")
Back to Journals » International Journal of Nanomedicine » Volume 17
Exosomal Vaccine Loading T Cell Epitope Peptides of SARS-CoV-2 Induces Robust CD8+ T Cell Response in HLA-A Transgenic Mice
Authors Shen AR, Jin XX, Tang TT, Ding Y, Liu XT, Zhong X, Wu YD, Han XL, Zhao GY, Shen CL , Lv LL, Liu BC
Received 31 March 2022
Accepted for publication 16 July 2022
Published 29 July 2022 Volume 2022:17 Pages 3325—3341
DOI https://doi.org/10.2147/IJN.S367494
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Phong A Tran
An-Ran Shen,1 Xiao-Xiao Jin,2 Tao-Tao Tang,1 Yan Ding,2 Xiao-Tao Liu,2 Xin Zhong,1 Yan-Dan Wu,2 Xue-Lian Han,3 Guang-Yu Zhao,3 Chuan-Lai Shen,2 Lin-Li Lv,1 Bi-Cheng Liu1
1Institute of Nephrology, Zhongda Hospital, Medical School of Southeast University, Nanjing, 210009, People’s Republic of China; 2Department of Microbiology and Immunology, Medical School of Southeast University, Nanjing, 210009, People’s Republic of China; 3State Key Laboratory of Pathogen and Biosecurity, Beijing Institute of Microbiology and Epidemiology, Beijing, 100071, People’s Republic of China
Correspondence: Lin-Li Lv; Bi-Cheng Liu, Institute of Nephrology, Zhongda Hospital, Medical School of Southeast University, Nanjing, 210009, People’s Republic of China, Tel +862583272512, Email [email protected]; [email protected]
Purpose: Current vaccines for the SARS-CoV-2 virus mainly induce neutralizing antibodies but overlook the T cell responses. This study aims to generate an exosomal vaccine carrying T cell epitope peptides of SARS-CoV-2 for the induction of CD8+ T cell response.
Methods: Thirty-one peptides presented by HLA-A0201 molecule were conjugated to the DMPE-PEG-NHS molecules, and mixed with DSPE-PEG to form the peptide-PEG-lipid micelles, then fused with exosomes to generate the exosomal vaccine, followed by purification using size-exclusion chromatography and validation by Western blotting, liquid nuclear magnetic resonance (NMR) test and transmission electron microscopy. Furthermore, the exosomal vaccine was mixed with Poly (I:C) adjuvant and subcutaneously administered for three times into the hybrid mice of HLA-A0201/DR1 transgenic mice with wild-type mice. Then, the epitope-specific T cell responses were detected by ex vivo ELISPOT assay and intracellular cytokine staining.
Results: The exosomal vaccine was purified from the Peak 2 fraction of FPLC and injected into the hybrid mice for three times. The IFN-γ spot forming units and the frequencies of IFN-γ+/CD8+ T cells were 10– 82-fold and 13– 65-fold, respectively, higher in the exosomal vaccine group compared to the Poly (I:C) control group, without visible organ toxicity. In comparison with the peptides cocktail vaccine generated in our recent work, the exosomal vaccine induced significantly stronger T cell response.
Conclusion: Exosomal vaccine loading T cell epitope peptides of SARS-CoV-2 virus was initially generated without pre-modification for both peptides and exosomes, and elicited robust CD8+ T cell response in HLA-A transgenic mice.
