")
Back to Journals » Journal of Experimental Pharmacology » Volume 13
Experimental Drugs for Panic Disorder: An Updated Systematic Review
Authors Caldirola D, Alciati A, Cuniberti F , Perna G
Received 8 February 2021
Accepted for publication 25 March 2021
Published 15 April 2021 Volume 2021:13 Pages 441—459
DOI https://doi.org/10.2147/JEP.S261403
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Daniela Caldirola,1,2 Alessandra Alciati,1– 3 Francesco Cuniberti,1,2 Giampaolo Perna1,2
1Department of Biomedical Sciences, Humanitas University, Milan, 20090, Italy; 2Department of Clinical Neurosciences, Villa San Benedetto Menni Hospital, Hermanas Hospitalarias, Como, 22032, Italy; 3Humanitas Clinical and Research Center, IRCCS, Milan, Rozzano, 20089, Italy
Correspondence: Daniela Caldirola
Department of Biomedical Sciences, Humanitas University, Via Rita Levi Montalcini 4, Pieve Emanuele, Milan, 20090, Italy
Email [email protected]
Abstract: Several effective pharmacological therapies for panic disorder (PD) are available, but they have some drawbacks, and unsatisfactory outcomes can occur. Expanding the variety of anti-panic medications may allow for improving PD treatment. The authors performed an updated systematic review of preclinical and clinical (Phase I–III) pharmacological studies to look for advances made in the last six years concerning novel-mechanism-based anti-panic compounds or using medications approved for nonpsychiatric medical conditions to treat PD. The study included seven published articles presenting a series of preclinical studies, two Phase I clinical studies with orexin receptor (OXR) antagonists, and two clinical studies investigating the effects of D-cycloserine (DCS) and xenon gas in individuals with PD. The latest preclinical findings confirmed and expanded previous promising indications of OXR1 antagonists as novel-mechanism-based anti-panic compounds. Translating preclinical research into clinical applications remains in the early stages. However, limited clinical findings suggested the selective OXR1 antagonist JNJ-61393115 may exert anti-panic effects in humans. Overall, OXR1 antagonists displayed a favorable profile of short-term safety and tolerability. Very preliminary suggestions of possible anti-panic effects of xenon gas emerged but need confirmation with more rigorous methodology. DCS did not seem promising as an enhancer of cognitive-behavioral therapy in PD. Future studies, including objective panic-related physiological parameters, such as respiratory measures, and expanding the use of panic vulnerability biomarkers, such as hypersensitivity to CO2 panic provocation, may allow for more reliable conclusions about the anti-panic properties of new compounds.
Keywords: novel drug, experimental therapy, orexin, D-cycloserine, xenon gas
Introduction
Panic disorder (PD) is a common disorder, with a lifetime prevalence of approximately 3.8% in general Western adult population.1,2 PD has a chronic course, a significant burden of psychiatric and medical comorbidities, and can have significant detrimental effects on daily life functioning and quality of life.3–5 PD is a heterogeneous disorder encompassing different phenomena, namely spontaneous panic attacks (PAs), the “core” of the disorder (ie, unexpected surges of somatic symptoms, such as accelerated heart rate and feelings of choking, accompanied by fear or discomfort), anticipatory anxiety, and panic-related maladaptive changes in behavior, which can lead to comorbid agoraphobia (AG) in approximately 70% of individuals with PD.6 Moreover, different phenomenological PD profiles may exist, possibly based on clinical symptoms and biological features.7 So far, the mechanism involved in PD is not clear-cut. The different clinical phenomena in PD are probably qualitatively distinct and related to diverse neural circuits and mechanisms.8,9 Some authors consider PAs as responses to threats related with body’s internal state. These threats may arise from multiple mechanisms, such as a decreased threshold for suffocation10 or the dysfunction of brain networks involved in defense reactions (eg, midbrain dorsal periaqueductal gray [DPAG]).11,12 Further developments postulate that an abnormal respiratory regulation, mainly related to hypersensitivity to chemoceptive stimuli (ie, carbon dioxide [CO2] and hydrogen ion [H+]) may be involved in both spontaneous and laboratory-induced PAs (ie, provoked by infusion of sodium lactate (NaLac) or inhalation of hypercapnic gas mixture). According to this view, PAs are different from fear reactions.13–15 We recently proposed that subtle imbalances in bodily homeostatic global functioning reduce global physical adaptability to internal changes and may lead to PAs.16
Conversely, scientific findings9,17–19 do not fully support the idea that PAs primarily arise from a malfunction of amygdala and limbic system resulting in a hyperactive “fear network”.20 Overall, according to these hypotheses, PAs may involve brain networks that modulate physiologic homeostatic processes, including the brainstem, hypothalamus, and insula. In contrast, the amygdala, limbic system, and higher brain areas (such as the prefrontal cortex) could be heavily implicated in panic-related phobias and anticipatory anxiety.17,21,22 In the treatment of PD, limited progress has been made for several years. Possible reasons include the uncertainty concerning panic pathophysiology, the fact that panic-related phenomena may be influenced by multiple neurotransmitters, and the lack of full understanding of action mechanisms of medications recommended for PD.23,24 The current guidelines recommend selective serotonin reuptake inhibitors (SSRIs) and the serotonin-norepinephrine reuptake inhibitor (SNRI)] venlafaxine, as first-line options; tricyclic antidepressants (TCAs) and benzodiazepines (BDZs) are second-line alternatives; other pharmacological options are available, but with limited scientific evidence. Finally, cognitive-behavioral therapy (CBT) is the first-line non-pharmacological intervention.25–27 Although the efficacy of recommended medications and CBT is well-established, many patients (approximately 20%–40%) did not achieve full remission, and the rates of relapse or persistence of subthreshold symptoms after termination of pharmacotherapy was substantial (up to 50% of patients).28 Furthermore, SSRIs, venlafaxine and TCAs are associated with several shortcomings, including delayed onset of action, possible rise in anxiety at the beginning of treatment, and several side effects, including weight gain or detrimental effects on sexual activity. BDZs have a fast onset of therapeutic effects but can cause sedation, fatigue, and memory/cognitive impairment; they also have a greater risk of tolerance, misuse, and dependence.25–27 Overall, this evidence suggests that unmet medical needs still exist in PD pharmacotherapy. Research over the last years has investigated the potential anti-panic effectiveness of medications already approved for other psychiatric disorders to address these issues. So, far, results are insufficient to support using different antidepressants (eg, nefazodone, mirtazapine, milnacipran, reboxetine, duloxetine), second-generation antipsychotics, or anticonvulsants in PD, as an alternative to the recommended medications.7,23,29–31 A recent 10-week open-label study found preliminary indications of clinical efficacy of vortioxetine in PD, which need further confirmation in studies with larger samples and a more rigorous methodology.32 Other attempts have involved compounds approved for medical uses not related to psychiatric conditions. The antibiotic D-cycloserine (DCS) received special attention as a possible enhancer of CBT effectiveness in PD due to its partial agonism at the glutamatergic N-methyl-D-aspartate (NMDA) receptor. However, more recent findings no longer supported its usefulness in this disorder.23 Finally, developing novel mechanism-based compounds has been considered an additional valuable option to identify more effective, tolerable, and faster-acting anti-panic treatments. In our previous review focused on this topic,33 as well as in a broader review of potential novel drugs for the whole group of anxiety disorders,34 compounds acting on glutamate and orexin systems seemed to emerge as the most encouraging options for future novel therapies for PD.
In the present systematic review, we updated our previous review33 by examining panic-related preclinical and clinical (Phase I–III) pharmacological studies to look for progress in the last six years. We were interested in assessing advance in developing novel mechanism-based anti-panic compounds and employing drugs authorized for nonpsychiatric uses to treat PD.
Methods
The systematic review followed the Cochrane Collaboration’s guidelines and we documented the results according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines35 (Figure 1, PRISMA flow diagram). We performed a search of peer-reviewed scientific literature, written in English, using PubMed, PsycINFO, and Embase databases from January 1, 2015, to December 31, 2020. We combined the search strategy of free-text terms and exploded MESH headings for the topics (“panic/panic disorder,” “preclinical trial/clinical trial/randomized controlled trial,” “experimental study/research,” and “novel/investigational/experimental drug or psychotropic drug). Additional articles were also searched using the reference lists of pertinent articles and reviews. Furthermore, we looked for pertinent findings from clinical trials using European and US trial registries36,37 (search term was “panic”). This protocol was not previously registered.
