")
Back to Journals » Cancer Management and Research » Volume 12
Exploration of the Potential Biomarkers of Papillary Thyroid Cancer (PTC) Based on RT2 Profiler PCR Arrays and Bioinformatics Analysis
Authors Peng Y, Zhang HW , Cao WH, Mao Y, Cheng RC
Received 17 June 2020
Accepted for publication 24 August 2020
Published 28 September 2020 Volume 2020:12 Pages 9235—9246
DOI https://doi.org/10.2147/CMAR.S266473
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Eileen O'Reilly
Ying Peng,1,2 Han-Wen Zhang,1,2 Wei-Han Cao,1,2 Ying Mao,1,3 Ruo-Chuan Cheng2
1Kunming Medical University of Yunnan Province, Kunming, Yunnan 650500, People’s Republic of China; 2Thyroid Disease Diagnosis and Treatment Center, First Affiliated Hospital of Kunming Medical University, Kunming, Yunnan 650032, People’s Republic of China; 3Thyroid Disease Diagnosis and Treatment Center, Second Affiliated Hospital of Kunming Medical University, Kunming, Yunnan 650032, People’s Republic of China
Correspondence: Ruo-Chuan Cheng Tel +86 13708467986
Email [email protected]
Background: Papillary thyroid carcinoma (PTC) has increased rapidly over recent years, and radiation, hormone effects, gene mutations, and others were viewed as closely related. However, the molecular mechanisms of PTC have not been cleared. Therefore, we intended to screen more accurate key genes and pathways of PTC by combining RT2 profiler PCR arrays and bioinformatics methods in this study.
Materials and Methods: RT2 profiler PCR arrays were firstly analyzed to identify differential expression genes (DEGs) in PTC. RT-qPCR were performed to verify the most significant differential expression genes. The TCGA database was used to further verify for expanded data. Enrichment analysis of Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) was analyzed. To construct the protein–protein interaction (PPI) network, we used STRING and Cytoscape to make module analysis of these DEGs.
Results: Sixteen differentially expressed genes were presented in RT2 profiler PCR arrays, including 13 down-regulated DEGs (DEGs) and three up-regulated DEGs (DEGs), while 13 stable DEGs were eventually verified. A total of 155 DEGs were presented in the TCGA database, including 82 up-regulated DEGs (DEGs) and 73 down-regulated DEGs (dDEGs). A total of 29 important genes were extracted after integrating these two results, GO and KEGG analyses were used to observe the possible mechanisms of action of these DEGs. The PPI network was constructed to observe hub genes. Prognostic analysis further demonstrated the involvement of these genes in the biological processes of PTC.
Conclusion: This study identified some potential molecular targets and signal pathways, which might help us raise our awareness of the mechanisms of PTC.
Keywords: papillary thyroid cancer, qiagen RT2 profiler PCR arrays, bioinformatics, DEGs, signal pathways
Introduction
In the past 30 years, the incidence of thyroid cancer (TC) has tripled, making it the fastest rising malignant solid tumor worldwide.1,2 The biological features of various subclasses of TC can vary from low potential to high potential forms, extreme aggressiveness, and lethality. Papillary thyroid cancer (PTC) accounts for 90% of all thyroid cancer cases,3 making it emergency to discover new biological and therapeutic targets for it. Although the treatment of PTC and its effect have been improved over recent years, the trauma caused by surgery and the recurrence of tumor are still intractable problems, bringing a certain life and economic burden to patients.
Generally, early diagnosis and management for PTC could improve patients’ prognosis.4 Therefore, it is of great significance to further explore the pathogenesis of PTC and find molecular targets with the potential for early diagnosis, early prevention, and early treatment. Following the advances in the identification of mutations in thyroid cancer, other genes (gene expression, miRNA) and epigenetic markers are expected to significantly improve the accuracy of thyroid nodule detection compared with currently available clinical tests.5,6 If this progress continues, future molecular tests are expected to be able to accurately predict the cancer risk of thyroid nodules. In addition, the development of these data can further inform specific treatment decisions, such as surgical coverage, multi-kinase, and specific kinase inhibitor therapies or combinations.7–9 The discovery of new gene mutations/rearrangements involved in the pathogenesis of thyroid cancer, such as BREAF and RAS, is expected to provide new and effective therapeutic targets.9,10
Along with the development of molecular biology technique, molecular diagnosis is further improved under the control of evidence-based evidence.11 High-throughput tools such as microarray and sequencing have been widely used to find molecular changes in the occurrence and development of diseases which are effective ways to detect the pathogenetic mechanism of diseases. For example, RT Profiler PCR array has been used by many scholars to explore the diagnostic targets of other tumors in recent years, and a lot of achievements have been made.12–14 Bioinformatics is a new discipline emerging with the launch of the human genome project,15 and has established a fundamental position in the life sciences. Mining and analyzing the massive data based bioinformatics also allow us to screen key genes or pathways associated with the diseases. To obtain a more accurate target for diagnosis and treatment of PTC, we combined the two analyses together in this study to screen relevant data and identify the DEGs that may play a role in PTC. In addition, we assessed the functions and roles of screened candidate genes.