Keywords: the severe acute respiratory syndrome coronavirus-2, exosomal vaccine, T cell, epitope, human leukocyte antigen-A
Graphical Abstract:
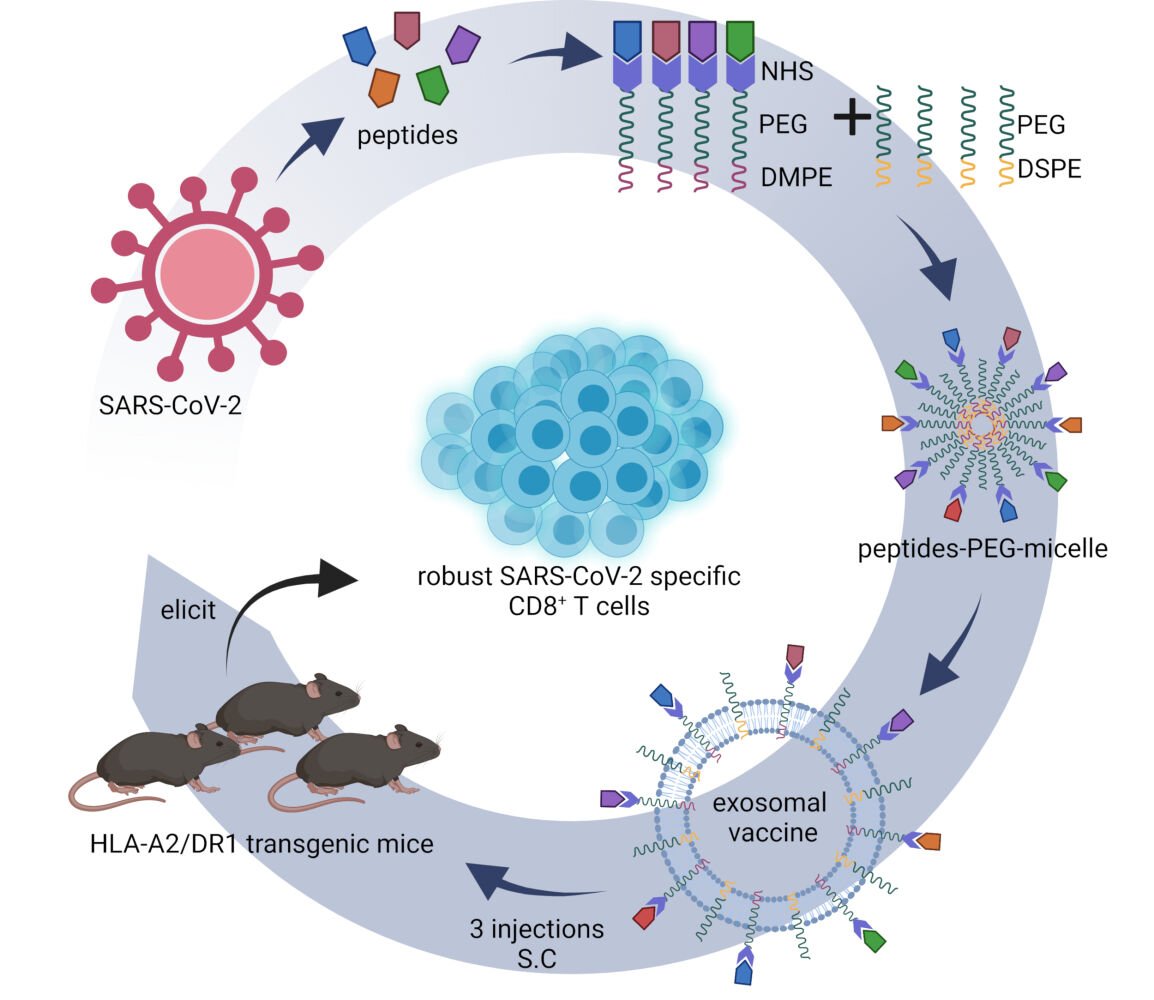
Introduction
Coronavirus disease 2019 (COVID-19) has been a worldwide pandemic since 2019 caused by the severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). The World Health Organization (WHO) has reported more than 527.60 million cases globally and nearly 6.29 million deaths as of June 1, 2022. Current vaccines aim to generate neutralizing antibodies to prevent SARS-CoV-2 infection and reduce severity upon re-infection by variants. However, the available data have shown a rapid decline of neutralizing antibodies after natural infection or vaccination,1–3 and the attenuation of neutralizing protection against the emerging variants.4–6 Increasing studies have demonstrated that antibodies and effector T cells play synergistic protection against SARS-CoV-2 infection.7–9 Moreover, the T cells play a critical role in long-term protection and defense against SARS-CoV-2 variants.10–12 Therefore, attentions have been shifted to the development of vaccines that induce T cell responses.13,14
Exosomes are extracellular vesicles generated by all cells, and they are of endosomal origin and in a size range of ~40 to 160 nm in diameter. Depending on the cell of origin, exosomes contain many constituents of a cell, including nucleic acids, proteins, lipids, and metabolites. They are mediators of near- and long-distance intercellular communication in health and disease, and affect various aspects of cell biology.15 They play a diverse role in cell-to-cell crosstalk and delivery of biological payloads, such as miRNAs,16 drugs,17 and proteins.18,19 Exosome-based immunotherapy against tumors20 and viral infections21 has shown immense clinical potential because of their ability to elicit strong host immune responses as well as useful pharmacological characteristics, such as high biocompatibility, low immunogenicity, low toxicity, and high stability in circulation.22,23 Furthermore, exosomes allow the cross-presentation of exogenous protein antigens to elicit CD8+ T cell responses.21 Although many approaches have been developed to introduce proteins or peptides into exosomes,25 most methods need the exosome surface or payload pre-modification. Loading multiple peptides or proteins into exosomes remains a challenge. In this study, we developed a novel self-assembly approach to load proteins or peptides of interest into the exosome membrane without payload or EV pre-modifications.
