")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 14
Exploring the Epigenetic Regulatory Role of m6A-Associated SNPs in Type 2 Diabetes Pathogenesis
Authors Chen M , Lin W , Yi J, Zhao Z
Received 15 August 2021
Accepted for publication 13 October 2021
Published 27 October 2021 Volume 2021:14 Pages 1369—1378
DOI https://doi.org/10.2147/PGPM.S334346
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Martin H Bluth
Miao Chen,1,2 Weimin Lin,1 Jianru Yi,1,2 Zhihe Zhao1,2
1State Key Laboratory of Oral Diseases & National Clinical Research Center for Oral Diseases, West China Hospital of Stomatology, Sichuan University, Chengdu, People’s Republic of China; 2Department of Orthodontics, West China Hospital of Stomatology, Sichuan University, Chengdu, People’s Republic of China
Correspondence: Jianru Yi; Zhihe Zhao Tel +86-28-85503040
; +86-28-85503645
Email [email protected]; [email protected]
Purpose: Genetic factors in type 2 diabetes (T2D) pathogenesis have been widely explored by the genome-wide association studies (GWAS), identifying a great amount of susceptibility loci. With the development of high-resolution sequencing, the N(6)-methyladenosine (m6A) RNA modification has been proved to be affected by genetic variation. In this study, we identified the T2D-associated m6A-SNPs from T2D GWAS data and explored the underlying mechanism of the pathogenesis of T2D.
Methods: We examined the association of m6A-SNPs with T2D among large-scale T2D GWAS summary statistics and further performed multi-omics integrated analysis to explore the potential role of the identified m6A-SNPs in T2D pathogenesis.
Results: Among the 15,124 T2D-associated m6A-SNPs, 71 of them reach the genome-wide significant threshold (5.0e-05). The leading SNP rs4993986 (C>G), which is located near the m6A modification site at the 3ʹ end of the HLA-DQB1 transcript, is expected to participate in the pathogenesis of T2D by influencing m6A modification to regulate the HLA-DQB1 expression.
Conclusion: The current study has suggested a potential correlation between m6A-SNPs and T2D pathogenesis and also provided new insights into the pathogenic mechanism of the T2D susceptibility loci identified by GWAS.
Keywords: type 2 diabetes, single nucleotide polymorphism, epigenetics, m6A, genome-wide association study
Introduction
With the continuous increase in obesity and life expectancy in recent decades, the global incidence of diabetes has also risen rapidly.1 Type 2 diabetes (T2D) is the most common type of diabetes, accounting for approximately 90% of all diabetes cases.2 T2D is a chronic non-infectious disease characterized by impaired islet beta-cell function, insulin resistance and the resultant hyperglycemia. Besides the common environmental factors, cumulative evidence indicates that genetic factors also play a decisive role in the occurrence and progression of T2D. Clarifying the involvement of genetic variants in the development of T2D is critical to understand its pathogenesis. Indeed, many endeavors have been made to investigate T2D susceptibility genes over the past years.3–5 Numerous susceptibility loci for T2D have been identified by a series of genome-wide association studies (GWAS).6–8
More than 400 genomic regions have been identified as affecting T2D.9 For example, variations in hematopoietically expressed Homeobox (HHEX), the Solute Carrier Family 30 Member 8 (SLC30A8), and the transcription factor-7-like2 (TCF7L2) gene regions were the earliest identified susceptibility loci in GWAS related to T2D. Most of these risk genes were related to glucose metabolism and lipid synthesis.10–12 Unfortunately, besides a small number of the published T2D-associated single nucleotide polymorphisms (SNPs) locating in protein coding regions, most of these SNPs are located in non-coding regions, making it more difficult to explain their biological effects.13,14
N6-methyladenosine (m6A) is the methylation of the adenosine at the sixth position of the nitrogen, which is the most common post-transcriptional RNA modification in eukaryotes. With the landmark study that identified the first m6A demethylase fat mass and obesity-associated protein (FTO) in 2011 and the transcriptome-wide profiling of m6A by high-throughput sequencing,15 precise mapping of m6A distribution on transcript has been achieved. These m6A peaks have a conserved modification motif RRACH (R for A or G, H for A, U or C).16 In mRNA transcripts, the recognition of modification sites by m6A methyltransferase is highly dependent on these m6A-specific sequences, while genetic variation may change these m6A-specific recognition sequences on local gene transcripts, thus affecting the modification and recognition of m6A. Through the combination of m6A sequencing and whole genome sequencing (WGS) of large-scale cohort, Xiong and Zhang et al proved that genetic variation is an effective driver of m6A modification, respectively.17,18 Recently, the correlations between m6A RNA methylation-related SNPs (m6A-SNPs) and hereditary diseases were widely explored.19–23 In studies of Mo et al, they identified a large number of m6A-SNPs affecting a series of phenotypes related to cardiovascular diseases, such as blood pressure, blood lipids and coronary heart disease.24–26 In the field of neurology, m6A-SNPs have been verified to be closely associated with stroke and multiple sclerosis.27,28 Besides, m6A-SNPs could also participate in cancer development. m6A-SNP rs178184 was shown to affect the occurrence of colon cancer by regulating the expression of NOVA1 gene.22 Our previous study proved that m6A RNA methylation-related SNPs might be involved in adiposity, which had a close causal relationship with T2D.29 These findings are crucial for a better understanding of the role of m6A modification in pathological changes caused by genetic variation. However, no study has been reported on the role of m6A-SNPs in T2D pathogenesis until now. In this study, we explored the association of m6A-SNPs with T2D and demonstrated the potential functions of these m6A-SNPs in T2D pathogenesis.
