")
Back to Journals » Journal of Experimental Pharmacology » Volume 13
Experimental Dopamine Reuptake Inhibitors in Parkinson’s Disease: A Review of the Evidence
Authors Müller T
Received 8 January 2021
Accepted for publication 3 March 2021
Published 29 March 2021 Volume 2021:13 Pages 397—408
DOI https://doi.org/10.2147/JEP.S267032
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Bal Lokeshwar
Thomas Müller
Department of Neurology, St. Joseph Hospital Berlin-Weissensee, Berlin, 13088, Germany
Correspondence: Thomas Müller
Department of Neurology, St. Joseph Hospital Berlin-Weissensee, Gartenstr. 1, Berlin, 13088, Germany
Tel +49 30 92790223
Fax +49 30 92790703
Email [email protected]
Abstract: Parkinson’s disease (PD) is the second most chronic neurodegenerative disorder worldwide. Deficit of monoamines, particularly dopamine, causes an individually varying compilation of motor and non-motor features. Constraint of presynaptic uptake extends monoamine stay in the synaptic cleft. This review discusses possible benefits of dopamine reuptake inhibition for the treatment of PD. Translation of this pharmacologic principle into positive clinical study results failed to date. Past clinical trial designs did not consider a mandatory, concomitant stable inhibition of glial monoamine turnover, i.e. with monoamine oxidase B inhibitors. These studies focused on improvement of motor behavior and levodopa associated motor complications, which are fluctuations of motor and non-motor behavior. Future clinical investigations in early, levodopa- and dopamine agonist naïve patients shall also aim on alleviation of non-motor symptoms, like fatigue, apathy or cognitive slowing. Oral levodopa/dopa decarboxylase inhibitor application is inevitably necessary with advance of PD. Monoamine reuptake (MRT) inhibition improves the efficacy of levodopa, the blood brain barrier crossing metabolic precursor of dopamine. The pulsatile brain delivery pattern of orally administered levodopa containing formulations results in synaptic dopamine variability. Ups and downs of dopamine counteract the physiologic principle of continuous neurotransmission, particularly in nigrostriatal, respectively mesocorticolimbic pathways, both of which regulate motor respectively non-motor behavior. Thus synaptic dopamine pulsatility overwhelms the existing buffering capacity. Onset of motor and non-motor complications occurs. Future MRT inhibitor studies shall focus on a stabilizing and preventive effect on levodopa related fluctuations of motor and non-motor behavior. Their long-term study designs in advanced levodopa treated patients shall allow a cautious adaptation of oral l-dopa therapy combined with a mandatory inhibition of glial monoamine turnover. Then the evidence for a preventive and beneficial, symptomatic effect of MRT inhibition on motor and non-motor complications will become more likely.
Keywords: dopamine reuptake, monoamines, monoamine reuptake inhibition, Parkinson’s disease; PD
Plain Language Summary
Dopamine reuptake inhibition extends the stay of dopamine in the synaptic left. This pharmacologic principle may improve dopamine substitution in patients with Parkinson’s disease (PD). Experimental researchers described promising positive outcomes on the efficacy of inhibition of dopamine-, respectively monoamine reuptake (MRT). The translation of these findings into corresponding clinical study results for the treatment of patients with PD failed in the past. Further clinical development of MRT inhibition was suspended. One of several reasons was the missing clinical research focus on the effects of MRT inhibition on non-motor symptoms, like fatigue or apathy. Mandatory inhibition of glial inhibition of monoamine metabolism is a hypothetical but essential precondition for the efficacy of MRT inhibitors. Aforementioned reflections shall be considered, if the efficacy of the MRT inhibitor is again investigated in patients with PD. Resurgence of clinical research is warranted on the efficacy of MRT inhibition as a promising therapy approach for patients with PD.
Introduction
Parkinson’s disease (PD), the world's second most chronic neurodegenerative disorder, is characterized by a decline of mainly nigrostriatal dopamine concentrations.1 Dopamine exerts its effects through different pathways in the brain under physiological conditions. The nigrostriatal one sends axons from the substantia nigra to the striatum and regulates movement. The mesocortical one originates from ventral tegmental brain regions, dominates the prefrontal cortex and is involved in learning, memory and executive function.2,3 The mesolimbic one projects from the ventral tegmental area of the midbrain and influences the ventral striatum, olfactory tubercle and parts of the limbic system. This route is associated with reward, emotion, and memory. The tubular infundibular pathway starts from the arcuate nucleus of the hypothalamus and projects to the median eminence of the hypothalamus. This one is involved in neuroendocrine regulation and wake-sleep cycle generation. Dopamine deficiency in PD mainly concerns the nigrostriatal route.4,5 It is responsible for the slowly evolving manifestation of disturbed motor behavior. The dopamine decline of other brain regions contributes to the onset of non-motor properties in PD.1–3 Therefore, dopamine substitution not only ameliorates the main motor symptoms akinesia, rigidity and tremor but also non-motor features of PD. This treatment concept was inaugurated in the early 1960s with the application of the blood brain barrier trespassing dopamine precursor l-3,4-dihydroxyphenylalanine (l-dopa) in PD patients.6–8 L-dopa is looked upon as the most efficacious and best tolerated PD drug. Oral l-dopa/dopa decarboxylase inhibitor (DDC-I) application is inevitably necessary with advance of PD. In the long term, chronic administration of oral l-dopa formulations in a fixed combination with inhibitors of the main metabolizing l-dopa enzymes results in onset of so-called motor complications.9 When the disease progresses, the duration of l-dopa response shortens in addition to the short plasma l-dopa half life. Fluctuations of plasma l-dopa plasma levels and of associated dopamine brain levels are not buffered any more within the dopaminergic nigrostriatal system. One consequence is onset of initial predictable “OFF”-periods. They are related to the intake of oral l-dopa formulations and characterized by reappearance of motor symptoms due to less intense stimulation of postsynaptic nigrostriatal neurons with dopamine.9 Dysregulation of gastrointestinal l-dopa transport further complicates oral l-dopa therapy. L-dopa is absorbed in the duodenal tract. The transfer of oral l-dopa formulations from the stomach to the duodenal tract depends on dopamine dependent negative feedback mechanisms. Too high l-dopa- and accordingly dopamine concentrations in the duodenal tract reduce the gastrointestinal motility.10–12 Transport of oral l-dopa containing formulations becomes more and more irregular and independent of previous intake particularly during higher and more frequent oral l-dopa dosing.13 The resulting erratic and delayed gastric emptying of oral l-dopa formulations in combination with further advance of PD causes unpredictability of “OFF”-intervals.10–12 At this stage, “OFF”-periods appear more sudden and independent of previous L-dopa intake. “OFF”-times are often compensated by additional intakes of soluble or inhaled l-dopa formulations or by an increase of l-dopa applications.9 Both strategies elevate the likelihood of transient too high dopamine levels in the synaptic cleft. As a result, an over stimulation of postsynaptic dopamine