In our recent work, we have validated 120 T cell epitopes from the envelope protein (E), membrane protein (M), nucleocapsid protein (N), spike glycoprotein (S), and RNA-dependent RNA polymerase (RdRp) of SARS-CoV-2.24 Here, we generated an exosomal vaccine carrying 31 T cell epitope peptides presented by human leukocyte antigen (HLA)-A0201 molecule, and induced SARS-CoV-2 specific CD8+ T cell responses in HLA-A0201/DR1 transgenic mice.
Materials and Methods
Human Blood Samples
The blood samples of healthy donors were collected from the Blood Component Preparation Section of Jiangsu Province Blood Center (Nanjing, Jiangsu province, China) in the form of a white blood cell filter disc on which the red blood cells (RBCs) have been filtered. Informed consent was waived for this study because the white blood cell filter discs were biological specimens collected from past clinical diagnosis and treatment, and their consent was obtained from Jiangsu Province Blood Center. The collection and use of human samples was approved by the Clinical Ethics Committee of Affiliated Zhongda Hospital of Southeast University, Nanjing, Jiangsu Province, China.
Experimental Mice
Ten-week-old female HLA-A*02:01/DR1 transgenic and H-2-β2m−/−/IAβ−/− C57BL/6 mice were from the Beijing Institute of Microbiology and Epidemiology. Ten-week-old male wild-type C57BL/6 mice were purchased from the Comparative Medicine Center of Yangzhou University (Yangzhou, China). The mice were housed in the specific pathogen-free Animal Centre of Southeast University (Nanjing, China). The hybrid mice of the HLA-A0201+/+/DR1+/+/H-2-β2m−/−/IAβ−/− C57BL/6 mice with the wild-type C57BL/6 mice were bred in house. Animal welfare and experimental procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals (Ministry of Science and Technology of China, 2006) and were approved by the Animal Ethics Committee of Southeast University, Nanjing, China.
Preparation of RBC-Derived Exosomes
Fresh blood was harvested from a white blood cell filter tray by washing it backwards with sterile phosphate-buffered saline (PBS). The RBC-derived exosomes were then prepared according to a previously described protocol with slight modifications.26 Briefly, RBCs were purified by density-gradient centrifugation using Ficoll-Paque and washed with PBS twice by centrifuging at 3000 g for 10 min at 4℃. Then, the RBCs were diluted 50 folds with PBS containing 1μM 4-bromo-calcimycin and were mildly shaken for 48 h at 4℃. Then, the RBC mixture was centrifuged at 3000 g for 20 min at 4℃. The supernatant was collected and centrifuged at 13,500 g for 30 min at 4℃ to remove large vesicles. The resulting supernatant was filtered with a 0.22 μm filter. Then, the supernatant was centrifuged at 200,000 g for 2 h at 4℃ in a XPN-optima-100 ultra-centrifuge (Beckman). The exosome pellet was harvested and resuspended in 100 μL of PBS and stored at –80℃.