Materials and Methods
Determination of m6A-SNPs for T2D
To identify the T2D-associated SNPs that probably affect RNA m6A modification, we retrieved a recently released large-scale T2D GWAS summary statistics among 81412 T2D cases and 370832 controls.6 The data is publicly available on the DIAGRAM website (http://diagram-consortium.org/index.html). We identified the potential m6A-SNPs by comparing the m6A-SNP list in m6AVar database with T2D summary statistics.30 The identified m6A-SNPs were annotated, and a preliminary screening was performed with a threshold of 5.0e-05 for subsequent analysis. GO enrichment analysis of m6A-SNPs passing threshold was performed through an online website metascape (http://metascape.org/gp/index.html).31
Integrative Analysis of T2D-Associated m6A-SNPs
It has been confirmed that m6A modification can affect the mRNA splicing, nuclear export, stability and degradation, thus affecting local gene transcription, which might be an important mechanism for m6A-SNPs to involve the pathogenesis of T2D. To investigate whether these genetic variants could lead to changes in gene transcription, we retrieved their expression quantitative trait loci (eQTL) effects through the HaploReg browser (http://pubs.broadinstitute.org/mammals/haploreg/haploreg.php).32 Further, we also searched for several other potential regulatory mechanisms of these SNPs on transcriptional regulation, such as altering protein binding and changing motif. The results are shown in Table 1 and Supplementary Table 1.
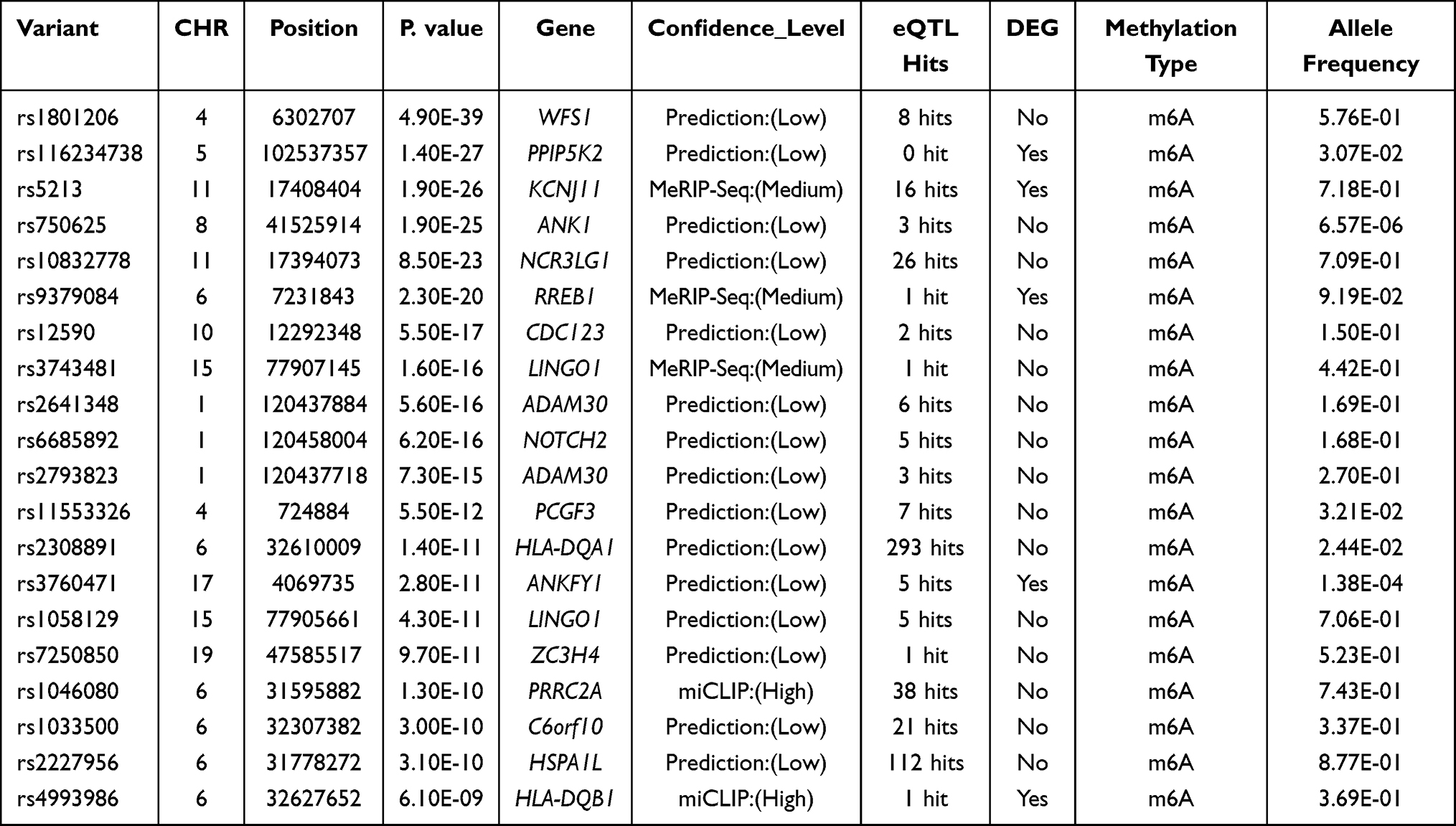 |
Table 1 The 20 Most Important m6A-SNPs Related to T2D |
Prediction of m6A Modification Near m6A-SNPs
Among the T2D-associated m6A-SNPs identified above, we predicted the potential m6A modification site on the transcript by inputting the reference and altered sequence to an online m6A modification prediction tool, SRAMP (http://www.cuilab.cn/sramp/).33 SRAMP is a website-based computational predictor for identifying mammalian RNA m6A modification sites. SRAMP includes three random forest classifiers and uses genomic sequence or cDNA sequence as input. To identify whether m6A-SNP could affect the nearby m6A modifications, the altered sequence and reference sequence were input into SRAMP, and the confidence of m6A modifications was predicted.
