")
Back to Journals » Journal of Experimental Pharmacology » Volume 12
Experimental Pharmacological Agents for the Treatment of Primary Biliary Cholangitis
Authors Floreani A
Received 12 September 2020
Accepted for publication 12 November 2020
Published 17 December 2020 Volume 2020:12 Pages 643—652
DOI https://doi.org/10.2147/JEP.S267375
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Bal Lokeshwar
Annarosa Floreani1,2
1University of Padova, Padova, Italy; 2Scientific Institute for Research, Hospitalization and Healthcare, Negrar, Verona, Italy
Correspondence: Annarosa Floreani
University of Padova, Padova, Italy
Tel +10393899418841
Email [email protected]
Abstract: The standard therapy for primary biliary cholangitis (PBC) is ursodeoxycholic acid (UDCA) which has shown to improve hepatic biochemistry, delay histological progression and improve transplant-free survival. Approximately 30– 40% of patients do not respond or are intolerant to UDCA. Obeticholic acid, a farnesoid X receptor (FXR) agonist is the only agent approved by the Food and Drug Administration for patients who do not respond to UDCA. Recently, combination therapy with UDCA and bezafibrate has been shown to improve biochemistry and both GLOBE and UK-PBC score in patients with an inadequate response to UDCA. More recently, new pharmacological agents have been included in Phase 2 and Phase 3 trials: PPAR agonists, non-bile acid FXR agonists, anti-NOX agents, immunomodulators and mesenchymal stem cells transplantation. This review gives an overview on the current experimental pharmacological agents employed in the treatment of PBC.
Keywords: PBC, treatment, ursodeoxycholic acid, obeticholic acid, fibrates, FXR agonists, PPAR agonists
Introduction
Primary biliary cholangitis (PBC) is a chronic cholestatic liver disease which can progresses to cirrhosis, liver failure and death.1 It involves predominantly more females than males with a F/M ratio of 9:1 in cohort series reporting epidemiology, natural history and clinical characteristics of the disease.1 The disease appears to be most common in Northern Europe and North America, but is relatively common in Southern Europe, Asia and Australia. The prevalence is variable between 2 to 58 patients per million people and the estimated incidence varies between 0.9 to 5.8 per 100.000 per year.2,3 Over the last 30 years PBC has been changed to a symptomatic disease characterized by symptoms of portal hypertension to a mild disease with a long natural history.4
The standard treatment for PBC is represented by ursodeoxycholic acid (UDCA) with a recommended dosage of 13–15 mg/Kg per day.5 Biochemical response to UDCA has been defined, based on observational studies of UDCA-treated patients. These studies reported that achievement of biochemical response (according to their specific criteria associated with an improved liver transplant (LT)-free survival as compared to patients with inadequate response.6–11 In a multicentre International Study of PBC patients treated with UDCA, bilirubin levels below the current upper limits of normal (ULN) were shown to be predictive of survival and 0.6 x ULN was established as the threshold from which point on the risk for liver transplant or death increases.12 It is also important to highlight that UDCA therapy improves LT-free survival in all patients with PBC both in those with early and advanced disease, as well as in patients not meeting accepted criteria for response to UDCA.13 These important findings suggest that even modest improvement in cholestatic biochemical parameters in PBC can translate into long-term benefit. Indeed, there is no definition a priori of the relationship between non-response to UDCA and survival. Two independent research groups: the Global PBC Study Group and the United Kingdom (UK)-PBC Consortium developed and externally validated the risk of progression in PBC in continuous prognostic models. The Globe score (www.globalpbc.com) was introduced in 2015, and was constructed using a derivation cohort of 2.488 patients, and a validation cohort of 1.634 UDCA-treated patients. The UK-PBC risk score (www.uk.pbc.com) was developed in the same year, in a nationwide cohort of 1.916 patients and validated in a cohort of 1.249 UDCA-treated patients. These models have been also validated in patients not taking UDCA, thus reflecting disease activity and stage expressed by biochemistry. There are two important differences in the two models that can be explained by the different endpoints used in the two scores. Indeed, Globe-PBC score takes into account LT and all-cause mortality, while the UK-PBC score considers LT and liver-related death. Interestingly, calculating the two models in the PBC POISE (Obeticholic Acid International Study of Efficacy) study population, both models demonstrated the potential use of these scores in individualizing risk prediction in PBC both in clinical practice and therapeutic trials.14 UDCA non-responders (which are estimated around 30–40% of treated patients), have lesser prognosis due to higher risk of disease progression, greater mortality risk, and plausibility to require liver transplantation.15
Thus, a second-line therapy for non-responders to UDCA has been proposed.
Obeticholic acid (OCA) is the only registered agent for second-line treatment in non responders to UDCA after one year of treatment or intolerant to UDCA. The decision to implement OCA includes at least one of the following biochemical values: i) ALP ≥1.67 x ULN, in Italy ALP threshold is 1.5 ULN; ii) total bilirubin >ULN but <2 x ULN. OCA is a synthetic derivative of chenodeoxycholic acid, agonist of farnesoid X receptor (FXR), which is a key nuclear receptor mainly expressed in the liver and gut which regulates bile acid synthesis preventing their toxic accumulation.16 Moreover, it acts promoting several functions including expression of bile salt export pump (BSEP) on the canalicular membrane of hepatocytes and controlling gene expression of organic solute transporter (OST) α-OSTβ (heteromeric transporters expressed mainly in the distal portion of the gut and bile ducts). Finally, FXR induces the expression of small heterodimer proteins, also known as nuclear receptor superfamily 0 which play a role in the transcriptional control of bile acid transport and metabolism.16 Consequently, OCA has several mechanisms of action: i) regulation of bile acid transport; ii) anti-inflammatory properties; and iii) anti-fibrotic mechanisms.17 Due to the induction of bile acid signalling pathway via fibroblast growth factor-19 (FGF-19), OCA has a more potent hepatoprotective effects than UDCA. OCA has been studied as monotherapy in an international randomized, double blind, placebo-controlled Phase 2 study in patients with PBC with the aim to assess the benefit of OCA in the absence of UDCA.18 Patients were randomized and dosed with placebo (n=23), OCA 10 mg (n=20) and OCA 50 mg (n=16) for 3 months. The primary endpoint was the percent change in ALP from the baseline to the end of double-blind Phase study. After 3 months patients were monitored through a 6-year open-label extension. ALP was significantly reduced in both OCA groups compared to placebo; moreover, OCA improved many biochemical parameters including GGT, alanine aminotransferase, conjugated bilirubin and immunoglobulins. Pruritus was the most adverse effect of OCA and was present in 15% of patients taking OCA 10 mg and in 38% of those taking 50 mg. OCA obtained FDA approval in 2016, on the basis of an international multicentre Phase III RCT of 216 patients.19 This trial demonstrated that ~59% of patients who did not respond to UDCA alone responded to OCA plus UDCA combination therapy and reached the clinical end-point (an alkaline phosphatase level of less than 1.67 times the upper limit of normal range, with a reduction of at least15% from baseline) during 12 months treatment. Thereafter, all patients were switched to receive OCA in an extension phase; 193 patients were treated during the open-label extension.20 In this 3-year interim analysis OCA was well tolerated and the performance of OCA was stable during this period. Moreover, a post hoc analysis of POISE trial demonstrated that OCA was associated with significant reductions in total and direct bilirubin, particularly with high baseline direct bilirubin.21 Thus, these results suggest substantial benefits of OCA in at-risk patients. Pruritus and fatigue were the most common adverse effects recorded in the POISE trial, being present in 77% and 33% respectively.21 Pruritus was scored as mild-to-moderate by the visual analogue scale and only 16 patients (8%) withdrew from the open-label extension phase because of pruritus. However, the majority of patients who complained of severe pruritus received also pruritus treatment respectively based on clinical judgment. A subgroup analysis study of liver biopsy collected from 17 patients at time of enrolment in the POISE trial until 3 years of treatment demonstrated that long-term OCA treatment was associated with improvement or stabilization of disease features, including ductular injury, fibrosis and collagen morphometry features.22 Despite the small sample size, this study indicates that most patients with an inadequate response to UDCA showed an amelioration or stabilization of multiple histological features of PBC after 3 years of OCA therapy.