receptors occurs and results in appearance of involuntary movements, termed as dyskinesia. These long-term side effects complicate the efficiency of oral l-dopa therapy. Nowadays, oral l-dopa formulations are administered with preponderant peripheral acting blockers (I) of peripheral dopa decarboxylase (DDC) and/or catechol-O-methyltransferase (COMT). DDC-I and COMT-I prolong the half life, decrease plasma fluctuations and thus improve the efficacy of l-dopa.9,14 Inhibition of the dopamine metabolizing monoamine oxidase B (MAO-B) enzyme activity in the brain also counteracts motor impairment in PD patients. All these therapeutic concepts help to spare oral l-dopa dosing.15,16 The future availability of subcutaneous l-dopa/DDC-I pump systems will noticeably reduce the issue of l-dopa associated fluctuations with related disturbances of motor- and non-motor behavior in PD.9,17 However, chronic L-dopa therapy has further still under recognized metabolic drawbacks. Only DDC constraint decreases the physiologic methylation potential, since L-dopa is consequently metabolized via COMT mainly. This reaction consumes methyl groups. This enzymatic process impairs the methyl group dependent detoxification potential and methyltransferases related processes. Both of them are important for many metabolic processes in humans, such as adequate gene function (Figure 1). Therefore supplementation with methyl group donating vitamins, i.e., B6, B12, folic acid, may be beneficial during long-term l-dopa application.18,19 A further drawback is the decrease of central free radical scavenging capacity due to neuronal dopamine turnover. One pathway is uptake and degradation by MAO and COMT in glial cells. Another route for dopamine released into the synapse is reuptake for reuse and store in presynaptic neuronal vesicles with dopamine transporter system (DAT). Dopamine out of these storage vesicles can easily undergo auto-oxidation via neuronal MAO-B. This process is associated with PD pathogenesis in terms of dopamine metabolism related oxidative stress generation and iron deposition.9,15 Iron accumulates in affected neurons, as iron plays a vital role in various physiological functions including DNA synthesis, mitochondrial respiration, and oxygen transport. Iron as the most abundant metal in the brain also acts as a cofactor for tyrosine hydroxylase (TH). This enzyme is the rate-limiting step in the synthesis of dopamine. Iron is also necessary for dopamine oxidation in neurons and microglia in the substantia nigra of PD patients.20 Increase of iron deposition accelerates neuronal dopamine turnover, but generates synthesis of reactive oxygen species. This so-called “oxidative stress” promotes the aggregation and misfolding of proteins, particularly α-synuclein. This process plays an eminent role in the pathophysiology of chronic neurodegeneration in PD.21–23 In summary, these possible metabolic consequences of too high l-dopa dosing, respectively dopamine overflow, may accelerate progression of PD, the general ageing process and facilitate onset of dementia and neuropathy.9 All these drawbacks of l-dopa therapy caused research on treatment alternatives for dopamine substitution. One concept was the development of dopamine agonists. They directly stimulate postsynaptic dopamine receptors more continuously in the brain and show a better affinity for the presynaptic located dopamine autoreceptor at the membrane of dopamine synthesizing neurons.24 Their side effect profile is inferior to l-dopa due to more frequent occurrence of nausea, dizziness, and edema. The neurotransmission of dopamine is also enhanced by decreased presynaptic release of glutamate with the MAO-B inhibitor safinamide. An alternative is the postsynaptic antagonism of glutamate receptors, located at postsynaptic dopaminergic neurons. This is most popular mode of action of amantadine. This compound also acts as a dopamine reuptake inhibitor (DRT-I).25,26 This review aims to discuss possible benefits of DRT-I’s for PD patients and putative research clinical research misconceptions of the past.
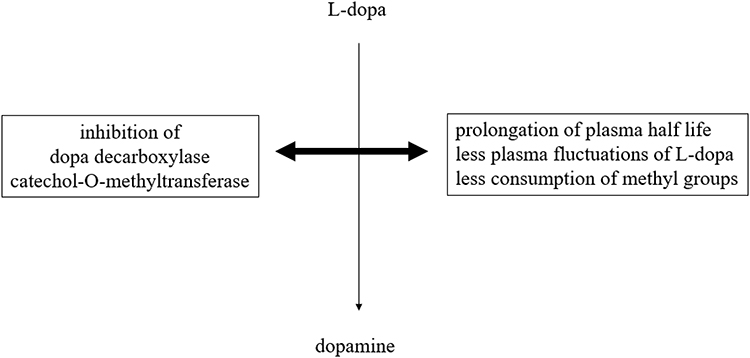 |
Figure 1 Simplified schematic illustration of peripheral l-dopa metabolic pathways in association with peripheral metabolic and pharmacokinetics effects. |
Methods
A systematic literature research was executed with the terms dopamine, reuptake, inhibitor and PD in the “Pubmed” database. A structured selection of references or studies was not performed.
Regulation of Dopamine Neurotransmission
The neuronal and glial mechanisms involved in dopamine transmission suggest, that an increase of the interval spent by dopamine in the synaptic cleft compensates the dopamine deficit in PD patients. This effect is achieved by neuronal and glial MAO-B- and mainly glial COMT-inhibition.15,27 Both modes of actions generate higher levels and longer presence of dopamine in the synaptic cleft due to the declined glial dopamine turnover (Figure 2). A further strategy is blocking of the presynaptic located dopamine autoreceptor activity, which acts as a sensor for neurotransmission under physiological conditions.5 This system modulates the activity of dopamine synthesizing neurons and controls dopamine synthesis, its release to the synaptic cleft and its reuptake. The presynaptic located dopamine transporter is the decisive protein for the dopamine transport within the cell following reuptake from the synaptic cleft. Too high synaptic dopamine levels cause a down regulation of presynaptic dopamine synthesis (Figure 2). It depends on the presynaptic dopamine generating enzyme activities.5 TH converts tyrosine to l-dopa. DDC metabolizes l-dopa to dopamine. Prolonged autoreceptor activation causes a down-regulation of TH following and leads to reduced filling of pre-synaptic dopamine vesicles.24,28 Additionally DAT activity, which is responsible for removal of dopamine from the synaptic cleft, goes up.5 The driving force for dopamine transporter mediated dopamine reuptake is the ion concentration gradient generated by the plasma membrane Na+K+ATPase. Too low dopamine concentrations enhance presynaptic dopamine generation and down regulate intracellular DAT activity. Accordingly, lack of dopamine reuptake induces an increase of extracellular dopamine levels.5
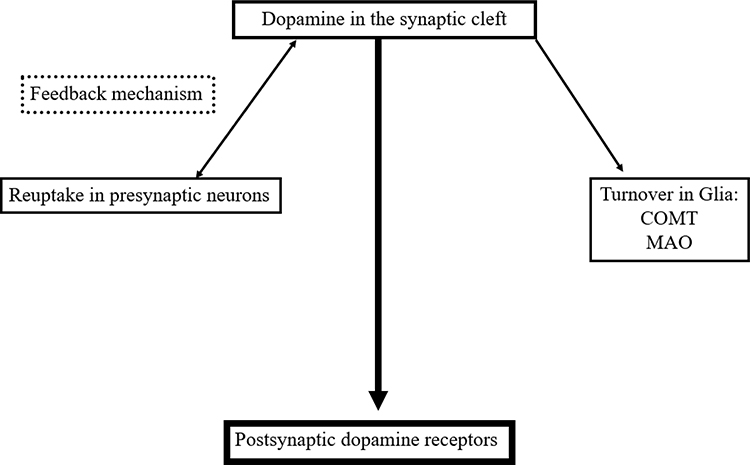 |
Figure 2 Simplified schematic drawing of consumption- and regulation mechanisms of dopamine in the synaptic cleft. Abbreviations: MAO, monamine oxidase; COMT, catechol-O-methyltransferase. |
Altered Motor- and Non-Motor Behavior in PD