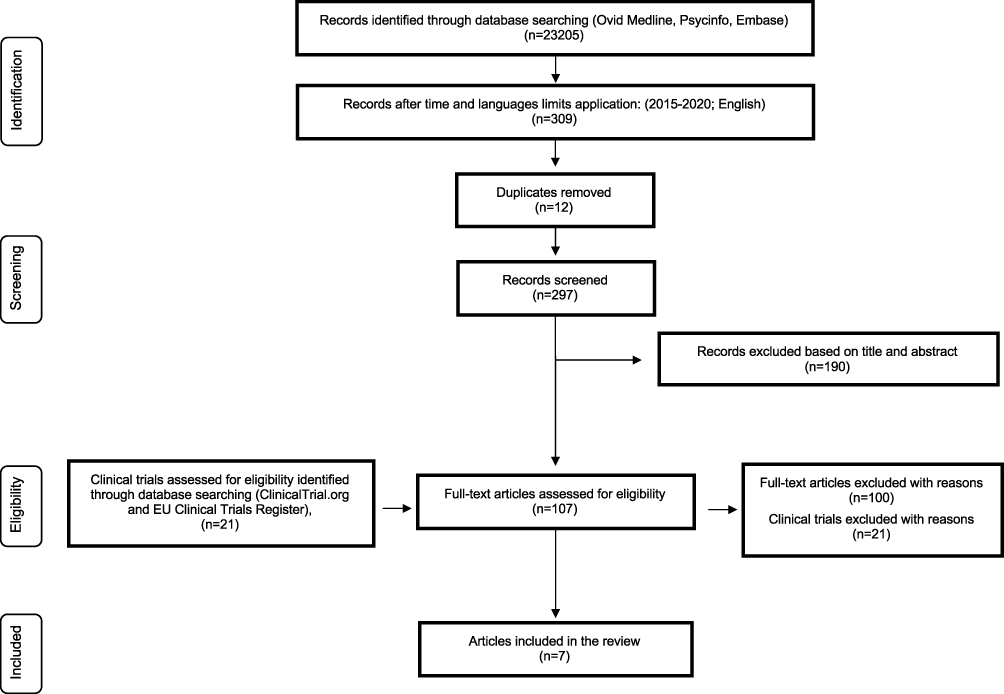 |
Figure 1 PRISMA flow diagram of study selection process.Notes: PRISMA figure adapted from Liberati A, Altman D, Tetzlaff J, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. Journal of Clinical Epidemiology. 2009;62(10). Creative Commons.35 |
Three of the study’s authors (DC, AA, and FC) independently carried out search and screening process; inconsistencies were discussed and resolved before proceeding.
Preclinical studies were included in the review if they used the most validated translational cross-species experimental models of induced PAs currently available, namely carbon dioxide (CO2) inhalation in naïve rats38 or sodium lactate (NaLac) infusion in panic-prone rats.39
Inclusion criteria for clinical studies admitted to the review were the following: Phase I to III pharmacological clinical trials; investigation of novel mechanism-based agents’ potential anti-panic effects or drugs already authorized for nonpsychiatric uses and differing in pharmacodynamics from drugs recommended for PD; adult participants (ie, ≥18 years of age); mentally healthy participants (in case of Phase I clinical trials) or participants with PD (as primary diagnosis) with or without agoraphobia (AG) (diagnostic screening in accordance to Refs. 6,40–44) (in case of Phase II/III clinical trials); use of the most validated experimental procedures to induce PAs in humans (eg, NaLac infusion or CO2 inhalation)38 when laboratory panicogenic challenges were planned; use of validated self- and/or clinician-reported measures. We only included articles with full-text availability. We excluded letters, commentaries, abstracts, reviews, case