")
Back to Journals » Infection and Drug Resistance » Volume 14
Expanding of ST11 Carbapenemase-Producing Klebsiella pneumoniae Subclones in a Chinese Hospital, Shenzhen, China
Authors Zhang M, Li J, Lu Y, Wu W, Wu J, Xu Y, Zhong Y, Liu S , Lin C, Xu S, Chen D, Gao X, Luo Y , Chen H, Liu X, Huang W
Received 3 January 2021
Accepted for publication 21 March 2021
Published 13 April 2021 Volume 2021:14 Pages 1415—1422
DOI https://doi.org/10.2147/IDR.S299478
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Min Zhang,1,2 Jia Li,3 Yuemei Lu,4 Weiyuan Wu,4 Jinsong Wu,4 Yanji Xu,1,5 Yuxin Zhong,1,2 Shiyi Liu,1,2 Chuchu Lin,6 Shuye Xu,3 Dandan Chen,1,2 Xuefeng Gao,7 Yutian Luo,8 Huaisheng Chen,8 Xueyan Liu,8 Wei Huang1,2
1Shenzhen People’s Hospital, Shenzhen Institute of Respiratory Diseases, Shenzhen, Guangdong, People’s Republic of China; 2Bacteriology & Antibacterial Resistance Surveillance Laboratory, Shenzhen People’s Hospital (The Second Clinical Medical College, Jinan University; The First Affiliated Hospital, Southern University of Science and Technology), Shenzhen, Guangdong, People’s Republic of China; 3Department of Nosocomial Infection Control, Bengbu Third People’s Hospital, Bengbu, People’s Republic of China; 4Department of Clinical Microbiology, Shenzhen People’s Hospital (The Second Clinical Medical College, Jinan University; The First Affiliated Hospital, Southern University of Science and Technology), Shenzhen, Guangdong, People’s Republic of China; 5Nanshan District Center of Chronic Disease Control, Shenzhen, People’s Republic of China; 6Department of Nosocomial Infection Control, Shenzhen People’s Hospital (The Second Clinical Medical College, Jinan University; The First Affiliated Hospital, Southern University of Science and Technology), Shenzhen, Guangdong, People’s Republic of China; 7Department of Gastroenterology and Hepatology, Shenzhen University General Hospital, Shenzhen, People’s Republic of China; 8Department of Intensive Care Unit, Shenzhen People’s Hospital (The Second Clinical Medical College, Jinan University; The First Affiliated Hospital, Southern University of Science and Technology), Shenzhen, Guangdong, People’s Republic of China
Correspondence: Wei Huang
Bacteriology & Antibacterial Resistance Surveillance Laboratory, Shenzhen Institute of Respiratory Diseases, Shenzhen People’s Hospital (The Second Clinical Medical College, Jinan University The First Affiliated Hospital, Southern University of Science and Technology), No. 1017 Dongmen North Road, Shenzhen People’s Hospital, Shenzhen, 518020, Guangdong, People’s Republic of China
Tel +86 7522942513
Fax +86 7522942514
Email [email protected]
Background: ST11 is the most prevalent sequence type of clinical Klebsiella pneumoniae in China.
Methods: We investigated the characteristics of the ST11 subclones using core genome multi-locus sequence typing (cgMLST). Ninety-three carbapenemase-producing K. pneumoniae isolates were collected at Shenzhen People’s Hospital. Then, whole-genome sequencing and cgMLST were used to discriminate apparent subclones within the ST11 group.
Results: We analyzed the prevalence and genetic relationships of these subclones. ST11 and K. pneumoniae carbapenemase (KPC-2) were the predominant genotype and carbapenemase, respectively, in the clinical carbapenemase-producing K. pneumoniae strains. cgMLST scheme genotyping divided the ST11 group into two clades across seven complex types (CTs). CT1313 was the most prevalent subclone. The deletion of galF and a high frequency of SNPs in genes associated with the stress- and SOS-responses were found in CT1291 and CT2405 over time, respectively.
Conclusion: Our results indicated that the subclones of the ST11 group had different patterns of prevalence. Highly discriminatory genotyping techniques, such as cgMLST scheme, should be used in further molecular epidemiology investigations.