PD patients suffer from a slowly increasing, unbalanced neurotransmission as consequence of chronic neurodegenerative processes. Not only dopamine deficiency but also deficits of other biogenic amines support manifestation of motor and associated non-motor symptoms.1 Typical early, non-motor PD signs are onset of apathy, anxiety, depression, concentration deficits, slowness of thinking and reaction, anhedonia, deterioration of cognitive abilities, sleep disturbances, fatigue and unspecific dull pain syndromes. All of them are not PD specific and may at least partially even occur before the initial transient onset of motor symptoms. They mainly reflect monoamine deficiencies in the nigrostriatal and mesocorticolimbic system.1 This early, so-called “prodromal” phase is an under recognized phenomenon of a slowly evolving chronic neurodegenerative process in the brain. It is also characterized by a probable compensatory decline of DAT activity.29–31 Diagnosis of PD is mostly made with the onset of motor symptoms. At this stage 70% of dopaminergic nigrostriatal neurons are already gone.1 The compensation capacity of the human brain, the so-called “neuroplasticity” phenomenon, is overwhelmed by the individual different neurotransmitter deficiencies and results in a heterogeneous onset of symptoms.1 Treatment with MAO-B inhibitors is mostly initiated at this stage particularly in PD patients with high life expectancy.15 They counteract the deficiency of biogenic amines. Hypothetically, inhibitors of MRT may complement this therapeutic approach. As a next step, dopamine substitution is initiated and adapted to the neurodegenerative process, i.e., with dopamine agonists. Long half life and affinity to postsynaptic dopamine receptors are the essential determinants of their efficacy. The affinity of dopamine agonists to the presynaptic dopaminergic autoreceptor is stronger than to the postsynaptic one.24 Dopamine agonists are also involved to a varying extent in stimulation of norepinephrine and serotonergic autoreceptors and postsynaptic receptors, i.e., piribedil or pramipexole.32,33 As consequence, the endogenous presynaptic synthesis of biogenic amines is down regulated. Thus it is rather unlikely to demonstrate clinical meaningful benefits on motor- and non-motor behavior in only treated dopamine agonist treated PD patients with inhibition of DAT or with MRT. Sooner or later with advance of PD, l-dopa has to be initiated and added to an already existing dopamine substitution regimen. L-dopa is the most efficacious and best tolerated compound for PD treatment. The risk for an at least transient, too much elevated dopaminergic neurotransmission goes up as a result of the fluctuating brain delivery of orally administered l-dopa. Onset of L-dopa dosing dependent motor complications is sometimes observed within months.34 This elevation of dopamine substitution with l-dopa may support manifestation of dopamine dysregulation syndromes, particularly in patients with certain preexisting and predisposing personality features.35,36 The most popular one is an increased susceptibility to addictive behavior, i.e. repeat l-dopa intake, punding, shopping, etc. Particularly, the dopaminergic pathways of the mesocorticolimbic system are looked upon as addiction generating reward centers. Thus, too high dopaminergic stimulation not only supports onset of dyskinesia in the motor system, it may also induce specific neuropsychiatric phenomena.35,36 Their intensity and frequency goes up with advance of PD, which is characterized by a diminishing dopamine buffering capacity and loss of autoreceptor function.24 Fluctuating l-dopa levels aggravate these interplay between motor- and non-motor symptoms in PD.24,37 Advanced PD patients better tolerate dyskinesia and associated non-motor features, i.e. irritability, mania or psychosis. They dislike dopamine deficiency associated with impaired motor behavior and features like depression, apathy and cognitive deficits.1 Therefore they ask for higher dosing of dopamine substituting compounds for alleviation of OFF-periods. Mostly they perform additional l-dopa/DDC-I intakes.38–40 Endogenous regulation of monoamine neurotransmission more and more looses its compensating and buffering capacity.5 Due to temporary, treatment related overflow of l-dopa and dopamine, l-dopa is also converted in serotonin generating neurons.5,41 Particular, neuronal dopamine reuptake- and intracellular dopamine transporter activity becomes irregular.5 Hypothetically, inhibitors of MRT may have a stabilizing and thus beneficial effect.5 Particularly, combination with novel extended release oral L-dopa capsules, such as Rytary® (formerly known as IPX066), may hypothetically provide a more continuous brain delivery of l-dopa.42,43 In conclusion, these considerations suggest that use and efficacy of DRT-I depend on specific stages of PD with their associated heterogeneous expression of motor and non-motor symptoms and the pharmacology of further dopamine substituting compounds.
Experimental and Clinical Research with DRT-I
Experimental PD models may mirror this complex interplay. They mainly focus on the loss of dopamine synthesizing nigral neurons. PD models with toxins, i.e., 6-OH-dopamine, rotenone or 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), induce PD like syndromes. The 6-OHDA acute model, although it is not progressive, has been successfully used to mimic the early versus late dopamine depletion stages in site specific brain regions. It was able to identify brain region specific neurobiological mechanisms that might be responsible for the complexity of symptom manifestation in PD. It may also suitable for a better future understanding of l-dopa-induced motor complications, such as dyskinesia.44–46 MPTP-, α-synuclein- and Parkin animal models recapitulate the progressive loss of dopamine neurons, the involvement of cortical and subcortical regions and other aspects of disease progression, even those involving the mesocorticolimbic system.47–49 These employed in vitro and in vivo PD models may mirror the slow progression of neuronal dying, which asks for continuous adaptation of dopamine substitution in the clinical practice of the maintenance of PD patients. Nevertheless translation of promising positive experimental research outcomes with DRT-I was complex, as i.e. co-administration of the various available PD drugs, i.e. MAO-B inhibitors, was only partially considered. This might have been one additional reason for the high failure risk in previous clinical trials with the various types of biogenic amine reuptake inhibitors.
Selective DRT-I
GBR-12,909 (vanoxerine; 1-[2-[bis-(4-fluorophenyl)methoxy]ethyl]-4-(3-phenylpropyl)piperazine dihydrochloride) and SEP-22,8791 have only been investigated in the 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridin (MPTP)-lesioned monkey PD model. Acute application of GBR-12,909 improved motor behavior in monotherapy in the MPTP-lesioned common marmoset. No dyskinesia were observed (Table 1).28,50
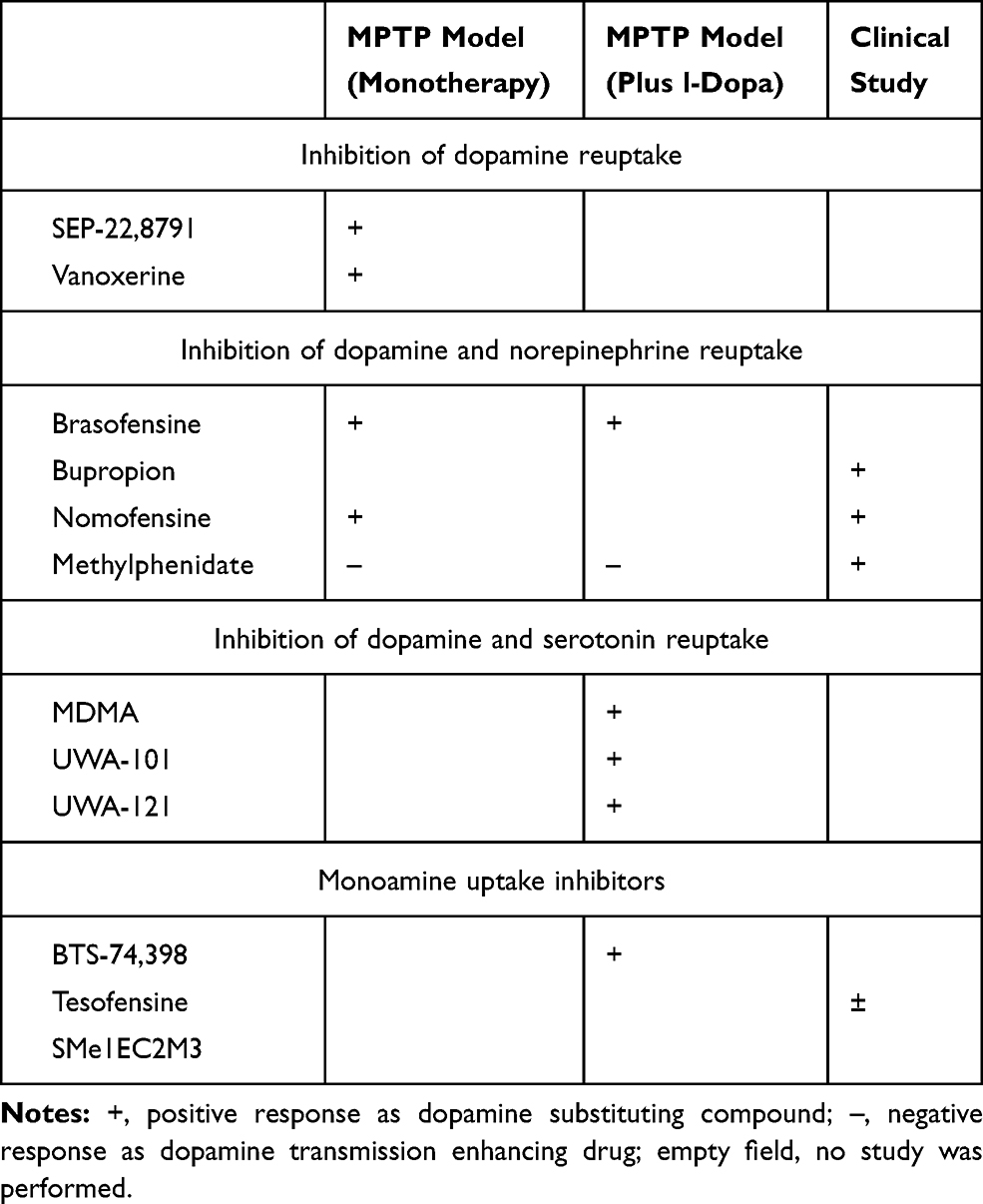 |
Table 1 Dopamine Substitution Enhancing Effects of Various Kinds of Uptake Inhibitors |
Constraint of Dopamine and Norepinephrine Uptake
The DRT-I brasofensine, bupropion, methylphenidate, and nomifensine additionally inhibit norepinephrine reuptake (NRT) activity. Brasofensine improved PD like motor symptoms in monotherapy and in combination with l-dopa. Under both conditions, dyskinesia onset was not observed.51 Nomifensine as monotherapy ameliorated motor behavior, whereas bupropion had no effect in the MPTP model on motor symptoms (Table 1).28
Clinical Trials
Dual DRT/NRT-I’s were tested in clinical PD studies. In a small trial, Brasofensine did not improve the effects of l-dopa on motor impairment. It ameliorated motor symptoms in early PD patients for a short interval. This positive effect did not last and was gone after 14 days.52,53 Bupropion improved PD symptoms. It may even exacerbate onset of dyskinesia as result of its dopamine neurotransmission enhancing efficacy.54–56 To date, it was repeatedly recommended as treatment of choice for depression in PD patients.57–59 Monotherapy with nomifensine showed positive effects in l-dopa naïve PD patients. Combination of nomifensine with l-dopa provided controversial outcomes.60–63 Methylphenidate was efficacious in combination with l-dopa. It was suggested as a treatment possibility for apathy and fatigue in PD patients (Table 1).4,64–67
Dual Inhibition of Dopamine and Serotonin Uptake
UWA-101 [2-(1,3-benzodioxol-5-yl)-1-cyclopropyl-N-methylethanamine], UWA-121 and S-3,4-methylenedioxymethamphetamine (MDMA) not only constrain DRT, but also the 5-hydroxytryptamine (serotonin, 5-HT) reuptake (SRT).68–70 To date, they were examined only in experimental PD models. They showed mild positive effects, such as extension of “ON”-time in PD patients with motor complications (Table 1).68–70