reports, and meta-analyses. We only included studies investigating D-cycloserine’s effects on PD that were published after February 28, 2018. We discussed in detail previous studies on this compound in our recent review.23 We did not include studies focused on treatment-resistant PD because this topic would require a dedicated review.
We only included clinical trials if they provided available results not already published as articles. After removing duplicates, we identified 297 published articles and 21 clinical trials. At the end of the entire process, we selected seven published articles suitable to be included in our review (Figure 1, PRISMA flow diagram).
Results
We included seven published articles, five of which encompassed a series of preclinical and Phase I clinical studies with antagonists of orexin receptors (Tables 1 and 2, respectively). The other two (Table 3) were clinical studies investigating the effects of D-cycloserine (randomized study) or xenon gas (open study), respectively, in individuals with PD.
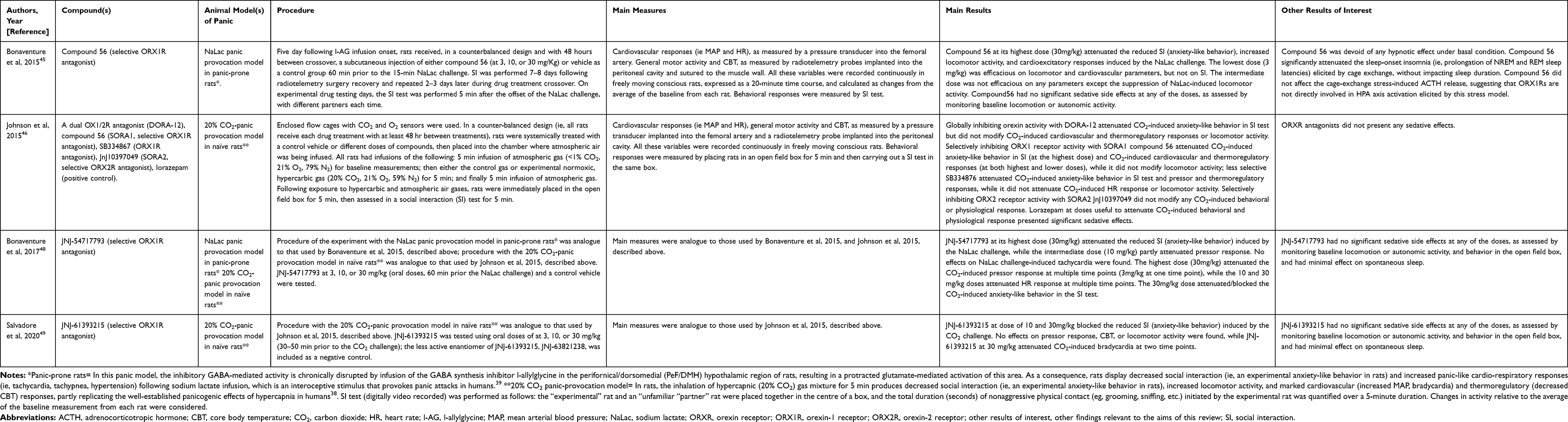 |
Table 1 Preclinical Studies with Orexin Receptor Antagonists |
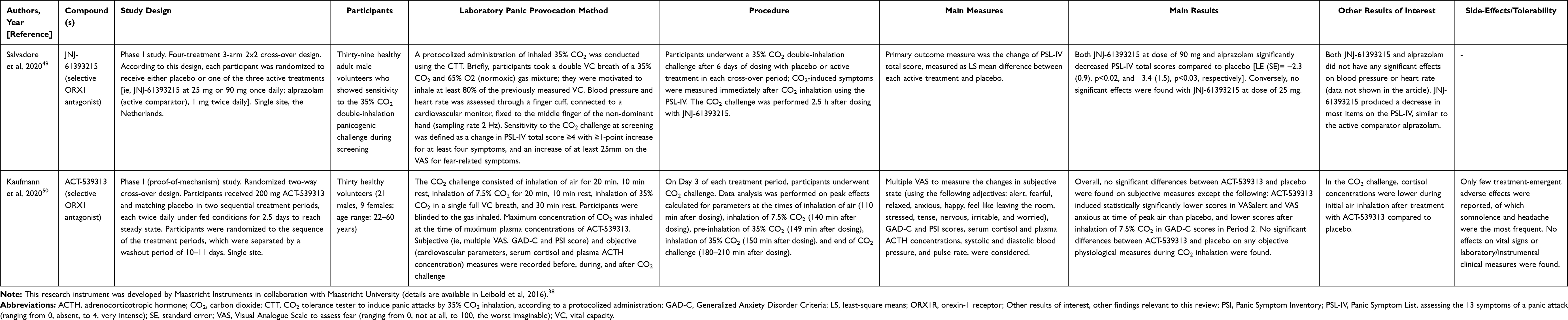 |
Table 2 Phase I Clinical Studies with Orexin Receptor Antagonists |
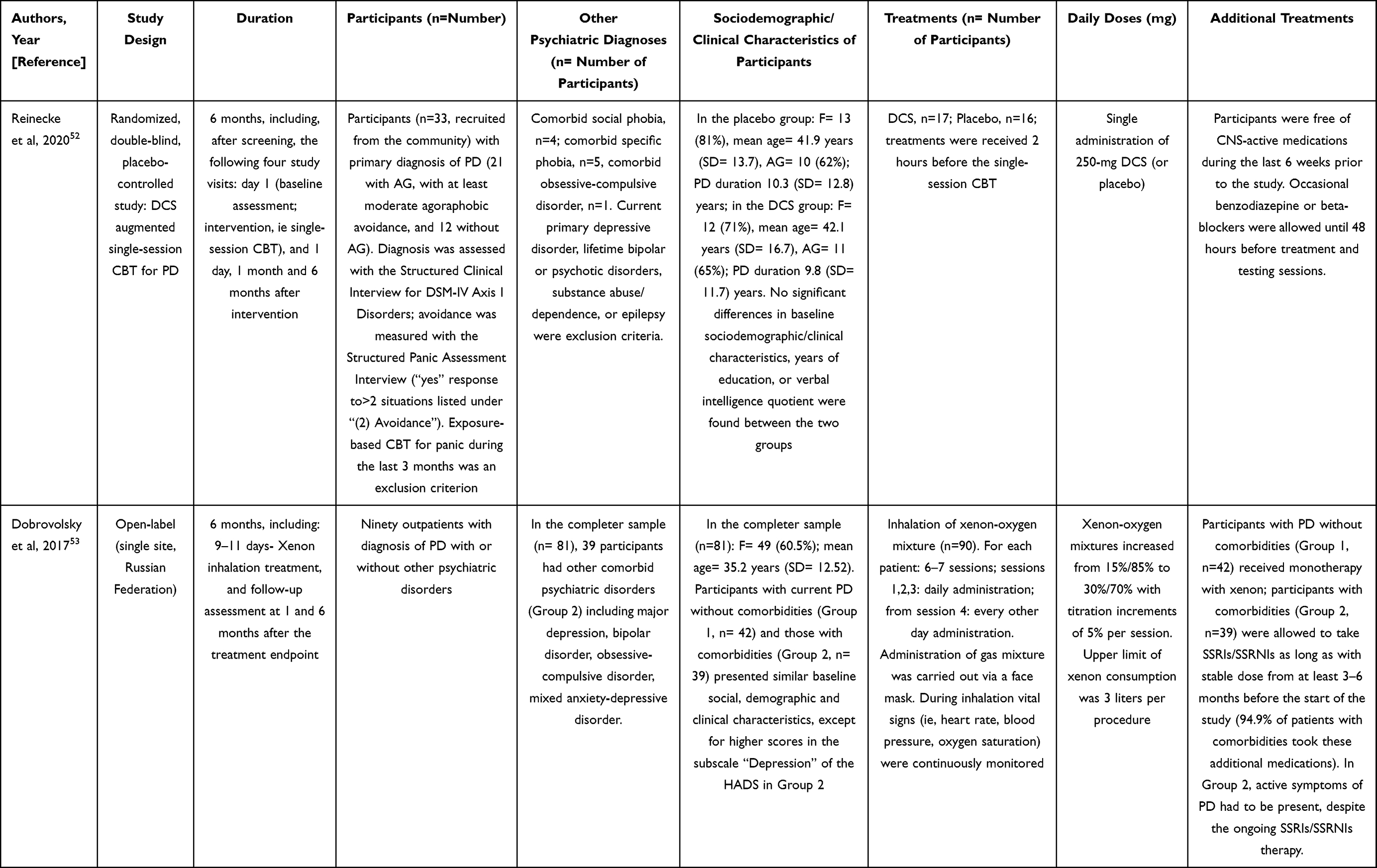 | 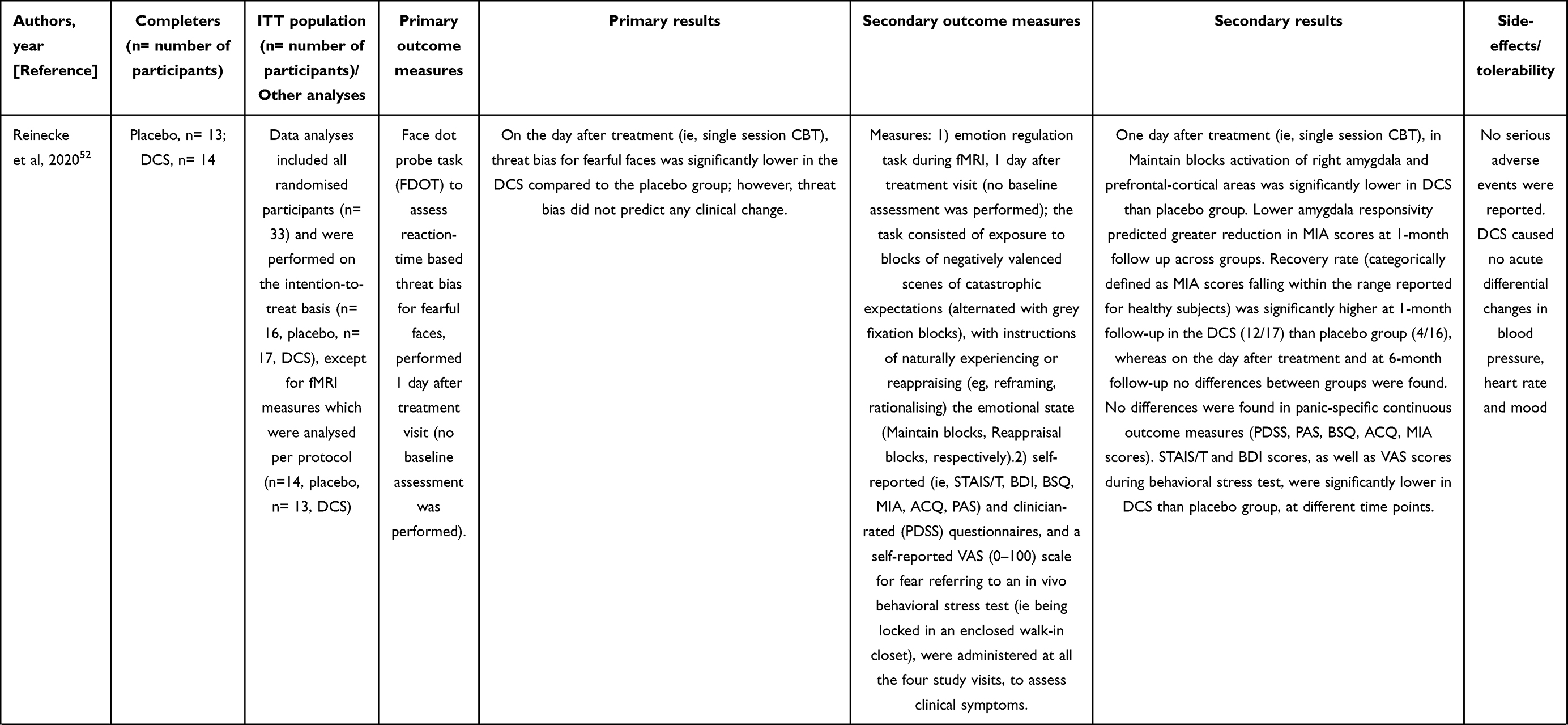 | 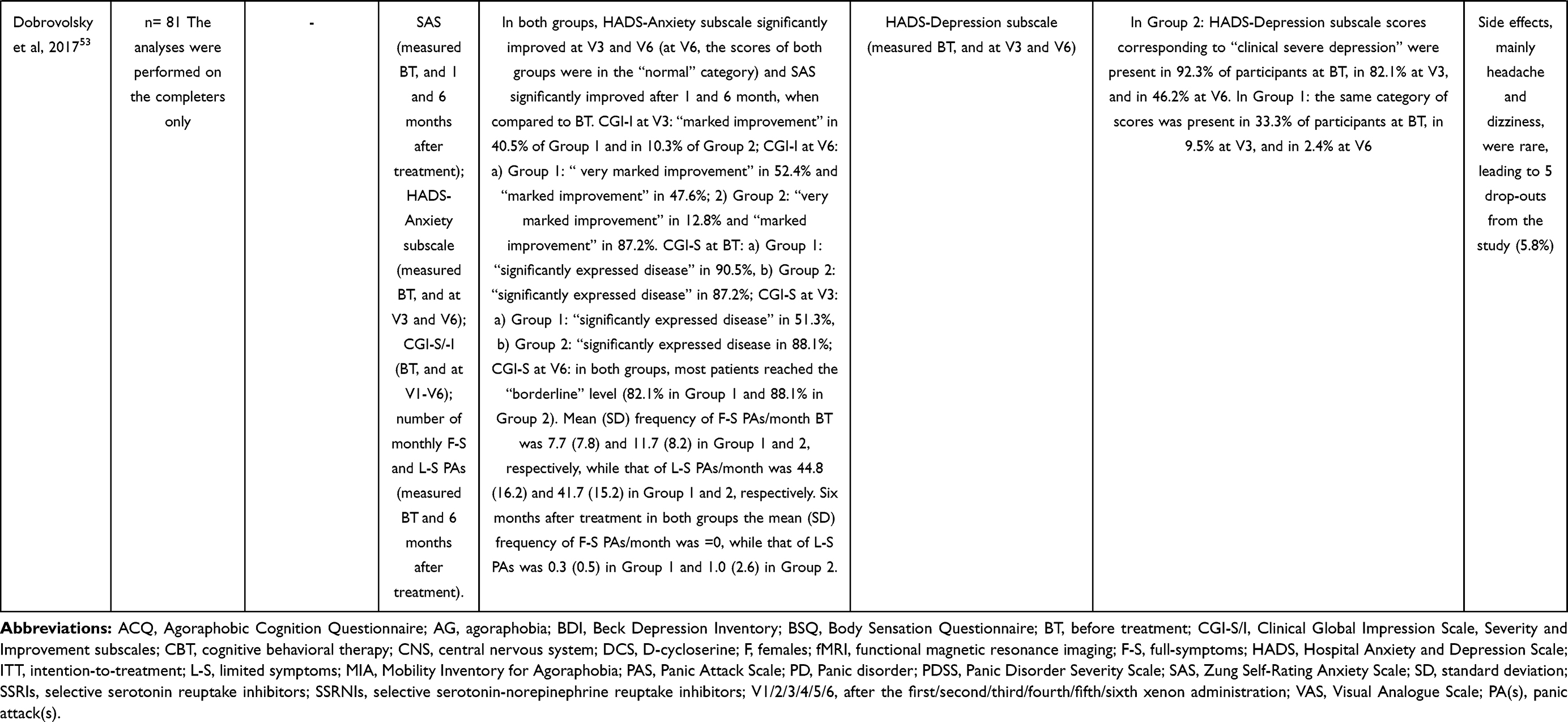 |
Table 3 Clinical Studies in Patients with Panic Disorder |
Antagonists of Orexin Receptors
In the following paragraphs, we reviewed preclinical (Table 1) and Phase I clinical studies (Table 2) assessing antagonists of orexin receptors as novel drugs with potential anti-panic properties.