Keywords: Klebsiella pneumoniae, whole genome sequencing, cgMLST, ST11, carbapenem-resistance
Introduction
Klebsiella pneumoniae is a common Gram-negative pathogen that causes opportunistic hospital-acquired infections worldwide. As the multiclass antimicrobial resistance (AMR) of K. pneumoniae is widespread in nosocomial settings, this pathogen has been recognized as an urgent threat to human health.1 Although carbapenem antibiotics are often used to treat drug-resistant bacteria, K. pneumoniae has acquired plasmid-encoded carbapenemases capable of inactivating most β-lactam antibiotics, including the carbapenems, thus conferring carbapenem-resistance.2
Carbapenemase-producing K. pneumoniae (CPKP) pose a daunting clinical challenge, as these bacteria cause invasive infections, with few therapeutic options and high mortality. More worryingly, the genetic diversity of CPKP appears to be increasing.3,4 The population structure of K. pneumoniae has been studied with several different genotyping techniques, including pulsed-field gel electrophoresis (PFGE), multi-locus VNTR analysis (MLVA), and multi-locus sequence typing (MLST).5 A standard MLST analysis, based on polymorphisms in seven K. pneumoniae housekeeping genes, found that the clonal complex 258 (CC258), which includes sequence type 258 (ST258) and its single-locus variants ST11 and ST340, dominated in CPKP isolates worldwide.6 In Asia, the dominant clone is ST11, accounting for up to 60% of Chinese CPKP isolates.7
Whole genome next-generation sequencing (WGS) has supported the reconstruction of many critical events in the evolutionary history of CPKP. Initial comparative sequencing efforts, focusing on ST258, confirmed and extended earlier observations based on capsule and standard MLST typing; these latter analyses, because they only queried seven genes, might underestimate clinically relevant differences in genetic content among isolates.8 Indeed, DeLeo et al demonstrated that ST258 included at least two clades, which are defined by differences in a 215-kb region that includes the cps locus.9 In addition, a PFGE analysis of 83 CPKP isolates showed that ST11 was subdivided into two distinct sub-lineages,10 while a phylogenetic analysis based on single-nucleotide polymorphisms (SNPs) among CPKP genomes partitioned 12 outbreak ST11 strains into three separate clades.11 It is thus likely that the ST11 group includes different sub-lineages, although the characteristics of these sub-lineages remain unknown.
A combination of WGS and core genome multi-locus sequence typing MLST (cgMLST) can analyze polymorphisms in thousands of genes in a core genome. cgMLST schemes include fixed sets of conserved genome-wide genes (2358 genes in the case of K. pneumoniae); alleles are used instead of single nucleotide polymorphisms (SNP) or concatenated sequences to mitigate the effects of recombination. Because this technique operates genome-wide and types thousands of genes, cgMLST combines the comparability of MLST and discriminatory power of PFGE. Here, we used cgMLST to investigate the population ecology of CPKP isolates collected over a two-year period at a large Chinese hospital.
Materials and Methods
Bacterial Isolates and Phenotypic Characterization
We collected consecutive non-replicate clinical isolates of carbapenem-resistant K. pneumoniae (CRKP) isolated between January 2016 and December 2017 from inpatients at the Shenzhen People’s Hospital, a medical center with 2500 beds in Luohu, Shenzhen, China. Species identification and antimicrobial susceptibility testing were performed with the VITEK-2 compact system (bioMérieux, Marcy-l’Étoile, France). Results were interpreted in accordance with guidelines published by the Clinical and Laboratory Standards Institute (CLSI; document M100-S26).12 The species identifications of all isolates were confirmed with matrix-assisted laser desorption/ionization mass spectrometry (MALDI-TOF-MS, bioMérieux, Marcy-l’Étoile, France). We used the modified Hodge test, performed according to CLSI 2016 recommendations, to identify the CPKP phenotype.12
Whole Genome Sequencing (WGS)