Differential Expression Analysis
For m6A-SNPs that showed eQTL signal, we further tried to determine whether the related gene expression of these T2D-associated m6A-SNPs genes in T2D was changed. Based on the transcriptomic data available in the gene expression omnibus (GEO) database, we displayed the expression level of the identified T2D-associated genes (m6A-SNPs showed eQTL signal). Three datasets GSE23343, GSE38642 and GSE78721 containing standardized T2D-associated gene expression signals were retrieved and downloaded from GEO database (http://www.ncbi.nlm.nih.gov/geo). Samples in GSE23343 were collected from 7 subjects with normal blood glucose levels and 10 patients with T2D. GSE38642 contained the gene expression profile in 63 islets from cadaver donors (including 9 diabetic and 54 non-diabetic). And GSE78721 contained gene expression level of adipose tissues from 63 patients suffering from T2D and 68 age/BMI matched normal glucose tolerance controls. Considering that multiple tests are used among the three datasets, we use the Bonferroni adjustment to set a more stringent threshold, p < 0.0167 (0.05/3) was set as a threshold for defining differential genes. Also, to prove that these gene expression differences were mediated by m6A-SNPs instead of the T2D phenotype, we downloaded the transcriptome data of tissues that are not closely related to the T2D phenotype as control sets, including enteroendocrine cells (GSE132831) and peripheral blood monocytes (GSE156061). We also explored the expression of these differential genes in control sets.
For single-cell RNA sequencing (scRNA-seq) data analysis, we downloaded the islet scRNA-seq data of high-fat diet-induced diabetic mice and healthy controls in GSE162512, and used the Seurat package to load the hdf5 format data for subsequent process.34 Based on the expression of classic cell markers, we defined the cell types and compared the expression of HLA-DQB1 (H2-Ab1) in different cell populations.
Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
A total of 8 db/db mice and 8 heterozygous mice at the age of 8 weeks were used. Male db/db mice were used as animal model of T2D, and the heterozygous mice were used as control. The islets of the mouse pancreas were isolated as previously described.35 In brief, the pancreatic tissue was cut into small pieces of 1~2 mm and digested in 1 mg/mL collagenase XI (Sigma) for 20 minutes. Ficoll solution was used to purify the islets by centrifugation. Total RNA of the obtained islets was extracted using Trizol reagent (Invitrogen), and then reversely transcribed to obtain stable cDNA using PrimeScriptTM RT reagent Kit with gDNA Eraser (TaKaRa Bio). The qRT-PCR was performed using SYBR Premix Ex Taq II (TaKaRa Bio) in Quant StudioTM 3 real-time fluorescent quantitative PCR instrument (ThermoFisher Scientific). Glyceraldehyde 3-phosphate dehydrogenase (Gapdh) was used as an internal reference to normalize the gene expression. The result was calculated using the 2−ΔΔCt method and expressed as a multiple change relative to Gapdh. The primer sequences were summarized in Supplementary Table 2. The animal studies were approved by the Animal Care and Ethics Committee of West China Hospital of Stomatology, Sichuan University. The animal experiments complied with the ARRIVE guidelines and were carried out in accordance with the National Institutes of Health guide for the care and use of Laboratory animals (NIH Publications No. 8023, revised 1978).
Results
Determination of T2D-Associated m6A-SNPs
We identified 15,124 m6A-SNPs and 16,420 potentially affected m6A sites (Figure 1). For these 15,124 T2D-associated m6A-SNPs identified, 11820, 2950 and 354 m6A-SNPs belong to the low, medium and high confidence categories, respectively. p < 5.0e-05 was defined as the significance threshold and 71 appeared to be associated with T2D (p < 5.0e-05) (Figure 2A). GO enrichment analysis showed that these 71 genes were mainly enriched in antigen processing and presentation related biological processes (Figure 2B). Among these m6A-SNPs, 56, 12 and 3 belong to the low, medium and high confidence intervals, respectively. 21 SNPs are non-synonymous, 20 are synonymous mutations, and 28 are located at untranslated regions (UTR) (Supplementary Table 1). As for other potential regulatory mechanisms of these m6A-SNPs, we found that 52 altered the motifs and 19 influenced the binding of regulatory proteins in different cell types from the ENCODE transcription factor ChIP-seq datasets (Supplementary Table 1).
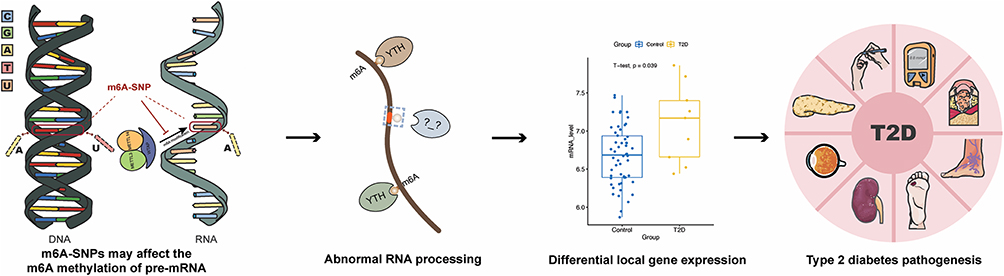 |
Figure 1 Design and main implementation steps of this study. |
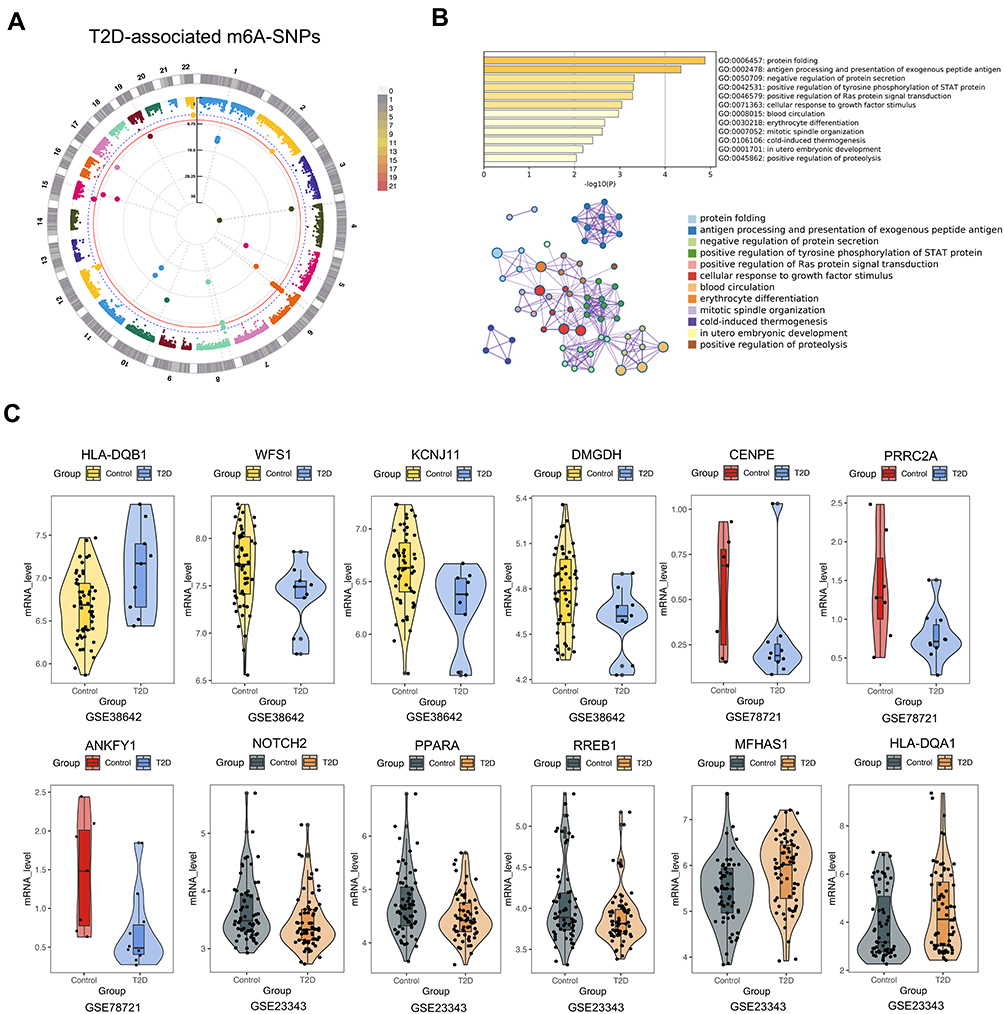 |