Preclinical Studies
One study45 characterized the orexin-1 receptor (OX1R) antagonist compound 56. The authors compared its properties to standard OX1R antagonists SB-408124, SB-334867, and GSK-1059865, in experiments on rodents. They demonstrated that the novel compound 56 has capability of penetrating into brain, and a very high and selective OX1R affinity and antagonism, without any hypnotic effect. Because compound 56 displayed better performance than the comparators, it was selected for the experiments described below. The authors evaluated compound 56 activity at different doses in two rat models of stress-induced hyperarousal, namely an external, exteroceptive threat (ie, cage exchange) and an internal, interoceptive body state threat (ie, intravenous NaLac panic provocation in panic-prone rats39). For the aim of this review, we focused on the results concerning the NaLac panic provocation model. Collectively, in this rat model of panic vulnerability, compound 56 significantly attenuated behavioral and cardiovascular reactions to intravenous NaLac infusion. No sedative side effects were identified throughout the procedure (details in Table 1).
To test the hypothesis that OX1R antagonists may play a prominent role in modulating panic-associated behavior and autonomic responses compared with orexin-2 receptor (OX2R) antagonists, other authors46 applied a 20% carbon dioxide (CO2) panic provocation model in naïve rats to identify and compared the potential anti-panic properties of the following three orexin- receptor (OXR) antagonists: compound 56 (SORA1, selective ORX1 antagonist), a dual OX1/2R antagonist (DORA-12), a close structural analog of suvorexant, to globally inhibit orexin activity, and OX2R antagonist JnJ10397049 (SORA2, selective ORX2 antagonist). Alongside these compounds, the benzodiazepine lorazepam was used as a positive control for panicolytic properties. Finally, the less selective ORX1 antagonist SB334867 (ie, it also has off-target affinities for non-orexin receptors) was tested, even though concerns regarding its hydrolytic instability47 make it less suitable as a candidate for future development. The authors found that the selective ORX1 antagonist was able to reduce both the 20% CO2-induced behavioral and cardiovascular response, while the dual OX1/2R antagonist diminished only the 20% CO2-induced behavior. Conversely, the selective OX2R antagonist reduced neither 20% CO2-induced behavior nor cardiovascular response. Contrary to lorazepam, orexin receptor antagonists did not present sedative side effects (details in Table 1).
A subsequent study48 reported a comprehensive pharmacological characterization of JNJ-54717793, a novel OX1R antagonist, and tested its potential anti-panic properties using both NaLac and 20% CO2 panic provocation models. The authors demonstrated JNJ-54717793’s capability of penetrating into brain, and its high and selective affinity and antagonism for OX1Rs, without significant impact on rats’ and mice’s spontaneous sleep. Overall, this compound attenuated NaLac- and 20% CO2-induced cardiovascular and behavioral responses, without sedative side effects (details in Table 1).
Finally, a recent article49 provided a translational evaluation of JNJ-61393215, a novel compound exerting selective antagonism at OX1Rs. The authors provided a comprehensive in vitro and in vivo pharmacological characterization of the compound. They evaluated JNJ-61393215’s potential anti-panic properties on panic-related responses induced by CO2 panic provocation models both in rats and humans. The following section describes the series of human studies. In vitro characterization showed that JNJ-61393215 had a high affinity, potency, and selectivity for OX1R. In vivo, in rats, the authors, using the same 20% CO2 panic provocation model used in the above-described studies, found that JNJ-61393215 could block anxiety-like behavioral response to CO2 inhalation, as measured during the social interaction test, and, only at its highest dose, attenuate CO2-induced cardiac response. The compound did not present apparent sedative side effects, and it minimally affected the sleep-wake state in baseline conditions (details in Table 1).
Phase I Clinical Studies
In the same article,49 the authors carried out a series of first-in-human conducted Phase I single and multiple ascending dose studies (SAD and MAD studies, respectively) to provide a pharmacological characterization of the novel selective OX1R antagonist JNJ-61393215, including its tolerability and safety. They also explored its potential anti-panic properties on panic responses induced by a 35% CO2 double-inhalation challenge (MAD study, part 2). All the studies were conducted in healthy (as defined based on clinical laboratory tests, physical and neurological examination at screening) adult male volunteers. We mainly focused on the results concerning the compound’s anti-panic properties, while we only briefly summarized below the main results of the SAD and MAD, part 1, studies. In the double-blind, randomized, placebo-controlled SAD study, 8 cohorts of participants (total number of participants: 72) received, under fasted conditions, single increasing (from 1 to 90 mg) oral doses of JNJ-61393215 or placebo. In the double-blind, randomized, placebo-controlled MAD study (part 1), 4 cohorts of participants (total number of participants: 32) received, under fasted conditions, multiple increasing doses of JNJ-61393215 (ie, 5, 15, 45, and 90 mg) or placebo. Collectively, the SAD and MAD part 1 studies demonstrated an acceptable safety profile of JNJ-61393215. The frequency of JNJ-61393215-related adverse events did not differ from placebo and the severity of the most common side effects (eg, headache, somnolence, dysgeusia) was mild. Even with the maximal doses administered (estimated receptor occupancy >95%), no somnolence occurred. Thirty-nine participants, who showed sensitivity to the 35% CO2 double-inhalation panicogenic challenge during screening, were included in a randomized study with two active compounds (ie, JNJ-61393215 and the active comparator alprazolam) and placebo. A four-treatment 3 arm 2x2 cross-over design was followed, with a duration of 7 days. The doses of JNJ-61393215 were 25 mg (estimated receptor occupancy >93%) or 90 mg (estimated receptor occupancy >98.5%) once daily, while 1 mg of alprazolam was administered twice daily (MAD study part 2, details in Table 2). After six days of active treatment or placebo, participants underwent a 35% CO2 double-inhalation challenge. Outcome measure, used to compare active treatment with matched placebo in each arm, was Panic Symptom List (PSL)-IV score, measuring CO2-induced-symptoms. Results were controlled for fixed and random effects, and baseline PSL-IV score was used as a covariate. Power calculation was carried out to identify an appropriate sample size, using the estimated PSL-IV total score significant changes. Both treatment with 90 mg of JNJ-61393215 and alprazolam significantly reduced CO2 induced-symptoms, whereas 25 mg of JNJ-61393215 did not exert significant effects. Neither JNJ-61393215 nor alprazolam exerted any effects on cardiovascular responses to CO2 challenge (data not shown in the article) (details in Table 2).
