")
Back to Journals » OncoTargets and Therapy » Volume 10
Expression of microRNA-30c via lentivirus vector inhibits the proliferation and enhances the sensitivity of highly aggressive ccRCC Caki-1 cells to anticancer agents
Authors Yang HL, Song EL, Shen GR, Zhu TH, Jiang TW, Shen H, Niu LP, Wang B, Lu ZY, Qian JP
Received 25 June 2016
Accepted for publication 29 November 2016
Published 2 February 2017 Volume 2017:10 Pages 579—590
DOI https://doi.org/10.2147/OTT.S115791
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Tohru Yamada
Honglin Yang,1,* Erlin Song,2,3,* Guorong Shen,1 Tonghua Zhu,1 Tingwang Jiang,4 Hao Shen,1 Liping Niu,1 Biao Wang,1 Zhaoyang Lu,5 Jianping Qian4
1Department of Laboratory Medicine, The first people's Hospital of Wujiang District, Suzhou, 2Department of Urinary Surgery, The First Affiliated Hospital of Harbin Medical University, 3Key Laboratory of Cardiovascular Medicine Research, Harbin Medical University, Ministry of Education, Harbin, 4Changshu Institution for Laboratory Medicine, Changshu, 5Department of Ultrasound Diagnosis, The first people's Hospital of Wujiang District, Suzhou, People's Republic of China
*These authors contributed equally to this work
Abstract: The clear cell renal cell carcinoma (ccRCC) is one of the most fatal urologic tumors, and the prognosis remains very poor for advanced or metastatic ccRCC. This study reveals the roles of microRNA (miR)-30c in regulating a highly aggressive ccRCC cell line proliferation by targeting MTA-1, which is a key mediator for human cancer metastasis. Results from quantitative real-time polymerase chain reaction showed that the expression of MTA-1, the target of miR-30c, was significantly higher in metastatic ccRCC specimens than in nonmetastatic ccRCC or nontumor specimens. Accordingly, endogenous miR-30c is at a much lower level in highly aggressive ccRCC Caki-1 cells than nontumor or ccRCC cell lines. Expression of miR-30c via lentivirus vector inhibits the proliferation, anchorage-independent growth, in vitro invasion or migration, or in vivo growth of Caki-1 cells by repressing MTA-1 protein expression. miR-30c also enhances the sensitivity of Caki-1 cells to anticancer agents, including sorafenib and paclitaxel. These data reveal the potential application of miR-30c and that its targeting gene, MTA-1, would be a potential target in metastatic ccRCC treatment.
Keywords: ccRCC, miR-30c, MTA-1, sorafenib, paclitaxel, Caki-1
Introduction
Clear cell renal cell carcinoma (ccRCC) accounts for about 3% of the cases of human malignancy and is the most common malignant tumor of adult kidney.1 It is also the second leading cause of cancer-related death among patients suffering from urologic cancers.2 At present, radical or partial nephrectomy is still the most effective treatment for local ccRCC.3 However, prognosis remains poor for advanced or metastatic ccRCCs because of low sensitivity to chemotherapy and radiotherapy.3 Recently, some small molecular kinase inhibitors, eg, sorafenib and sunitinib, have evolved rapidly during clinical application.4 Unfortunately, the risk of adverse events and disparate clinical benefits limits clinical benefits of these drugs for treatment of advanced and metastatic ccRCCs.4 Therefore, it is valuable to examine whether ccRCC resistance to chemotherapy or radiotherapy is due to highly aggressive features.
A series of studies showed that human pro-oncogene MTA-1 is aberrantly expressed during the metastatic and aggressive process of human cancers, such as breast, lung, liver, ovarian, and prostate cancers.5 A high level of MTA-1 is associated with proliferation, angiogenesis, and, especially in cancer cells, invasiveness or metastasis. MTA-1 can promote the epithelial–mesenchymal transition (EMT) process by repressing E-cadherin transcription.6 It also promotes tumorigenesis and metastasis by up-regulating TGF-β signaling activity.6 MTA-1 participates in antitumor drugs resistance of breast cancer.7 A high level of MTA-1 suggests development of resistance to tamoxifen.8 Although some evidence showed that MTA-1 mediates proliferation or metastasis and could be a therapeutic target in human cancers, potential roles or applications of MTA-1 in ccRCC remain poorly defined. It has been considered that MTA-1 could be a master regulator of cancer cells metastasis or chemotherapeutic resistance. It is valuable to reveal the roles of MTA-1 in advanced or metastatic ccRCCs.
MicroRNAs (miRNAs or miRs), which is a series of noncoding RNA transcripted by RNA pol III, participate in human cancer regulation by targeting 3′UTR of mRNA sequences.9 Aberrant expression of miRs would participate in the proliferation, survival, and metastasis of many types of human cancers.10 Recently, some miRs have been demonstrated to be tumor suppressors. Expression of miR-122, 34a, 452, 125b, 148a, 137 or let-7 would inhibit the proliferation of cancer cells and enhance the sensitivity of cancer cells to antitumor agents.10–12 Thus, it is necessary to identify and reveal miR targeted at MTA-1. In the present work, we used TargetScan and miRanda to predict potential miRs targeting MTA-1. Among these miRs, a low level of miR-30c expression has been identified in some cancers, eg, lung cancer, ovarian cancer, stomach cancer, breast cancer, and bladder cancer.13 Moreover, Heinzelmann et al14 showed that miR-30c would be one of the miRs related to metastasis and poor prognosis in clear cell renal cell carcinoma. Kong et al13 also revealed the relationship between ERα, miR-30c, and MTA-1 in endometrial cancer. Thus, it is valuable to declare whether miR-30c modulates highly aggressive ccRCC cell line proliferation and metastasis via MTA-1 and the potential application of miR-30c in the ccRCC MDR (Multidrug resistance) process. The results showed that a high level of MTA-1 is associated with the metastatic ccRCC and that endogenous miR-30c expression is inversely associated with MTA-1. Next, common highly aggressive ccRCC model Caki-1 cells were used. Overexpression of miR-30c via lentivirus vector significantly inhibits the proliferation, colony formation, anchorage-independent growth, invasion, and migration. Moreover, miR-30c also enhances the sensitivity of Caki-1 cells to antitumor agents.