Figure 2 Identification of T2D related m6A-SNPs. (A) The identified T2D related m6A-SNPs with genome-wide significance are displayed in the Manhattan plot. P < 5.0e−05 was set as the suggestive threshold, and p < 5.0e-08 was set as the genome-wide threshold. (B) GO enrichment analysis of the T2D related m6A-SNPs (p < 5.0e-05). (C) Gene expression differences between T2D patients and controls in islet (GSE38642), adipose (GSE78721), and liver tissue (GSE23343). |
Identification of Differentially Expressed Genes
For the 56 T2D-associated m6A-SNPs with eQTL signals, we analyzed whether the related gene expression changed in published datasets about the gene expression of the tissues from T2D patients and controls. The etiology of T2D is complicated, and the abnormal function of organs related to glucose metabolism may lead to blood glucose homeostasis disorders. For instance, liver dysfunction will lead to a decline in glycogen synthesis capacity, resulting in an imbalance in blood glucose homeostasis.36,37 And the excessive accumulation of adipose tissue, especially visceral adipose tissue, can lead to chronic inflammation and promote the progression of T2D.38,39 Also, the dysfunction and loss of β-cells that secrete insulin in the endocrine pancreas can also cause T2D.40 Therefore, the transcriptomic data of these three tissues were selected in the GEO database to validate the expression change of the m6A-SNPs related genes in T2D patients. We found that 11 of these genes were differentially expressed in at least one dataset (Supplementary Table 1). Then, the expression level of representative genes with medium or high confidence were selected to be shown (Figure 2C). Islet beta-cells can secrete insulin to regulate blood glucose, and impaired beta-cell function can lead to hyperglycemia caused by insufficient insulin secretion. In GSE38642, HLA-DQB1 (p = 0.00365124) and KCNJ11 (p = 0.00538383) were found to be differentially expressed in islet tissue. Insulin resistance caused by obesity also leads to T2D. PPARA (p = 0.00839) and MFHAS1 (p = 0.00067) were found to be abnormally expressed in GSE78721 (adipose tissue). In addition, the liver can respond to blood glucose concentration and regulate the secretion of insulin or glucagon, thereby regulating the homeostasis of glucose. In GSE23343 (liver tissue), RREB1 (p = 0.0107675) was differentially expressed. Therefore, we found that 11 m6A-SNP-related genes were differentially expressed between T2D patients and healthy controls. The abnormal expression of these genes may be involved in the pathogenesis of T2D. In the control sets (enteroendocrine cells and peripheral blood monocytes), we found that 8 of 11 were also differentially expressed in at least one control set (Supplementary Table 3). Therefore, we believe that the differential expression of these genes is caused by m6A modification and not related to T2D phenotype.
To validate whether the top m6A-SNP-related genes showed expression changes during T2D pathogenesis, we detected the expression of these genes in the pancreatic islets of db/db mice and heterozygous control by qRT-PCR. The results showed that the expression of 8 genes was found to be significantly different between the diabetic group and control. Although the expression of Mfhas1, Ppara and Rreb1 did not show a significant difference, their average expression levels were the same as the trend of these genes in GEO datasets (Supplementary Figure 1). In short, the results of qRT-PCR proved that the expression of these m6A-SNP-related genes changed under the diabetic condition.
Integrative Analysis of T2D-Associated m6A-SNPs
To further elucidate the potential functional mechanisms of the identified 71 T2D-associated m6A-SNPs, we further investigated whether the 71 T2D-associated m6A-SNPs were related to the expression level of local genes and the underlying functional mechanism. In all, 56 T2D-related m6A-SNPs displayed eQTL signals related to local genes expression. Among these m6A-SNPs, 19 of them might change the binding of transcription factors in different cell lines, and 52 of them might change motifs (Table 1 and Supplementary Table 1). From a transcriptional influence standpoint, the leading SNP rs4993986 was located at the 3ʹ-UTR of HLA-DQB1 with DNaseI hypersensitivity and ENCODE transcription factor clusters binding signals. RNA-binding protein (RBP) analysis revealed that the RBP PABPC1 showed a binding region to rs4993986 (Figure 3A). According to the results of SRAMP prediction, the original m6A modification peak disappeared (labeled in red) when the altered gene sequence was input (Figure 3B), and the RNA secondary structure prediction showed that rs4993986 (C>G) was located nearby the m6A modification site (Figure 3C). In short, m6A-SNPs may affect the expression of local genes at the transcriptional level through the above-mentioned multiple pathways.