Genomic DNA was extracted from each isolate using the Sodium Dodecyl Sulfate (SDS) method,13 and quantified using a Qubit (Thermo Fisher, Waltham, MA, USA). The DNA library was constructed using an insert size of 350 bp. A-tailed, paired-end adapters were ligated to the DNA fragments and sequenced on an Illumina HiSeq 2500-PE150 platform (Illumina, San Diego, CA, USA). We used fastp to perform quality control and the preprocessing of fastq to remove adaptor sequences, low-quality reads (phred quality < Q15), low-complexity reads (30%), and polyG/polyX tails.14 Genome assembly was performed using de novo SPAdes Genome Assembler (version 3.12.0).15 We identified the antimicrobial resistance genes and virulence factors in the isolates by scanning the genome contigs against the ResFinder and VFDB databases using ABRicate (version 0.8.7). We performed capsular typing on the assembled sequences with Kaptive (version 0.5.1).16
Genomic Characterization
Analysis of the genome sequences for MLST, cgMLST genotyping and minimum spanning trees construction were performed with Ridom SeqSphere+ (version 5.1.0) (Ridom GmbH, Munster, Germany), based on the cgMLST scheme, very closely related genomes (ie, those ≤15 alleles distance) were “lumped” together as a complex type (CT).17 Core genome alignment, recombination detection/filtration, and phylogenetic reconstruction were performed with the Harvest suite (version 1.2), Spine (version 0.3.2) and AGEnt (version 0.3.1) were used to compare the genomes of isolates.18,19 We annotated the results with Prokka (version 1.13.3).20
Viability in Serum
The K. pneumoniae clinical isolates P1291-19, P1291-20, P2405-8, and P2405-9 were used for the serum viability experiment. Bacteria from frozen stocks were cultured in Luria-Bertani (LB) broth overnight at 37°C with shaking (220 rpm). Overnight cultures were diluted 1/100 and re-cultured until the optical density at 600 nm (OD600) was 0.6–0.8. We adjusted the cultures with LB medium to concentrations of 2×106 colony-forming units (CFU)/mL. Cultures were then inoculated with 2×105 CFU in 3 mL LB medium with or without 20% human serum (Lablead), and re-cultured. We removed 100 µL aliquots of each culture at 30 min, 60 min, 90 min, and 120 min after inoculation. Aliquots were diluted and plated on LB agar. Survival was expressed as the ratio of CFUs in the serum-added LB to the CFUs in unsupplemented LB.
Statistical Methods
Pairs of experimental groups were compared using the two-sample Student’s t test or the Mann–Whitney U-test if the requirements of the t test were not met. We considered P < 0.05 statistically significant. All analyses were performed using GraphPad Prism (GraphPad Software Inc., San Diego, CA, USA).
Results
Non-repetitive CRKP isolates collected from patients with confirmed K. pneumoniae infections were frozen and stored by the clinical microbiology laboratory at the Shenzhen People’s Hospital. We confirmed CPKP in 93 of these isolates. The dominant carbapenemase was KPC-2 (84 isolates), followed by IMP-4 (5 isolates), NDM-1 (3 isolates), KPC-9 (2 isolates), and OXA-23 (1 isolate) (Supplementary Figure S1). OXA-23 carbapenemase was identified in isolate that also carried both IMP-4 and KPC-9.
Unsurprisingly, MLST analysis showed that ST11 was the most prevalent lineage among the CPKP strains (83 isolates), followed by ST3427 (3 isolates). The other ST types were represented by only a single isolate each (Figure 1A). Classical MLST analysis identified only 9 STs, while cgMLST identified 15 CTs. The most common CTs were CT1313 (33 isolates), CT1291 (20 isolates), and CT2410 (10 isolates), followed by CT2405 (9 isolates), CT2445 (6 isolates), CT2418 (4 isolates), and CT2379 (3 isolates). Eight other CT types were identified in only one isolate each (Figure 1B). Four of the identified ST types were previously undescribed (http://bigsdb.pasteur.fr/klebsiella/klebsiella.html), as were 14 CT types (https://www.cgmlst.org/ncs). Seven CT types were included in the ST11 group (Figure 1C). CT1313, the most prevalent ST11 subclone, was isolated throughout the two-year study period. The first strains of CT2405 (P2405-1), CT2410 (P2410-1), and CT2418 (P2418-1) were isolated more than six months after the start of the study period, while the first strain of CT2445 (P2445-1) was isolated just two months before the end of the study period (Figure 2A).