In another sub-analysis study, the effects of OCA on established non-invasive measures of liver fibrosis and outcome, namely the AST to platelet ratio (APRI) and transient elastography (TE) were investigated.23 APRI was significantly reduced from baseline to double-blind month 12 in both OCA-treated groups compared to placebo-treated groups. Similarly, APRI score was significantly reduced during the open-label extension phase. During double-blind and open-label phases, while not significant, the OCA 10 mg group had mean reductions in liver stiffness, while both OCA 5–10 mg and placebo groups had mean increases in liver stiffness.23
The first description of real-world effectiveness of OCA included 64 Canadian patients with PBC with incomplete response or intolerant to UDCA which were treated with OCA for a median 13.1 months.24 Of the whole cohort, 44 patients met POISE-inclusion criteria, 39% of whom (N=17) had 12-month biochemical assessment. In this group 18% of patients (3/17) met the 12-month POISE primary endpoint. However, considering 19-month observation period, 9/21 (43%) achieved this target. Overall, significant reductions in ALP, GGT, transaminases and IgM were reported in the whole cohort. Worsening or new onset of pruritus was documented in 26 patients (41%) and was the cause for OCA discontinuation in 5 of them. Other reasons for OCA discontinuation were skin rash in 2 patients, hepatotoxicity in 2 and incomplete response after 1 year of therapy in the remaining 2 patients.
Fibric acid derivatives are anti-lipidemic agents proposed as second-line therapy for PBC due to their anti-cholestatic, anti-inflammatory and anti-fibrotic effects. Fibric acid derivatives (fibrates) are agonists of the peroxisome proliferator-activated receptors (PPAR), which belong to the superfamily of nuclear receptors. PPAR is known to exist in 3 isoforms: α, β/δ, and γ. These isoforms are encoded by distinct genes and have different patterns of distribution. Fenofibrate is the PPAR-agonist which stimulates the transcription and protein expression of multidrug resistance protein 3 (MDR3) and increases the biliary excretion of phosphatidylcholine.25 Furthermore, it increases biliary secretion of phosphatidylcholine improving biomarkers of cholestasis. Bezafibrate has been reported to act as a dual PPARα and -β and pregnane X receptor (PXR) agonist.26 The BEZURSO trial (Phase 3 Study of Bezafibrate in Combination With UDCA) is the first ever placebo-controlled trial of a fibrate in PBC. In this 24-month study, second-line use of bezafibrate in addition to continued UDCA resulted in a rate of complete biochemical response significantly higher than that achieved with placebo and UDCA.27 This effect was associated with a parallel improvement in symptoms and surrogate markers of fibrosis. Side effects of fibric acid derivatives include increased creatinine levels, heartburn, and transient elevation of transaminases. Gallstone formation (possibly due to repression of bile acid synthesis) and paradoxical hypercholesterolemia have been reported in patients with PBC treated with clofibrate,28 but were not confirmed during treatment with fenofibrate or bezafibrate.
A comparative effect of second line therapy with OCA or fibrates has been evaluated in a multicentre retrospective study including 277 PBC patients from 30 centres in Spain.29 Sixty-five patients received OCA (5 mg) and 201 fibrates (85% bezafibrate 400 mg, 16% fenofibrate 200 mg) and 11 OCA and fibrates. Fibrates were associated with higher decrease in ALP, while OCA with higher transaminases improvement. Another study presenting the comparative efficacy data of OCA vs bezafibrate n 59 patients treated with OCA or bezafibrate was presented at an AASLD meeting in Boston 2019.30 i There were no significant differences in the incidence of ascites, varices, variceal bleeding and number of patients listed for liver transplantation between the two groups. Overall percentage ALP reductions were greater following bezafibrate vs OCA treatment (P<0.001). Ten percent of bezafibrate-treated patients developed bilirubin elevation greater than OCA-treated patients (10% vs 3%, P=n.s.). Both these studies highlight real world data of OCA and fibrates and offer important points for future trial designs in PBC.
The additive effects of fibric acid derivatives combined with OCA were studied in a multicentre retrospective cohort of patients with PBC.31 Fifty patients were given a combination of OCA (5–10 mg/day), fibrates (bezafibrate 400 mg/day or fenofibrate 200 mg/day) and UDCA (13–15 mg/day). Triple therapy was associated with a significant fall in ALP level compared to dual therapy and with an odds ratio for ALP normalization of 5.5 (95% CI: 1.8–17.1, P=0.003).