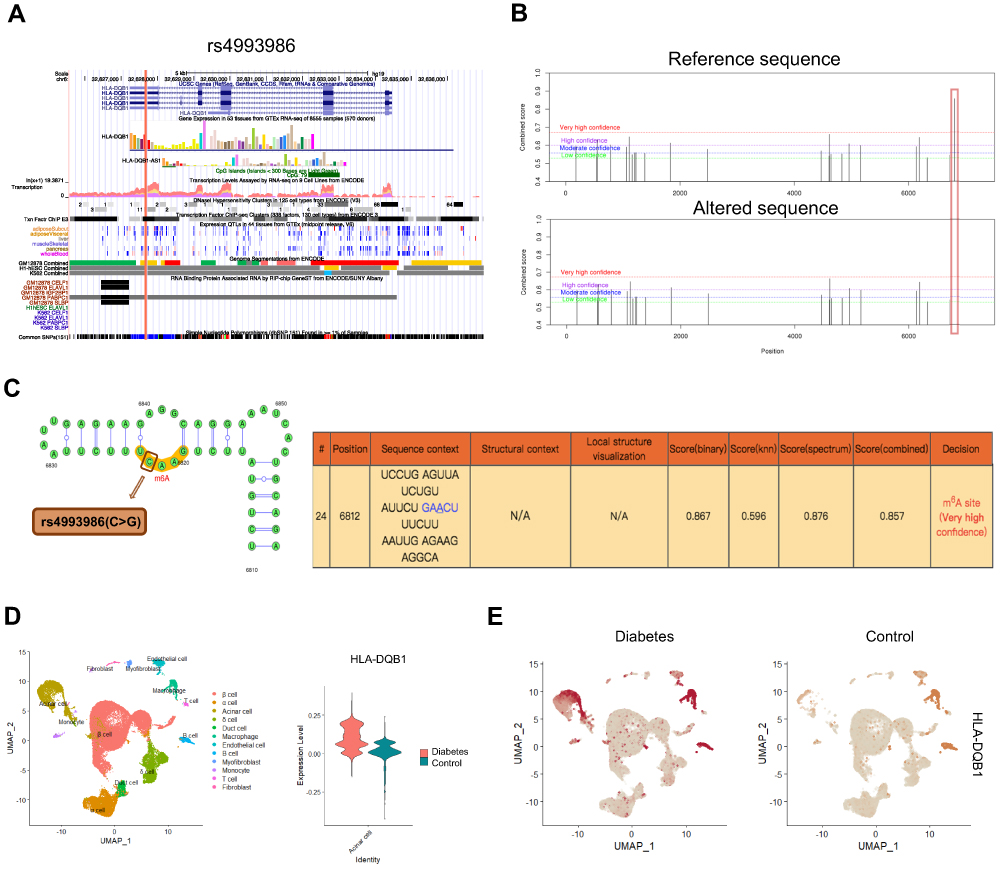 |
Figure 3 Integrated analysis of the leading m6A-SNP rs4993986. (A) The protein coding region of rs4993986 in HLA-DQB1 gene sequence shows high transcriptional activity and DNaseI hypersensitivity. RNA-binding protein analysis revealed that rs4993986 exhibited a binding region to the RBP PABPC1. (B) Prediction of m6A modification on the HLA-DQB1 transcript (ENST00000434651.6) on the SRAMP website. (C) The secondary structure of HLA-DQB1 transcript and the position of rs4993986 near the m6A modification site. (D) The expression of HLA-DQB1 (H2-Ab1) was identified in the mouse pancreas scRNA-seq data. Violin plot shows that HLA-DQB1 expression is increased in diabetic islet acinar cells. (E) HLA-DQB1 expression of different cell types in islet of diabetes and control group. |
Furthermore, in the scRNA-seq dataset of islet from diabetic mice induced by high-fat diets, we found that HLA-DQB1 was highly expressed in two cell populations (macrophages and acinar cells). The expression of HLA-DQB1 was increased in the acinar cells of diabetic group (Figure 3D and E), which is consistent with the up-regulation of HLA-DQB1 expression in diabetic patient islet.
Discussion
It has been found that more than 170 RNA modifications are involved in physiological processes and disease progression,41 among which m6A modification is considered to be the most abundant modification in mRNA.42 m6A modification has been reported to exert an important role in blood glucose homeostasis. Conditional knockout of the m6A methyltransferase Mettl14 led to decreased insulin secretion in β-cells and blood glucose intolerance.43,44 In addition, the hepatocyte-specific knockout of Mettl3, which is also a key component of the m6A methyltransferase complex, improved the mouse hepatic insulin sensitivity and inhibited the fatty acid metabolism.45 However, no studies have yet found the contribution of m6A-SNPs to the pathogenesis of T2D. In the current study, a great amount of T2D-associated m6A-SNPs have been identified, which are verified by other published datasets at genes expression level.