Triple Blocking of DRT, NRT, and SRT
BTS-74,398 [1-([1-(3,4-dichlorophenyl)cyclobutyl]-2-(3-diaminethylaminopropylthio)ethanone] and tesofensine are nonselective monoamine transporter inhibitors. BTS-74,398 was positive in the MPTP model. This drug did not further enhance the effects of l-dopa co-administration. Tesofensine was only applied in clinical trials to date. Sustained effects were not reported in monotherapy following an initial improvement of PD symptoms. There were mild effects in more advanced PD patients (Table 1).71,72
DRT-I in PD: Current Situation
Following the more or less disappointing clinical results,41,52,53,71–73 research on the concept of MRT inhibition in PD did not go on. Moreover the effects of brasofensine and tesofensine in monotherapy seemed to fade over time. This effect further scrutinized the potential use of these agents in PD patients.53,62,71,73 To date, the main focus of DRT-I research was to demonstrate an improved motor behavior similar to the findings with the MPTP model. Researchers also aimed to develop DRT-I as dyskinesia preventing or -ameliorating therapies in conjunction with chronic oral l-dopa/DDC-I application. One did not consider the following hypothesis. Blocking of MRT activity in presynaptic neurons with subsequent longer maintenance of monoamines in the synaptic cleft may be compensated by an enzymatic up regulation of glial monoamine turnover via MAO and COMT in glial cells (Figure 2).5,15 This theory is in line with the clinical observation of the fading symptomatic effect of brasofensine and tesofensine in monotherapy in PD patients (Table 1).
Use of DRT-I: Future Aspects in Early PD Patients
The former clinical research concept for the DRT-I use should be reconsidered in PD patients. These compounds may probably only add an additional benefit combined with a mandatory MAO-B inhibitor application in early, l-dopa- and dopamine agonist naïve PD patients. These clinical trials should also aim on alleviation of non-motor symptoms like depression, fatigue, apathy or deterioration of cognitive abilities in the prodromal or early phase of PD.2,3,74 Since cognitive deficits manifest in the early phases of PD with impaired DAT activity, dopamine neurotransmission enhancing agents may particularly improve these non-motor PD symptoms. Another therapeutic concept of a dopamine neurotransmission enhancing effect is provided by central glial COMT inhibition of dopamine turnover, i.e., with tolcapone. Prior pilot trial outcomes demonstrated beneficial effects of tolcapone on depression.75,76 Small clinical studies also described an improved cognition and cortical information processing following tolcapone application in patients and healthy volunteers. Dopamine increase via central COMT inhibition relative selectively improved prefrontal physiologic efficiency and thus cognitive behaviour, in particularly apathy and motivation.77–79 Central COMT inhibition is more regionally restricted in contrast to the effects of biogenic amine stimulants, such as amphetamine, which enhance transmission of dopamine, norepinephrine and 5-HT in widespread regions of the brain.77–79 Findings from experimental human studies report an improvement of in particular apathy and related features similar to fMRI findings obtained from healthy volunteers.77–79 Therefore a triple combination with a centrally acting COMT-inhibitor would complement the aforementioned concept for early l-dopa- and dopamine agonist naïve patients. Aim is to maximize the effects of endogenous available monoamines, particularly dopamine. However the already tested dual combination of the MAO-B inhibitor selegiline and the central active COMT-inhibitor tolcapone did not provide a relevant symptomatic effect in previously untreated PD patients. One may hypothesize, that a hypothetical up regulation of DRT activity was responsible for this negative outcome.80 Use of the currently only available central COMT-I tolcapone is restricted worldwide due to rare onset of fatal hepatic failures in a dosage of 200 mg t.i.d. However, tolcapone application with a lower dose, i.e., 200 mg o.i.d., may be enough for central COMT-I.81,82 Thus, such a triple concept may be tested with available PD compounds, such as combination of 200 mg tolcapone plus rasagiline 1 mg and retarded release amantadine, i.e., one 193-mg tablet.83 Amantadine is mainly looked upon as a NMDA antagonist as the most popular mode of action, but it also has dopamine reuptake inhibiting properties.83 Resembling concepts were already proposed in PD patients with early PD onset, to delay application of L-dopa and dopamine agonists.84,85 Reasons were the quality of life limiting side effect profile of dopamine agonists, which cause nausea, edema and dizziness, and the still ongoing neurotoxicity debate on early l-dopa use in PD patients with long life expectancy.86–90 The Levodopa in EArly Parkinson’s disease (LEAP) study recently again showed that L-dopa/DDC-I facilitates progression of PD. Patients with longer L-dopa/carbidopa exposure showed a faster rate of change in their Unified Parkinson’s Disease Rating Scale scores in the second study phase compared those with a later initiation of 300 mg L-dopa/carbidopa daily intake.91 This controversially discussed trial is worthy of criticism. 39% of the delayed start patients earlier changed to the early start l-dopa cohort, but the exact moments and concomitant dopamine replacement therapies with dopamine agonists or amantadine were not described, respectively analyzed in detail.9,90,92
DRT-I in l-Dopa Treated PD Patients
Application of oral l-dopa/DDC-I formulations causes l-dopa plasma fluctuations and pulsatile l-dopa brain bioavailability following the blood barrier transfer. Peripheral COMT-inhibition reduces these ups and downs of l-dopa in plasma to a certain extent. Pulsatility of l-dopa counteracts the physiologic principle of continuous dopaminergic stimulation. Too high l-dopa levels are metabolized to dopamine by abundant enzymatic decarboxylation in presynaptic dopamine and serotonin generating neurons. This mechanism is mandatory for further existing buffering strategies, which are dopamine reuptake from the synaptic cleft or neuronal and glial MAO-B - and glial COMT mediated dopamine turnover. The interplay between these various buffering strategies is overwhelmed by repeat pulsatile l-dopa delivery. Long-term and repeat l-dopa overuse facilitates generation of oxidative stress via the neuronal MAO-B metabolism pathway of dopamine, which is known to accelerate progression of PD in addition to aggravation of motor complications.15,16 To date, clinical trials with MRT inhibitors (MRT-I) only focused on symptomatic benefits in l-dopa treated PD within relative short study durations. However, introduction of oral L-dopa therapy in conjunction with stable inhibition of COMT, MAO-B and MRT may provide less pulsatile l-dopa delivery and allow a stable, individually adapted and reduced l-dopa dosing. This triple combination may prevent or delay onset of motor complications. A resembling concept was investigated in the failed STRIDE (STalevo Reduction In Dyskinesia Evaluation) STRIDE-PD study. This trial aimed to demonstrate a later onset of dyskinesia, in PD patients with a need for oral l-dopa/DDC-I therapy as an adjunct to their existing dopamine substitution therapy. L-dopa initiation was performed with l-dopa/carbidopa/entacapone or with l-dopa/carbidopa.93–95 This study was negative due to design failures, i.e. a standardized and mandatory up titration of the l-dopa containing formulations.