The effect of fibric acid derivatives on pruritus deserves a separate discussion. Their benefit against this symptom has been widely reported. Interestingly, a double blind, randomized, placebo-controlled trial with the aim to assess the effects of bezafibrate on pruritus (FITCH) has been conducted in 70 patients with PBC, PSC and secondary sclerosing cholangitis with moderate to severe pruritus.32 The primary endpoint was ≥50% reduction of pruritus (scored on visual analogue scale). Bezafibrate (400 mg/day) led 45%, placebo 11% to the primary endpoint (P=0.003). Fibric acid derivatives relieve cholestasis-associated pruritus by autotaxin-independent mechanism (Kremer). However, this important action in ameliorating pruritus ensures fibric acid derivates to be employed as second line therapy in PBC in case of moderate-severe pruritus. As they act also as reducing cholesterol, a consideration should be made for the subgroup of patients with PBC and the atherosclerotic profile (namely hypercholesterolemia with low levels of high-density lipoprotein [HDL]) in whom fibric acid derivates could be employed to prevent the risk for cardiovascular events.
Actually, novel experimental agents are known for their interesting mechanisms of action, ie, bile acid modulation, antifibrotic, anti-inflammatory effect, and immunomodulation. Table 1 summarizes the RCTs trials still ongoing.
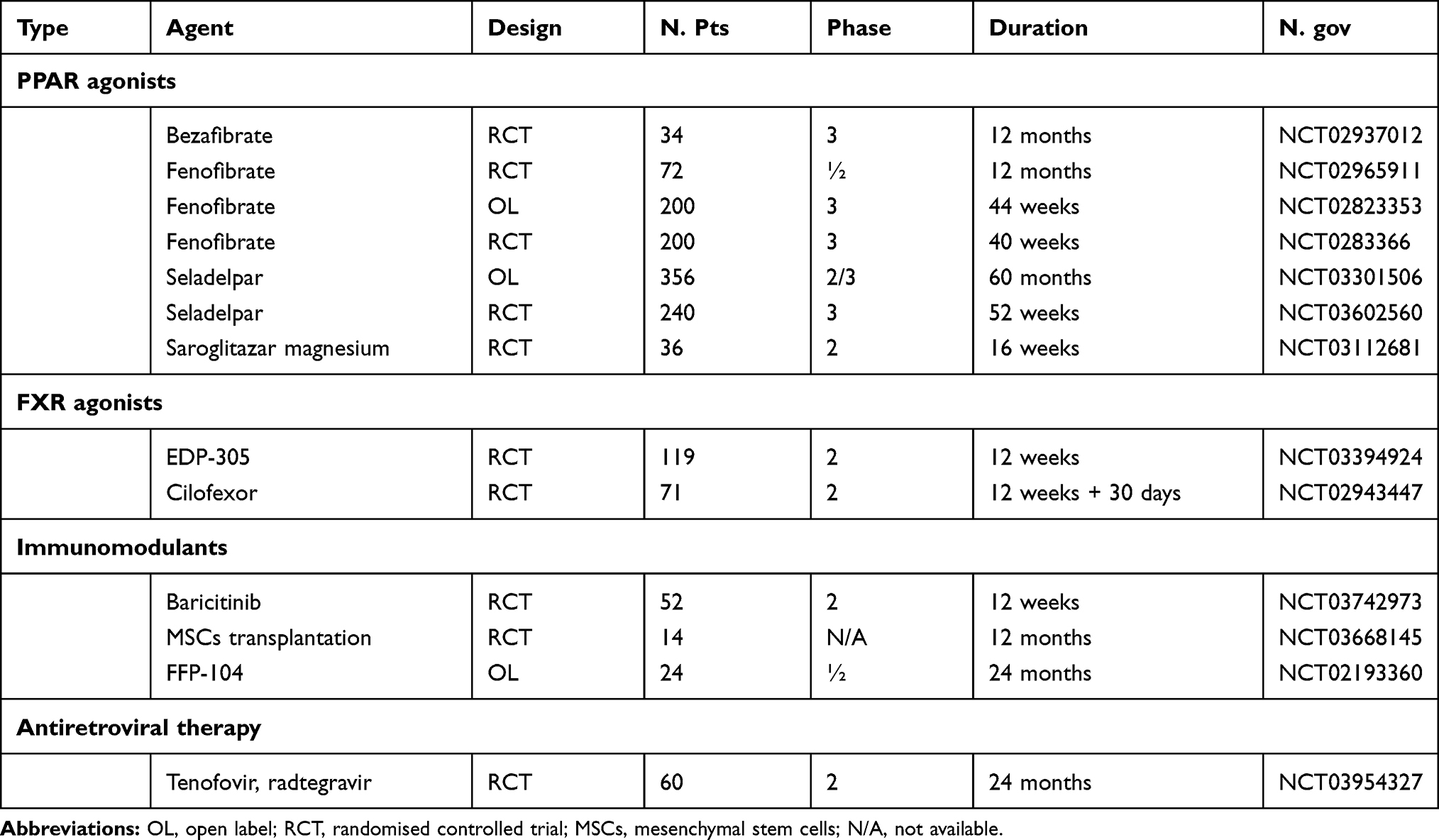 |
Table 1 Ongoing Controlled Trials with Experimental Agents in PBC |
Non-Bile Acid FXR Agonists
Tropifexor is a highly potent non-bile acid FXR agonist that has shown efficacy in animal models of cholestasis and non-alcoholic steatohepatitis (NASH), mainly in reducing fibrosis.33 In a Phase 2 study PBC patients with an inadequate response to UDCA were randomized in cohorts (to receive 30 µg, 60 µg or 90 µg of tropifexor once daily or matching placebo for 4 weeks.34 A planned interim analysis was conducted in cohort 3 (90g). There was a brisk decrease in GGT, ALP, ALT and AST with 72% reduction in GGT at 90 µg at day 28. Reduction of HDL of 33% and 26% occurred at doses 60 mg and 90 mg respectively and returned to normal by the end of the study. There was no increase in total or LDL cholesterol. These results showed a good tolerability of tropifexor which is a potential future agent for PBC.34
Cilofexor, a non-steroidal FXR agonist, has been tested in a trial involving 71 patients with PBC non-responders to UDCA (NCT02943447). They were randomized to receive either cilofexor 30 mg, cilofexor 100 mg or placebo once a day for 12 weeks. Patients who received 100 mg dose achieved a significant median reduction in ALT 8–13.8%, P=0.05), GGT 8–47.7%, P<0.001), CRP 8–33.6%, P=0.03) and primary bile acids (−30.5%, P=0.0.008). Focusing ALP performance, a reduction greater than 25% was obtained in 17% and 18% of patients in the 100mg and 30 mg cilofexor groups respectively vs 0% of the placebo group. The major side-effect was pruritus which was more common in patients treated with high dose cilofexor than in those who received 30 mg. Promising results from a Phase 3 trial in patients with PSC have been reported.35
EDP-305 is a potent FXR agonist which contains steroid and non-steroid components without the carboxylic acid group normally present in other classes of FXR agonists and natural bile acids. A 12-week randomized double blind, placebo-controlled study (NCT03394924, INTREPID Study) was conducted to evaluate the safety, tolerability and efficacy in subjects with PBC and inadequate response to UDCA. The primary end-point was to evaluate the proportion of subjects with at least 20% reduction in ALP or normalization of ALP at week 12. After randomization, 31 patients were included in arm 1 (1 mg EDP-305), 28 in arm 2 (2.5 mg) and 9 in the placebo arm. In the intent-to treat (ITT) analysis EDP-305 1 mg and 2.5 mg resulted in 45% and 46% reduction respectively, compared to 11% in placebo group. Five patients in the 2.5 mg arm experienced severe pruritus. The most common side effects included besides pruritus, gastrointestinal-related symptoms (nausea, vomiting, diarrhoea), headache and dizziness. Pruritus was present in 51% of the subjects in the 2.5 mg arm compared to less than 10% in the 1 mg arm.