m6A modification of RNA can be recognized by different m6A readers, which may improve the stability of RNA transcripts (eg IGFBPs),46 or promote the degradation of transcripts (eg YTHDF2).47 RNA methylation has been reported to regulate the entire mRNA metabolism process, including RNA stability, splicing, nuclear transport, translation, and degradation, thereby affecting the key biological functions in the regulation of different cellular processes.48,49 Therefore, the expression of genes corresponding to m6A-SNPs may be affected. Experimental epitranscriptomic study conducted by Tian and his colleagues proved that overexpression of the rs8100241[A] (an m6A-SNP, G>A) allele significantly increased the level of ANKLE1 m6A and promoted the expression of ANKLE1 protein compared with that of rs8100241[G] allele.50 To validate whether the screened m6A-SNP-related genes were involved in the diabetes pathogenesis, we retrieved the GEO database and selected three tissues (liver, adipose, and pancreatic islets) from T2D patients to explore the expression of m6A-SNP-related genes under diabetic status. 11 m6A-SNP related genes were differentially expressed between T2D patients and the controls in at least one dataset. Also in diabetic mice, the expression trend of the m6A-SNPs related genes was verified by qRT-PCR. HLA-DQB1 encoded by the leading m6A-SNP rs4993986 was significantly upregulated in islets of diabetic patients and islet acinar cells in diabetic mice. With a minor allele frequency of 0.3691, rs4993986 was considered as a common SNP. Human leukocyte antigen (HLA) class II molecules are the gene loci found to be most highly associated with autoimmune diabetes mellitus.51 Increased expression of HLA-DQB1 results in beta cell failure mediated by autoimmunity.52 The structural changes of the HLA-DQB1 gene also participated in T2D pathogenesis,53 indicating that genetic variations in HLA-DQB1 locus were closely related to the occurrence of T2D. To determine the cell-type specific contribution of these m6A-SNPs, we further analyzed the scRNA-seq data of islet tissue in diabetic mice, and found that HLA-DQB1 was abnormally expressed in acinar cells under diabetic condition, which might participate in the progression of diabetes.
In order to avoid a large number of false-positive results, traditional GWAS usually sets a strict significance threshold (usually p < 5.0e-08) to screen for susceptibility loci.54 With the traditional significance threshold, the moderately correlated variations found in GWAS data can be ignored, and further research is needed to determine if they are involved in the disease.55 Thus, a relatively loose threshold of p < 5.0e-05 was set to avoid missing some valuable SNPs in this study.26 Exploring the potential functions of identified m6A-SNPs as multifunctional variants is essential for the study of T2D pathogenesis. In addition to affecting m6A modification, genetic variation could also alter gene expression by affecting transcriptional regulation factor binding, changing motif and RNA secondary structure. Therefore, the biological role of m6A-SNP should be considered as a multifunctional variation. By querying the HaploReg browser, we found several other potential regulatory mechanisms for these m6A-SNPs to affect gene transcription. For instance, rs4993986 was located in the DNaseI hypersensitivity cluster region, which may affect the binding of four proteins and alter six motifs. rs4993986 had a binding region with RBP PABPC1, which could be recruited to m6A modification sites by the m6A reader protein IGFBP2, and increased the transcript stability.56 Therefore, rs4993986 should be considered as a multifunctional variant involved in the pathogenesis of T2D. In addition, the possible m6A methylation site is predicted by a sequence of the HLA-DQB1 transcript on the SRAMP website. A m6A modification peak with a very high confidence at the 3ʹ end of the HLA-DQB1 transcript near the SNP rs4993986 is predicted, suggesting that rs4993986 is likely to affect the modification of m6A methylation of this site.
In conclusion, we made the first endeavor to explore the relationship between T2D and m6A-SNPs in this study. 71 m6A-SNPs were potentially associated with T2D and the potential functions were identified. For example, rs4993986 has been shown to be located near a m6A modification at the 3ʹ end of the HLA-DQB1 transcript, which may participate in the pathogenesis of T2D by altering HLA-DQB1 expression. Gene therapy targeting HLA-DQB1 locus and m6A demethylation enzymes may alleviate the progression of T2D. Also, there are still several limitations in the current study. We speculated that m6A-SNPs participated in T2D pathogenesis by affecting local gene expression. However, m6A modification has also been proved to affect translation process.57 Therefore, the validation of the screened m6A-SNPs at protein expression level has yet to be performed. As the exploration of RNA epigenetic regulatory networks increases, more m6A modifications in non-coding regions will be detected. Genetic variations near these sites may also be considered as m6A-SNPs to affect the pathogenesis of T2D, thus more m6A-SNPs that may affect non-coding regions remain to be identified. Further study targeting these m6A-SNPs to alleviate the development of T2D may become a new direction.
Data Sharing Statement
The data that support the results of this study are available from the corresponding author upon reasonable requirements. The m6ASNPs data can be downloaded from the m6AVar database (http://m6avar.renlab.org/). T2D GWAS data can be publicly obtained from http://diagram-consortium.org/index.html. The public gene expression data GSE23343, GSE38642, GSE78721, GSE132831 and GSE156061 can be downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). scRNA-seq data of diabetic mouse islet can be downloaded from GSE162512.