Material and methods
Plasmids and lentivirus (LV) construction
Inhibitor of miR-30c was obtained from Invitrogen Company (Carlsbad, CA, USA). The full length sequence of MTA-1 which contains 3′UTR sequence was obtained from the National Center for Biotechnology Information (NCBI) database. For the preparation of MTA-1 mutated vectors, mutated sequence (UGUUUAC to GACGGGU) of miR-30c target sites located in MTA-1 mRNA 3′-UTR was introduced. MTA-1 vector containing mutated full length of MTA-1 were synthesized by chemical synthesis (Gene Ray Company, Shanghai, People’s Republic of China) and cloned into pcDNA3.1 plasmids. The lentivirus (LV) was prepared following the methods described in our previous work.15 Briefly, the full length sequence of miR-30c was cloned into lv-ef1a-IRES-EGFP vectors by chemical synthesis. Then, lv-ef1a-miR30c-IRES-EGFP or lv-ef1a-IRES-EGFP (control) plasmid was co-transfected with D8.91 and pVSV-G into 293T cells, respectively.
RNA isolation and quantitative real-time polymerase chain reaction (qRT-PCR)
The kits to examine miR-30c were obtained from Invitrogen Company. RNA samples isolation and qRT-PCR were performed following the methods described previously.16 The relative RNA levels were normalized to the expression of β-Actin and performed using the ΔΔCt method. The primers for MTA-1 were forward primer 5′-CGC TGA CCA GCA TCA TTG AGT-3′; reverse primer 5′- TGG TTC GGA TTT GGC TTG TTA-3′.
Western blot analysis
Antibodies against MTA-1 and GAPDH were obtained from Santa Cruz Biotechnology (Santa Cruz Biotech, CA, USA). IgG antibodies conjugated with the horseradish peroxidase (HRP) were obtained from Sigma (St Louis, MO, USA). Western blot analysis was performed using standard protocols.17 In brief, total protein samples were performed by SDS-PAGE and trans-printed to polyvinylidene fluoride (PVDF) membranes (EMD Millipore, Billerica, MA, USA). The membranes were then blocked with 5% BSA in TBST buffer and incubated for 2 h at 37°C with primary antibodies. Next, membranes were incubated with the HRP-conjugated secondary antibodies. Finally, the blot was developed with enhanced chemiluminescence reagents (Pierce, Thermo Fisher Scientific, Waltham, MA, USA) by X-ray films.
Anchorage-independent growth
Caki-1 cells, which were infected with control (LV-control) or miR-30c (LV-miR-30c), were plated into six-well plates (1,000 cells per well) (Corning Incorporated, Corning, NY, USA), with a bottom layer of 0.7% low melting temperature agar in Dulbecco’s Modified Eagle’s Medium (DMEM) and a top layer of 0.25% agar in DMEM. Colony number was scored after 3 to 4 weeks of growth.18
Trans-well invasion assay (in vitro migration and invasion)
Caki-1 cells, which were infected with control or miR-30c, were analyzed by trans-well assays performed in 24-well plates chamber (Corning Incorporated) fitted with a polyethylene terephthalate filter membrane with 8-μm pores. Next, the membrane’s undersurface was preincubated by 30 μL ECM (Extracellular matrix) from Sigma-Aldrich Co., mixed with serum-free RPMI 1640 in 1:5 dilution for 4 h at 37°C. The top chambers of the trans-wells were filled with 0.1 mL of cells (5×105 cells per mL) in serum-free medium, and the bottom chambers were filled with 0.3 mL of RPMI 1640 containing 10% FBS. The cells were incubated in the trans-wells at 37°C in 5% CO2 for 12 h. For migration assay, the cells were incubated in chambers without an ECM coating.
In vivo experiments
Caki-1 cells, which were infected with control or miR-30c, was injected into 4- to 6-week-old nude mice (5×105 per animal, 8 mice per group) [29]. The tumor volumes were measured every 5 days by length and width. Volumes of tumor were calculated as (width2 × length)/2. The Animal Experiment Committee of The first people's Hospital of Wujiang District, Suzhou, People's Republic of China, approved all protocols for treating animals, and in vivo studies were carried out in accordance with the UK Animals (Scientific Procedures) Act, 1986, and associated guidelines.
Chemotherapeutic drugs and inhibition rate experiments
Sorafenib was obtained from Meilun Biology Technology Co, LTD (Dalian, Liaoning province, People’s Republic of China); paclitaxel was obtained from Sigma-Aldrich Co.. For inhibition analysis, the cells were treated with the chemotherapeutic drugs for 48 h and then analyzed by MTT assays. The concentrations of sorafenib and paclitaxel are shown in Table 1. The absorbance was measured using a multifunctional microplate reader at 490 nm. The inhibition rate = (OD 490 control group − OD 490 administration group)/(OD 490 control group − OD 490 blank group) ×100%.19,20
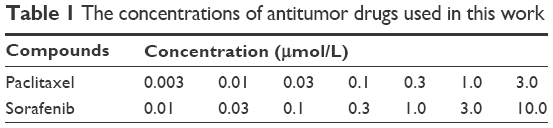 | Table 1 The concentrations of antitumor drugs used in this work |
Ethics statement
Our studies are in compliance with the Helsinki Declaration and aim to declare the function and the underlying molecular mechanisms of MTA-1 in highly aggressive ccRCC regulation. The clinical specimens were preserved by our lab as described previously. Samples were used only for quantitative RT-PCR for miR analysis. The certification for tissue collection and study protocol was described in our previous work. In addition, methods did not relate to the clinical trial or methods. Cell lines used were obtained from the typical biologic sample preservation center in China and have been described.
Statistical analysis
The results were represented as the average from triplicate experiments and expressed as the mean ± standard deviation. Results from Western blot were analyzed by Alpha Innotech software (San Leandro, CA, USA). The relative expression was calculated as follows: (indicated group expression level/loading control expression level)/(control group expression level/loading control expression level).21 The associations between categorical variables were assessed using the chi-square test or the Fisher’s exact test. Bonferroni correction with or without two-way analysis of variance (ANOVA) was performed to determine the statistical significance among the groups (SPSS 17.0 software, SPSS Inc., Chicago, IL, USA).
Results
MTA-1/miR-30c expresses highly in metastatic ccRCC specimens and cell lines
To examine the baseline expression of miR-30c and MTA-1 in ccRCC tissues, quantitative RT-PCR was performed. The expression of MTA-1 mRNA was evaluated in samples of ccRCC rather than in the adjacent nontumor specimens, and the highest level of MTA-1 was detected in metastatic specimens (Figure 1A and C). In parallel, the expression of miR-30c is inversely associated with MTA-1 (Figure 1B and D).