Conclusion
Experimental pharmacology showed some evidence that inhibition of dopamine reuptake may be beneficial for the balance of the dopamine deficit in PD models.73 The translation of these positive experimental results into corresponding clinical trial outcomes in PD patients failed or study programs were stopped due to probable missing commercial prospects of success.73 Approval as a PD drug was and is still mostly related to a benefit on motor behavior or reduction of “OFF-intervals” or dyskinesia. Therefore, a detailed evaluation of putative positive responses on specific non-motor symptoms, i.e., apathy in early PD patients, is rarely performed to date.37 Clinical MRT-I trial designs did not consider the complex interplay between enzymatic inhibition of MAO-B and COMT in association with constraint of MRT in PD.73 Instead, the main objective was to ameliorate motor symptoms in monotherapy or to improve l-dopa associated motor complications in advanced PD patients.73 MRT-I studies in l-dopa treated PD patients only focused on central actions in the brain. Possible peripheral modes of actions related with dopamine reuptake inhibition were not investigated. As an example, one is the feedback mechanism of gastric emptying velocity, which is regulated by l-dopa, respectively dopamine. It contributes to delayed and erratic duodenal l-dopa absorption and thus to the unpredictability of motor complications.11 As aforementioned, stimulation of the dopamine autoreceptor as sensor for too high dopamine concentrations in the synaptic cleft reduces endogenous synthesis of dopamine.5 It also supports dopamine removal and transport via the neuronal DAT.5 A resembling dopamine receptor mediated pattern exists for regulation of gastrointestinal velocity. Thus, high duodenal l-dopa respectively dopamine concentrations delay gastric emptying.96 Accordingly, the transport of oral l-dopa/DDC-I formulations to the duodenal main gastrointestinal absorption site of l-dopa slows down.11 It warrants further research, whether DRT-I have an effect on gastric emptying velocity similar to the peripheral dopamine receptor antagonist domperidone.13,97,98 Aforementioned reflections shall be considered, if study designs on the efficacy of SMe1EC2M3, a novel MRT-I with dopamine reuptake inhibiting properties, are planned in PD patients.99 A future approval of SMe1EC2M3 is very likely, when this drug demonstrates symptomatic, motor symptoms enhancing effects in PD as initial step of the clinical development. Then as a second step, one may consider to perform long-term trials with a combination of DRT-I plus MAO-B inhibitor plus dopamine oxidation inhibitors, such as potent iron chelators, as promising future therapeutic concept to slow down progression of PD.20 One may perform a concomitant assessment with DAT function imaging techniques for intra individual comparisons at different disease stages, such as the early, prodromal diagnostic -, early drug naïve – and advanced one with and without freezing of gait, similar to older investigations.34,100–102 Such a design is supported by experimental findings with DRT-I use. They showed the DRI efficacy in α-synucleinopathy models with DAT trafficking.103–106 In this regard it is noteworthy, that α-synuclein/DAT complexes occurred in postmortem PD brains and experimental PD models including primates.100,102,103,107 The pathological α-synuclein interplay with its synaptic partner synapsin III selectively boosted the locomotor activity in a α-synuclein PD model, while this effect was lost upon synapsin III gene silencing with co-administration of the MRT-I methylphenidate.108,109 In conclusion, a resurgence of clinical research on the efficacy of MRT-I for the treatment of PD patients will be an interesting option for the future drug development in PD.
Disclosure
The author reports no conflicts of interest in this work.
References
1. Przuntek H, Müller T, Riederer P. Diagnostic staging of Parkinson’s disease: conceptual aspects. J Neural Transm (Vienna). 2004;111(2):201–216. doi:10.1007/s00702-003-0102-y
2. De Leonibus E, Verheij MM, Mele A, Cools A. Distinct kinds of novelty processing differentially increase extracellular dopamine in different brain regions. Eur J Neurosci. 2006;23(5):1332–1340. doi:10.1111/j.1460-9568.2006.04658.x
3. De Leonibus E, Pascucci T, Lopez S, Oliverio A, Amalric M, Mele A. Spatial deficits in a mouse model of Parkinson disease. Psychopharmacology (Berl). 2007;194(4):517–525. doi:10.1007/s00213-007-0862-4
4. Pagonabarraga J, Kulisevsky J. Apathy in Parkinson’s disease. Int Rev Neurobiol. 2017;133:657–678.
5. Sulzer D, Cragg SJ, Rice ME. Striatal dopamine neurotransmission: regulation of release and uptake. Basal Ganglia. 2016;6:123–148.
6. Cotzias GC, Papavasiliou PS, Gellene R. Modification of Parkinsonism–chronic treatment with L-dopa. N Engl J Med. 1969;280(7):337–345. doi:10.1056/NEJM196902132800701
7. Foley P, The L-DOPA story revisited. Further surprises to be expected? J Neural Transm Suppl. 2000;(60):1–20. doi:10.1007/978-3-7091-6301-6_1
8. Birkmayer W, Hornykiewicz O. The effect of l-3,4-dihydroxyphenylalanine (=DOPA) on akinesia in parkinsonism. Parkinsonism Relat Disord. 1998;4(2):59–60. doi:10.1016/S1353-8020(98)00013-3
9. Müller T. Pharmacokinetics and pharmacodynamics of levodopa/carbidopa cotherapies for Parkinson’s disease. Expert Opin Drug Metab Toxicol. 2020;16(5):403–414. doi:10.1080/17425255.2020.1750596
10. Hardoff R, Sula M, Tamir A, et al. Gastric emptying time and gastric motility in patients with Parkinson’s disease. Mov Disord. 2001;16(6):1041–1047. doi:10.1002/mds.1203
11. Müller T, Erdmann C, Bremen D, et al. Impact of gastric emptying on levodopa pharmacokinetics in Parkinson disease patients. Clin Neuropharmacol. 2006;29(2):61–67. doi:10.1097/00002826-200603000-00001
12. Müller T. Pharmacokinetic considerations for the use of levodopa in the treatment of Parkinson disease: focus on levodopa/carbidopa/entacapone for treatment of levodopa-associated motor complications. Clin Neuropharmacol. 2013;36(3):84–91. doi:10.1097/WNF.0b013e31828f3385
13. Schuurkes JA, Van Nueten JM. Is dopamine an inhibitory modulator of gastrointestinal motility? Scand J Gastroenterol Suppl. 1981;67:33–36.