Materials and Methods
Human Tissue Samples
Human tissue samples (The total number of samples was eight) were collected from patients with thyroid cancer from the First Affiliated Hospital of Kunming Medical University. Based on the findings from hematoxylin and eosin staining of sections for pathological diagnosis and histological types,16 two groups were included, papillary thyroid carcinoma (PTC with stageI17) and healthy controls (histologically identified as normal thyroid tissue at a distance of more than 2 cm from the edge of the cancer).18 All tissue specimens were immediately frozen and transferred to the Kunming Institute of Biology, after which they were used to extracted total RNA. The patients characteristics are presented in Table 1.
 |
Table 1 The Characteristics of Patients. All Patients Were Excluded Other Tumors or Diseases and Only Diagnosed as Papilliary Thyroid Cancer (PTC). All Patients Were PTC with Stage I |
RT2 Profiler PCR Arrays Test
According to the manufacturer’s directions, total RNA was extracted from the tissues with Trizol reagent (Thermo Fisher Scientific, Inc., USA). A spectrophotometer was used to identify RNA quality and the RNA quality control parameters OD260/280 were between 1.8–2.0. Reverse transcription was performed by using the All-in-One™ First-Strand cDNA Synthesis kit (GeneCopoeia, USA) under the following reaction conditions: 42°C for 60 minutes and 70°C for 5 minutes. The cDNA was used on the real-time RT2 Profiler PCR Array (QIAGEN, Cat. no. PAHS-033Z) in combination with RT2 SYBR®Green qPCR Mastermix (Cat. no. 330,529). Each array plate contained one set of 96 wells for testing. Genomic DNA contamination, reverse transcription, and positive PCR controls were included in each 96-well set on each plate. Glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) was used as the assay reference gene. CT values were derived to an Excel file to build a table of CT values, which is then uploaded onto the data analysis web portal at http://www.qiagen.com/geneglobe. Samples contained controls and test groups. CT values were normalized based on Automatic selection from full panel of reference genes.
Quantitative Real-Time-PCR
Total RNA was extracted from the tissues with Trizol reagent (Thermo Fisher Scientific, Inc., USA). A spectrophotometer was used to identify RNA quality and the RNA quality control parameters OD260/280 were between 1.8–2.0. RT was performed using the All-in-One™ First-Strand cDNA Synthesis kit (GeneCopoeia, USA) under the following reaction conditions: 42°C for 60 minutes and 70°C for 5 minutes. Quantitative real-time-PCR was carried out with SYBR® Premix Ex TaqTM II (GeneCopoeia, USA) on an Applied 7000 Real-Time PCR System (Applied Biosystem, USA). Samples were repeatedly amplified for 40 cycles. The housekeeping gene GAPDH was selected as an internal control for mRNA abundance. Fold changes in the levels of target gene mRNAs were determined using the formula 2−ΔΔCt. The primers presented in Table 2 were used.
 |
Table 2 Primers Used by RT-qPCR to Verify the Most Significant Genes |
Datasets and Data Preprocessing
Download gene expression RNAseq sequencing data (FPKM) and clinical data (Phenotype) of GDC TCGA-THCA in the TCGA database19
(https://xenabrowser.net/). A total of 559 samples were collected, including 501 tumor samples and 58 normal samples. Using R packages limma package20 (Version 3.10.3, http://www.bioconductor.org/packages/2.9/bioc/html/limma.html) provides the classical bayesian method, differential expression analysis was performed according to the cancer vs paracancer group. The threshold for the DEGs was set as P-value<0.05 and |log2 foldchange (FC)|>2.
GO and KEGG Analysis
To analyze the functions of DEGs, respectively on the Gene Ontology21 functional annotation and KEGG22 pathway, enrichment analysis was carried out by using R package clusterProfiler23 (version: 3.8.1, http://bioconductor.org/packages/release/bioc/html/clusterProfiler.html) for the above differences in Gene in accordance with the cut mRNA. Significance threshold P-value<0.05 was considered as a significant enrichment result.
Construction of a PPI Network
To investigate the possible hub genes/proteins that might play a significant role in the biological process, the interaction relationship between gene-coding proteins was predicted and analyzed in combination with STRING24 (Version: 11.0, http://www.string-db.org/) database. The input gene set is the difference mRNA obtained above, and the species is homo. A confidence score >0.4 was set, which may filter out the critical module.
Prognosis Analysis of Significance Genes
The TCGA-GEIPA database was used to select THCA data collection, and on the basis of median expression level, the significance genes were divided into high and low expression groups, respectively, for OS (Overall Survival) and DFS (Disease-free survival) analysis. The genes with P<0.05 were found to be the key genes for prognosis.