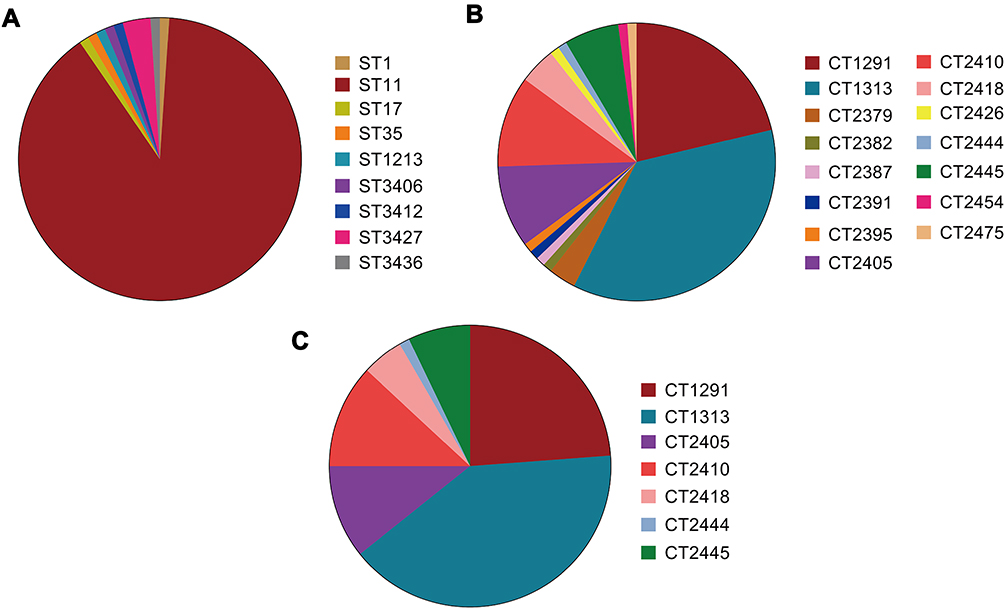 |
Figure 1 Distribution of genotypes among carbapenemase-producing strains of Klebsiella pneumoniae strains. Genotype distribution of isolates based on (A) multi locus sequence typing (MLST) analysis and (B) core genome multi locus sequence typing (cgMLST) scheme. (C) Distribution of CT types within the ST11 group. Abbreviations: ST, sequence type; CT, complex type. |
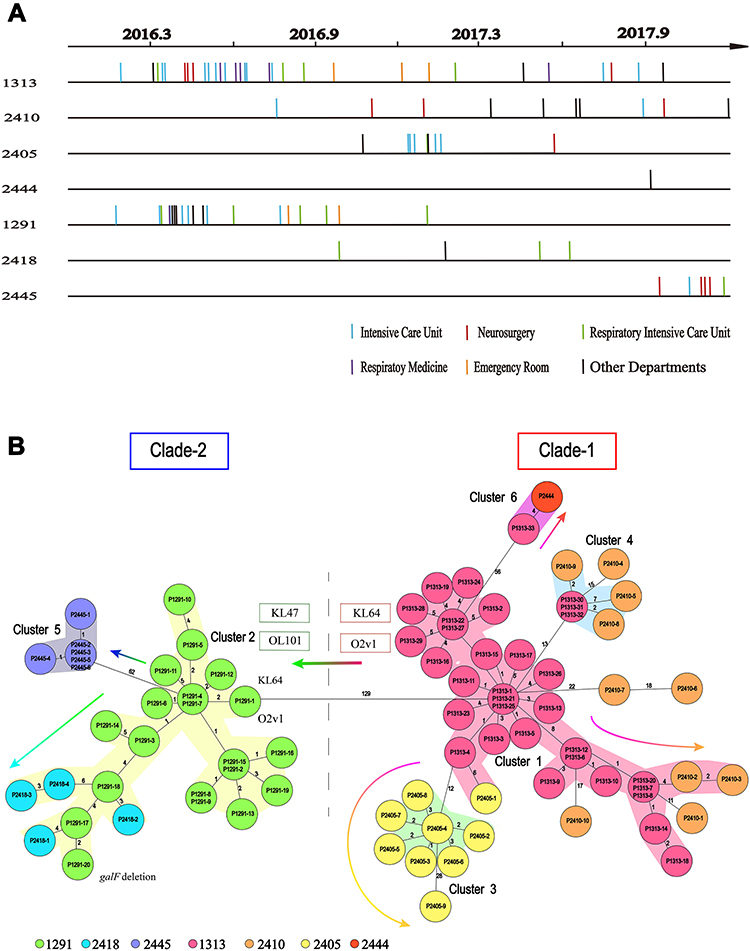 |
Figure 2 Timelines of the CT type isolates in the ST11 group, and the evolutionary relationships among these isolates. (A) Sampling dates for each isolate. Vertical lines represent the isolates in each CT group. There are 33, 10, 9, 1, 20, 4 and 6 isolates in the CT1313 (P1313-1 to P1313-33), CT2410 (P2410-1 to P2410-10), CT2405 (P2405-1 to P2405-9), CT2444 (P2444), CT1291 (P1291-1 to P1291-20), CT2418 (P2418-1 to P2418-4), and CT2445 (P2445-1 to P2445-6) group, respectively. (B) Minimum spanning tree based on cgMLST scheme analysis of the different CT types in the ST11 group of carbapenemase-producing K. pneumoniae. Each circle represents the isolates belonging to the corresponding CT type, based on the sequence analysis of 2358 cgMLST target genes. The code in each circle is the case serial number. The numbers on the branches represent the number of alleles between each pair of genes. The cluster distance threshold is 15. The arrows indicate evolution direction. |
A previous study demonstrated that ST258 K. pneumoniae fell into two distinct genetic clades based on a phylogenetic analysis of the SNPs in the core genome; the genetic differences between the two clades were due to divergences in genes associated with capsule polysaccharide biosynthesis.9 To identify the relationships among the different CT groups within ST11, we constructed minimum spanning trees for all ST11 isolates based on the cgMLST scheme. The previous study was consistent with the minimum spanning tree based on the cgMLST allelic profiles of ST11 K. pneumoniae recovered here. Two genetic clades were identified in the ST11 lineage: CT1313, CT2410, CT2405, and CT2444 clustered together as clade-1; CT1291, CT2418, and CT2445 clustered together as clade-2.