14. Muhlack S, Herrmann L, Salmen S, Müller T. Fewer fluctuations, higher maximum concentration and better motor response of levodopa with catechol-O-methyltransferase inhibition. J Neural Transm. 2014;121(11):1357–1366. doi:10.1007/s00702-014-1213-3
15. Riederer P, Müller T. Monoamine oxidase-B inhibitors in the treatment of Parkinson’s disease: clinical-pharmacological aspects. J Neural Transm (Vienna). 2018;125(11):1751–1757. doi:10.1007/s00702-018-1876-2
16. Przuntek H, Conrad B, Dichgans J, et al. SELEDO: a 5-year long-term trial on the effect of selegiline in early Parkinsonian patients treated with levodopa. Eur J Neurol. 1999;6(2):141–150. doi:10.1111/j.1468-1331.1999.tb00007.x
17. Ramot Y, Nyska A, Maronpot RR, et al. Ninety-day local tolerability and toxicity study of ND0612, a novel formulation of levodopa/carbidopa, administered by subcutaneous continuous infusion in minipigs. Toxicol Pathol. 2017;45(6):764–773. doi:10.1177/0192623317729891
18. Klostermann F, Jugel C, Müller T, Marzinzik F. Malnutritional neuropathy under intestinal levodopa infusion. J Neural Transm (Vienna). 2012;119(3):369–372. doi:10.1007/s00702-011-0689-3
19. Müller T, Laar TV, Cornblath DR. Peripheral neuropathy in Parkinson’s disease: levodopa exposure and implications for duodenal delivery. Parkinsonism Relat Disord. 2013;19(5):501–507. doi:10.1016/j.parkreldis.2013.02.006
20. Billings JL, Gordon SL, Rawling T, et al. l-3,4-dihydroxyphenylalanine (l-DOPA) modulates brain iron, dopaminergic neurodegeneration and motor dysfunction in iron overload and mutant alpha-synuclein mouse models of Parkinson’s disease. J Neurochem. 2019;150(1):88–106. doi:10.1111/jnc.14676
21. Sian-Hulsmann J, Riederer P. The role of alpha-synuclein as ferrireductase in neurodegeneration associated with Parkinson’s disease. J Neural Transm (Vienna). 2020;127(5):749–754. doi:10.1007/s00702-020-02192-0
22. Sun Y, Pham AN, Hider RC, Zheng H, Waite TD. Effectiveness of the iron chelator CN128 in mitigating the formation of dopamine oxidation products associated with the progression of Parkinson’s disease. ACS Chem Neurosci. 2020;11(21):3646–3657. doi:10.1021/acschemneuro.0c00557
23. Zhou ZD, Lan YH, Tan EK, Lim TM. Iron species-mediated dopamine oxidation, proteasome inhibition, and dopaminergic cell demise: implications for iron-related dopaminergic neuron degeneration. Free Radic Biol Med. 2010;49(12):1856–1871. doi:10.1016/j.freeradbiomed.2010.09.010
24. Ekesbo A, Rydin E, Torstenson R, Sydow O, Laengstrom B, Tedroff J. Dopamine autoreceptor function is lost in advanced Parkinson’s disease. Neurology. 1999;52(1):120–125. doi:10.1212/WNL.52.1.120
25. Fletcher EA, Redfern PH. The effect of amantadine on the uptake of dopamine and noradrenaline by rat brain homogenates. J Pharm Pharmacol. 1970;22(12):957–959. doi:10.1111/j.2042-7158.1970.tb08486.x
26. Heimans RL, Rand MJ, Fennessy MR. Effects of amantadine on uptake and release of dopamine by a particulate fraction of rat basal ganglia. J Pharm Pharmacol. 1972;24(11):875–879. doi:10.1111/j.2042-7158.1972.tb08906.x
27. Schendzielorz N, Oinas JP, Myohanen TT, Reenila I, Raasmaja A, Mannisto PT. Catechol-O-methyltransferase (COMT) protein expression and activity after dopaminergic and noradrenergic lesions of the rat brain. PLoS One. 2013;8(4):e61392. doi:10.1371/journal.pone.0061392
28. Hansard MJ, Smith LA, Jackson MJ, Cheetham SC, Jenner P. Dopamine, but not norepinephrine or serotonin, reuptake inhibition reverses motor deficits in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated primates. J Pharmacol Exp Ther. 2002;303(3):952–958. doi:10.1124/jpet.102.039743
29. Giordano N, Iemolo A, Mancini M, et al. Motor learning and metaplasticity in striatal neurons: relevance for Parkinson’s disease. Brain. 2018;141(2):505–520. doi:10.1093/brain/awx351
30. Longhena F, Faustini G, Missale C, Pizzi M, Bellucci A. Dopamine transporter/ alpha-synuclein complexes are altered in the post mortem caudate putamen of Parkinson’s disease: an in situ proximity ligation assay study. Int J Mol Sci. 2018;19(6):19. doi:10.3390/ijms19061611
31. Berg D, Godau J, Seppi K, et al. The PRIPS study: screening battery for subjects at risk for Parkinson’s disease. Eur J Neurol. 2013;20(1):102–108. doi:10.1111/j.1468-1331.2012.03798.x
32. Millan MJ, Maiofiss L, Cussac D, Audinot V, Boutin JA, Newman-Tancredi A. Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. I. A multivariate analysis of the binding profiles of 14 drugs at 21 native and cloned human receptor subtypes. J Pharmacol Exp Ther. 2002;303(2):791–804. doi:10.1124/jpet.102.039867
33. Thobois S. Proposed dose equivalence for rapid switch between dopamine receptor agonists in Parkinson’s disease: a review of the literature. Clin Ther. 2006;28(1):1–12. doi:10.1016/j.clinthera.2005.12.003
34. Fahn S, Oakes D, Shoulson I, et al. Levodopa and the progression of Parkinson’s disease. N Engl J Med. 2004;351:2498–2508.
35. Witjas T, Kaphan E, Azulay JP, et al. Nonmotor fluctuations in Parkinson’s disease: frequent and disabling. Neurology. 2002;59(3):408–413. doi:10.1212/WNL.59.3.408
36. Witjas T, Eusebio A, Fluchere F, Azulay JP. Addictive behaviors and Parkinson’s disease. Rev Neurol (Paris). 2012;168(8–9):624–633. doi:10.1016/j.neurol.2012.06.014
37. Müller T, Öhm G, Eilert K, et al. Benefit on motor and non-motor behavior in a specialized unit for Parkinson’s disease. J Neural Transm (Vienna). 2017;124(6):715–720. doi:10.1007/s00702-017-1701-3
38. de Chazeron I, Durif F, Chereau-Boudet I, et al. Compulsive eating behaviors in Parkinson’s disease. Eat Weight Disord. 2019;24(3):421–429. doi:10.1007/s40519-019-00648-1
39. Delpont B, Lhommee E, Klinger H, et al. Psychostimulant effect of dopaminergic treatment and addictions in Parkinson’s disease. Mov Disord. 2017;32(11):1566–1573. doi:10.1002/mds.27101
40. Martini A, Dal LD, Edelstyn NMJ, Salgarello M, Lugoboni F, Tamburin S. Dopaminergic neurotransmission in patients with Parkinson’s disease and impulse control disorders: a systematic review and meta-analysis of PET and SPECT studies. Front Neurol. 2018;9:1018. doi:10.3389/fneur.2018.01018
41. Melamed E, Hefti F, Wurtman RJ. Nonaminergic striatal neurons convert exogenous L-dopa to dopamine in parkinsonism. Ann Neurol. 1980;8(6):558–563. doi:10.1002/ana.410080603