Statistical Analysis
Independent Student’s t-tests were used for statistical analyses between two groups in RT2 Profiler PCR Arrays and quantitative real-time-PCR. Experimental values are described as the mean±standard deviation, P-values<0.05 indicated a significant difference between two groups.
Besides, evaluating from the different multiple and significance level performed differential expression analysis in TGCA data. The threshold for the DEGs was set as P<0.05 and |log2 foldchange (FC)|>2. Evaluating from the different multiple and significance levels performed differential expression analysis. The threshold for the DEGs was set as P<0.05 and |log2 foldchange (FC)|>2.
Results
Screen Out DEGs Through RT2 Profiler PCR Arrays Test
To explore whether PTC has similar targets with other tumors, PTC and control tissues were firstly analyzed to identify DEGs by using RT2 Profiler PCR Arrays Test (the number of samples was two). Compared with control, a total of 16 DEGs were identified, consisting of three uDEGs and 13 dDEGs in PTC tissues (Figure 1A, the threshold for the DEGs was set as |log2 foldchange (FC)|>2. Compared with the Normal group, P<0.05 indicated that the difference was statistically significant). The data is also analyzed by picturing plots, the scatter plot and heat map correspondingly (Figure 1B and C). To verify the most significant genes, RT-qPCR were performed on more clinical samples (the number of samples was six) and verified 13 stable DEGs eventually (Figure 1D, the threshold was set as |log2 foldchange (FC)|>2. Compared with the normal group, *P<0.05, **P<0.01, ns presented non-significant, P>0.05).
 |
Figure 1 DEGs between PTC and normal through RT2 profiler PCR arrays test. (A) A total of 16 DEGs were identified out, consisting of three uDEGs and 13 dDEGs in PTC tissues. The ordinate is a multiple of difference and the threshold for the DEGs was set as |log2 foldchange (FC)|>2. Compared with the Normal group, P<0.05 indicated that the difference was statistically significant. (B) The DEGs’ expression levels of the two group are extracted to draw a scatter plot. Yellow plots above the line represent higher expression genes, blue plots under the line represent lower expression genes. (C) Two groups of differential genes intersect and merge gene expression calorimetry maps. Based on the expression levels of the DEGs in each group, a logarithm of 2 is taken. Using systematic clustering (Hierarchical Cluster), the overall clustering result of the sample is finally obtained. Red means higher expression, blue means lower expression. (D) The expression levels of the two genes are extracted to draw a heat map. Red represents high expression in the sample, and green represents low expression in the sample. (E) RT-qPCR were performed on more clinical samples to verify these most significant DEGs, eventually 13 stable DEGs were screened out. The horizontal coordinate represents the multiple of difference and and the threshold was set as |log2 foldchange (FC)|>2. Compared with the Normal group, |
Identification of DEGs in PTC by Using Public Database
To screen the significant genes in PTC, we then mined and analyzed data in the TCGA database. Gene expression RNAseq sequencing data (FPKM) and clinical data (Phenotype) of GDC TCGA-THCA were downloaded for analysis between PTC patients and control samples, with P<0.05 and |log2 foldchange (FC)|>2 as criteria. Compared with the control group, a total of 155 DEGs was identified from the database, consisting of 82 uDEGs and 73 dDEGs in PTC (Figure 2A). Moreover, the top 10 uDEGs and top 10 dDEGs have been screened out, respectively (Figure 2B and C).
 |
Figure 2 DEGs between PTC and normal by mining and analyzing data in the TCGA database. (A) A total of 155 DEGs were identified, consisting of 82 uDEGs and 73 dDEGs in PTC tissues; DEGs between PTC and normal intersect and merge gene expression calorimetry maps. Using systematic clustering (Hierarchical Cluster), the overall clustering result of the sample is finally obtained. Red means higher expression, green means lower expression. (B) The top 10 uDEGs and top 10 dDEGs have been further screened out, respectively. A similar gene expression calorimetry map was drawn. Red means higher expression, green means lower expression. (C) Volcanic map of the top10 uDEGs and top 10 dDEGs in PTC group. P<0.05 was corrected according to the threshold value described in the method. DEGs with significant differential expression are indicated by red dots (up-regulation) and green dots (down-regulation). Genes without significant differential expression are indicated by black dots. |
DEGs Functional Analysis and PPI Network Construction
To estimate the functions of identified DEGs, enrichment analysis of GO and KEGG were analyzed. GO analysis showed that the uDEGs and dDEGs were enriched in various biological processes, cellular components, and molecular function (Figure 3A and B). KEGG pathway analysis revealed that the DEGs were enriched in pathways in thyroid hormone synthesis, tyrosine metabolism, and mineral absorption (Figure 3C and D). A total of 185 interaction pairs of the DEGs were identified using the STRING database. A PPI network with 113 nodes of the DEGs was constructed based on these pairs (Figure 3E).