Finally, a Phase I double-blind, placebo-controlled, randomized MAD study (study 1) assessed pharmacokinetics and pharmacodynamics of the orally active, reversible, brain penetrant selective OXR1 antagonist ACT-539313.50 Tolerability and safety of the compound were also examined. Furthermore, the authors conducted a preliminary Phase I study aimed to evaluate its potential anti-panic properties using a CO2 challenge consisting of consecutive inhalation of air, 7.5% CO2 and 35% CO2-single breath (study 2). For this review, we mainly focused on study 2, while we only briefly summarized below the main results of study 1. Both studies were conducted in physically and mentally healthy adult volunteers.
Study 1 included 28 healthy participants receiving, under fed conditions, multiple-ascending oral doses of ACT-539313 (up to 200 mg two times every day). A comprehensive test battery was used to assess the possible pharmacodynamic impact of the compound on central nervous system (CNS) functions. A small decrease in saccadic peak velocity and unstable tracking performance was found, without dose-dependency or significant impairment in vigilance or visuomotor performance. The most frequently reported side effect was somnolence, at a dose of 200 mg, while severe adverse events were not found.
Study 2 (details in Table 2) had a randomized, two-way cross-over design and included 30 female and male participants. A dose of 200 mg of ACT-539313 was used. Power calculation was carried out to identify an appropriate sample size, using the estimated Visual Analog Scale (VAS) for anxiety changes induced by 7.5% CO2-inhalation.51 Overall, ACT-539313 and placebo did not significantly differ in subjective panic-related measures or objective parameters during CO2 inhalation.
Limited indications of potential general anxiolytic properties of ACT-539313 were reported, as shown by reduced anxiety during air inhalation and generalized anxiety symptoms during 7.5% CO2 inhalation, under ACT-539313 compared with placebo (details in Table 2).
Clinical Studies of Drugs Authorized for Nonpsychiatric Uses
In the following paragraphs, we reviewed clinical studies assessing effectiveness in patients with PD of the antibiotic D-cycloserine (DCS) used in tuberculosis treatment and the general anesthetic Xenon gas (Table 3).
D-Cycloserine
D-cycloserine (DCS) was recently investigated in a randomized, double-blind, placebo-controlled augmentation study, conducted in a small sample of participants with PD (with and without agoraphobia), recruited from a general population.52 The aims were to evaluate whether 250 mg of DCS, administered in a single oral dose, had capability of influencing fear-related neurocognitive markers. Participants received DCS two hours before single-session cognitive-behavioral therapy (CBT). The primary outcome was threat bias for fearful faces (as measured with reaction-time), while the secondary outcome was amygdala reactivity to threat. These evaluations were performed one day after the treatment visit. Other secondary outcomes included clinical symptoms changes, as measured with multiple self- and clinician-administered psychometric questionnaires throughout 6-month follow-up. Randomization sequence (generated by an automated random number generator), masking, treatment allocation and distribution were planned to guarantee concealment during the study. An expert clinical psychologist delivered the single-session CBT, lasting about 60 minutes, including cognitive preparation and psychoeducation, exposure to fear-provoking situations and bodily sensations, and cognitive debriefing. Were recruited 16 participants per group to achieve 70% power in detecting a difference (effect size, d= 0.8; p= 0.05) in threat bias between placebo and DCs. DCS-group presented lower threat bias for fearful faces and amygdala reactivity to threat than the placebo group, as measured the day after treatment. Recovery of agoraphobic avoidance after 1 month of treatment was greater in DCS-group than placebo-group, whereas significant differences between the two groups disappeared during 6-months follow-up. The two groups did not significantly differ in continuous panic-specific clinical measures at any time point of the study (details in Table 3).
Xenon Gas
The effectiveness of xenon gas inhalations in a sample of participants with PD without (group 1) and with (group 2) other psychiatric comorbidities was evaluated in a 6 month-open-label study.53 Participants received 6–7 inhalations at sub-anesthetic concentrations. Xenon-oxygen mixture was delivered through a face mask, and each inhalation lasted 2.5–4 minutes, with concomitant continuous monitoring of cardiovascular parameters and oxygen saturation. In both groups, general anxiety symptoms, as measured with the anxiety-related sub-scores of the Hospital Anxiety and Depression Scale (HADS-A), significantly decreased after 3 xenon inhalations. By the end of treatment, all participants were in the “norm” HDS-A category (at the beginning of treatment, all participants were in the “clinically severe anxiety” category). Similarly, anxiety symptoms, as measured with Zung Self-Rating anxiety scale, significantly decreased from scores corresponding to “high level of anxiety” at the beginning of treatment to scores corresponding to “no anxiety” in group 1 and “minimum degree of anxiety” in group 2, at both 1 and 6 months after the end of the treatment. Without statistical analyses, descriptive reporting was provided about Clinical Global Impression (CGI) Scale scores and panic attacks (PAs). Severity and Improvement subscales of CGI Scale showed similar severity of PD in both groups at the beginning of treatment, greater improvement after 3 xenon inhalations in group 1, and a certain persistence of this tendency at the final time point. At 6-months follow-up, PAs were absent in both groups, while the frequency of limited-symptom PAs per month was very low (0.3, SD = 0.46 in group 1; 1.0, SD = 2.64 in group 2). No explanation about PA assessment methodology was reported. Overall, xenon was well tolerated. In four out of the five participants who dropped out because of headache and dizziness, mild organic brain disease of vascular origin was found, making them more sensitive to xenon side effects (details in Table 3).
Discussion
We reviewed preclinical and clinical (Phase I–III) pharmacological studies of the last 6 years. We considered whether advances were made in developing novel mechanism-based anti-panic compounds or in employing drugs already authorized for nonpsychiatric uses and differing in pharmacodynamics from drugs recommended for PD. A limitation of our review is that the protocol was not previously registered.
We found encouraging research on anti-panic properties of orexin receptor antagonists and interesting preliminary findings on xenon gas, while DCS did not appear of interest to increase the effectiveness of CBT in PD.
Studies of Novel Mechanism-Based Compounds
The latest preclinical research supported selective orexin-1 receptor (OX1R) antagonists as promising novel mechanism-based anti-panic compounds, confirming and expanding previous results.33 In contrast, selective orexin-2 receptor (OX2R) antagonism was not associated with significant anti-panic effects.
The reviewed preclinical studies collectively demonstrated that different selective OX1R antagonists significantly reduced behavioral and cardiovascular responses to the most validated translational cross-species experimental models of induced PAs, namely CO2 inhalation in naïve rats38 or NaLac infusion in panic-prone rats.39 In contrast, the dual OX1/2R antagonist DORA-12 attenuated only the 20% CO2-induced behavior. This effect was considered primarily related to its OX1R antagonism because, in the same study, a selective OX2R antagonist reduced neither 20% CO2-induced behavior nor cardiovascular response.46 Moreover, OX1R antagonists, in contrast to benzodiazepines, did not cause apparent sedative side effects during procedures in any study. However, all the reviewed preclinical studies’ main general limitation was the lack of assessment of respiratory parameters. Thereby it remained unresolved whether the tested OX1R antagonists were capable of modifying CO2-/NaLac-induced panic-related respiratory response. The involvement of respiratory symptoms and function in human panic is well-established7,54 and panic-related CO2-induced respiratory response was demonstrated in mice.38 Furthermore, a recent animal study associated exaggerate ventilation after CO2 inhalation in a model of “PD-like” respiratory phenotype, induced by neonatal maternal separation in rats, with disturbance of estradiol modulation of OX neurons in females. This dysregulation disinhibited OX neurons and increased OX-A levels in the hypothalamus, while an OX1R antagonist reduced the hyperventilation in response to CO2.55 Therefore, future preclinical studies with OX1R antagonists should include a respiratory assessment to better characterize and support the anti-panic properties of OXR1 antagonists. Indeed, to enlarge in preclinical research, the array of scientifically grounded, objective panic-related variables, such as respiratory parameters, may increase the translational validity of results and reduce the risk of bias related to the inferential assessment of behavior, whose translational validity may be more uncertain and questionable.
Attempts of translating preclinical research on selective OX1R antagonists to potential clinical applications resulted in only two recent Phase I published studies,49,50 yielding conflicting results.