Characterization of Exosomes
The size and counts of the RBC-derived exosomes were assessed by nanoparticle tracking analysis (ZetaView, PMX 110, Particle Metrix, Inning am, Ammersee, Germany). The morphology analysis of the exosomes was performed by first diluting them with PBS and loading them onto the carbon-coated copper grids for 3–5 min. Then, the grids with the exosome samples were stained with 2% phosphotungstic acid for 1–2 min. Excess liquid was removed with a filter paper, and the grids were dried at room temperature. Then, the morphology of the exosomes was acquired using the transmission electron microscope (TEM, Hitachi HT 7700, Japan) at 120 kV. The exosome protein levels were quantified using the BCA protein assay kit (KeyGEN Biotech, Nanjing, China). Western blot analysis was performed with exosome-specific protein markers, such as CD63, CD9, and Alix, using specific antibodies.
Preparation of BSA-PEG-Micelles
We dissolved 1,2-Dimyristoyl-sn-glycero-3-phosphoethanolamine (DMPE)-polyethylene glycol (PEG)-NHS (MW: 4257.98, customized in Ponsure Biological, Shanghai) and BSA (66.4 KDa, BioFroxx Germany) in DMSO at a 1:3 molar ratio with 1% triethylamine (TEA, Aladdin, Shanghai) overnight at room temperature to generate BSA-conjugated DMPE-PEG (BSA-PEG-DMPE). Then, we added HEPES buffer (10mM HEPES, pH 7.4) to dilute the concentration of DMSO below 1%, and 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine (DSPE)-PEG (MW: 2805.497, Ponsure Biological) was supplemented at a 1:1 molar ratio with the DMPE-PEG-NHS pre-existed in the system and maintained for 15 min at 60℃. The free BSA molecules were removed by ultrafiltration using Vivaspin tubes (100KD MWCO, Millipore). The BSA quantity in the BSA-PEG micelles was measured using the BCA protein assay kit (KeyGEN Biotech). The BSA-PEG micelles were stored at 4℃ for use within a period of 2 weeks.
Preparation of BSA-PEG-Lipid-Exosomes
The BSA-PEG micelles were redissolved at 60℃ for 10 min followed by ultrasonic vibration twice (5s/time) with 20-μm amplitude to reduce the micelle size. RBC-exosomes were centrifuged at 1000 g for 10 min at 4℃, and the pellet was removed. The supernatant was mixed with the BSA-PEG-micelles at a 1:1 ratio based on the protein concentrations and was incubated in a water bath for 2 h at 40℃. The resulting solution with the BSA-PEG-lipid-exosomes was then cooled to 4℃ followed by instant purification.
Purification of BSA-PEG-Lipid-Exosomes by Size-Exclusion Chromatography
Smartarose CL-4B (SEC0081, Smart-Lifesciences Biotechnology Co. Ltd., Changzhou, China) was loaded onto the SXK16/40 column (Smart-Lifesciences) according to the manufacturer’s instructions. The column was equilibrated with PBS and stored in 20% alcohol at 4℃. The exosome-micelle mixture was purified by fast protein liquid chromatography (FPLC) system (Pharmacia Fine Chemicals, Uppsala, Sweden) with the SXK16/40 Smartarose CL-4B filtration column. The samples were eluted at a speed of 1 mL/min, and 3 mL fractions were collected. The protein levels of the fractions were analyzed with a UV spectrophotometer at 280 nm. The chromatography data were acquired and analyzed using the UNICORN software (version 7.1, GE Life Sciences). Then, the protein samples from each fraction were separated on a 12% SDS-PAGE and analyzed by Western blotting using mouse anti-BSA antibodies and mouse anti-human CD63 mAb to identify the peak fractions containing the BSA-PEG-micelles.