 |
Figure 3 TCGA-DEGs functional were analyzed. In order to understand the biological processes, cell components, and molecular functions involved in DEGs. The GO gene enrichment analysis of DEGs can be obtained as shown (A, B). In addition, to understand the metabolic pathway, signal pathway, and other information of these DEGs, KEGG enrichment analysis of the DEGs was obtained (C, D), where Ratio refers to the number of differential genes enriched in the pathway in the pathway, Ratio to the number of all genes in this pathway. The larger the ratio, the greater the degree of enrichment. (A) GO analysis of uDEGs; (B) GO analysis of dDEGs. (C) KEGG analysis of uDEGs; (D) KEGG analysis of dDEGs. (E) Networks were constructed, in which nodes that share the same cluster identity are typically close to each other, the clustering was as shown above. |
Nine Duplicate Genes Were Found by Comparative Analysis
To further determine the potential targets of PTC, we accomplished a comparative analysis between the TCGA database and RT2 Profiler PCR Arrays test results. Among 13 differential expression genes that screened in RT2 Profiler PCR Arrays test, nine genes were also found in the TCGA database. The analysis of this nine genes expression in the TCGA database is as follows, while only four genes showed similar differences (Figure 4A).
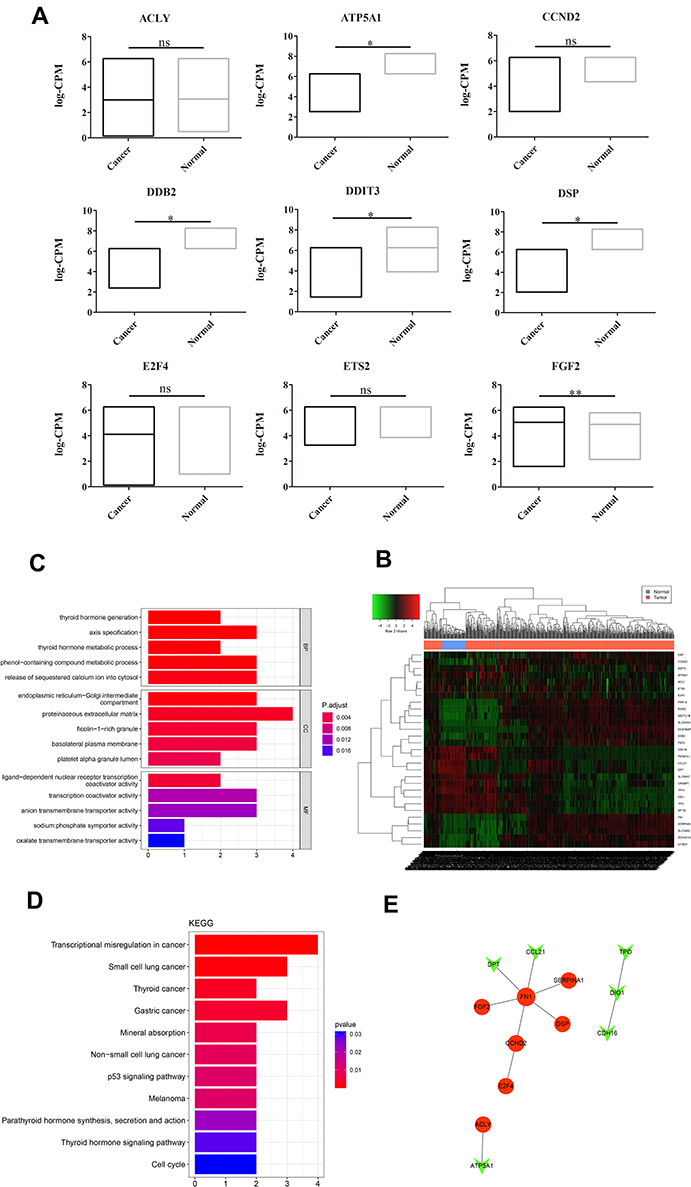 |
Figure 4 A comparative analysis between RT2 profiler PCR arrays test and TCGA database and eventually 29 genes functional were analyzed. (A) Among the differential expression genes in RT2 Profiler PCR Arrays test, ninegenes were also found in TCGA. The ordinate represents the |log2 foldchange (FC)| of the DEGs. Compared with the Normal group, *P<0.05, **P<0.01, ns presented non-significant, P>0.05. However, their expression analyzed results were not identical. ETS2, E2F4, CCND2, and ACLY showed no significant differential expression between PTC and normal based the TCGA database. (B) Eventually 29 genes were screened out and a heatmap was presented to show their expressions. Besides, GO enrichment analysis and KEGG analysis for these genes were also performed to explore their potential mechanism. (C) GO analysis of these genes; (D) KEGG analysis of these genes. (E) Network were constructed, in which nodes that share the same cluster identity are typically close to each other. |
Functional Analysis and PPI Network Construction of Duplicate Genes and Top 10 DEGs
To further verify the functions of the potential targets in PTC, we integrated the nine duplicate genes and the identified top 10 DEGs (10 uDEGs and 10 dDEGs, respectively) (Figure 4B, the threshold was set as |log2 foldchange (FC)|>2. Compared with the Normal group, *P<0.05, **P<0.01, ns presented non-significant, P>0.05). Enrichment analysis of GO and KEGG were analyzed as above. GO analysis showed that these genes were enriched in various biological processes, cellular components, and molecular function (Figure 4C). KEGG pathway analysis indicated that these genes were enriched in various pathways, especially in transcriptional misregulation in cancer (Figure 4D). A total of 10 interaction pairs of these genes were identified using the STRING database. A PPI network with 13 nodes of these genes was constructed based on these pairs (Figure 4E).