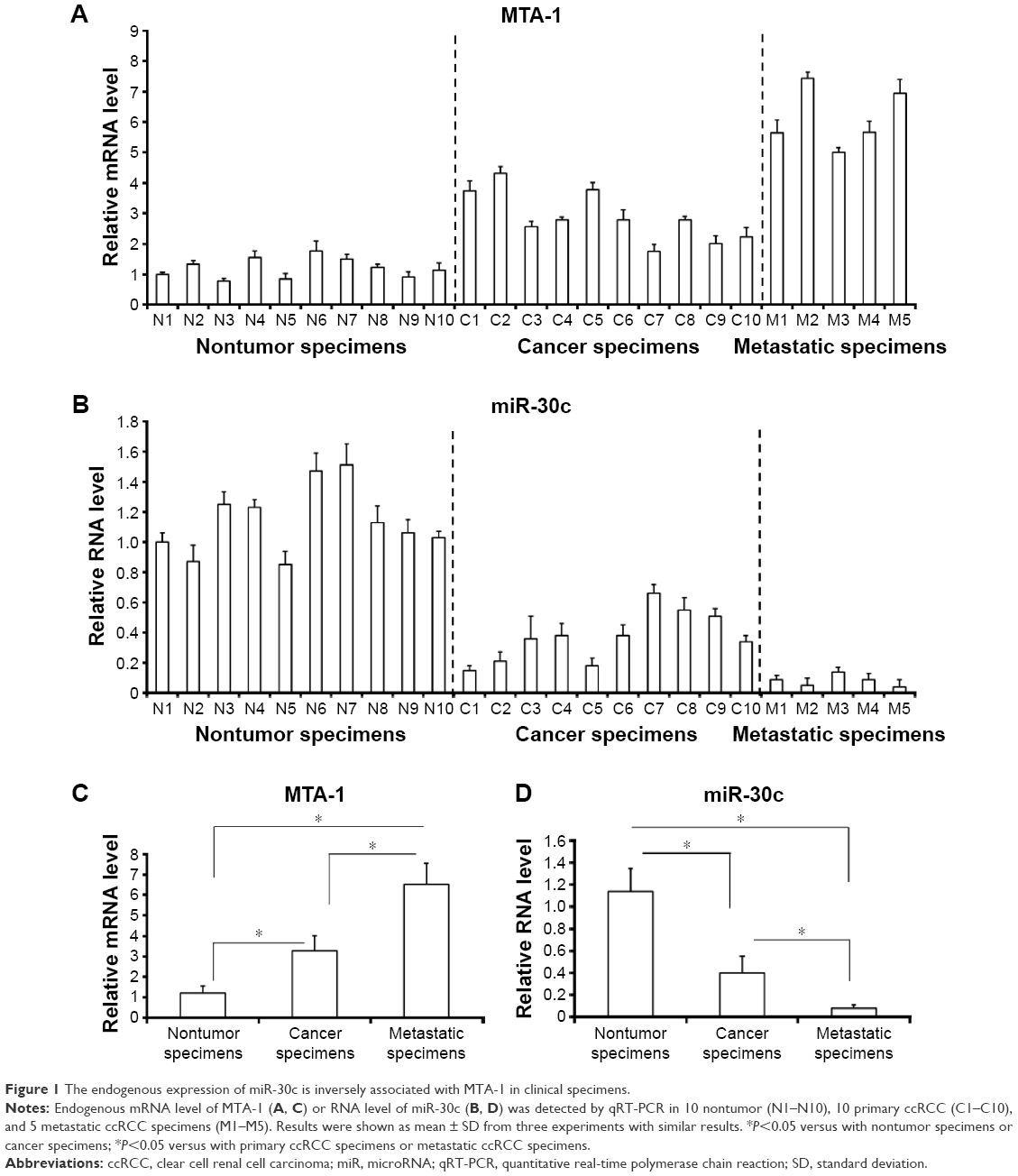 | Figure 1 The endogenous expression of miR-30c is inversely associated with MTA-1 in clinical specimens. |
Next, the endogenous expression of MTA-1/miR-30c was examined in ccRCC or nontumor kidney cell lines. Four different cell lines, including two nontumor kidney cell lines HKC and HEK293, as well as ccRCC cell line 786-O and a highly aggressive cell line Caki-1, were examined by qRT-PCR. As shown in Figure 2, the mRNA level of MTA-1 was markedly elevated in ccRCC cell lines compared to the HKC or HEK293 cell line (Figure 2A and B). The highest expression of MTA-1 was identified in Caki-1 cell (Figure 2A and B). In addition, the expression of miR-30c was inversely associated with MTA-1 in cells (Figure 2C and D). These results indicated that MTA-1 would participate in ccRCC metastasis and that the low level of miR-30c would contribute to the aberrant expression of MTA-1.
 | Figure 2 Expression of miR-30c is inversely associated with MTA-1 in kidney or ccRCC cell lines. |
miR-30c inhibits the ability of Caki-1 cells to proliferate and anchorage-independent growth
To elucidate the effect of MTA-1 in Caki-1 cells proliferation, we generated miR-30c expressing lentivirus (LV) vectors. Caki-1 cells, which were infected with control (LV-control) or miR-30c (LV-miR-30c), were harvested for MTT assays. As shown in Figure 3A and B, the expression of MTA-1, a target of miR-30c, was significantly reduced after LV-miR-30c infection. The proliferation of Caki-1 cells infected with miR-30c is much lower than cells infected empty vectors (LV-control) or the parental cells (Figure 3C). There was no significant difference between parental cells (negative control) and LV-control infected cells (Figure 3C).