Liquid Nuclear Magnetic Resonance Test
In order to identify the peak fraction containing BSA-PEG-lipid-exosome, all of the three chromatography peak fractions were diluted in PBS, concentrated by ultrafiltration to 1 mL and subjected to liquid nuclear magnetic resonance test (NMR) (600 MHz, Advance III 400HD, Bruker, Germany) for identifying PEG, hydrolytic NHS, and DMPE molecules in the BSA-PEG-DMPE macromolecules. The spectral peaks for PEG, hydrolytic NHS groups and DMPE were identified at 3.6 ppm, 2.6 ppm, and 1.17 ppm, respectively.
Western Blotting
The samples from RBC-derived exosomes, BSA-PEG-micelles, and the peak fractions for the BSA-PEG-lipid-exosomes from size-exclusion chromatography were analyzed by Western blotting. The protein concentrations of all samples were estimated using the BCA assay kit (KeyGEN Biotech). Then, equal amounts of protein were separated on a 10% SDS-PAGE and transferred to the PVDF membrane (Millipore) using a high quality wet protein transfer system (L00686C eBLOT L1, Genscript, Nanjing, China). The membranes were then blocked with 5% skim milk powder in TBST buffer for 1 h at RT. The blots were then incubated overnight at 4℃ with the primary antibodies including mouse anti-BSA (1:2000, Proteintech), mouse anti-human CD63 (1:1000, Santa Cruz), mouse anti-human CD9 (1:1000, Proteintech), or mouse anti-human Alix (1:1000, Santa Cruz). Subsequently, the blots were incubated with HRP-conjugated goat anti-mouse IgG antibodies (1:5000, Cell signaling) for 1 h at RT. Then, the blots were developed using the Enhanced chemiluminescence detection kit (BL520A, Biosharp).
Preparation and Purification of Peptide-PEG-Lipid-Exosomes
We screened and validated 120 T cell epitope peptides from SARS-CoV-2 proteins in the previous study.24 Among these, 31 validated epitope peptides (VEPs) (9- or 10-mer) presented by the HLA-A0201 molecules were dissolved individually in DMSO at a concentration of 10 μL/mg/peptide and mixed together with 1 mg per peptide before use. We used the same procedure used for generating the BSA-PEG-lipid-exosome complex to generate the peptide-PEG-lipid-exosome complex. DMPE-PEG-NHS and the mixture of VEPs were dissolved in DMSO at a 1:3 molar ratio with 1% TEA overnight at RT to form the peptides-PEG-DMPE complex. Then, HEPES buffer was added to dilute the DMSO concentration below 1%. Then, DSPE-PEG was added at a 1:1 molar ratio to the pre-existing peptides-DMPE-PEG-NHS complex and maintained for 15 min at 60℃. The free peptides were removed, and the protein levels in the resultant peptides-PEG-micelles were quantified using the BCA assay kit and stored at 4℃ for use in the following 2 weeks. Then, the peptides-PEG-micelles were redissolved, ultrasonicated to reduce the micelle size, and co-incubated with the RBC-derived exosomes at a 1:1 ratio in a water bath for 2 h at 40℃. The resulting solution was cooled to 4℃ followed by instant purification by the FPLC system with the SXK16/40 Smartarose CL-4B filtration column. Finally, the fractions of the second peak were collected, concentrated in normal saline by ultrafiltration, and used as the peptides-PEG-lipid-exosome complex.
Immunization of the HLA-A2 Transgenic Mice with Peptides-PEG- Lipid-Exosomes
Eight-week-old hybrid C57BL/6 mice of the HLA-A0201+/+/DR1+/+/H-2-β2m−/−/I-Aβ−/− genotype and wild-type C57BL/6 mice were randomly divided into two groups (5 male mice per group) and vaccinated with peptides-PEG-lipid-exosome/Poly (I:C) (Vaccine group) and Poly (I:C)/normal saline (Control group), respectively. Each mouse was vaccinated subcutaneously three times on days 0, 7, and 21. The peptides-PEG-lipid-exosomes (containing 1.64 mg peptides) complex was mixed with 500 μg of Poly (I:C) (HMW VacciGrade, InvivoGen) and injected into five mice in the vaccine group (around 10 μg peptide/mouse). Each mouse in the control group was injected with 100 μg of Poly (I:C) in normal saline. Seven days after the final injection (day 28), the splenocytes of the mice were prepared to evaluate the epitope peptide-specific T cell responses.