42. Margolesky J, Singer C. Extended-release oral capsule of carbidopa-levodopa in Parkinson disease. Ther Adv Neurol Disord. 2018;11:1756285617737728. doi:10.1177/1756285617737728
43. Nausieda PA, Hsu A, Elmer L, et al. Conversion to IPX066 from standard levodopa formulations in advanced Parkinson’s disease: experience in clinical trials. J Parkinsons Dis. 2015;5(4):837–845. doi:10.3233/JPD-150622
44. Calabrese V, Di MA, Marino G, et al. Rapamycin, by inhibiting mTORC1 signaling, prevents the loss of striatal bidirectional synaptic plasticity in a rat model of L-DOPA-induced dyskinesia. Front Aging Neurosci. 2020;12:230. doi:10.3389/fnagi.2020.00230
45. Eshraghi M, Ramirez-Jarquin UN, Shahani N, et al. RasGRP1 is a causal factor in the development of l-DOPA-induced dyskinesia in Parkinson’s disease. Sci Adv. 2020;6:eaaz7001. doi:10.1126/sciadv.aaz7001
46. Guttler C, Altschuler J, Tanev K, et al. Levodopa-induced dyskinesia are mediated by cortical gamma oscillations in experimental parkinsonism. Mov Disord. 2020. doi:10.1002/mds.28403
47. Costa C, Sgobio C, Siliquini S, et al. Mechanisms underlying the impairment of hippocampal long-term potentiation and memory in experimental Parkinson’s disease. Brain. 2012;135(6):1884–1899. doi:10.1093/brain/aws101
48. Decressac M, Mattsson B, Lundblad M, Weikop P, Bjorklund A. Progressive neurodegenerative and behavioural changes induced by AAV-mediated overexpression of alpha-synuclein in midbrain dopamine neurons. Neurobiol Dis. 2012;45(3):939–953. doi:10.1016/j.nbd.2011.12.013
49. Durante V, de Iure A, Loffredo V. Alpha-synuclein targets GluN2A NMDA receptor subunit causing striatal synaptic dysfunction and visuospatial memory alteration. Brain. 2019;142(5):1365–1385. doi:10.1093/brain/awz065
50. Hansard MJ, Smith LA, Jackson MJ, Cheetham SC, Jenner P. Dopamine reuptake inhibition and failure to evoke dyskinesia in MPTP-treated primates. Eur J Pharmacol. 2002;451(2):157–160. doi:10.1016/S0014-2999(02)02268-9
51. Pearce RK, Smith LA, Jackson MJ, Banerji T, Scheel-Kruger J, Jenner P. The monoamine reuptake blocker brasofensine reverses akinesia without dyskinesia in MPTP-treated and levodopa-primed common marmosets. Mov Disord. 2002;17(5):877–886. doi:10.1002/mds.10238
52. Frackiewicz EJ, Jhee SS, Shiovitz TM, et al. Brasofensine treatment for Parkinson’s disease in combination with levodopa/carbidopa. Ann Pharmacother. 2002;36(2):225–230. doi:10.1345/aph.1A152
53. Yu P. Brasofensine NeuroSearch. Curr Opin Investig Drugs. 2000;1:504–507.
54. Cheng WC, Liu CM, Hsieh MH, Hwang TJ. Bupropion-related parkinsonism and dystonia. J Clin Psychopharmacol. 2009;29(6):616–618. doi:10.1097/JCP.0b013e3181c07a3f
55. Goetz CG, Tanner CM, Klawans HL. Bupropion in Parkinson’s disease. Neurology. 1984;34(8):1092–1094. doi:10.1212/WNL.34.8.1092
56. Vegda M, Panda S. Bupropion-induced dystonia in a patient with parkinson’s disease. J Mov Disord. 2020;13(3):241–243. doi:10.14802/jmd.20046
57. Benincasa D, Pellicano C, Fanciulli A, Pontieri FE. Bupropion abates dopamine agonist-mediated compulsive behaviors in Parkinson’s disease. Mov Disord. 2011;26(2):355–357. doi:10.1002/mds.23395
58. Raskin S, Durst R. Bupropion as the treatment of choice in depression associated with Parkinson’s disease and it’s various treatments. Med Hypotheses. 2010;75(6):544–546. doi:10.1016/j.mehy.2010.07.024
59. Zaluska M, Dyduch A. Bupropion in the treatment of depression in Parkinson’s disease. Int Psychogeriatr. 2011;23(2):325–327. doi:10.1017/S1041610210001687
60. Bedard P, Parkes JD, Marsden CD. Nomifensine in Parkinson’s disease. Br J Clin Pharmacol. 1977;4(Suppl 2):187S–190S. doi:10.1111/j.1365-2125.1977.tb05751.x
61. Park DM, Findley LJ, Teychenne PF. Nomifensine in parkinsonism. Br J Clin Pharmacol. 1977;4(Suppl 2):185S–186S. doi:10.1111/j.1365-2125.1977.tb05750.x
62. Park DM, Findley LJ, Hanks G, Sandler M. Nomifensine: effect in Parkinsonian patients not receiving levodopa. J Neurol Neurosurg Psychiatry. 1981;44(4):352–354. doi:10.1136/jnnp.44.4.352
63. Teychenne PF, Park DM, Findley LJ, Rose FC, Calne DB. Nomifensine in parkinsonism. J Neurol Neurosurg Psychiatry. 1976;39(12):1219–1221. doi:10.1136/jnnp.39.12.1219
64. Kapur A. Is methylphenidate beneficial and safe in pharmacological cognitive enhancement? CNS Drugs. 2020;34:1045–1062.
65. Lou JS. Fatigue in Parkinson’s disease and potential interventions. NeuroRehabilitation. 2015;37(1):25–34. doi:10.3233/NRE-151238
66. Nutt JG, Carter JH, Carlson NE. Effects of methylphenidate on response to oral levodopa: a double-blind clinical trial. Arch Neurol. 2007;64(3):319–323. doi:10.1001/archneur.64.3.319
67. Shen Y, Huang JY, Li J, Liu CF. Excessive daytime sleepiness in parkinson’s disease: clinical implications and management. Chin Med J (Engl). 2018;131(8):974–981. doi:10.4103/0366-6999.229889
68. Huot P, Johnston TH, Gandy MN, et al. The monoamine re-uptake inhibitor UWA-101 improves motor fluctuations in the MPTP-lesioned common marmoset. PLoS One. 2012;7(9):e45587. doi:10.1371/journal.pone.0045587
69. Huot P, Johnston TH, Lewis KD, et al. UWA-121, a mixed dopamine and serotonin re-uptake inhibitor, enhances L-DOPA anti-parkinsonian action without worsening dyskinesia or psychosis-like behaviours in the MPTP-lesioned common marmoset. Neuropharmacology. 2014;82:76–87. doi:10.1016/j.neuropharm.2014.01.012
70. Johnston TH, Millar Z, Huot P, et al. A novel MDMA analogue, UWA-101, that lacks psychoactivity and cytotoxicity, enhances l-DOPA benefit in parkinsonian primates. FASEB J. 2012;26(5):2154–2163. doi:10.1096/fj.11-195016
71. Hauser RA, Salin L, Juhel N, Konyago VL. Randomized trial of the triple monoamine reuptake inhibitor NS 2330 (tesofensine) in early Parkinson’s disease. Mov Disord. 2007;22(3):359–365. doi:10.1002/mds.21258
72. Rascol O, Poewe W, Lees A, et al. Tesofensine (NS 2330), a monoamine reuptake inhibitor, in patients with advanced Parkinson disease and motor fluctuations: the ADVANS study. Arch Neurol. 2008;65(5):577–583. doi:10.1001/archneur.65.5.577
73. Huot P, Fox SH, Brotchie JM. Dopamine reuptake inhibitors in parkinson’s disease: a review of nonhuman primate studies and clinical trials. J Pharmacol Exp Ther. 2016;357(3):562–569. doi:10.1124/jpet.116.232371
74. Cools R. Dopaminergic modulation of cognitive function-implications for L-DOPA treatment in Parkinson’s disease. Neurosci Biobehav Rev. 2006;30:1–23.
