNA topoisomerase II in cultured human cells. Cancer Res. 1998;48(11):3230–3235. | ||
Ludvíková M, Holubec L Jr, Ryska A, Topolcan O. Proliferative markers in diagnosis of thyroid tumors: a comparative study of MIB-1 and topoisomerase II-a immunostaining. Anticancer Res. 2005;25(3A):1835–1840. | ||
Beauvais DM, Ell BJ, McWhorter AR, Rapraeger AC. Syndecan-1 regulates alphavbeta3 and alphavbeta5 integrin activation during angiogenesis and is blocked by synstatin, a novel peptide inhibitor. J Exp Med. 2009;206(3):691–705. | ||
Rapraeger AC. Synstatin: a selective inhibitor of the syndecan-1-coupled IGF1R-αvβ3 integrin complex in tumorigenesis and angiogenesis. FEBS J. 2013;280(10):2207–2215. | ||
Baghy K, Horváth Z, Regős E, et al. Decorin interferes with platelet-derived growth factor receptor signaling in experimental hepatocarcinogenesis. FEBS J. 2013;280(10):2150–2164. | ||
Liang S, Xu JF, Cao WJ, Li HP, Hu CP. Human decorin regulates proliferation and migration of human lung cancer A549 cells. Chin Med J (Engl). 2013;126(24):4736–4741. | ||
Horváth Z, Kovalszky I, Fullár A, et al. Decorin deficiency promotes hepatic carcinogenesis. Matrix Biol. 2014;35:194–205. | ||
Jim Leu SJ, Sung JS, Chen MY, et al. The matricellular protein CCN1 suppresses lung cancer cell growth by inducing senescence via the p53/p21 pathway. J Cell Biochem. 2013;114(9):2082–2093. | ||
Nadir Y, Brenner B. Heparanase multiple effects in cancer. Thromb Res. 2014;133 Suppl 2:90–94. | ||
Zhao H, Liu H, Chen Y, et al. Oligomannurarate sulfate, a novel heparanase inhibitor simultaneously targeting basic fibroblast growth factor, combats tumor angiogenesis and metastasis. Cancer Res. 2006;66(17):8779–8787. | ||
Ishida K, Hirai G, Murakami K, et al. Structure-based design of a selective heparanase inhibitor as an antimetastatic agent. Mol Cancer Ther. 2004;3(9):1069–1077. | ||
Tímár J, Diczházi C, Bartha I, et al. Modulation of heparan-sulphate/chondroitin-sulphate ratio by glycosaminoglycan biosynthesis inhibitors affects liver metastatic potential of tumor cells. Int J Cancer. 1995;62(6):755–761. | ||
Pogány G, Jeney A, Timár J, Major J, Lapis K. Modulation of glycoconjugate biosynthesis by 5-hexyl-2′-deoxyuridine in highly metastatic Lewis lung carcinoma cells. Neoplasma. 1990;37(5):501–510. | ||
Jeney A, Timár J, Pogány G, et al. Glycosaminoglycans as novel target in antitumor therapy. Tokai J Exp Clin Med. 1990;15(2–3):167–177. | ||
Park CP, Bissel MJ, Barcellos-Hoff MH. The influence of the microenvironment on the malignant phenotype. Mol Med Today. 2000;8:324–329. | ||
Heino J, Massagué J. Transforming growth factor-beta switches the pattern of integrins expressed in MG-63 human osteosarcoma cells and causes a selective loss of cell adhesion to laminin. J Biol Chem. 1989;264(36):21806–21811. | ||
Sethi T, Rintoul RC, Moore SM, et al. Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: a mechanism for small cell lung cancer growth and drug resistance in vivo. Nat Med. 1999;5(6):662–668. | ||
Hayashido Y, Kitano H, Sakaue T, et al. Overexpression of integrin αv facilitates proliferation and invasion of oral squamous cell carcinoma cells via MEK/ERK signaling pathway that is activated by interaction of integrin αvβ8 with type I collagen. Int J Oncol. 2014;45(5):1875–1882. | ||