ELISPOT and Intracellular IFN-γ Staining
The T cell epitope-specific responses in the primed mice were evaluated by IFN-γ ELISPOT. Briefly, the 31 VEPs restricted by the HLA-A0201 molecules were reconstituted in a PBS buffer containing DMSO before use at a storage concentration of 2 mg/mL. For the IFN-γ ELISPOT assay, the VEPs were grouped into eight pools according to their protein source, acidity and alkalinity characteristics. For the intracellular IFN-γ staining, the 31 VEPs were grouped into five pools according to their protein source.
ELISPOT microplates (96 wells, PVDF membrane, Merck & Millipore) were coated with anti-IFN-γ capture mAb (BD Biosciences) at 4℃ overnight according to the manufacturer’s instructions. The spleen cells (2 × 105 cells/100μL/well) from each of the primed mice were seeded into each well together with a single peptide pool (2 μg/well for each peptide), PHA (10 μg/mL as positive control), irrelevant T cell epitope peptides as non-specific control (HLA-A2-restricted AFP158-166 and A24-restricted AFP424-432 at 2 μg/well) or PBS without peptide as negative control. These mixtures were incubated for 20 h at 37℃ and 5% CO2. Then, the plates were incubated with a biotinylated anti-IFN-γ detecting antibody (BD) for 2 h at RT. Then, the plates were incubated with streptavidin-conjugated HRP (BD) for 1 h at RT. Finally, AEC solution (BD) was used to develop the color. The spots were imaged and enumerated with a Mabtech IRISTM Elispot & FluoroSpot Reader (Mabtech, Nacka Strand, Sweden).
In addition, the spleen cells from each of the primed mice were incubated with a single peptide pool (20 μg/mL for each peptide), PHA (10 μg/mL), irrelevant epitope peptides (AFP158-166 and AFP424-432, 20 μg/mL for each peptide), or PBS without peptide for 16 h in a 48-well plate with serum-free RPMI-1640 medium at 37℃ and 5% CO2. Then, the cells were co-cultured with the Brefeldin A (BFA)/monensin mixture (2.5 μL/well, MultiSciences, Hangzhou, China) for another 6 h. The cells were then harvested, washed, blocked with anti-mouse CD16/CD32 antibodies for 20 min at 4℃, and stained with FITC-labeled anti-CD3 and PE-labeled anti-CD8 antibodies for 30 min at 4℃. After washing, the cells were fixed and permeabilized using Fix&Perm kit (MultiSciences) according to the manufacturer’s protocol. The fixed cells were further incubated with APC-anti-mouse IFN-γ (clone XMG1.2, BD) for another 30 min at 4℃ and analyzed by flow cytometry in a BD FACS Calibur (BD Bioscience). The frequencies of IFN-γ+ cells in the CD3+/CD8+ populations were calculated.
Hematoxylin-Eosin Staining
Twenty-eight days after primary immunization, mice were sacrificed, and the heart, liver, lung, and kidney tissues were harvested and fixed in 4% paraformaldehyde overnight. The individual lobes of the biopsy organs were placed in the processing cassettes, dehydrated with a serial alcohol gradient, and embedded in paraffin wax blocks. Five-micrometer-thick tissue sections were cut, dewaxed in xylene, rehydrated through decreasing concentrations of ethanol, and washed into PBS. Then, the sections were stained with hematoxylin and eosin, and photographed.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 7 (GraphPad, La Jolla, CA, USA). Two-tailed unpaired t-test was used to compare the data between control group and vaccine group. p < 0.05 was considered significant.