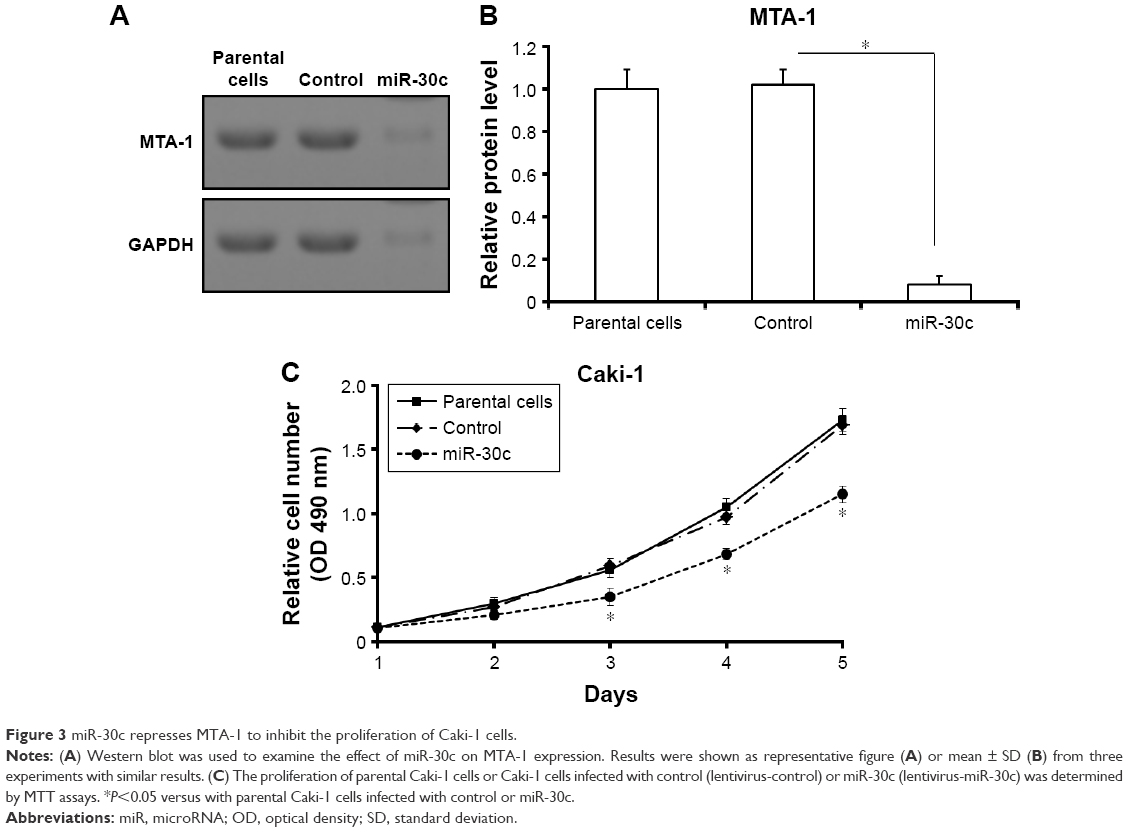 | Figure 3 miR-30c represses MTA-1 to inhibit the proliferation of Caki-1 cells. |
Next, the soft agar methods were performed to test the anchorage-independent growth of Caki-1 cells. Overexpression of miR-30c significantly inhibited the anchorage-independent growth of Caki-1 cells (Figure 4). Taken together, these data showed that down-regulation of MTA-1 expression via miR-30c inhibits proliferation or anchorage-independent growth of Caki-1 cells.
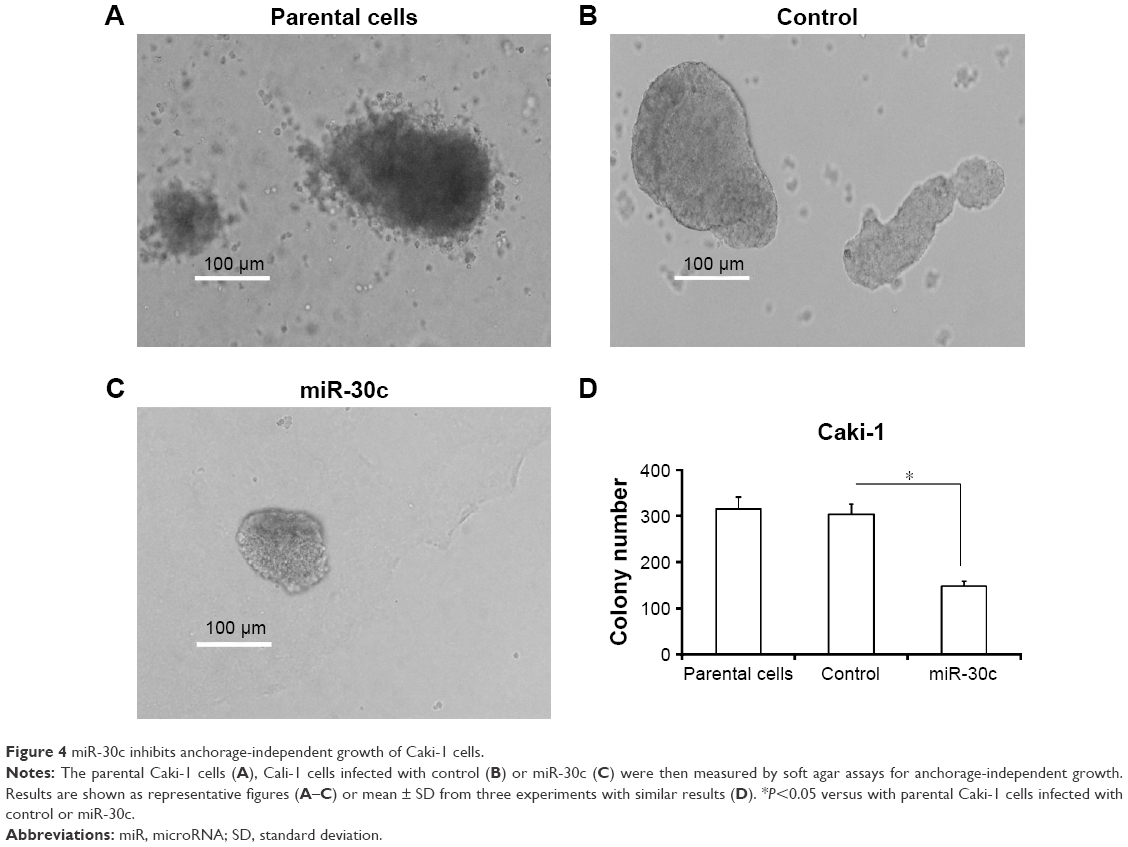 | Figure 4 miR-30c inhibits anchorage-independent growth of Caki-1 cells. |
miR-30c inhibits in vitro invasion and migration of Caki-1 cells
To further explore the roles of miR-30c in Caki-1 cells, we examined in vitro invasion and migration by trans-well methods. Infection of miR-30c decreased the ability of Caki-1 cells to invade as well as migrate (Figure 5A and B) compared with control. The inhibition rate for relative invasion or migration cell number of Caki-1 was 84.20% (for invasion) or 79.41% (for migration), respectively. These results indicated that MTA-1 may be a key regulator of ccRCC metastasis and that decreasing MAT-1 expression via miR-30c significantly inhibits in vitro invasion or migration of Caki-1 cells.