Böger C, Warneke VS, Behrens HM, et al. Integrins αvβ 3 and α vβ 5 as prognostic, diagnostic, and therapeutic targets in gastric cancer. Gastric Cancer. Epub 2014 Oct 15. | ||
Shield K, Riley C, Quinn MA, Rice GE, Ackland ML, Ahmed N. Alpha2beta1 integrin affects metastatic potential of ovarian carcinoma spheroids by supporting disaggregation and proteolysis. J Carcinog. 2007;6:11. | ||
Ruffini F, Graziani G, Levati L, Tentori L, D’Atri S, Lacal PM. Cilengitide downmodulates invasiveness and vasculogenic mimicry of neuropilin 1 expressing melanoma cells through the inhibition of αvβ5 integrin. Int J Cancer. 2015;136(6):E45–E58. | ||
Élez E, Kocáková I, Höhler T, et al. Abituzumab combined with cetuximab plus irinotecan versus cetuximab plus irinotecan alone for patients with KRAS wild-type metastatic colorectal cancer: the randomised phase I/II POSEIDON trial. Ann Oncol. 2015;26(1):132–140. | ||
Zhou J, Chen Y, Lang JY, Lu JJ, Ding J. Salvicine inactivates beta 1 integrin and inhibits adhesion of MDA-MB-435 cells to fibronectin via reactive oxygen species signaling. Mol Cancer Res. 2008;6(2):194–204. | ||
Habibi D, Ogloff N, Jalili RB, et al. Borrelidin, a small molecule nitrile-containing macrolide inhibitor of threonyl-tRNA synthetase, is a potent inducer of apoptosis in acute lymphoblastic leukemia. Invest New Drugs. 2012;30(4):1361–1370. | ||
Williams TF, Mirando AC, Wilkinson B, Francklyn CS, Lounsbury KM. Secreted threonyl-tRNA synthetase stimulates endothelial cell migration and angiogenesis. Sci Rep. 2013;3:1317. | ||
Bissell MJ, Weaver VM, Lelièvre SA, Wang F, Petersen OW, Schmeichel KL. Tissue structure, nuclear organization, and gene expression in normal and malignant breast. Cancer Res. 1999;59(7 Suppl):1757s–1764s. | ||
Provisor AJ, Ettinger LJ, Nachman JB, et al. Treatment of nonneoplastic osteosarcoma of the extremity with preoperative and postoperative chemotherapy: a report from the Children’s Cancer Group. J Clin Oncol. 1997;15(1):76–84. | ||
Sanderson RD. Heparan sulfate proteoglycans in invasion and metastasis. Semin Cell Dev Biol. 2001;12(2):89–98. | ||
Kouniavsky G, Khaikin M, Zvibel I, et al. Stromal extracellular matrix reduces chemotherapy-induced apoptosis in colon cancer cell lines. Clin Exp Metastasis. 2002;19(1):55–60. | ||
Rintoul RC, Sethi T. Extracellular matrix regulation of drug resistance in small-cell lung cancer. Clin Sci (Lond). 2002;102(4):417–424. | ||
Anninga JK, Gelderblom H, Fiocco M, et al. Chemotherapeutic adjuvant treatment for osteosarcoma: where do we stand? Eur J Cancer. 2011;47(16):2431–2445. | ||
Fuks Z, Vlodavsky I, Andreeff M, McLoughlin M, Haimovitz-Friedman A. Effects of extracellular matrix on the response of endothelial cells to radiation in vitro. Eur J Cancer. 1992;28A(4–5):725–731. | ||
Truong HH, Xiong J, Ghotra VP, et al. β1 integrin inhibition elicits a prometastatic switch through the TGFβ-miR-200-ZEB network in E-cadherin-positive triple-negative breast cancer. Sci Signal. 2014;7(312):ra15. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.