Ethics Approval and Informed Consent
The animal studies were approved by the Animal Care and Ethics Committee of West China Hospital of Stomatology, Sichuan University (WCHSIRB-D-2018-057), in compliance with institutional guidelines for the care and use of animals.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by grants from the National Natural Science Foundation of China (grant no. 81801018, 81771048), Research and Development Program, West China Hospital of Stomatology Sichuan Univercity (RD-03-202012), and Sichuan Science and Technology Program (grant no. 2020YFS0170).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Spracklen CN, Horikoshi M, Kim YJ, et al. Identification of type 2 diabetes loci in 433,540 East Asian individuals. Nature. 2020;582(7811):240–245. doi:10.1038/s41586-020-2263-3
2. Hu C, Jia W. Diabetes in China: epidemiology and genetic risk factors and their clinical utility in personalized medication. Diabetes. 2018;67(1):3–11. doi:10.2337/dbi17-0013
3. Lawlor N, Stitzel ML. (Epi)genomic heterogeneity of pancreatic islet function and failure in type 2 diabetes. Mol Metab. 2019;27:S15–S24. doi:10.1016/j.molmet.2019.06.002
4. Fuchsberger C, Flannick J, Teslovich TM, et al. The genetic architecture of type 2 diabetes. Nature. 2016;536(7614):41–47. doi:10.1038/nature18642
5. Alonso LC. T2D risk genes: exome sequencing goes straight to the source. Cell Metab. 2019;30(1):10–11. doi:10.1016/j.cmet.2019.06.010
6. Mahajan A, Wessel J, Willems SM, et al. Refining the accuracy of validated target identification through coding variant fine-mapping in type 2 diabetes. Nat Genet. 2018;50(4):559–571. doi:10.1038/s41588-018-0084-1
7. Suzuki K, Akiyama M, Ishigaki K, et al. Identification of 28 new susceptibility loci for type 2 diabetes in the Japanese population. Nat Genet. 2019;51(3):379–386. doi:10.1038/s41588-018-0332-4
8. Mahajan A, Taliun D, Thurner M, et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet. 2018;50(11):1505–1513. doi:10.1038/s41588-018-0241-6
9. Sirdah MM, Reading NS. Genetic predisposition in type 2 diabetes: a promising approach toward a personalized management of diabetes. Clin Genet. 2020;98(6):525–547. doi:10.1111/cge.13772
10. Horikoshi M, Hara K, Ito C, Nagai R, Froguel P, Kadowaki T. A genetic variation of the transcription factor 7-like 2 gene is associated with risk of type 2 diabetes in the Japanese population. Diabetologia. 2007;50(4):747–751. doi:10.1007/s00125-006-0588-6
11. Pascoe L, Tura A, Patel SK, et al. Common variants of the novel type 2 diabetes genes CDKAL1 and HHEX/IDE are associated with decreased pancreatic beta-cell function. Diabetes. 2007;56(12):3101–3104. doi:10.2337/db07-0634
12. Sladek R, Rocheleau G, Rung J, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445(7130):881–885. doi:10.1038/nature05616
13. Hrdlickova B, de Almeida RC, Borek Z, Withoff S. Genetic variation in the non-coding genome: involvement of micro-RNAs and long non-coding RNAs in disease. Biochim Biophys Acta. 2014;1842(10):1910–1922. doi:10.1016/j.bbadis.2014.03.011
14. Tak YG, Farnham PJ. Making sense of GWAS: using epigenomics and genome engineering to understand the functional relevance of SNPs in non-coding regions of the human genome. Epigenetics Chromatin. 2015;8(1):57. doi:10.1186/s13072-015-0050-4
15. Jia G, Fu Y, Zhao X, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7(12):885–887. doi:10.1038/nchembio.687
16. Dominissini D, Moshitch-Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201–206. doi:10.1038/nature11112
17. Xiong X, Hou L, Park YP, Molinie B, Gregory RI, Kellis M. Genetic drivers of m(6)A methylation in human brain, lung, heart and muscle. Nat Genet. 2021;53(8):1156–1165. doi:10.1038/s41588-021-00890-3
18. Zhang Z, Luo K, Zou Z, et al. Genetic analyses support the contribution of mRNA N(6)-methyladenosine (m(6)A) modification to human disease heritability. Nat Genet. 2020;52(9):939–949. doi:10.1038/s41588-020-0644-z
19. Chai T, Tian M, Yang X, Qiu Z, Lin X, Chen L. Genome-wide identification of RNA modifications for spontaneous coronary aortic dissection. Front Genet. 2021;12:696562. doi:10.3389/fgene.2021.696562
20. Mo XB, Zhang YH, Lei SF. Genome-wide identification of m(6) A-associatedSNPs as potential functional variants for bone mineral density. Osteoporos Int. 2018;29(9):2029–2039. doi:10.1007/s00198-018-4573-y
21. Sun X, Dai Y, Tan G, Liu Y, Li N. Integration analysis of m(6) A-SNPsand eQTLs associated with sepsis reveals platelet degranulation and staphylococcus aureus infection are mediated by m(6)A mRNA methylation. Front Genet. 2020;11:7. doi:10.3389/fgene.2020.00007
22. Zhao H, Jiang J, Wang M, Xuan Z. Genome-wide identification of m6A-associated single-nucleotide polymorphisms in colorectal cancer. Pharmgenomics Pers Med. 2021;14:887–892. doi:10.2147/pgpm.S314373
23. Zhu R, Tian D, Zhao Y, Zhang C, Liu X. Genome-wide detection of m(6) A-associated genetic polymorphisms associated with Ischemic stroke. J Mol Neurosci. 2021;71(10):2107–2115. doi:10.1007/s12031-021-01805-x
24. Mo X, Lei S, Zhang Y, Zhang H. Genome-wide enrichment of m(6) A-associated single-nucleotide polymorphisms in the lipid loci. Pharmacogenomics J. 2018;19(4):347–357. doi:10.1038/s41397-018-0055-z
25. Mo X-B, Lei S-F, Zhang Y-H, Zhang H. Examination of the associations between m6A-associated single-nucleotide polymorphisms and blood pressure. Hypertens Res. 2019;42(10):1582–1589. doi:10.1038/s41440-019-0277-8
26. Mo XB, Lei SF, Zhang YH, Zhang H. Detection of m(6) A-associatedSNPs as potential functional variants for coronary artery disease. Epigenomics. 2018;10(10):1279–1287. doi:10.2217/epi-2018-0007