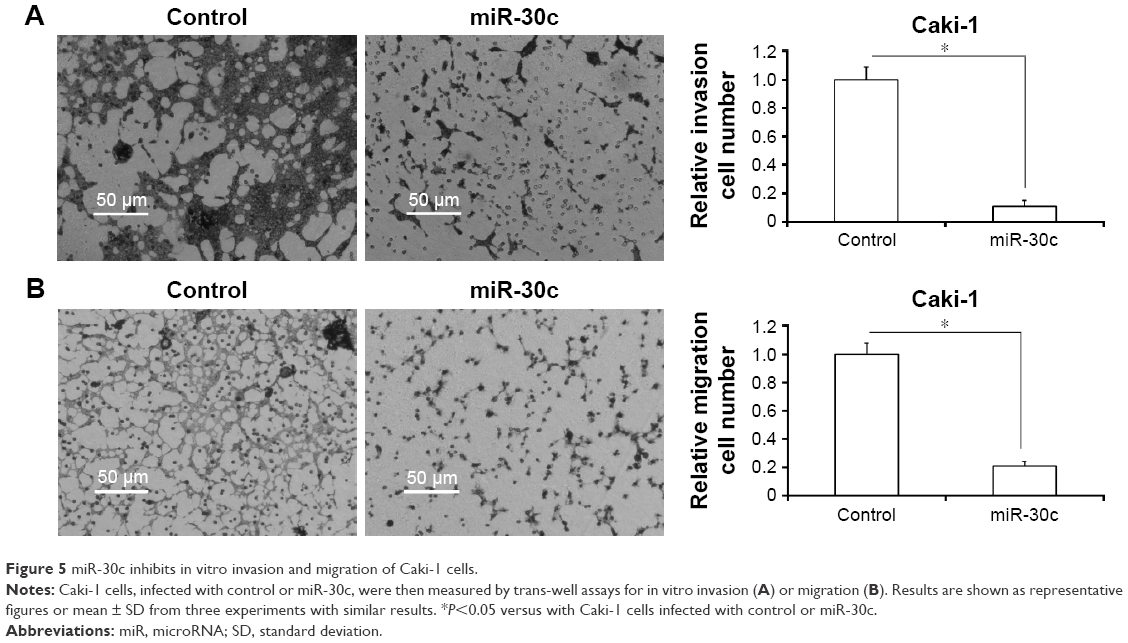 | Figure 5 miR-30c inhibits in vitro invasion and migration of Caki-1 cells. |
miR-30c attenuates in vivo growth of Caki-1 cells in nude mice mode
Next, in vivo growth of Caki-1 cells was examined. As shown in Figure 6, infection of miR-30c decreased the in vivo growth of Caki-1 cells compared with control. These results further confirmed the effect of miR-30c on ccRCC cells proliferation.
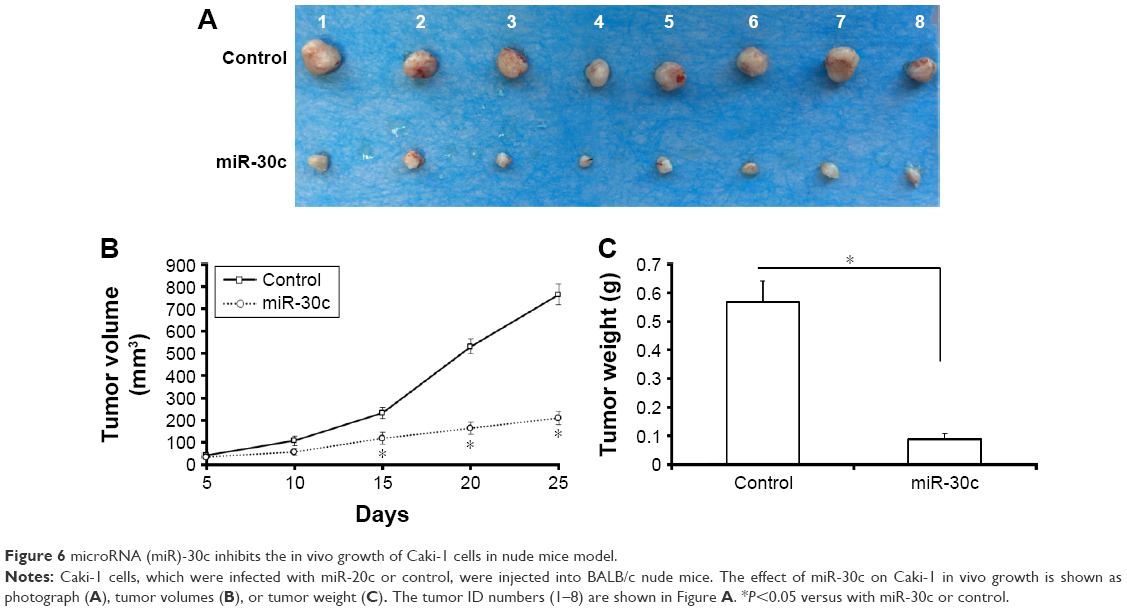 | Figure 6 microRNA (miR)-30c inhibits the in vivo growth of Caki-1 cells in nude mice model. |
miR-30c inhibits Caki-1 cells proliferation via MTA-1
To further reveal the specificity of miR-30c on Caki-1 proliferation, the inhibitor of miR-30c or mutated-MTA-1 vector (MTA-1Mut) was also used (Figure 7). miR-30c reduced the expression of MTA-1 but not mutated MTA-1 (Figure 7B and C). Furthermore, co-transfection inhibitor of miR-30c or MTA-1 mutation almost blocked the effect of miR-30c in Caki-1 cells (Figure 7D and E). Taken together, these results suggested that miR-30c attenuates Caki-1 cells proliferation by targeting MTA-1.