Identified Genes Expression Characteristics and Prognostic Association Analysis
To analyze the association of these identified genes with PTC prognostic, we choose the TCGA-GEIPA database. The THCA dataset was selected, and these identified genes were assigned to two groups according to the median expression amount, high and low expression, respectively. DFS (Disease-free survival) and OS (Overall Survival) analysis was conducted, and the genes with P-value<0.05 were viewed to be the key genes for prognosis. The results presented that TFF3, CRABP1, and TPO showed a strong correlation with DFS, while METTL7B, RXRG, PRR15, ETS2, and CCND2 showed a strong correlation with OS. The prognostic association analysis and the expression status of these genes are summarized in Table 3.
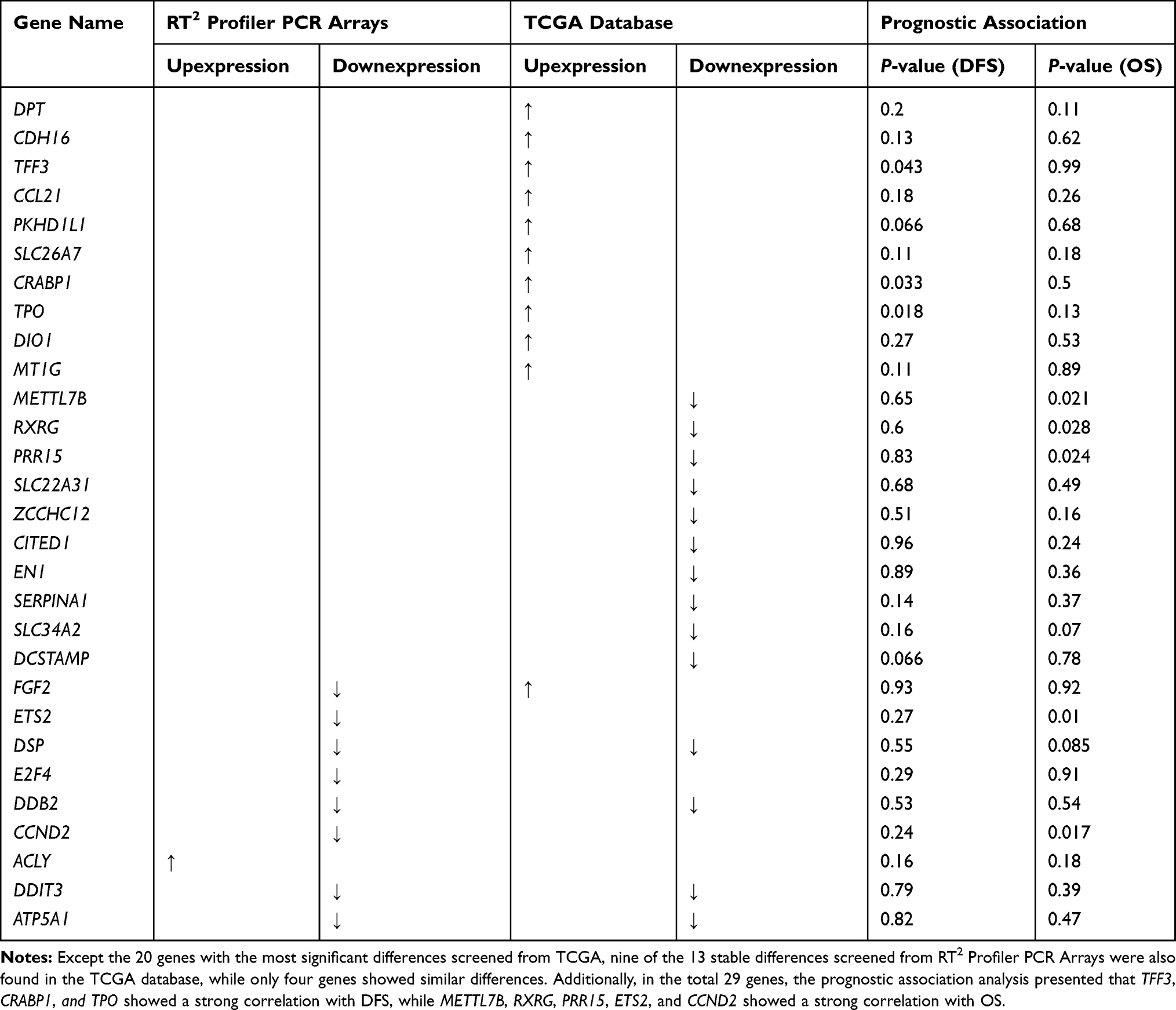 |
Table 3 Prognostic Association Analysis and the Expression Status of the Genes |
Discussion
In recent years, great progress has been made in genomics research on PTC,25–27 but the specific mechanism was still not clear.28,29 Actually, future molecular tests are expected to be able to accurately predict the cancer risk of thyroid nodules and inform specific treatment decisions.30,31 Bioinformatics is a new discipline emerging with the launch of the human genome project.15 In this process, a large number of database resources of genome and protein structure and sequence have been generated, which has promoted the development of bioinformatics. Effective use of various bioinformatics tools, such as databases, software, and other information resources, will have a huge impact on the development of disease prevention, diagnosis, treatment, and new drug development from the perspective of molecular biology, which benefited exploring PTC’s molecule targets. Besides, RT Profiler PCR array has also been used by many scholars to explore the diagnostic targets of other tumors and a lot of achievements have been made, which can provide a rich reference for the subsequent in-depth research on the diagnosis and treatment of PTC. Therefore, it is meaningful to screen out more accurate differential genes through combining this two methods. Furthermore, previous studies using expression data have focused on DEGs to identify potential genes involved in PTC,32–35 and few people have paid attention to the interaction of related genes. Network analysis groups interactive genes into one module in which genes are enriched for specific biological pathways. Hence to further investigate the functional relevance of the identified genes in disease, functional annotation and enrichment analyses were very important.36 In this study, in addition to combining the two methods to screen out more accurate differential gene expression analysis, a total of 29 DEGs screened by these two methods were used to establish a co-expression network for PTC samples. Exploring gene–gene interactions among these networks may help to identify pathways of dissonance in the disease process. This approach enhances the ability to detect functional genes, altering their crosstalk with other genes, not just their expression levels.
In the present study, RT2 Profiler PCR Array tests were used to analyze whether PTC has some similar targets with other tumors (Figure 1A). Additionally, data mining and bioinformatics analysis were also performed to identify the DEGs from patients with PTC and healthy controls (Figure 2A) and enrichment analysis of GO and KEGG were analyzed to present the possible mechanism of the DEGs (Figure 3A–D). To identify the possible highly correlated target genes involved in the pathogenesis of PTC, we integrated the bioinformatics analysis top 10 DEGs (uDEGs and dDEGs, respectively), and the duplicate nine genes between RT2 Profiler PCR Array test and bioinformatics analysis (Figure 4C). These genes are shown in detail in Table 2. Enrichment analysis of GO and KEGG were analyzed to further present the possible mechanism of these genes. A high degree of connectivity in the PPI network of these genes were also shown in Figure 4E.