One study49 found that the selective OX1R antagonist JNJ-61393115 significantly decreased, in a sample of healthy men, panic symptoms induced by a double vital capacity 35% CO2 inhalation. This is a validated procedure to provoke a response meeting the criteria of a PA in individuals not suffering from PAs or PD.38 These results were partly consistent with the authors’ favorable effects with the same compound on the 20% CO2 induced panic-like responses in rats.49 The study’s strength was to have included only participants who had shown sensitivity to the same 35% CO2 panicogenic challenge during screening to decrease possible risk of “floor effect” that could have undermined the identification of anti-panic activity of the compound. Unfortunately, the efficacy of JNJ-61393115 on panic-related CO2-induced physiological responses in humans remains to be established because the respiratory response to CO2 inhalation was not assessed. The apparent lack of effects on cardiovascular parameters was difficult to interpret due to the marked inter-individual variability in these parameters that emerged during the procedure.49 Another limitation was that the changes in ratings on the Visual Analog Scale (VAS) for fear during CO2 challenge were not used as outcome measures. As VAS for fear is usually included in criteria used to identify CO2-induced PAs,38 the lack of this measure may partly weaken the conclusions about the compound’s anti-panic effects. Finally, the results’ generalizability to females should also be assessed in the light of preclinical and clinical evidence for higher orexin system expression in females.56
Bearing in mind these shortcomings, the preliminary effects of this compound on CO2-induced panic symptoms, along with its favorable brief-term profile of safety and tolerability, suggest that this novel compound deserves further investigation in clinical studies, possibly involving patients with PD.
Conversely, a different selective OX1R antagonist (ACT-539313), tested in the other published Phase I study, including also a small number of females,50 did not display any specific anti-panic effect, while it exhibited potential general anxiolytic properties. However, this proof-of–mechanism study presented two main limitations that may have compromised detection of the compound’s potential anti-panic effects. The first limitation was that using an ad-hoc sequential inhalation procedure, including a single inhalation of 35% CO2 that is not fully validated to induce PAs in healthy individuals. The second is a possible floor effect related to the lack of screening participants based on sensitivity to CO2.
This body of pharmacological research on OX1R antagonists is consistent with current knowledge concerning putative connections between the orexin (OX) system, OXR1s, and panic pathophysiology. Excitatory OX neuropeptides originate from the precursor prepro-OX, which is produced by perifornical (Pef) and lateral hypothalamic neurons, and act via the OX1Rs and OX2Rs. The OX system modulates several different biological processes, including chemoreception and cardiorespiratory and behavioral responses to increased CO2 or decreased H+ that are particularly relevant to PAs.46 Several studies associated panic-related behavior and the cardiovascular response of naïve rats exposed to 20% CO2 inhalation, or panic-prone rats to NaLac infusion, with increased cellular activity within hypothalamic OX neurons and cardiorespiratory brainstem circuits. Conversely, in prepro-OX knockout rats, NaLac-induced panic-like responses were blocked, which reduced ventilatory response to inspired CO2. The latter effect was also obtained when the rostral medullary raphes of naïve rats were infused with an ORX1 antagonist.57–60 In addition to the midbrain raphe, OX1Rs are localized in other brain areas that are putatively implicated in human PAs and defensive/emotional responses to an internal threat, such as locus coeruleus, brainstem cardiorespiratory nuclei, and periaqueductal gray, and they are also located in the limbic system.60,61 Conversely, OX2Rs are mainly localized in wake-promoting systems, such as histaminergic system,46 and therefore are not specifically relevant to panic. Notably, important concerning translational studies, conservation between human and rat forms of OX-A and OX1R is very high, namely 100 and 94%, respectively.46 Finally, preliminary studies in humans found associations between orexin receptor genetic polymorphisms and PD with AG,62,63 and increased OX in the cerebrospinal fluid (CSF) of individuals with panic symptoms.59 Long-term therapy with the SSRI sertraline, a recommended anti-panic drug, reduced CSF OX levels in depressed patients, whereas treatment with bupropion, an antidepressant without anti-panic activity, did not.64
In conclusion, the ability of OX1R antagonists to decrease hypersensitivity to CO2/NaLac (a biomarker of human panic7,16) in animal models encouraged future development of these compounds as potential novel treatment for PAs in patients with PD. Although very limited, preliminary clinical findings supported this possibility, suggesting that compounds such as JNJ-61393115 deserve further clinical investigation. We did not find ongoing clinical trials with OXR1 antagonists for PD. In contrast, a pilot, randomized placebo-controlled clinical trial with the dual OX1/2R antagonist suvorexant, an approved medication to treat insomnia, is active to assess its effects on OX blood levels and response to 35% CO2 challenge in individuals with PD (ClinicalTrials.gov Identifier: NCT02593682).
Finally, OX1R antagonists have been also involved in attenuating fear-conditioning processes and enhancing of fear extinction processes.65–67 These compounds may have potential additional therapeutic effects in PD, acting on conditioned responses and panic-related phobias.7,16
Studies of Drugs Authorized for Nonpsychiatric Uses
Interesting perspectives concerning possible anti-panic properties of inhalations of xenon gas at sub-anesthetic concentrations arose from a 6 months-open-label study involving a moderately large group of patients with PD with/without psychiatric comorbidity.53 Xenon gas is an approved anesthetic, which does not produce metabolites. Xenon decreases excitatory neurotransmission by exerting inhibitory effects on multiple sites, including the glutamatergic NMDA receptor (on which xenon competes with the glycine) and other sites, such as AMPA-, 5-HT3-, and nicotinic receptors.68
The procedure used in the study rapidly reduced the global clinical severity of PD and general anxiety symptoms, resulting in the absence of PAs at 6 months after treatment, with a favorable profile of safety and tolerability. The study suffered from several limitations, including the open design, the descriptive reporting without statistical analyses of some outcome measures, the lack of definition of methods used to assess PAs during the trial. Moreover, it remained unclear to what extent the significant decrease in anxiety and panic symptoms in the sub-group of patients presenting psychiatric comorbidities was attributable to the concomitant reduction of other-than-panic type of psychiatric symptoms. Therefore, although promising, the results should be considered preliminary and provisional and require confirmation in studies applying a more rigorous methodology. Consistently, a double-blind, randomized, placebo-controlled clinical trial of xenon inhalation in patients with PD has been planned (ClinicalTrials.gov Identifier: NCT04432155), although no patients have been recruited yet.
The potential anti-panic properties of xenon gas are coherent with its pharmacodynamic features, mainly reducing glutamatergic neurotransmission. Glutamate modulates multiple brain circuits implicated in panic responses and defensive reactions, such as the PeF hypothalamic area, periaqueductal gray, and the orexin system. Moreover, it influences several panic-related neurotransmitters, such as serotonin and noradrenaline.39,69 For panic-like reactions to occur in panic-prone rats, glutamate receptors had to be activated.39 In humans, a link between panic and imbalance of glutamatergic and GABAergic systems was proposed, and associations between PD and polymorphisms in genes encoding the enzyme catalyzing the conversion of glutamate into GABA (ie, the glutamate decarboxylases) were found.70,71 Consequently, modulation of the glutamatergic system may be a favorable approach for treatment of PAs. Finally, differently from other NMDA receptor blockers such as ketamine, the xenon mechanism of action on the glycine site of the NMDA receptor68 does not produce psychotomimetic effects, thereby making it particularly suitable for translation to clinical psychiatric settings.
Results of the latest randomized placebo-controlled clinical study with DCS52 were consistent with other recent discouraging findings concerning its efficacy as a potential enhancer of CBT for PD, which we reviewed elsewhere.23 DCS, an approved antibiotic for tuberculosis, received attention in clinical psychiatric research because in preclinical studies this compound promoted extinction of conditioned fear, probably by exerting a partial agonism at the glutamatergic NMDA receptor.72
Unfortunately, the administration of DCS before a single-session CBT intervention did not differ significantly from placebo in improving any panic-specific continuous clinical measure throughout the entire 6 months-study we reviewed. A greater clinical recovery of agoraphobic avoidance at 1-month follow-up in the subsample treated with DCS was found, but the advantage disappeared at subsequent follow-up. As the sample size was very small, a type-II error cannot be excluded. However, this medication did not appear of interest as an augmentation strategy of CBT in PD treatment.
Conclusion
In conclusion, the latest preclinical findings expanded previous promising indications of OXR1 antagonists as novel-mechanism-based anti-panic compounds. Attempts of translating preclinical research to potential clinical applications are still at an early stage, as only two published Phase I studies were available, providing mixed results. However, although very limited, some encouraging clinical findings suggested the selective OXR1 antagonist JNJ-61393115 may exert anti-panic effects in humans. Overall, this body of research supports the usefulness of further clinical research on OXR1 antagonists. Very preliminary suggestions of xenon gas’s possible anti-panic effects in patients with PD emerged but need confirmation. Conversely, at present DCS does not seem promising as a CBT enhancer in PD.