PPAR Agonists
Elafibranor (ELA), a dual PPAR α/δ agonist has been recently evaluated in a Phase 2, double-blind, placebo-controlled study in PBC.36 Forty-five patients with PBC and inadequate response to UDCA were enrolled to receive ELA 80 mg quarterly a day, or ELA 120 mg quarterly a day, or placebo. The primary end-point was defined as the percentage of ALP change. ELA demonstrated a significant decrease in mean ALP at week 12 (−48% in the arm with 80 mg and −41% in the arm with 120 mg (P<0.001). The composite end-point (ALP <1.67 x ULN plus ALP decrease >15% plus total bilirubin <ULN) was reached in 67% and 79% of patients respectively. Moreover, ELA-treated patients showed improvement in lipid markers, reduction in inflammatory markers (IgM, CRP, haptoglobin, fibrinogen) and decrease in C4 (an intermediate of bile acid synthesis). Both doses of ELA were well tolerated, and pruritus improved in 24% of patients treated with 80% and in 49% in those treated with 120 mg. These beneficial effects make ELA a promising novel treatment candidate for PBC.
Seladelpar, a selective PPAR-δ agonist, was tested in a 12-week double-blind, randomized, placebo-controlled Phase 2 trial.37 Seventy patients with inadequate response or intolerance to UDCA were randomly assigned to placebo, seladelpar 50 mg/day, or seladelpar 200 mg/day. The primary end-point was the percentage of change in ALP from baseline over 12 weeks. During recruitment, 3 patients treated with seladelpar developed fully reversible asymptomatic grade 3 alanine transferase increase, thus the study was terminated early.
Nevertheless, the results of seladelpar in terms of efficacy, safety and tolerability have been presented at EASL Annual meeting 2020 in a 1-year, Phase 2, open label uncontrolled dose-finding study.38 One hundred and nineteen patients received oral daily doses of 2.5 or 10 mg seladelpar; after 12 weeks doses could be increased up to 10 mg based on biochemical response. Over 1 year seladelpar appeared safe, well tolerated and did not induce pruritus. Four patients discontinued seladelpar due to adverse events (two of them were considered related to the drug: heartburn grade 1 and transaminase elevation grade 2). The composite end-point (ALP <1.67 x ULN, −15% reduction in ALP, total bilirubin <ULN) was met in 53% in 5/10 mg and 69% in 10 mg; 14% of patients in 5/10 mg and 33% in 10 mg normalised ALP. Actually, the results of Phase 3 trial with seladelpar in PBC are ongoing (ENHANCE study). Interim analysis of ENHANCE study have been discussed at AASLD Liver Meeting November 2020. Two hundred and sixty-five patients with PBC and with inadequate response (or intolerant) to UDCA were randomized to placebo, seladelpar 5 mg or seladelpar 10 mg once a day. The primary endpoint was a reduction in ALP level <1.67 x the ULN with at least a 15% decrease from baseline and a normal level of total bilirubin after 52 weeks. Due to the early termination of the study the primary outcome measure was amended prior to the data base lock to a 3-month interpoint which was reached by 167 patients. Seladelpar was well tolerated and achieved the primary composite outcome with highly statistical significance in 78.2% of patients in the 10 mg group (n=55) and in 57.1% in the 5 mg group (n=56) compared to 12.5% on placebo (n=56), P<0.0001. Seladelpar also demonstrated a strong dose-dependent reduction in pruritus assessed by numerical rating scale. These results suggest that seladelpar may be a candidate for second line therapy in PBC.
Targeting FGF19
FGF19 is a hormone that acts directly on the liver to suppress expression of CYP7A1, the key enzyme that catalyses the first and rate-limiting step in the pathway of bile acid synthesis.39 NGM282 (Aldafermin), an engineered analogue of FGF19 has been evaluated in a 28 day-multicentre, randomized, double-blind Phase 2 trial in PBC.40 Forty-five patients with PBC who had an inadequate response to UDCA were randomly assigned to subcutaneous daily doses of either NGM282 at 0.3 mg (n=14), 3 mg (n=16) or placebo (n=15). The primary endpoint was a change in ALP from baseline after 28 days of treatment. At the end of the study ALP was significantly reduced with NGM282 treatment at both 0.3 mg and 3 mg vs placebo; 50% of patients receiving NMGM282 0.3 and 46% of those receiving 3 mg achieved 15% of greater reduction in ALP levels from baseline compared with 7% receiving placebo. Most adverse events were grade 1 to grade 2 in severity, with gastrointestinal disorders more frequent in the treatment group vs placebo. Thus, the tolerability profile was acceptable, but future studies are warranted to assess the durability of biochemical response and to verify whether the efficacy is related to a real improvement of robust clinical outcomes, such as decrease in decompensation or death.
Targeting the NADPH Oxidase (NOX) Enzymes
NOX enzymes play a role in multiple processes such as fibrosis, inflammation, pain processing, cancer development and neurodegeneration. Anti-NOX agents have shown to prevent the development of fibrosis in an experimental model of sclerosing cholangitis.41 Moreover, a Phase 2 trial with GKT831 (setanaxib) has been conducted in 111 patients with PBC stratified into 3 arms: placebo (N.37), GKT831 400 mg once a day (N.38) and GKT831 400 mg twice a day (N.36).42 The primary end point was change in GGT at week 24 and secondary end points were changes in ALP, liver stiffness evaluated with fibroscan, and quality of life (QoL). GKT831 achieved rapid, dose and time dependent reductions in markers of cholestasis; in particular, a greater reduction in GGT (29% for GKT831 400 mg twice a day vs 8% placebo, P<0.01) in patients with higher baseline GGT, suggesting that GKT831 may also benefit patients with more advance disease. The treatment was well tolerated, and no treatment interruption or discontinuations due to pruritus or fatigue were recorded. Following these positive results, the PBC Phase 3 trial is being planned.