As shown in minimum spanning trees, six clusters were separated by a mean pairwise allelic distance of 15 alleles. Combining the isolation timeline (Figure 2A), the position of CT1313 was consistent with its possible role as a progenitor of CT2410, CT2405, and CT2444, while the position of CT1291 was consistent with its possible role as a progenitor of CT2418 and CT2445 (arrows in Figure 2B). All ST11 isolates in clade-1 examined here had a K-locus (KL) of KL64. Most strains in clade-2 were KL47, except for the first CT1291 strain P1291-1 (KL64). All ST11 strains in clade-1 examined here had an O-locus (OL) of O2v1. Most strains in clade-2 were OL101, except for P1291-1 (O2v1). Changes of KL and OL type suggest that CT1291 is a subclone expanding from CT1313.
We compared the genomes of CT1291 and CT2405 isolates, respectively, and found, intriguingly, that the strain P1291-20 (sampling date Feb 2, 2017) was lacking galF (Figure 3A), a gene present in the genomes of all other strains in this CT group (P1291-1 through P1291-19). The phylogenetic trees based on the SNPs in core genomes showed that a zone with a high frequency of SNPs in strain P2405-9 compared to other strains in CT2405 group. This region included the genes yciE, rrrD, smc, lexA, and dinI (Figure 3B; Supplementary Table S1).
 |
Figure 3 The phylogenetic trees of CT1291 and CT2405. Visualization of the core genomes of (A) CT1291 and (B) CT2405, as aligned with the Harvest suite. The leaves of the reconstructed phylogenetic tree (left) are paired with the corresponding row in the multiple sequence alignment. P1291-20 and P2405-9 have been selected (left: highlighted in aqua; right: highlighted in black). A SNP density plot (lilac line) reveals the phylogenetic signature of several clades. The light gray regions flanking the operon indicate unaligned sequences. The blue dotted box indicates the zone with a high frequency of SNPs and the corresponding genes in P2405-9. |
To determine whether the CT1291 and CT2405 strains were associated with a less stress-resistant phenotype, we compared the viability of these strains in human serum. The viability of P1291-20 and P2405-9 in 20% serum was lower than that of other isolates from the same CT groups (Figure 4).