By the enrichment analysis of GO, we found that these genes were associated with various biological functions. For biological process (BP), genes were enriched in thyroid hormone generation, axis specification, thyroid hormone metabolic process, phenol-containing compound metabolic process, release of sequestered calcium ion into cytosol. For cellular components (CC), genes were enriched in the endoplasmic reticulum-Golgi intermediate, compartment, proteinaceous extracellular matrix, ficolin-1-rich granule, basolateral plasma membrane, platelet alpha granule lumen. Besides, for molecular function (MF), our results presented that genes were enriched in ligand-dependent nuclear receptor transcription, which seemed to be a little different from other researches,37 this may be because the total number of genes we screen out is relatively small. However, ligand-dependent nuclear receptor transcription was once proved to mediate the transcription to enhance transcription of target genes in cancer,38 which provides evidence that this process may play an important role in PTC.
KEGG pathway analysis indicated that these genes were enriched in pathways in cancer, which is similar to other researches.36 In our results, the transcriptional misregulation pathway seemed to be especially different, which corresponds to our GO analysis results which presented that it may exert important functions in PTC. And a PPI network with 13 nodes of these genes was further constructed. Additionally, prognostic association analysis presented that TFF3, CRABP1, and TPO showed a strong correlation with DFS, while METTL7B, RXRG, PRR15, ETS2, and CCND2 showed a strong correlation with OS. Theses genes are worth investigating further.
Our study has several limitations. Firstly, not all genes related to PTC reported in previous genetic studies have been identified. This study aimed to screen out new and more reliable PTC targets by combining this two tools, and emphasized the impact of gene–gene interaction on tumor pathological processes, providing a new perspective for the detection of potential functional genes. However, due to the relatively limited data obtained from the RT2 Profiler PCR Arrays and TCGA database, further studies are needed under a larger sample size. Secondly, our conclusions are based solely on analysis of gene expression profiles, the exact effects of identified genes need to be confirmed by biological experiments. Therefore, future validation experiments are warranted to examine the results. However, validation and enrichment analysis were used in this study to identify the identified genes. Certain genes, such as PINX1, CCND2, and FGF2, have been demonstrated to be involved in PTC pathology by prior functional and experimental studies.39–41
In summary, the potential molecular targets of PTC were preliminarily studied in this study. Firstly, RT2 Profiler PCR Arrays were used to screen out targets that were similar to other tumors, and then the DEGs in PTC were selected through the mining and analysis of the TCGA database. By combining the results of these two processes, 29 genes were eventually screened for further analysis. Using prognostic correlation analysis, some of these genes were predicted to be significantly correlated with the final outcome of PTC. In a word, our study found some new novel genes and pathways (especially transcriptional misregulation in cancer) were identified that may exert important functions in PTC. These results contribute to the understanding of the genetic basis of PTC and provide novel insights into the identification of potential functional genes in PTC.