Compared to our previous review on this topic,7 advances have been made, including introducing CO2 panicogenic challenge in preclinical research to increase the translational validity of the results; unfortunately, respiratory parameters, very relevant to panic, have not been considered yet. Likewise, in future clinical studies, including objective panic-related physiological parameters, such as cardiorespiratory ones, and expanding the use of 35% CO2 panic provocation challenge, may allow for more reliable conclusions about the anti-panic properties of new compounds than using the sole clinical measures. Furthermore, considering patients with PD’s phenomenological profiles, based on clinical symptoms and biological features,7 may help researchers understand if certain compounds may be particularly suitable for patients with specific clinical and/or physiological features. However, since we are unlikely to have newly approved PD medication soon, increasing efforts to develop a more personalized use of the recommended anti-panic drugs, based on each individual patient’s features, may allow for improvement of PD’s outcomes in a relatively brief time.7,23
Acknowledgments
The authors would like to thank Enago (www.enago.com) for the English language review.
Disclosure
Daniela Caldirola, Alessandra Alciati, Francesco Cuniberti, and Giampaolo Perna are scientific consultants for Medibio LTD. Giampaolo Perna has served as consultant for Lundbeck and Pfizer. The author reports no other potential conflicts of interest in this work.
References
1. Kessler RC, Petukhova M, Sampson NA, Zaslavsky AM, Wittchen HU. Twelve-month and lifetime prevalence and lifetime morbid risk of anxiety and mood disorders in the United States. Int J Methods Psychiatr Res. 2012;21(3):169–184. doi:10.1002/mpr.1359
2. de Jonge P, Roest AM, Lim CCW, et al. Cross-national epidemiology of panic disorder and panic attacks in the world mental health surveys. Depress Anxiety. 2016;33(12):1155–1177. doi:10.1002/da.22572
3. Skapinakis P, Lewis G, Davies S, Brugha T, Prince M, Singleton N. Panic disorder and subthreshold panic in the UK general population: epidemiology, comorbidity and functional limitation. Eur Psychiatry. 2011;26(6):354–362. doi:10.1016/j.eurpsy.2010.06.004
4. Batelaan N, Smit F, de Graaf R, van Balkom A, Vollebergh W, Beekman A. Economic costs of full-blown and subthreshold panic disorder. J Affect Disord. 2007;104(1–3):127–136. doi:10.1016/j.jad.2007.03.013
5. Brettschneider C, Bleibler F, Hiller TSS, et al. The allocation of resources in the care for patients with panic disorder in Germany: an excess cost analysis informing policy and science. Cost Eff Resour Alloc. 2019;17(1):9. doi:10.1186/s12962-019-0177-4
6. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders.
7. Caldirola D, Perna G. Toward a personalized therapy for panic disorder: preliminary considerations from a work in progress. Neuropsychiatr Dis Treat. 2019;15:1957–1970. doi:10.2147/NDT.S174433
8. Perna G. Understanding anxiety disorders: the psychology and the psychopathology of defence mechanisms against threats. Riv Psichiatr. 2013;48(1):73–75. doi:10.1708/1228.13618
9. Johnson PL, Federici LM, Shekhar A. Etiology, triggers and neurochemical circuits associated with unexpected, expected, and laboratory-induced panic attacks. Neurosci Biobehav Rev. 2014;46(P3):429–454. doi:10.1016/j.neubiorev.2014.07.027
10. Klein DF. False suffocation alarms, spontaneous panics, and related conditions: an integrative hypothesis. Arch Gen Psychiatry. 1993;50(4):306–317. doi:10.1001/archpsyc.1993.01820160076009
11. Graeff FG, Del-Ben CM. Neurobiology of panic disorder: from animal models to brain neuroimaging. Neurosci Biobehav Rev. 2008;32(7):1326–1335. doi:10.1016/j.neubiorev.2008.05.017
12. Canteras NS, Graeff FG. Executive and modulatory neural circuits of defensive reactions: implications for panic disorder. Neurosci Biobehav Rev. 2014;46(P3):352–364. doi:10.1016/j.neubiorev.2014.03.020
13. Preter M, Klein DF. Lifelong opioidergic vulnerability through early life separation: a recent extension of the false suffocation alarm theory of panic disorder. Neurosci Biobehav Rev. 2014;46(P3):345–351. doi:10.1016/j.neubiorev.2014.03.025
14. Perna G, Caldirola D, Bellodi L. Panic disorder: from respiration to the homeostatic brain. Acta Neuropsychiatr. 2004;16(2):57–67. doi:10.1111/j.0924-2708.2004.0080.x
15. Esquivel G, Schruers KR, Maddock RJ, Colasanti A, Griez EJ. Acids in the brain: a factor in panic? J Psychopharmacol. 2010;24(5):639–647. doi:10.1177/0269881109104847
16. Perna G, Caldirola D. Is panic disorder a disorder of physical fitness? A heuristic proposal. F1000Research. 2018;7:294. doi:10.12688/f1000research.12788.1
17. Paul ED, Johnson PL, Shekhar A, Lowry CA. The Deakin/Graeff hypothesis: focus on serotonergic inhibition of panic. Neurosci Biobehav Rev. 2014;46(P3):379–396. doi:10.1016/j.neubiorev.2014.03.010
18. Dresler T, Guhn A, Tupak SV, et al. Revise the revised? New dimensions of the neuroanatomical hypothesis of panic disorder. J Neural Transm. 2013;120(1):3–29. doi:10.1007/s00702-012-0811-1
19. Feinstein JS, Buzza C, Hurlemann R, et al. Fear and panic in humans with bilateral amygdala damage. Nat Neurosci. 2013;16(3):270–272. doi:10.1038/nn.3323
20. Gorman JM, Kent JM, Sullivan GM, Coplan JD. Neuroanatomical hypothesis of panic disorder, revised. Am J Psychiatry. 2000;157(4):493–505. doi:10.1176/appi.ajp.157.4.493
21. Perna G, Guerriero G, Brambilla P, Caldirola D. Panic and the Brainstem: clues from neuroimaging studies. CNS Neurol Disord Drug Targets. 2014;13(6):1049–1056. doi:10.2174/1871527313666140612112923
22. Goossens L, Leibold N, Peeters R, et al. Brainstem response to hypercapnia: a symptom provocation study into the pathophysiology of panic disorder. J Psychopharmacol. 2014;28(5):449–456. doi:10.1177/0269881114527363
23. Caldirola D, Alciati A, Riva A, Perna G. Are there advances in pharmacotherapy for panic disorder? A systematic review of the past five years. Expert Opin Pharmacother. 2018;19(12):1357–1368. doi:10.1080/14656566.2018.1504921
24. Caldirola D, Alciati A, Dacco S, Micieli W, Perna G. Relapse prevention in panic disorder with pharmacotherapy: where are we now? Expert Opin Pharmacother. 2020;21(14):1699–1711. doi:10.1080/14656566.2020.1779220
25. American Psychiatric Association. Practice Guideline for the Treatment of Patients with Panic Disorder.
26. National Institute for Health and Care Excellence. Generalised anxiety disorder and panic disorder (with or without agoraphobia) in adults: management. NICE. 2011. Available from:https://www.nice.org.uk/guidance/cg113.
27. Bandelow B, Michaelis S, Wedekind D. Treatment of anxiety disorders. Dialogues Clin Neurosci. 2017;19(2):93–107.
28. Perna G, Caldirola D. Management of treatment-resistant panic disorder. Curr Treat Options Psychiatry. 2017;4(4):371–386. doi:10.1007/s40501-017-0128-7
29. Bandelow B. Current and novel psychopharmacological drugs for anxiety disorders. Adv Exp Med Biol. 2020;1191:347–365. doi:10.1007/978-981-32-9705-0_19
30. Zugliani MM, Cabo MC, Nardi AE, Perna G, Freire RC. Pharmacological and neuromodulatory treatments for panic disorder: clinical trials from 2010 to 2018. Psychiatry Investig. 2019;16(1):50–58. doi:10.30773/pi.2018.12.21.1
31. Perna G, Alessandra A, Raffaele B, et al. Is there room for second-generation antipsychotics in the pharmacotherapy of panic disorder? A Systematic review based on PRISMA guidelines. Int J Mol Sci. 2016;17(4):551. doi:10.3390/ijms17040551
32. Shah A, Northcutt J. An open-label, flexible dose adaptive study evaluating the efficacy of vortioxetine in subjects with panic disorder. Ann Gen Psychiatry. 2018;17. doi:10.1186/s12991-018-0190-6.