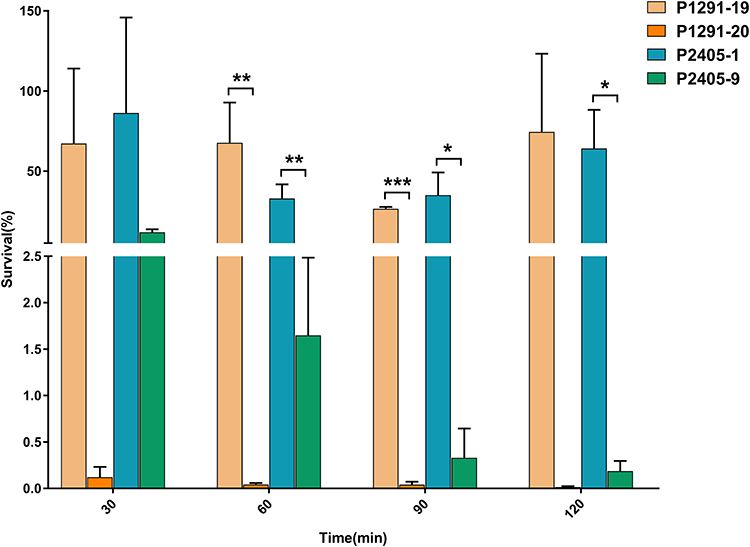 |
Figure 4 The viability of isolates in serum. P1291-19 and P2405-1 were randomly selected, and the viability of each of these isolates in serum was compared to that of the last isolate in each corresponding CT group (P1291-20 and P2405-9, respectively) (n=3, *P<0.05, **P<0.01, ***P<0.001). |
Discussion
The increasing prevalence of CRKP is a major public health concern. The production of carbapenemases is the primary mechanism by which CRKP resists antibiotic treatment. Transmission of these resistance genes can be mediated by horizontal transfer via transposons or plasmids, or by clonal spread.21 We found that ST11 was the most common clone associated with the most common KPC-2 carbapenemase in the largest medical center in Shenzhen between 2016 and 2017. This was consistent with a previous study in China and other Asian countries.22 However, the P2445-4 strain in this study carried both OXA-23 and KPC-2 carbapenemase, which is uncommon in K. pneumoniae. The existence of OXA-23 among CRKP clinical isolates in Egypt was also reported recently.23 This suggests that we should be alert to the spread of OXA-23 in K. pneumoniae, which is only prevalent in Acinetobacter baumannii.23 However, MLST schemes corresponding to CPKP strains have remained imprecisely defined.9,10 cgMLST analysis may eliminate spurious MLST associations. Although this typing method needs a commercial software SeqSphere+, which may suggest less robustness, the high resolution makes it become easier to eliminate unnecessary interference in the complicated analysis process. Previous studies have used cgMLST to clarify the origin and evolution of community-acquired Staphylococcus aureus and to identify separate Acinetobacter baumannii outbreaks.24,25
A cgMLST scheme for K. pneumoniae was developed and evaluated in 2017.26 Although cgMLST analysis suggested that epidemic ST11 might have several sub-clones, the properties of these sub-clones were unknown. Here, we identified several different CT types within the ST11 group, and showed that the subclones of ST11 differed with respect to prevalence.
However, even with the high resolution of cgMLST, we still found some significant differences between the genomes of the strains with same CT-type. The galF gene was highlighted by comparing the genomes of isolates in CT1291 group. High-pressure liquid chromatography (HPLC) analyses indicated that the reaction catalyzed by GalF was associated with CPS biosynthesis, and that GalF was responsible for CPS translocation and surface assembly.27 Previous studies using mutants (Δwca and Δatf) showed that silencing galF reduced or eliminated CPS production compared with NTUH-K2044.27,28 Genetic differences within the CT2405 group result from a zone with a high frequency of SNPs; this zone includes genes involved in the stress-response (yciE) and the SOS-response (rrrD, smc, lexA and dinI). Thus, these SNPs may have the potential to affect the prevalence of K. pneumoniae.