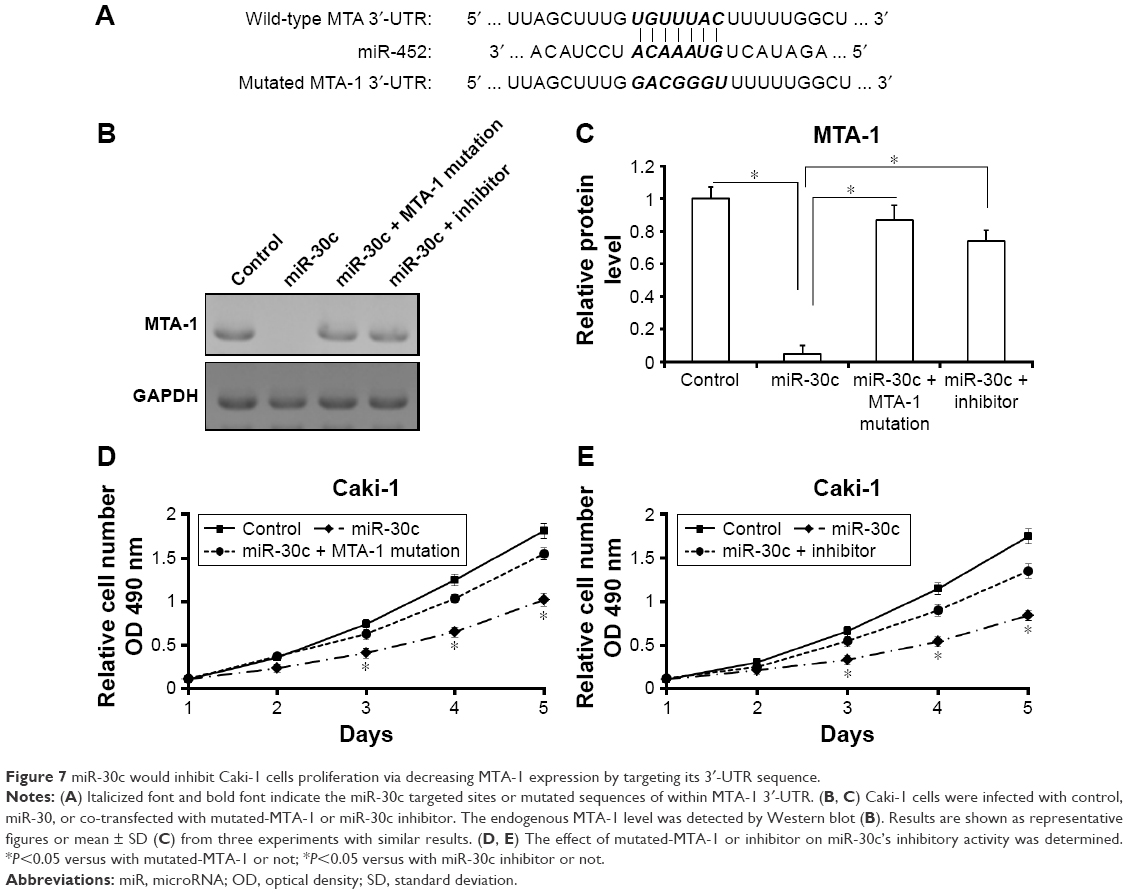 | Figure 7 miR-30c would inhibit Caki-1 cells proliferation via decreasing MTA-1 expression by targeting its 3′-UTR sequence. |
miR-30c enhances the sensitivity of Caki-1 cells to antitumor agents
As shown in Figure 8, sorafenib and paclitaxel inhibited Caki-1 cells’ survival in a dose-dependent manner. The inhibition rates (IR) of sorafenib and paclitaxel on Caki-1 cells were significantly enhanced after miR-30c infection. The 50% inhibiting effect concentration values correspondingly decreased from 4.48±0.33 μmol/L to 0.87±0.074 μmol/L, or from 0.79±0.11 μmol/L to 0.082±0.01 μmol/L, respectively (Table 2). Transfection of miR-30c inhibitor or MTA-1 mutation almost blocked the activity of miR-30c.
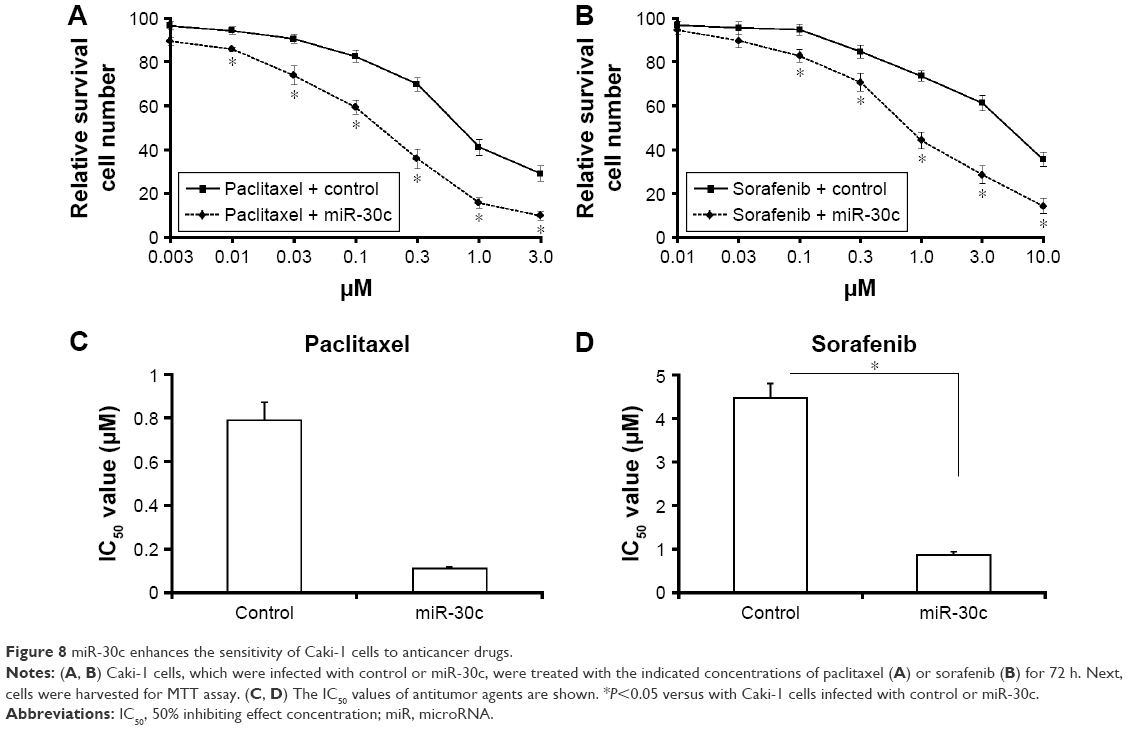 | Figure 8 miR-30c enhances the sensitivity of Caki-1 cells to anticancer drugs. |
 | Table 2 miR-30c enhances the sensitivity of anticancer drugs |
Then, the colony formation was examined (Figure 9). Infection of miR-30c significantly enhanced the inhibitory effect of paclitaxel (Figure 9A) and sorafenib (Figure 9B) on Caki-1 colony formation. Infection of miR-30c inhibitor or MTA-1 mutation almost blocked the activity of miR-30c on Caki-1 colony formation. These data indicated that MTA-1 would participate in the chemotherapy of ccRCC and that down-regulation of MTA-1 via miR-30c enhances the sensitivity of Caki-1 cells to antitumor agents.