Immunomodulators
Rituximab was evaluated in two open-label studies in PBC patients with incomplete response to UDCA, but both studies showed a limited efficacy, although some impressive reduction in ALP was noticed.43,44 Rituximab was also found ineffective for treatment of fatigue in a Phase 2 randomized controlled trial in PBC.45
Anti-CXCL10 is a human monoclonal antibody directed against chemokine (C-X-C motif) ligand 10 implicated in the recruitment of inflammatory T cells into the liver. In PBC patients, serum levels of CXCL10 are greater as is the frequency of peripheral blood cells expressing CXCR3 compared to healthy controls.46 Within the liver, CXCR3+ cells are detectable in PBC, especially on CD4+cells in the portal areas and the damaged bile ducts. Interestingly, in situ hybridization of PBC liver samples demonstrated CXCL10 message in hepatocytes surrounded by infiltrating monocytes but not in bile ducts.47 The safety and efficacy of an anti-CXCL10 (NI-0801) was assessed in an open-label Phase 2a study including 29 patients with PBC with an inadequate response to UDCA.48 Patients received i.v. administration of NI-0801 (10 mg/Kg) every 2 weeks and were followed up for 3 months after the last infusion. No serious side-effects were reported, but the most common adverse events included headaches (52%), pruritus (34%), fatigue (24%) and diarrhoea (21%). The study was terminated because no clinically significant improvements in liver tests were observed.48
Ustekinumab, a monoclonal antibody that specifically binds IL-12 and IL-23 has been investigated in a multicentre, open label trial including an inadequate response to UDCA, but the results were disappointing, showing a moderate decrease in ALP.49
Abatacept (CTL4-Ig) was ineffective in an open-label trial in achieving biochemical responses associated to an improvement of clinical outcomes.50
Baricitinib is a Janus kinase (JAK) inhibitor (and more specifically a selective JAK1 and JAK2 inhibitor. Janus kinase are a family of four protein tyrosin kinase (JAK1, KAK2, JAK3 and tyrosin kinase 2) that play a role in cytokine signal transduction. Baricitinib is approved in US and Europe for treatment of rheumatoid arthritis. A clinical trial (NCT03742973) with Baricitinib (LY3009104) is currently ongoing in PBC patients who do not respond or unable to take UDCA.
FFP-104, an anti-CD40 monoclonal antibody is a new agent which is expected to interfere with the normal immune reaction underlying the disease. In particular, CD40 plays an important role in the efficient activation of T-cells by antigen presenting cells, fibroblasts and other non-lymphoid cells. A Phase 2 trial including PBC patients is currently ongoing (NCT02193360).
Mesenchymal stem cells (MSCs) transplantation has been proposed for end-stage PBC as alternative to liver transplantation.51 In fact, MSCs can modulate and repair injured tissue by modulating immune response through different mechanisms including cell-to-cell interaction and secretion of paracrine factors.52 The first clinical trial on PBC was conducted in China (NCT01662973). Seven patients with an incomplete response to UDCA were included with the aim to evaluate the safety and efficacy of umbilical cord-derived MSCs.53 All patients well tolerated the procedure, with no side-effects after 48 weeks of follow-up. The treatment led to a significant decrease in both serum ALP and GGT, whereas the other biochemical parameters did not change significantly. A second study was conducted thereafter by the same group utilizing the MSCs from allogenic donors of patient’s family members.54 The efficacy of this procedure was evaluated at 1, 3, 6 and 12 months of follow-up. Biochemical parameters, such as transaminases, GGT and IgM significantly improved and histological fibrosis remained stable. However, further studies are required to recommend the use of MSCs therapy in PBC. Actually, a new study is ongoing (NCT03668145).
Antiretroviral Therapy
A Canadian research group hypothesized a viral involvement triggering PBC, after finding serological reactivity to retrovirus.55 Consequently, a multicentre, double-blind, cross-over, placebo-controlled trial with antiretroviral therapy in PBC was performed (NCT01614405). The enrolment was limited to 13 patients because most patients were intolerant to lopinavir-ritonavir (LPRr). Patients were randomized to daily combination tenofovir-emtricitabine (TDF/FTC) and LPRr.56 A significant reduction in ALP by 25% (P<0.05) was observed, but a great limitation was represented by the frequency of side-effects. Actually, a new trial warranting the search for better tolerated combinations is ongoing (NCT03954327).
Targeting Pruritus
The guideline-approved anti-pruritic strategies (cholestyramine, rifampicin, naltrexone, sertraline) are often ineffective in amelioration pruritus in PBC. Actually, new agents targeting pruritus in cholestasis have been developed.
Ileal Bile Acid Transporter Inhibitors (IBATs)
The main goal of IBATs is to improve pruritus in cholestasis by decreasing retained circulating bile acids. Several compounds altering ileal reabsorption of bile acids have been proposed.
A Phase 2 study assessed the efficacy and safety of maralixabat, a selective, ileal apical, sodium-dependent, bile acid inhibitor in PBC patients with pruritus (CLARITY study).57 Patients were randomized 2:1 to maralixabat (10 or 20 mg/day) or placebo for 13 weeks in combination with UDCA (when tolerated). The primary endpoint was change in Adult Itch Reported Outcome average sum score from baseline to the end of the study. Adverse events (gastrointestinal disorders being the most frequent ones) were reported in 78.6% of patients treated with maralixabat and in 50% in those treated with placebo. Despite improvement from baseline risk scores, no significant difference was observed in the magnitude of effect between the drug and placebo.
Linerixabat (GSK2330672) was tested in a Phase 2 cross-over, randomized placebo trial in 21 patients during two consecutive 14-day treatment period in a crossover sequence.58 The primary endpoints were safety and tolerability. The secondary endpoints were changes in pruritus scores using the 0 to 10 numerical rating scale, PBC-40 itch domain score and 5-D itch scale (NCT01899703) and changes in bile acids. Linerixabat was generally well tolerated, but diarrhoea was the most common adverse effect. The percentage changes of itch scores were −57% in the numerical rating scale, −31% in the PBC-40 itch domain and −35% in the 5-D itch scale. Compared to placebo the differences were statistically significant. A larger Phase 2 study including 147 patients is still ongoing to confirm the beneficial effect on itching and further assess the drug tolerability (NCT02966834).