Our timeline also revealed the genetic relationships among the different CT groups, with the exception of CT1313 and CT1291. Based on the KL and OL type results, we inferred that CT1291 might have emerged from CT1313, because the isolate P1291-1 has the same KL and OL type as CT1313 and other clade-1 isolates. Thus, in our hospital, CT1313 was the progenitor of ST11 CPKP and the subclone expanding is ongoing.
Conclusion
In summary, we confirmed the heterogeneity of the ST11 group using cgMLST scheme genotyping. With the discriminatory power of this technique, we revealed the genomic relationship of ST11 subclones, and characterized these isolates with respect to prevalence. Our findings highlight the necessity of accurately classifying subclone within ST11 type in the future molecular epidemiology investigation.
Data Sharing Statement
The genome sequences were deposited in the GenBank, under BioProject PRJNA517992.
Ethics Statement
This study was conducted in accordance with the Declaration of Helsinki. The research protocol was approved by the research ethics committee of Shenzhen People’s Hospital (reference 257 number LL-KY-2019002). The research ethics committee of Shenzhen People’s Hospital did not require written informed consent from participants because no personal information about patients was requested.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was funded by the International Collaborative Research Fund (GJHZ20180413181716797) and Free Inquiry Fund (JCYJ20180305163929948) of Shenzhen Science and Technology Innovation Commission to WH.
Disclosure
The authors state that they have no conflicts of interest.
References
1. Centers for Disease Control and Prevention. Antibiotic resistance threats in the United States; 2013. Available from: http://www.cdc.gov/drugresistance/threat-report-2013/.
2. Jeon JH, Lee JH, Lee JJ, et al. Structural basis for carbapenem-hydrolyzing mechanisms of carbapenemases conferring antibiotic resistance. Int J Mol Sci. 2015;16(12):9654–9692. doi:10.3390/ijms16059654
3. Nordmann P, Cuzon G, Naas T. The real threat of Klebsiella pneumoniae carbapenemase-producing bacteria. Lancet Infect Dis. 2009;9(4):228–236. doi:10.1016/S1473-3099(09)70054-4
4. Gomez-Simmonds A, Greenman M, Sullivan SB, et al. Population structure of klebsiella pneumoniae causing bloodstream infections at a New York City Tertiary Care Hospital: diversification of multidrug-resistant isolates. J Clin Microbiol. 2015;53(7):2060–2067. doi:10.1128/JCM.03455-14
5. Diancourt L, Passet V, Verhoef J, Grimont PA, Brisse S. Multilocus sequence typing of Klebsiella pneumoniae nosocomial isolates. J Clin Microbiol. 2005;43(8):4178–4182. doi:10.1128/JCM.43.8.4178-4182.2005
6. Chen L, Mathema B, Chavda KD, et al. Carbapenemase-producing Klebsiella pneumoniae: molecular and genetic decoding. Trends Microbiol. 2014;22(12):686–696. doi:10.1016/j.tim.2014.09.003
7. Zhang R, Liu L, Zhou H, et al. Nationwide surveillance of clinical carbapenem-resistant Enterobacteriaceae (CRE) strains in China. EBioMedicine. 2017;19:98–106. doi:10.1016/j.ebiom.2017.04.032
8. Snitkin ES, Zelazny AM, Thomas PJ, et al. Tracking a hospital outbreak of carbapenem-resistant Klebsiella pneumoniae with whole-genome sequencing. Sci Transl Med. 2012;4(148):148ra116. doi:10.1126/scitranslmed.3004129
9. DeLeo FR, Chen L, Porcella SF, et al. Molecular dissection of the evolution of carbapenem-resistant multilocus sequence type 258 Klebsiella pneumoniae. Proc Natl Acad Sci U S A. 2014;111(13):4988–4993. doi:10.1073/pnas.1321364111
10. Lu MC, Tang HL, Chiou CS, et al. Clonal dissemination of carbapenemase-producing Klebsiella pneumoniae: two distinct sub-lineages of Sequence Type 11 carrying blaKPC-2 and blaOXA-48. Int J Antimicrob Agents. 2018;52(5):658–662. doi:10.1016/j.ijantimicag.2018.04.023
11. Jiang Y, Wei Z, Wang Y, et al. Tracking a hospital outbreak of KPC-producing ST11 Klebsiella pneumoniae with whole genome sequencing. Clin Microbiol Infect. 2015;21(11):1001–1007. doi:10.1016/j.cmi.2015.07.001
12. CLSI. Performance standards for antimicrobial susceptibility testing; twenty-sixth informational supplement. CLSI document M100-S26. Clinical and Laboratory Standards Institute. 2016.