 | Figure 9 miR-30c up-regulates the inhibitory activity of paclitaxel and sorafenib on Caki-1 cell colony formation. |
Discussion
In this work, our data identified that MTA-1 would be a key regulator of metastatic ccRCC proliferation of metastasis, and revealed the proliferation inhibiting roles of miR-30c in metastatic ccRCC by targeting MTA-1. This notion is supported by results that decreased expression of MTA-1 via miR-30c significantly attenuated the proliferation or anchorage-independent growth of Caki-1 cells. As highly aggressive ccRCC cell lines, infection of miR-30c disrupted the migration and invasion of Caki-1. Moreover, miR-30c also enhanced the sensitivity of Caki-1 cells to antitumor agents. Therefore, MTA-1 may be an interesting novel target for the treatment of highly aggressive ccRCC, and expression of miR-30c would be a potential strategy for ccRCC treatment.
The ccRCC is the most common malignance of adult kidney, and the overall survival of ccRCC is highly variable, ranging from 1 to 10 years.22 It has been confirmed that patients with advanced stage or metastatic ccRCC may have a poor prognosis owing to insensitive chemotherapy or radiotherapy.22 There is therefore an urgent need to identify the detailed mechanisms of or novel options for ccRCC treatment. Our previous work showed that KLF4 expression was lower in ccRCC tissues and that patients with weakly or negatively stained KLF4 had larger tumor size and worse prognosis.15 Another work also revealed aberrant expression of Tubulin cofactor A (TBCA) in ccRCC, and revealed its function as a novel positive regulator of ccRCC progression, invasion, and metastasis. Exploration of the mechanisms suggested that TBCA could function via modulating cytoskeleton integration and influencing cell cycle progress.3 Although these data enhanced our knowledge about ccRCC, more work is needed to declare mechanisms for insensitive chemotherapy or radiotherapy during ccRCC treatment.
MTA-1 is one of the key regulators of human cancer metastasis. It would promote invasion, metastasis, and angiogenesis and associate poor clinical outcome tumor by up-regulating the expression of fibronectin, MMP2, or MMP9.5,23 In addition, MTA-1 would also be a positive regulator of cancer cells proliferation.5,24 MTA-1 can also enhance growth of nasopharyngeal carcinoma cells by promoting G1/S-phase transition.24 Yao et al5 reviewed the related research works and indicated that MTA-1 would modulate the cell proliferation and G1/S-phase transition via Cyclin D1 and P21.5 In this work, up-regulation of MTA-1 can be detected both in metastatic ccRCC cell lines and in specimens. The expression of miR-30c, which targets to MTA-1, is inversely associated with MTA-1. Infection of miR-30c inhibits Caki-1 cells proliferation, anchorage-independent growth, in vitro invasion, and migration by repressing MTA-1. These results revealed the functions of MTA-1 in highly aggressive ccRCC, and expression of miR-30c via lentivirus vectors would be a potential approach in ccRCC treatment. In addition, Kaur et al6 showed that MTA-1 can promote the EMT process by repressing E-cadherin transcription.6 It also promotes tumorigenesis and metastasis by up-regulating TGF-β signaling activity.6 Human cancer cells may develop recurrence or resistance to therapies via the EMT process. It has been shown that MTA-1 participates in antitumor drugs resistance of breast cancer.7 A high level of MTA1 suggests development of resistance to tamoxifen.8 Therefore, identification of the effects of MTA-1/miR-30c on ccRCC EMT and related signaling pathways, eg, TGFβ/Smads, will be future work. Further, a large cohort study to reveal the expression of mTA-1/miR-30c in ccRCC is also needed.
It has been well known that ccRCC, especially highly aggressive or metastatic ccRCC, is resistant to conventional chemotherapy.25 Although small molecular kinase inhibitors, eg, sunitinib and sorafenib, are foremost choices for patients suffering from advanced or metastatic ccRCCs, these treatments rarely produce complete responses, and curative effects are not observed.25 Thus, further work is needed to address the appropriate biomarkers and therapeutic targets of ccRCC. Some important genes, eg, MDR-1, VHL, or Notch-1, would participate in regulation of ineffective chemotherapy.26–29 Gururaj et al8 reported that MTA-1 would mediate the resistance of ERα negative breast cancer cells to antitumor agents. In the present study, we found that down-regulation of MTA-1 expression via miR-30c significantly enhanced the sensitivity of Caki-1 cells to antitumor drugs. Moreover, there are still some other pathways involved in chemotherapeutic or radiotherapeutic resistance, eg, Notch-1 or MDR-1 (multidrug resistance 1). Notch-1 signaling pathway mediates cancer cell-fate decision via pro-survival, antiapoptosis, or EMT process.16 MDR-1 and some other resistance protein, eg, BCRP, are transcripted by some nuclear receptors, eg, PXR or CAR.19 Thus, it is necessary to identify the effect of MTA-1 on Notch-1 or interaction between MTA-1 and PXR or CAR, in future.
Conclusion
In this work, we reported that MTA-1 may be an important regulator of ccRCC metastasis or chemotherapy resistance. Infection of miR-30c inhibited the proliferation and enhanced the sensitivity of highly aggressive ccRCC Caki-1 cells to anticancer agents by repressing MTA-1 expression. In light of this, we showed that expression of miR-30c could participate in the epigenetic downmodulation of MTA-1 and would guide further development of novel therapies to benefit ccRCC patients.
Acknowledgments
The authors thank Dr Peng Zhang at the State Key Laboratory of Kidney Diseases Department of Urology, PLA General Hospital, Beijing, People’s Republic of China, for his helpful advice. This study was supported by The Science and Technology Development Project of Changshu (CS201417).
Author contributions
HLY, ELS, GRS and THZ carried out experiments. ELS and TWJ drafted the manuscript. HS, LPN, BW and ZYL participated in experiments. JPQ performed the statistical analysis. TWJ designed the study. All authors made substantial contributions to the design and conception; acquisition, analysis or interpretation of data; took part in revising the manuscript; gave final approval of the version to be published; and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Disclosure
The authors report no conflicts of interest in this work.