Finally, A4250 (odevixibat) was tested in terms of tolerability and efficacy in an open label Phase 2 study in 9 patients with PBC and pruritus (NCT02360852).59 Patients were treated with either 0.75 mg (n=4) or 1.5 mg (n=5) of A4250 for 4 weeks. All 9 patients reported a remarkable improvement in pruritus assessed by visual analogue scale, 5-Ditch scale and the pruritus domain of the PBC-40 questionnaire. Unfortunately, tolerability was scarce, since 5 patients withdrew the study due to abdominal pain and diarrhoea.
Targeting Fatigue
Fatigue is a complex symptom characterized by feeling of exhaustion, lethargy, discomfort and limiting the quality of life. It is not clear if it is an extra-hepatic symptom of hepatic inflammation or a manifestation of an extra-hepatic disease. Moreover, the probability that liver transplantation improve fatigue in advanced PBC is roughly 50%.60 There is currently no licensed therapy for fatigue in PBC. Prescribed exercise therapy warrants further evaluation, particularly to assess whether the bioenergetic abnormalities seen in peripheral muscle can be modified following exercise. A pilot study demonstrated a greater improvement of muscle pH in PBC patients than healthy controls, and also an amelioration of fatigue, social, and emotional symptoms following an exercise program.61
An open study using modafinil, a central nervous system active agent effective for the treatment of daytime somnolence in the context of narcolepsy, was used in 21 patients with PBC suffering of daytime somnolence and fatigue.62 Modafinil was started at a dose of 100 mg/day and was titrated according to tolerability and response. Only 14 patients were able to tolerate the treatment for the full 2-month period. In those patients modafinil therapy was associated with improvement in excessive daytime somnolence and associated fatigue. Suggestion from this trial was to design a placebo-controlled study to confirm the effectiveness of modafinil in reducing fatigue in PBC patients.
Interestingly, in an experimental model of cholestatic liver disease induced by bile duct ligation, early OCA administration improved cognitive impairment (abnormal activity, short-term memory and spatial recognition.63
The first randomized controlled trial of treatment of fatigue in PBC was performed in Newcastle upon Tyne in UK (NCT2376335).45 In this Phase 2 trial 57 patients with PBC and moderate to severe fatigue were randomized to receive two doses of either rituximab (1000 mg) or placebo. The primary outcome measure was fatigue severity assessed using PBC-40 domain at 3 months. The rationale for choosing rituximab was a reported improvement of fatigue associated to a number of autoimmune conditions, including Sjogren’s syndrome that shows an association with PBC. Rituximab, however, failed to show an improvement of fatigue in patients with PBC.
Conclusions
New pharmacological agents have been included in Phase 2 and Phase 3 trials for PBC patients non responders to first line therapy with UDCA. Besides OCA which is the first drug to be registered in many countries, fibrates appear to be promising to ameliorate biochemistry and symptoms of the disease. Several types of agents: PPAR agonists, non-bile acid FXR agonists, anti-NOX agents, immunomodulators and mesenchymal stem cells transplantation are currently included in ongoing trial. The majority of them have shown beneficial effects on biochemical endpoints. No data are available on robust endpoints, such as transplant-free survival. The treatment landscape for PBC is probably a combination treatment with multiple agents targeting different mechanism of pathogenesis.
Disclosure
Annarosa Floreani has received consultancy fees during the last two years from Intercept. The author reports no other conflicts of interest in this work.
References
1. Carey EJ, Ali AH, Lindor KD. Primary biliary cirrhosis. Lancet. 2015;386:1565–1575. doi:10.1016/S0140-6736(15)00154-3
2. Marchall HU, Henriksson I, Lindenberg S, et al. Incidence, prevalence, and outcome of primary biliary cholangitis in a nationwide Swedish population-based cohort. Sci Rep. 2019;9:1–8.
3. Baldursdottir TR, Bergmann OM, Jonasson JG, et al. The epidemiology and natural history of primary biliary cirrhosis: a nationwide publication-based study. Eur J Gastroenterol Hepatol. 2012;24:824–830. doi:10.1097/MEG.0b013e328353753d
4. Murillo Perez CF, Goet JC, Lammers WJ, et al. Milder disease stage in patients with primary biliary cholangitis over a 44-year period: a changing natural history. Hepatology. 2018;67:1920–1930. doi:10.1002/hep.29717
5. European Association for the Study of the Liver. EASL clinical practice guidelines: the diagnosis and management of patients with primary biliary cholangitis. J Hepatol. 2017;67:145–172.
6. Pares A, Caballeria L, Rodes J. Excellent long-term survival in patients with primary biliary cirrhosis and biochemical response to ursodeoxycholic acid. Gastroenterology. 2006;130:715–720. doi:10.1053/j.gastro.2005.12.029
7. Corpechot C, Abenavoli L, Rabahi N, et al. Biochemical response to ursodeoxycholic acid and long-term prognosis in primary biliary cirrhosis. Hepatology. 2008;48:871–877. doi:10.1002/hep.22428
8. Kuiper EM, Hansen BE, de Vries RA, et al. Improved prognosis in patients with primary biliary cirrhosis that have a biochemical response to ursodeoxycholic acid. Gastroenterology. 2009;136:1281–1287. doi:10.1053/j.gastro.2009.01.003
9. Kumagi T, Guindi M, Fischer SE, et al. Baseline ductopenia and treatment response predict long-term histological progression in primary biliary cirrhosis. Am J Gastroenterol. 2010;105:2186–2194. doi:10.1038/ajg.2010.216
10. Corpechot C, Chazouilleres O, Poupon R. Early primary biliary cirrhosis: biochemical response to treatment and prediction of long-term outcome. J Hepatol. 2011;55:1361–1367. doi:10.1016/j.jhep.2011.02.031
11. Lammers WJ, van Buuren HR, Hirschfield GM, et al. Levels of alkaline phosphatase and bilirubin are surrogate end points of outcomes of patients with primary biliary cirrhosis: an international follow-up study. Gastroenterology. 2014;147:1338–1349. doi:10.1053/j.gastro.2014.08.029
12. Murillo Perez CF, Harms MH, Lindor KD, et al. Goals of treatment for improved survival in primary biliary cholangitis: treatment target should be bilirubin within the normal range and normalization of alkaline phosphatase. Am J Gastroenterol. 2020;115:1066–1074.