33. Perna G, Schruers K, Alciati A, Caldirola D. Novel investigational therapeutics for panic disorder. Expert Opin Investig Drugs. 2015;24(4):491–505. doi:10.1517/13543784.2014.996286
34. Sartori SB, Singewald N. Novel pharmacological targets in drug development for the treatment of anxiety and anxiety-related disorders. Pharmacol Ther. 2019;204:107402. doi:10.1016/j.pharmthera.2019.107402
35. Liberati A, Altman DG, Tetzlaff J, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. J Clin Epidemiol. 2009;62(10):e1–e34. doi:10.1016/j.jclinepi.2009.06.006
36. European Medicines Agency. EU Clinical Trials Register. Available from: https://www.clinicaltrialsregister.eu/.
37. US National Library of Medicine. ClinicalTrials.gov. Available from: http://clinicaltrials.gov/.
38. Leibold NK, Van Den Hove DLAA, Viechtbauer W, et al. CO2 exposure as translational cross-species experimental model for panic. Transl Psychiatry. 2016;6(9):e885–e885. doi:10.1038/tp.2016.162
39. Johnson PL, Shekhar A. An animal model of panic vulnerability with chronic disinhibition of the dorsomedial/perifornical hypothalamus. Physiol Behav. 2012;107(5):686–698. doi:10.1016/j.physbeh.2012.03.016
40. World Health Organisation. International Statistical Classification of Diseases and Related Health Problems.
41. World Health Organisation. ICD-10: International Statistical Classification of Diseases and Related Health Problems: Tenth Revision.
42. World Health Organisation. International Statistical Classification of Diseases and Related Health Problems.
43. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders.
44. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders
45. Bonaventure P, Yun S, Johnson PL, et al. A selective orexin-1 receptor antagonist attenuates stress-induced hyperarousal without hypnotic effects. J Pharmacol Exp Ther. 2015;352(3):590–601. doi:10.1124/jpet.114.220392
46. Johnson PL, Federici LM, Fitz SD, et al. Orexin 1 and 2 Receptor Involvement in CO2-INDUCED panic-associated behavior and autonomic responses. Depress Anxiety. 2015;32(9):671–683. doi:10.1002/da.22403
47. McElhinny CJ, Lewin AH, Mascarella SW, Runyon S, Brieaddy L, Carroll FI. Hydrolytic instability of the important orexin 1 receptor antagonist SB-334867: possible confounding effects on in vivo and in vitro studies. Bioorganic Med Chem Lett. 2012;22(21):6661–6664. doi:10.1016/j.bmcl.2012.08.109
48. Bonaventure P, Dugovic C, Shireman B, et al. Evaluation of JNJ-54717793 a novel brain penetrant selective orexin 1 receptor antagonist in two rat models of panic attack provocation. Front Pharmacol. 2017;8(JUN):1–13. doi:10.3389/fphar.2017.00357
49. Salvadore G, Bonaventure P, Shekhar A, et al. Translational evaluation of novel selective orexin-1 receptor antagonist JNJ-61393215 in an experimental model for panic in rodents and humans. Transl Psychiatry. 2020;10:1. doi:10.1038/s41398-020-00937-9
50. Kaufmann P, Ort M, Golor G, Kornberger R, Dingemanse J. Multiple-dose clinical pharmacology of the selective orexin-1 receptor antagonist act-539313. Prog Neuropsychopharmacol Biol Psychiatry. 2020;(xxxx):110166. doi:10.1016/j.pnpbp.2020.110166
51. Diaper A, Osman-Hicks V, Rich AS, et al. Evaluation of the effects of venlafaxine and pregabalin on the carbon dioxide inhalation models of generalised anxiety disorder and panic. J Psychopharmacol. 2013;27(2):135–145. doi:10.1177/0269881112443742
52. Reinecke A, Nickless A, Browning M, Harmer CJ. Neurocognitive processes in D-cycloserine augmented single-session exposure therapy for anxiety: a randomized placebo-controlled trial. Behav Res Ther. 2020;129(September2019):103607. doi:10.1016/j.brat.2020.103607
53. Dobrovolsky A, Ichim TE, Ma D, Kesari S, Bogin V. Xenon in the treatment of panic disorder: an open label study. J Transl Med. 2017;15(1):137. doi:10.1186/s12967-017-1237-1
54. Meuret AE, Rosenfield D, Wilhelm FH, et al. Do unexpected panic attacks occur spontaneously? Biol Psychiatry. 2011;70(10):985–991. doi:10.1016/j.biopsych.2011.05.027
55. Tenorio-Lopes L, Fournier S, Henry MS, Bretzner F, Kinkead R. Disruption of estradiol regulation of orexin neurons: a novel mechanism in excessive ventilatory response to CO2 inhalation in a female rat model of panic disorder. Transl Psychiatry. 2020;10:1. doi:10.1038/s41398-020-01076-x
56. Grafe LA, Bhatnagar S. The contribution of orexins to sex differences in the stress response. Brain Res. 2020;1731:145893. doi:10.1016/j.brainres.2018.07.026
57. Johnson PL, Fitz SD, Hollis JH, et al. Induction of c-Fos in ’panic/defence’-related brain circuits following brief hypercarbic gas exposure. J Psychopharmacol. 2011;25(1):26–36. doi:10.1177/0269881109353464
58. Johnson PL, Samuels BC, Fitz SD, Lightman SL, Lowry CA, Shekhar A. Activation of the orexin 1 receptor is a critical component of CO 2-mediated anxiety and hypertension but not bradycardia. Neuropsychopharmacology. 2012;37(8):1911–1922. doi:10.1038/npp.2012.38
59. Johnson PL, Truitt W, Fitz SD, et al. A key role for orexin in panic anxiety. Nat Med. 2010;16(1):111–115. doi:10.1038/nm.2075
60. Nattie E, Li A. Respiration and autonomic regulation and orexin. In: Progress in Brain Research. Vol. 198. Elsevier B.V.;2012:25–46. doi:10.1016/B978-0-444-59489-1.00004-5
61. Johnson PL, Samuels BC, Fitz SD, et al. Orexin 1 receptors are a novel target to modulate panic responses and the panic brain network. Physiol Behav. 2012;107(5):733–742. doi:10.1016/j.physbeh.2012.04.016
62. Annerbrink K, Westberg L, Olsson M, et al. Panic disorder is associated with the Val308Iso polymorphism in the hypocretin receptor gene. Psychiatr Genet. 2011;21(2):85–89. doi:10.1097/YPG.0b013e328341a3db
63. Gottschalk MG, Richter J, Ziegler C, et al. Orexin in the anxiety spectrum: association of a HCRTR1 polymorphism with panic disorder/agoraphobia, CBT treatment response and fear-related intermediate phenotypes. Transl Psychiatry. 2019;9(1):75. doi:10.1038/s41398-019-0415-8
64. Salomon RM, Ripley B, Kennedy JS, et al. Diurnal variation of cerebrospinal fluid hypocretin-1 (Orexin-A) levels in control and depressed subjects. Biol Psychiatry. 2003;54(2):96–104. doi:10.1016/S0006-3223(02)01740-7
65. Abreu AR, Molosh AI, Johnson PL, Shekhar A. Role of medial hypothalamic orexin system in panic, phobia and hypertension. Brain Res. 2020;1731. doi:10.1016/j.brainres.2018.09.010.
66. Soya S, Sakurai T. Orexin as a modulator of fear-related behavior: hypothalamic control of noradrenaline circuit. Brain Res. 2020;1731:146037. doi:10.1016/j.brainres.2018.11.032
67. Soya S, Takahashi TM, McHugh TJ, et al. Orexin modulates behavioral fear expression through the locus coeruleus. Nat Commun. 2017;8:1. doi:10.1038/s41467-017-01782-z
68. Zhou C. General anesthesia mediated by effects on ion channels. World J Crit Care Med. 2012;1(3):80. doi:10.5492/wjccm.v1.i3.80
69. Harvey BH, Shahid M. Metabotropic and ionotropic glutamate receptors as neurobiological targets in anxiety and stress-related disorders: focus on pharmacology and preclinical translational models. Pharmacol Biochem Behav. 2012;100(4):775–800. doi:10.1016/j.pbb.2011.06.014
70. Hettema JM, An SS, Neale MC, et al. Association between glutamic acid decarboxylase genes and anxiety disorders, major depression, and neuroticism. Mol Psychiatry. 2006;11(8):752–762. doi:10.1038/sj.mp.4001845
71. Long Z, Medlock C, Dzemidzic M, Shin YW, Goddard AW, Dydak U. Decreased GABA levels in anterior cingulate cortex/medial prefrontal cortex in panic disorder. Prog Neuro Psychopharmacol Biol Psychiatry. 2013;44:131–135. doi:10.1016/j.pnpbp.2013.01.020
72. Hofmann SG. D-cycloserine for treating anxiety disorders: making good exposures better and bad exposures worse. Depress Anxiety. 2014;31(3):175–177. doi:10.1002/da.22257
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.