References
Zhang N, Wu Y, Gong J, et al. Germline genetic variations in PDZD2 and ITPR2 genes are associated with clear cell renal cell carcinoma in Chinese population. Oncotarget. Epub January 14, 2016. | ||
Nuerrula Y, Rexiati M, Liu Q, Wang YJ. Differential expression and clinical significance of serum protein among patients with clear-cell renal cell carcinoma. Cancer Biomark. 2015;15:485–491. | ||
Zhang P, Ma X, Song E, et al. Tubulin cofactor A functions as a novel positive regulator of ccRCC progression, invasion and metastasis. Int J Cancer. 2013;133:2801–2811. | ||
Escudier B, Eisen T, Stadler WM, et al; TARGET Study Group. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356:125–134. | ||
Yao Y, Feng S, Xiao M, Li Y, Yang L, Gong J. MTA1 promotes proliferation and invasion in human gastric cancer cells. Onco Targets Ther. 2015;8:1785–1794. | ||
Kaur E, Gupta S, Dutt S. Clinical implications of MTA proteins in human cancer. Cancer Metastasis Rev. 2014;33:1017–1024. | ||
Tuncay-Cagatay S, Cimen I, Savas B, Banerjee S. MTA-1 expression is associated with metastasis and epithelial to mesenchymal transition in colorectal cancer cells. Tumour Biol. 2013;34:1189–1204. | ||
Gururaj AE, Holm C, Landberg G, Kumar R. Breast cancer-amplified sequence 3, a target of metastasis-associated protein 1, contributes to tamoxifen resistance in premenopausal patients with breast cancer. Cell Cycle. 2006;5:1407–1410. | ||
Qin H, Sha J, Jiang C, et al. miR-122 inhibits metastasis and epithelial-mesenchymal transition of non-small-cell lung cancer cells. Onco Targets Ther. 2015;8:3175–3184. | ||
Ma D, Jia H, Qin M, et al. MiR-122 Induces radiosensitization in non-small cell lung cancer cell line. Int J Mol Sci. 2015;16:22137–22150. | ||
Zhang Y, Han L, Pang J, Wang Y, Feng F, Jiang Q. Expression of microRNA-452 via adenoviral vector inhibits non-small cell lung cancer cells proliferation and metastasis. Tumour Biol. 2016;37:8259–8270. | ||
Chen Y, Feng F, Gao X, et al. MiRNA153 reduces effects of chemotherapeutic agents or small molecular kinase inhibitor in HCC cells. Curr Cancer Drug Targets. 2015;15:176–187. | ||
Kong X, Xu X, Yan Y, et al. Estrogen regulates the tumour suppressor MiRNA-30c and its target gene, MTA-1, in endometrial cancer. PLoS One. 2014;9:e90810. | ||
Heinzelmann J, Henning B, Sanjmyatav J, et al. Specific miRNA signatures are associated with metastasis and poor prognosis in clear cell renal cell carcinoma. World J Urol. 2011;29:367–373. | ||
Song E, Ma X, Li H, et al. Attenuation of krüppel-like factor 4 facilitates carcinogenesis by inducing g1/s phase arrest in clear cell renal cell carcinoma. PLoS One. 2013;8:e67758. | ||
Jia H, Yang Q, Wang T, et al. Rhamnetin induces sensitization of hepatocellular carcinoma cells to a small molecular kinase inhibitor or chemotherapeutic agents. Biochim Biophys Acta. 2016;1860:1417–1430. | ||
Leonov G, Shah K, Yee D, Timmis J, Sharp TV, Lagos D. Suppression of AGO2 by miR-132 as a determinant of miRNA-mediated silencing in human primary endothelial cells. Int J Biochem Cell Biol. 2015;69:75–784. | ||
Ma H, Yao Y, Wang C, et al. Transcription factor activity of estrogen receptor α activation upon nonylphenol or bisphenol A treatment enhances the in vitro proliferation, invasion, and migration of neuroblastoma cells. Onco Targets Ther. 2016;9:3451–3463. | ||
Zhao J, Bai Z, Feng, F, et al. Cross-talk between EPAS-1/HIF-2α and PXR signaling pathway regulates multi-drug resistance of stomach cancer cell. Int J Biochem Cell Biol. 2016;72:73–88. | ||
Feng F, Lu YY, Zhang F, et al. Long interspersed nuclear element ORF-1 protein promotes proliferation and resistance to chemotherapy in hepatocellular carcinoma. World J Gastroenterol. 2013;19:1068–1078. | ||
Yang FQ, Zhang HM, Chen SJ, Yan Y, Zheng JH. MiR-506 is down-regulated in clear cell renal cell carcinoma and inhibits cell growth and metastasis via targeting FLOT1. PLoS One. 2015;10:e0120258. | ||
Cano-González A, López-Rivas A. Opposing roles of TGF-β and EGF in the regulation of TRAIL-induced apoptosis in human breast epithelial cells. Biochim Biophys Acta. 2016;1863:2104–2114. | ||
Song Q, Zhang H, Wang M, et al. MTA1 promotes nasopharyngeal carcinoma growth in vitro and in vivo. J Exp Clin Cancer Res. 2013;32:54. | ||
Shaikh T, Handorf EA, Murphy CT, et al. Contemporary trends in the utilization of radiotherapy in patients with renal cell carcinoma. Urology. 2015;86:1165–1173. | ||
Zhou L, Liu XD, Sun M, et al. Targeting MET and AXL overcomes resistance to sunitinib therapy in renal cell carcinoma. Oncogene. 2016;35:2687–2697. | ||
Walsh N, Larkin A, Kennedy S, et al. Expression of multidrug resistance markers ABCB1 (MDR-1/P-gp) and ABCC1 (MRP-1) in renal cell carcinoma. BMC Urol. 2009;9:6. | ||
Cautain B, de-Pedro N, Schulz C, et al. Identification of the Lipodepsipeptide MDN-0066, a novel inhibitor of VHL/HIF pathway produced by a new pseudomonas species. PLoS One. 2015;10:e0125221. | ||
Sjölund J, Johansson M, Manna S, et al. Suppression of renal cell carcinoma growth by inhibition of Notch signaling in vitro and in vivo. J Clin Invest. 2008;118:217–228. | ||
Santoni M, Rizzo M, Burattini L, Berardi R, Carteni G, Cascinu S. Novel agents, combinations and sequences for the treatment of advanced renal cell carcinoma: when is the revolution coming? Curr Cancer Drug Targets. 2013;13:313–325. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.