13. Harm MH, van Buuren HR, Corpechot C, et al. Ursodeoxycholic acid treatment and liver transplantation-free survival in patients with primary biliary cholangitis. J Hepatol. 2019;71:357–365. doi:10.1016/j.jhep.2019.04.001
14. Harm MH, Carbone M, Lammers WJ, et al. Clinical application of the Globe score and UK-PBC score in a real world trial cohort of patients with primary sclerosing cholangitis. Hepatol Commun. 2018;19:683–692.
15. Selmi C, Bowlus CL, Gershwin ME, et al. Primary biliary cirrhosis. Lancet. 2011;377:1600–1609. doi:10.1016/S0140-6736(10)61965-4
16. Gerussi A, Lucà M, Cristoferi L, et al. New therapeutic targets in autoimmune cholangiopathies. Frontiers Med. 2020. doi:10.3389/fmed.2020.00117
17. Modica S, Petruzzelli M, Bellafante E, et al. Selective activation of nuclear bile acid receptor FXR in the intestine protects mice against cholestasis. Gastroenterology. 2012;142:335–336. doi:10.1053/j.gastro.2011.10.028
18. Kowdley K, Luketic V, Chapman R, et al. A randomized trial of obeticholic acid monotherapy in patients with primary biliary cholangitis. Hepatology. 2018;67:1890–1902. doi:10.1002/hep.29569
19. Nevens F, Andreone P, Mazzella G, et al. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med. 2016;375:631–643. doi:10.1056/NEJMoa1509840
20. Trauner M, Nevens F, Shifmann ML, et al. Long-term efficacy and safety of obeticholic acid for patients with primary biliary cholangitis: a 3-year results of an international open-label extension study. Lancet Gastroenterol Hepatol. 2019;4:445–453.
21. Pares A, Shiffman M, Vargas V, et al. Reduction and stabilization of bilirubin with obeticholic acid treatment in patients with primary biliary cholangitis. Liver Int. 2020;40:1121–1129. doi:10.1111/liv.14429
22. Bowlus CL, Pockros PJ, Kremer AE, et al. Long-term obeticholic acid therapy improves histological endpoints in patients with primary biliary cholangitis. Clin Gastroenterol Hepatol. 2020;18:1170–1178. doi:10.1016/j.cgh.2019.09.050
23. Hirschfield GM, Floreani A, Trivedi PJ, et al. Long-term effect of obeticholic acid on transient elastography and AST to platelet ratio index in patients with PBC. Gut. 2017;66:A98–A99.
24. Roberts SB, Ismail M, Kanagalingam G, et al. Real-world effectiveness of obeticholic acid in patients with primary biliary cholangitis. Hepatol Commun. 2020;4:1332–1345. doi:10.1002/hep4.1518
25. Ghonem NS, Ananthanarayanan M, Soroka CJ, et al. Peroxisome proliferator-activated receptor alpha activates human multidrug resistance transporter 3/ATP-binding cassette protein subfamily B4 transcription and increases rat biliary phosphatidylcholine secretion. Hepatology. 2014;59:1030–1042. doi:10.1002/hep.26894
26. Honda A, Ikegami T, Nakamuta M, et al. Anticholestatic effects of bezafibrate in patients with primary biliary cirrhosis treated with ursodeoxycholic acid. Hepatology. 2013;57:1931–1941. doi:10.1002/hep.26018
27. Corpechot C, Chazouilleres O, Rousseau A, et al. A placebo-controlled trial of bezafibrate in primary biliary cholangitis. N Engl J Med. 2018;378:2171–2181. doi:10.1056/NEJMoa1714519
28. Summerfield JA, Elias E, Sherlock S. Effects of clofibrate in primary biliary cirrhosis: hypercholesterolemia and gallstones. Gastroenterology. 1975;69:998–1000. doi:10.1016/S0016-5085(19)32419-9
29. Reig A, Alvarez-Navascucs C, Vergara Gomez M, et al. Comparative effects of second-line therapy with obeticholic acid or fibrates in primary biliary cholangitis patients. J Hepatol. 2020;73:
30. Culver E, Hayden J, Thornburn D, et al. Obeticholic acid and bezafibrate in primary biliary cholangitis: a comparative evaluation of efficacy through real world clinical practice. Hepatology. 2019;suppl.1:770A.
31. Sorel P-A, Lam L, Carrat F, et al. Additive beneficial effects of fibrates combined with obeticholic acid in the treatment of patients with primary biliary cholangitis and inadequate response to second-line therapy. Hepatology. 2019. late-breaking abstracts (LP6).
32. De Vries E, Bolier R, Goet J, et al. Fibrates for itch in fibrosing cholangiopathies: a double blind, randomized, placebo, controlled trial. Gastroenterology. 2020. doi:10.1053/j.gastro.2020.10.001
33. Tully DC, Rucker PV, Chianelli D, et al. Discovery of tropifexor (LJN452), a highly potent non-bile acid FXR agonist for the treatment of cholestatic liver diseases and non-alcoholic steatohepatitis (NASH). J Med Chem. 2017;60:9960–9973. doi:10.1021/acs.jmedchem.7b00907
34. Schramm C, Hirschfield G, Mason AL, et al. Early assessment of safety and efficacy of tropifexor, a potent non-bile acid FXR agonist, in patients with primary biliary cholangitis: an interim analysis of an ongoing phase 2 study. J Hepatol. 2018;suppl. 1(S 103):LB0–007.
35. Trauner M, Gulamhusein A, Hameed B, et al. The non-steroidal farnesoid X receptor agonist cilofexor (GS-9674) improves markers of cholestasis and liver injury in patients with primary sclerosing cholangitis. Hepatology. 2019;70:788–801. doi:10.1002/hep.30509
36. Schattenberg J, Pares A, Kowdley KV, et al. Elafibranor, a peroxisome proliferator-activated receptor alpha and delta agonist demonstrates favourable efficacy and safety in patients with primary biliary cholangitis and inadequate response to ursodeoxycholic acid treatment. J Hepatol. 2019;suppl 1S:LBO–02.
37. Jones D, Boudes PF, Swain MG, et al. Seladelpar (MBX-8025) a selective PPAR-delta agonist, in patients with primary biliary cholangitis with an inadequate response to ursodeoxycholic acid; a double blind, randomized, placebo-controlled, phase 2, proof-of-concept study. Lancet Gastroenterol Hepatol. 2017;2:716–726. doi:10.1016/S2468-1253(17)30246-7
38. Levy C, Bowlus C, Neff G, et al. Durability of treatment response after 1 year of therapy with seladelpar in patients with primary biliary cholangitis (PBC): final results of an international phase 2 study. J Hepatol. 2020;73:
39. Russell DW. Fifty years of advances in the bile acid synthesis and metabolism. J Lipid Res. 2009;50:S120–S125. doi:10.1194/jlr.R800026-JLR200
40. May MJ, Wigg AJ, Leggett BA, et al. NGM282 for treatment of patients with primary biliary cholangitis: a multicenter, randomized, double-blind, placebo-controlled trial. Hepatol Commun. 2018;2:1–14.