27. Mo XB, Lei SF, Qian QY, Guo YF, Zhang YH, Zhang H. Integrative analysis revealed potential causal genetic and epigenetic factors for multiple sclerosis. J Neurol. 2019;266(11):2699–2709. doi:10.1007/s00415-019-09476-w
28. Mo XB, Lei SF, Zhang YH, Zhang H. Integrative analysis identified IRF6 and NDST1 as potential causal genes for Ischemic stroke. Front Neurol. 2019;10:517. doi:10.3389/fneur.2019.00517
29. Lin W, Xu H, Yuan Q, Zhang S. Integrative genomic analysis predicts regulatory Role of N (6)-methyladenosine-associated SNPs for adiposity. Front Cell Dev Biol. 2020;8:551. doi:10.3389/fcell.2020.00551
30. Zheng Y, Nie P, Peng D, et al. m6AVar: a database of functional variants involved in m6A modification. Nucleic Acids Res. 2018;46(D1):D139–d145. doi:10.1093/nar/gkx895
31. Zhou Y, Zhou B, Pache L, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10(1):1523. doi:10.1038/s41467-019-09234-6
32. Ward LD, Kellis M. HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 2016;44(D1):D877–81. doi:10.1093/nar/gkv1340
33. Zhou Y, Zeng P, Li YH, Zhang Z, Cui Q. SRAMP: prediction of mammalian N6-methyladenosine (m6A) sites based on sequence-derived features. Nucleic Acids Res. 2016;44(10):e91. doi:10.1093/nar/gkw104
34. Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36(5):411–420. doi:10.1038/nbt.4096
35. Li DS, Yuan YH, Tu HJ, Liang QL, Dai LJ. A protocol for islet isolation from mouse pancreas. Nat Protoc. 2009;4(11):1649–1652. doi:10.1038/nprot.2009.150
36. De Silva NMG, Borges MC, Hingorani AD, et al. Liver function and risk of type 2 diabetes: bidirectional Mendelian randomization study. Diabetes. 2019;68(8):1681–1691. doi:10.2337/db18-1048
37. Wewer Albrechtsen NJ, Pedersen J, Galsgaard KD, et al. The liver-α-cell axis and type 2 diabetes. Endocr Rev. 2019;40(5):1353–1366. doi:10.1210/er.2018-00251
38. Desjardins EM, Steinberg GR. Emerging role of AMPK in Brown and Beige Adipose Tissue (BAT): implications for obesity, insulin resistance, and type 2 diabetes. Curr Diab Rep. 2018;18(10):80. doi:10.1007/s11892-018-1049-6
39. Petersen MC, Shulman GI. Mechanisms of insulin action and insulin resistance. Physiol Rev. 2018;98(4):2133–2223. doi:10.1152/physrev.00063.2017
40. Eguchi K, Nagai R. Islet inflammation in type 2 diabetes and physiology. J Clin Invest. 2017;127(1):14–23. doi:10.1172/jci88877
41. Frye M, Harada BT, Behm M, He C. RNA modifications modulate gene expression during development. Science. 2018;361(6409):1346–1349. doi:10.1126/science.aau1646
42. Li Y, Wang J, Huang C, Shen M, Zhan H, Xu K. RNA N6-methyladenosine: a promising molecular target in metabolic diseases. Cell Biosci. 2020;10(1):19. doi:10.1186/s13578-020-00385-4
43. De Jesus DF, Zhang Z, Kahraman S, et al. m(6)A mRNA methylation regulates human β-cell biology in physiological states and in type 2 diabetes. Nat Metab. 2019;1(8):765–774. doi:10.1038/s42255-019-0089-9
44. Men L, Sun J, Luo G, Ren D. Acute deletion of METTL14 in β-cells of adult mice results in glucose intolerance. Endocrinology. 2019;160(10):2388–2394. doi:10.1210/en.2019-00350
45. Xie W, Ma LL, Xu YQ, Wang BH, Li SM. METTL3 inhibits hepatic insulin sensitivity via N6-methyladenosine modification of Fasn mRNA and promoting fatty acid metabolism. Biochem Biophys Res Commun. 2019;518(1):120–126. doi:10.1016/j.bbrc.2019.08.018
46. Huang H, Weng H, Sun W, et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20(3):285–295. doi:10.1038/s41556-018-0045-z
47. Zhao BS, Wang X, Beadell AV, et al. m(6)A-dependent maternal mRNA clearance facilitates zebrafish maternal-to-zygotic transition. Nature. 2017;542(7642):475–478. doi:10.1038/nature21355
48. Chao Y, Li HB, Zhou J. Multiple functions of RNA methylation in T cells: a review. Front Immunol. 2021;12:627455. doi:10.3389/fimmu.2021.627455
49. Willbanks A, Wood S, Cheng JX. RNA epigenetics: fine-tuning chromatin plasticity and transcriptional regulation, and the implications in human diseases. Genes. 2021;12(5):627. doi:10.3390/genes12050627
50. Tian J, Ying P, Ke J, et al. ANKLE1 N6-methyladenosine-related variant is associated with colorectal cancer risk by maintaining the genomic stability. Int J Cancer. 2019;146(12):3281–3293. doi:10.1002/ijc.32677
51. Frommer L, Flesch BK, König J, Kahaly GJ. Amino acid polymorphisms in Hla class II differentiate between thyroid and polyglandular autoimmunity. J Clin Endocrinol Metab. 2020;105(6):Jun. doi:10.1210/clinem/dgz164
52. Onengut-Gumuscu S, Chen WM, Robertson CC, et al. Type 1 diabetes risk in African-Ancestry participants and utility of an Ancestry-Specific Genetic Risk Score. Diabetes Care. 2019;42(3):406–415. doi:10.2337/dc18-1727
53. Mayer A, Fabien N, Gutowski MC, et al. Contrasting cellular and humoral autoimmunity associated with latent autoimmune diabetes in adults. Eur J Endocrinol. 2007;157(1):53–61. doi:10.1530/eje-07-0060
54. Sham PC, Purcell SM. Statistical power and significance testing in large-scale genetic studies. Nat Rev Genet. 2014;15(5):335–346. doi:10.1038/nrg3706
55. Mo XB, Lu X, Zhang YH, Zhang ZL, Deng FY, Lei SF. Gene-based association analysis identified novel genes associated with bone mineral density. PLoS One. 2015;10(3):e0121811. doi:10.1371/journal.pone.0121811
56. Zhang L, Wan Y, Zhang Z, et al. IGF2BP1 overexpression stabilizes PEG10 mRNA in an m6A-dependent manner and promotes endometrial cancer progression. Theranostics. 2021;11(3):1100–1114. doi:10.7150/thno.49345
57. Han X, Wang M, Zhao YL, Yang Y, Yang YG. RNA methylations in human cancers. Semin Cancer Biol. 2020. doi:10.1016/j.semcancer.2020.11.007
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.