Ethics Affirming
This research has met all the guidelines outlined in the Declaration of Helsinki and was allowed by Kunming Medical University First Affiliated Hospital’s Ethical Committee ((2020) L no. 2). All patients included in this study provided informed oral consent. The informed verbal consent process was approved by the ethics committee of Kunming Medical University First Affiliated Hospital [(2020) L no. 2].
Funding
This research was supported by the Association Foundation Program of Yunnan Science and Technology Department and Kunming Medical University (grant number 2018FE001(−168)).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Davies L, Welch HG. Current thyroid cancer trends in the United States. JAMA Otolaryngol Head Neck Surg. 2014;140(4):317–322. doi:10.1001/jamaoto.2014.1
2. Kitahara CM, Sosa JA. The changing incidence of thyroid cancer. Nat Rev Endocrinol. 2016;12(11):646–653. doi:10.1038/nrendo.2016.110
3. Zhao J, Zhirong L, Chen Y, et al. MicroRNA-766 inhibits papillary thyroid cancer progression by directly targeting insulin receptor substrate 2 and regulating the PI3K/Akt pathway. Int J Oncol. 2019;54(1):315–325. doi:10.3892/ijo.2018.4615
4. Fagin JA, Wells SA. Biologic and clinical perspectives on thyroid cancer. N Engl J Med. 2016;375(11):1054–1067. doi:10.1056/NEJMra1501993
5. Giordano TJ. Genomic hallmarks of thyroid neoplasia. Annu Rev Pathol. 2018;13(1):141–162. doi:10.1146/annurev-pathol-121808-102139
6. Liang J, Cai W, Feng D, et al. Genetic landscape of papillary thyroid carcinoma in the Chinese population. J Pathol. 2017.
7. Haugen BR, Alexander EK, Bible KC, et al. 2015 American thyroid association management guidelines for adult patients with thyroid nodules and differentiated thyroid cancer: the american thyroid association guidelines task force on thyroid nodules and differentiated thyroid cancer. Thyroid. 2016;26(1):1–133. doi:10.1089/thy.2015.0020
8. Tirrò E, Martorana F, Romano C, et al. Molecular alterations in thyroid cancer: from bench to clinical practice. Genes. 2019;10(9):709. doi:10.3390/genes10090709
9. Lee SE, Hwang TS, Choi YL, et al. Molecular profiling of papillary thyroid carcinoma in korea with a high prevalence of BRAF(V600E) mutation. Thyroid. 2017;27(6):802–810. doi:10.1089/thy.2016.0547
10. Paulson VA, Shivdasani P, Angell TE, et al. Noninvasive follicular thyroid neoplasm with papillary-like nuclear features accounts for more than half of “carcinomas” harboring RAS mutations. Thyroid. 2017;27(4):506–511. doi:10.1089/thy.2016.0583
11. Labourier E, Shifrin A, Busseniers AE, et al. Molecular testing for miRNA, mRNA, and DNA on fine-needle aspiration improves the preoperative diagnosis of thyroid nodules with indeterminate cytology. J Clin Endocrinol Metab. 2015;100(7):2743–2750. doi:10.1210/jc.2015-1158
12. Schildgen V, Warm M, Brockmann M, et al. Oncotype DX breast cancer recurrence score resists inter-assay reproducibility with RT2-profiler multiplex RT-PCR. Sci Rep. 2019;9(1):20266. doi:10.1038/s41598-019-56910-0
13. Ma GT, Lee SK, Park KK, et al. Artemisinin-daumone hybrid inhibits cancer cell-mediated osteolysis by targeting cancer cells and osteoclasts. Cell Physiol Biochem. 2018;49(4):1460–1475. doi:10.1159/000493449
14. Roberts J, Mehta R, Curran I, Raju J. Dietary acrylamide exposure in F344 rats and colon tumor-bearing nude nu/nu mice: dataset of gene expression of cancer pathway targets and methylation status of tumor suppressor genes in colon mucosae and tumors. Data Brief. 2019;27:104763. doi:10.1016/j.dib.2019.104763
15. Binhua T. Recent advances of deep learning in bioinformatics and computational biology. Front Genet. 2019;26(10):214.