75. Fava M, Rosenbaum JF, Kolsky AR, et al. Open study of the catechol-O-methyltransferase inhibitor tolcapone in major depressive disorder. J Clin Psychopharmacol. 1999;19(4):329–335. doi:10.1097/00004714-199908000-00008
76. Moreau JL, Borgulya J, Jenck F, Martin JR. Tolcapone: a potential new antidepressant detected in a novel animal model of depression. Behav Pharmacol. 1994;5(3):344–350. doi:10.1097/00008877-199406000-00012
77. Apud JA, Weinberger DR. Treatment of cognitive deficits associated with schizophrenia: potential role of catechol-O-methyltransferase inhibitors. CNS Drugs. 2007;21(7):535–557. doi:10.2165/00023210-200721070-00002
78. Apud JA, Mattay V, Chen J, et al. Tolcapone improves cognition and cortical information processing in normal human subjects. Neuropsychopharmacology. 2007;32(5):1011–1020. doi:10.1038/sj.npp.1301227
79. Fava M, Rosenbaum JF, Kolsky AR, et al. Open study of the catechol-O-methyltransferase inhibitor tolcapone in major depressive disorder. J Clin Psychopharmacol. 2007;22(4):329–335.
80. Hauser RA, Molho E, Shale H, Pedder S, Dorflinger EE. A pilot evaluation of the tolerability, safety, and efficacy of tolcapone alone and in combination with oral selegiline in untreated Parkinson’s disease patients. Tolcapone De novo study group. Mov Disord. 1998;13(4):643–647. doi:10.1002/mds.870130406
81. Männisto PT, Kaakkola S. Catechol-O-methyltransferase (COMT): biochemistry, molecular biology, pharmacology, and clinical efficacy of the new selective COMT inhibitors. Pharmacol Rev. 1999;51(4):593–628.
82. Müller T. Catechol-O-methyltransferase inhibitors in Parkinson’s disease. Drugs. 2015;75(2):157–174. doi:10.1007/s40265-014-0343-0
83. Müller T, Kuhn W, Möhr JD. Evaluating ADS5102 (amantadine) for the treatment of Parkinson’s disease patients with dyskinesia. Expert Opin Pharmacother. 2019;20(10):1181–1187. doi:10.1080/14656566.2019.1612365
84. Rinne UK. Treatment of early Parkinson’s disease. Parkinsonism Relat Disord. 2000;7(1):59–62. doi:10.1016/S1353-8020(00)00040-7
85. Silver DE, Ruggieri S. Initiating therapy for Parkinson’s disease. Neurology. 1998;50:S18–S22. doi:10.1212/WNL.50.6_Suppl_6.S18
86. Fahn S. Does levodopa slow or hasten the rate of progression of Parkinson’s disease? J Neurol. 2005;252(Suppl 4):IV37–IV42. doi:10.1007/s00415-005-4008-5
87. Hinz M, Stein A, Cole T. The Parkinson’s disease death rate: carbidopa and vitamin B6. Clin Pharmacol. 2014;6:161–169. doi:10.2147/CPAA.S70707
88. Müller T. Detoxification and antioxidative therapy for levodopa-induced neurodegeneration in Parkinson’s disease. Expert Rev Neurother. 2013;13:707–718. doi:10.1586/ern.13.50
89. Müller T, Trommer I, Muhlack S, Mueller BK. Levodopa increases oxidative stress and repulsive guidance molecule A levels: a pilot study in patients with Parkinson’s disease. J Neural Transm (Vienna). 2016;123(4):401–406. doi:10.1007/s00702-016-1519-4
90. Leal RM, Rascol O, Ferreira JJ. The “long and winding road” of the disease-modifying effects of levodopa has not ended yet. Mov Disord. 2020;35(3):397–399. doi:10.1002/mds.27961
91. Verschuur CVM, Suwijn SR, Boel JA, et al. Randomized delayed-start trial of levodopa in Parkinson’s disease. N Engl J Med. 2019;380(4):315–324. doi:10.1056/NEJMoa1809983
92. Rma DB, Clarke CE, Espay AJ, Fox SH, Lang AE. Initiation of pharmacological therapy in Parkinson’s disease: when, why, and how. Lancet Neurol. 2020;19(5):452–461. doi:10.1016/S1474-4422(20)30036-3
93. Müller T. Entacapone. Expert Opin Drug Metab Toxicol. 2010;6(983–993):983–993. doi:10.1517/17425255.2010.502167
94. Stocchi F, Rascol O, Kieburtz K, et al. Initiating levodopa/carbidopa therapy with and without entacapone in early Parkinson disease: the STRIDE-PD study. Ann Neurol. 2010;68(1):18–27. doi:10.1002/ana.22060
95. Warren OC, Kieburtz K, Rascol O, et al. Factors predictive of the development of Levodopa-induced dyskinesia and wearing-off in Parkinson’s disease. Mov Disord. 2013;28(8):1064–1071. doi:10.1002/mds.25364
96. Levein NG, Thorn SE, Wattwil M. Dopamine delays gastric emptying and prolongs orocaecal transit time in volunteers. Eur J Anaesthesiol. 1999;16(4):246–250. doi:10.1097/00003643-199904000-00006
97. Barone JA. Domperidone: a peripherally acting dopamine 2-receptor antagonist. Ann Pharmacother. 1999;33(4):429–440. doi:10.1345/aph.18003
98. Shuto K, Shiozaki Z, Kojima T, Tanaka M. Antagonism of KW-5338 (domperidone) against emesis and depression of intestinal motility induced by L-DOPA. J Pharmacobiodyn. 1980;3(12):709–714. doi:10.1248/bpb1978.3.709
99. Koprdova R, Csatlosova K, Durisova B, et al. Electrophysiology and behavioral assessment of the new molecule SMe1EC2M3 as a representative of the future class of triple reuptake inhibitors. Molecules. 2019;24(23):24. doi:10.3390/molecules24234218
100. Chu Y, Muller S, Tavares A, et al. Intrastriatal alpha-synuclein fibrils in monkeys: spreading, imaging and neuropathological changes. Brain. 2019;142(11):3565–3579. doi:10.1093/brain/awz296
101. Schapira AH, McDermott MP, Barone P, et al. Pramipexole in patients with early Parkinson’s disease (PROUD): a randomised delayed-start trial. Lancet Neurol. 2013;12(8):747–755. doi:10.1016/S1474-4422(13)70117-0
102. Whone AL, Watts RL, Stoessl AJ, et al. Slower progression of Parkinson’s disease with ropinirole versus levodopa: the REAL-PET study. Ann Neurol. 2003;54(1):93–101. doi:10.1002/ana.10609
103. Bellucci A, Navarria L, Falarti E, et al. Redistribution of DAT/alpha-synuclein complexes visualized by “in situ” proximity ligation assay in transgenic mice modelling early Parkinson’s disease. PLoS One. 2011;6(12):e27959. doi:10.1371/journal.pone.0027959
104. Chadchankar H, Ihalainen J, Tanila H, Yavich L. Decreased reuptake of dopamine in the dorsal striatum in the absence of alpha-synuclein. Brain Res. 2011;1382:37–44. doi:10.1016/j.brainres.2011.01.064
105. Lee FJ, Liu F, Pristupa ZB, Niznik HB. Direct binding and functional coupling of alpha-synuclein to the dopamine transporters accelerate dopamine-induced apoptosis. FASEB J. 2001;15(6):916–926. doi:10.1096/fj.00-0334com
106. Wersinger C, Prou D, Vernier P, Sidhu A. Modulation of dopamine transporter function by alpha-synuclein is altered by impairment of cell adhesion and by induction of oxidative stress. FASEB J. 2003;17(14):2151–2153. doi:10.1096/fj.03-0152fje
107. Bellucci A, Collo G, Sarnico I, Battistin L, Missale C, Spano P. Alpha-synuclein aggregation and cell death triggered by energy deprivation and dopamine overload are counteracted by D2D3 receptor activation. J Neurochem. 2008;106(2):560–577. doi:10.1111/j.1471-4159.2008.05406.x
108. Faustini G, Longhena F, Varanita T, et al. Synapsin III deficiency hampers alpha-synuclein aggregation, striatal synaptic damage and nigral cell loss in an AAV-based mouse model of Parkinson’s disease. Acta Neuropathol. 2018;136(4):621–639. doi:10.1007/s00401-018-1892-1
109. Faustini G, Longhena F, Bruno A, et al. Alpha-synuclein/synapsin III pathological interplay boosts the motor response to methylphenidate. Neurobiol Dis. 2020;138:104789. doi:10.1016/j.nbd.2020.104789
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.