41. Nishio T, Hu R, Koyama Y, et al. Activated hepatic stellate cells and portal fibroblasts contribute to cholestatic liver fibrosis in MDR2 knockout mice. J Hepatol. 2019;71:573–585. doi:10.1016/j.jhep.2019.04.012
42. Dalekos GN, Invernizzi P, Nevens F, et al. Efficacy of GKT831 in patients with primary biliary cholangitis and inadequate response to ursodeoxycholic acid: interim efficacy results of a phase 2 clinical trial. J Hepatol. 2019;70:e1–e2. doi:10.1016/S0618-8278(19)30002-7
43. Tsuda M, Moritoki Y, Lian ZX, et al. Biochemical and immunologic effects of rituximab in patients with primary biliary cirrhosis and incomplete response to ursodeoxycholic acid. Hepatology. 2012;55:512–521.
44. Myers RP, Swain MG, Lee SS, et al. B-cell depletion with rituximab in patients with primary biliary cirrhosis refractory to ursodeoxycholic acid. Am J Gastroenterol. 2013;108:933–941.
45. Khanna A, Jopson L, Howel D, et al. Rituximab is ineffective for treatment of fatigue in primary biliary cholangitis: a phase 2 randomized controlled trial. Hepatology. 2019;70:1646–1657. doi:10.1002/hep.30099
46. Chuang YH, Lian ZX, Cheng CM, et al. Increased levels of chemokine receptor CXCR3 and chemokines IP-10 and MIG in patients with primary biliary cirrhosis and their first degree relatives. J Autoimmun. 2005;25:
47. Nishioji K, Okanoue T, Itoh Y, et al. Increase of chemokine interferon-inducible protein-10 (IP-10) in the serum of patients with autoimmune liver diseases and increase of its mRNA expression in hepatocytes. Clin Exp Immunol. 2001;123:271–279. doi:10.1046/j.1365-2249.2001.01391.x
48. De Graaf KL, Lapeyre G, Guilhot F, et al. NI-0801, an anti-chemokine (C-X-C motif) ligand 10 antibody in patients with primary biliary cholangitis and an incomplete response to ursodeoxycholic acid. Hepatol Commun. 2018;2:492–503. doi:10.1002/hep4.1170
49. Hirschfield GM, Gerschwin ME, Strauss R, et al. Ustekinumab for patients with primary biliary cholangitis who have an inadequate response to ursodeoxycholic acid: a proof-of-concept study. Hepatology. 2016;64:189–199. doi:10.1002/hep.28359
50. Bowlus CL, Yang G-X, Liu CH, et al. Therapeutics trials of biologics in primary biliary cholangitis: an open-label study of abatacept and review of the literature. J Autoimmun. 2019;101:26–34. doi:10.1016/j.jaut.2019.04.005
51. Arsenijevic A, Harrell CR, Fellabaum C, Volarevic V. Mesenchimal stem cells as new therapeutic agents for the treatment of primary biliary cholangitis. Anal Cell Pathol. 2017;2017:7492836. doi:10.1155/2017/7492836
52. Alfaif M, Eom YW, Newsome PN, Baik SK. Mesenchymal stromal cell therapy for liver diseases. J Hepatol. 2018;68:1272–1285.
53. Wang L, Li J, Liu H, et al. A pilot study of umbilical cord-derived mesenchymal stem cell transfusion in patients with primary biliary cirrhosis. J Gastroenterol Hepatol. 2013;28(suppl.1):85–92. doi:10.1111/jgh.12029
54. Wang L, Han Q, Chen H, et al. Allogenic bone marrow mesenchymal stem cell transplantation in patients with UDCA-resistant primary biliary cirrhosis. Stem Cell Dev. 2014;23:2482–2489. doi:10.1089/scd.2013.0500
55. Mason A, Xu L, Guo L, et al. Detection of retroviral antibodies in primary biliary cirrhosis and other idiopathic biliary disorders. Lancet. 1998;35:1620–1624. doi:10.1016/S0140-6736(97)10290-2
56. Lytvyak E, Hosomani I, Montano-Loza AJ, et al. Randomized clinical trial: combination antiretroviral therapy with tenofovir-emtricitabine and lopinavir-ritonavir in patients with primary biliary cholangitis. Can Liver J. 2019. doi:10.3138/canlivj.2018-0020
57. Mayo MJ, Pockros PJ, Jones D, et al. A randomized, controlled, phase 2 study of maralixabat in the treatment of itching associated with primary biliary cholangitis. Hepatol Commun. 2019;3:365–381.
58. Hegade VS, Kendrick SF, Dobbins RL, et al. Effect of ileal bile acid transporter inhibitor GSK23330672 on pruritus in primary biliary cholangitis: a double-blind, randomised, placebo-controlled, crossover, phase 2a study. Lancet. 2017;389:1114–1123. doi:10.1016/S0140-6736(17)30319-7
59. Al-Dury S, Wahlström A, Wahlin S, et al. Pilot study with IBAT inhibitor A4250 for the treatment of cholestatic pruritus in primary biliary cholangitis. Sci Rep. 2018;8:6658. doi:10.1038/s41598-018-25214-0
60. Zenouzi R, Weiler-Normann C, Lohse AW. Is fatigue in primary biliary cirrhosis cured by transplantation? J Hepatol. 2013;59:418–419. doi:10.1016/j.jhep.2013.05.037
61. Hollingsworth K, Newton JL, Taylor R, et al. Pilot study of peripheral muscle function in primary biliary cirrhosis: potential implications for fatigue pathogenesis. Clin Gastroenterol Hepatol. 2008;6:1041–1048. doi:10.1016/j.cgh.2008.04.013
62. Jones DEJ, Newton JL. An open study of modafinil for the treatment of daytime somnolence and fatigue in primary biliary cirrhosis. Aliment Pharmacol Ther. 2007;25:471–476. doi:10.1111/j.1365-2036.2006.03223.x
63. Millar B, Richardson C, McKay K, et al. Obeticholic acid therapy improves cognitive decline in cholestatic liver disease. J Hepatol. 2017;66:FR396. doi:10.1016/S0168-8278(17)31070-X
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.