16. Tallini G, Biase DD, Repaci A, et al. What’s new in thyroid tumor classification, the 2017 World Health Organization classification of tumours of endocrine organs. Thyroid FNA Cytol. 2019.
17. Amin MB, American Joint Committee on Cancer and American Cancer Society. AJCC Cancer Staging Manual.
18. Jeffus S. Histology for pathologists, 4th edition. Am J Surg Pathol. 2014;38(4):582. doi:10.1097/PAS.0000000000000175
19. Goldman M, Craft B, Brooks A, et al. The UCSC xena platform for public and private cancer genomics data visualization and interpretation. bioRxiv. 2019;326470.
20. Smyth GK. Limma: linear models for microarray data. In: Bioinformatics and Computational Biology Solutions Using R and Bioconductor. New York: Springer New York; 2005:397–420.
21. Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–29. doi:10.1038/75556
22. Kanehisa M, Goto S, Kyoto KEGG. Encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi:10.1093/nar/28.1.27
23. Yu G, Wang L-G, Han Y, et al. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287. doi:10.1089/omi.2011.0118
24. Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2014;1003.
25. Agrawal N, Akbani R, Aksoy B, Cancer Genome Atlas Research Network, corp-author. Integrated genomic characterization of papillary thyroid carcinoma. Cell. 2014;159(3):676–690. doi:10.1016/j.cell.2014.09.050
26. Handkiewicz-Junak D, Swierniak M, Rusinek D, et al. Gene signature of the post-chernobyl papillary thyroid cancer. Eur J Nucl Med Mol Imaging. 2016;43(7):1267–1277. doi:10.1007/s00259-015-3303-3
27. Ye L, Zhou X, Huang F, et al. The genetic landscape of benign thyroid nodules revealed by whole exome and transcriptome sequencing. Nat Commun. 2017;8(1):15533. doi:10.1038/ncomms15533
28. Ringel MD, Giordano TJ. Molecular pathogenesis of thyroid neoplasia. Surg Thyroid Parathyroid Glands. 2021;181–185.
29. Carr F. Thyroid cancer. Cancer. 2019.
30. Manzella L, Massimino M, Stella S, et al. Activation of the IGF axis in thyroid cancer: implications for tumorigenesis and treatment. Int J Mol Sci. 2019;20(13):3258. doi:10.3390/ijms20133258
31. Massimino M, TIRRÒ ELENA, Stella S, et al. Effect of combined epigenetic treatments and ectopic nis expression on undifferentiated thyroid cancer cells. Anticancer Res. 2018;38(12):6653–6662. doi:10.21873/anticanres.13032
32. Ø F, Bruland O, Akslen LA, et al. Gene expression in poorly differentiated papillary thyroid carcinomas. Thyroid. 2006;16(2):161–175. doi:10.1089/thy.2006.16.161
33. Qiu J, Zhang W, Xia Q, et al. RNA sequencing identifies crucial genes in papillary thyroid carcinoma (PTC) progression. Exp Mol Pathol. 2016;100(1):151–159. doi:10.1016/j.yexmp.2015.12.011
34. Qu T, Li YP, Li XH, Chen Y. Identification of potential biomarkers and drugs for papillary thyroid cancer based on gene expression profile analysis. Mol Med Rep. 2016;14(6):5041–5048. doi:10.3892/mmr.2016.5855
35. Zhu X, Yao J, Tian W. Microarray technology to investigate genes associated with papillary thyroid carcinoma. Mol Med Rep. 2015;11(5):3729–3733. doi:10.3892/mmr.2015.3180
36. Zeng-Xin A, Chen Y-C, Jun-Min L, et al. Identification of potential functional genes in papillary thyroid cancer by co-expression network analysis. Oncol Lett. 2018;16(4):4871–4878. doi:10.3892/ol.2018.9306
37. Liu Y, Gao S, Jin Y, et al. Bioinformatics analysis to screen key genes in papillary thyroid carcinoma. Oncol Lett. 2020;19(1):195–204. doi:10.3892/ol.2019.11100
38. Stewart MD, Wong J. Nuclear receptor repression: regulatory mechanisms and physiological implications. Prog Mol Biol Transl Sci. 2009;87:235–259.
39. Jihoon K, Kanghee H, Jin KH, et al. The clinical significance of PINX1 expression in papillary thyroid carcinoma. Hum Pathol. 2018;81:176–183. doi:10.1016/j.humpath.2018.07.004
40. Leone V, D’Angelo D, Rubio I, et al. MiR-1 is a tumor suppressor in thyroid carcinogenesis targeting CCND2, CXCR4, and SDF-1alpha. J Clin Endocrinol Metab. 2011;96(9):E1388–E1398. doi:10.1210/jc.2011-0345
41. Huang JK, Ma L, Song WH, et al. LncRNA-MALAT1 promotes angiogenesis of thyroid cancer by modulating tumor-associated macrophage FGF2 protein secretion. J Cell Biochem. 2017;118(12):4821–4830. doi:10.1002/jcb.26153
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.