13. Chen WP, Kuo TT. A simple and rapid method for the preparation of gram-negative bacterial genomic DNA. Nucleic Acids Res. 1993;21(9):2260. doi:10.1093/nar/21.9.2260
14. Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–i890. doi:10.1093/bioinformatics/bty560
15. Antipov D, Korobeynikov A, McLean JS, Pevzner PA. hybridSPAdes: an algorithm for hybrid assembly of short and long reads. Bioinformatics. 2016;32(7):1009–1015. doi:10.1093/bioinformatics/btv688
16. Wyres KL, Wick RR, Gorrie C, et al. Identification of Klebsiella capsule synthesis loci from whole genome data. Microb Genom. 2016;2:e000102. doi:10.1099/mgen.0.000102
17. Junemann S, Sedlazeck FJ, Prior K, et al. Updating benchtop sequencing performance comparison. Nat Biotechnol. 2013;31(4):294–296. doi:10.1038/nbt.2522
18. Treangen TJ, Ondov BD, Koren S, Phillippy AM. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 2014;15(11):524. doi:10.1186/s13059-014-0524-x
19. Ozer EA, Allen JP, Hauser AR. Characterization of the core and accessory genomes of Pseudomonas aeruginosa using bioinformatic tools Spine and AGEnt. BMC Genomics. 2014;15(1):737. doi:10.1186/1471-2164-15-737
20. Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–2069. doi:10.1093/bioinformatics/btu153
21. Munoz-Price LS, Quinn JP. The spread of Klebsiella pneumoniae carbapenemases: a tale of strains, plasmids, and transposons. Clin Infect Dis. 2009;49(11):1739–1741. doi:10.1086/648078
22. Yang J, Ye L, Guo L, et al. A nosocomial outbreak of KPC-2-producing Klebsiella pneumoniae in a Chinese hospital: dissemination of ST11 and emergence of ST37, ST392 and ST395. Clin Microbiol Infect. 2013;19(11):E509–E515. doi:10.1111/1469-0691.12275
23. El-Badawy MF, El-Far SW, Althobaiti SS, Abou-Elazm FI, Shohayeb MM. The First Egyptian Report showing the co-existence of bla NDM-25, bla OXA-23, bla OXA-181, and bla GES-1 among carbapenem-resistant K. pneumoniae Clinical isolates genotyped by BOX-PCR. Infect Drug Resist. 2020;13:1237–1250. doi:10.2147/IDR.S244064
24. Strauss L, Stegger M, Akpaka PE, et al. Origin, evolution, and global transmission of community-acquired Staphylococcus aureus ST8. Proc Natl Acad Sci U S A. 2017;114(49):E10596–E10604. doi:10.1073/pnas.1702472114
25. Higgins PG, Prior K, Harmsen D, Seifert H. Development and evaluation of a core genome multilocus typing scheme for whole-genome sequence-based typing of Acinetobacter baumannii. PLoS One. 2017;12(6):e0179228. doi:10.1371/journal.pone.0179228
26. Zhou H, Liu W, Qin T, Liu C, Ren H. Defining and evaluating a core genome multilocus sequence typing scheme for whole-genome sequence-based typing of Klebsiella pneumoniae. Front Microbiol. 2017;8:371. doi:10.3389/fmicb.2017.00371
27. Ho JY, Lin TL, Li CY, et al. Functions of some capsular polysaccharide biosynthetic genes in Klebsiella pneumoniae NTUH K-2044. PLoS One. 2011;6(7):e21664. doi:10.1371/journal.pone.0021664
28. Peng D, Li X, Liu P, et al. Transcriptional regulation of galF by RcsAB affects capsular polysaccharide formation in Klebsiella pneumoniae NTUH-K2044. Microbiol Res. 2018;216:70–78. doi:10.1016/j.micres.2018.08.010
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.