Results
Preparation and Characterization of RBC-Derived Exosomes
The purification protocol of exosomes from human RBCs by ultracentrifugation is shown in Figure 1A. Majority (75%) of the RBC-derived exosomes were 50 −150 nm in diameter (average diameter: 128 nm) (Figure 1B). The exosomes displayed typical round shape in the TEM images (Figure 1C). Western blot analysis showed that the exosome markers, namely, Alix, CD63 and CD9 (Figure 1D).
 |
Figure 1 Preparation and characterization of RBC-derived exosomes. Exosomes were extracted from RBCs and characterized. (A) The diagram of exosomes preparation; (B) The size distribution and quantity of exosomes identified by nanoparticle tracking analysis; (C) The morphology of exosomes measured by TEM; (D) Protein markers (Alix, CD63, and CD9) of exosomes identified using Western blotting. |
Generation and Purification of BSA-PEG-Lipid-Exosomes
The NHS group-conjugated lipid-PEG (DMPE-PEG-NHS) complexes were prepared according to the manufacturer’s instructions. The NHS group forms covalent bonds with the COOH groups of proteins or peptides. This enables DMPE (lipid) to complex with any peptide or protein of interest. The BSA-PEG-lipid micelles or peptides-PEG-lipid micelles were allowed to insert into the lipid bilayers of the exosomes by self-assembly and structure relaxation caused by high temperature (Figure 2).
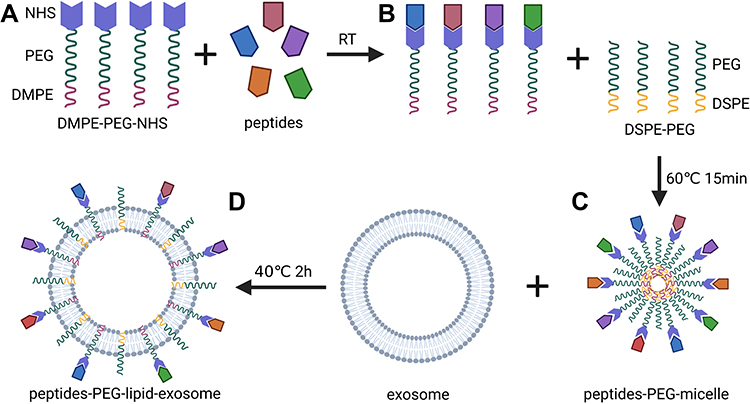 |
Figure 2 The diagram of Peptides-PEG-lipid-exosome generation. DMPE-PEG lipid molecules were conjugated with NHS group, then covalently bind to peptides of interest (A), and form the micelles with DSPE-PEG (B), and finally self-assembly insert into the lipid bilayer of exosome during co-incubation for 2 h at 40℃ (C) to form the peptides-PEG-lipid-exosome (D). |
The feasibility of this novel approach in generating peptides-PEG-lipid-exosome complexes was first tested by preparing BSA-PEG-lipid-exosome complexes. The molar ratio between BSA and DPME-PEG-NHS was optimized by testing 1:1, 1:1.5, 1:3, and 1:4 ratios as shown in Table 1. In order to calculate the BSA loading capacity and loading efficiency in the BSA-PEG-micelles, the conjugated BSA with DMPE in BSA-PEG micelle solution and the free BSA in the filtered solution was quantified using BCA kit, respectively. The results showed that the BSA loading capacity was highest (85 μg BSA was conjugated with 2 mg DMPE-PEG-NHS) at 1:3 molar ratio of DMPE-PEG-NHS with BSA (Figure 3A). The loading efficiency was 91.84% at 1:3 molar ratio and then decreased rapidly when the BSA input increased (Figure 3B). Then, the RBC-exosomes were mixed with BSA-PEG-micelles in a 1:1 ratio based on the protein concentrations. The mixture was incubated in a water bath for 2 h at 40℃. The BSA-PEG-lipid-exosomes solution was then cooled to 4℃ and instantly purified using size-exclusion chromatography.
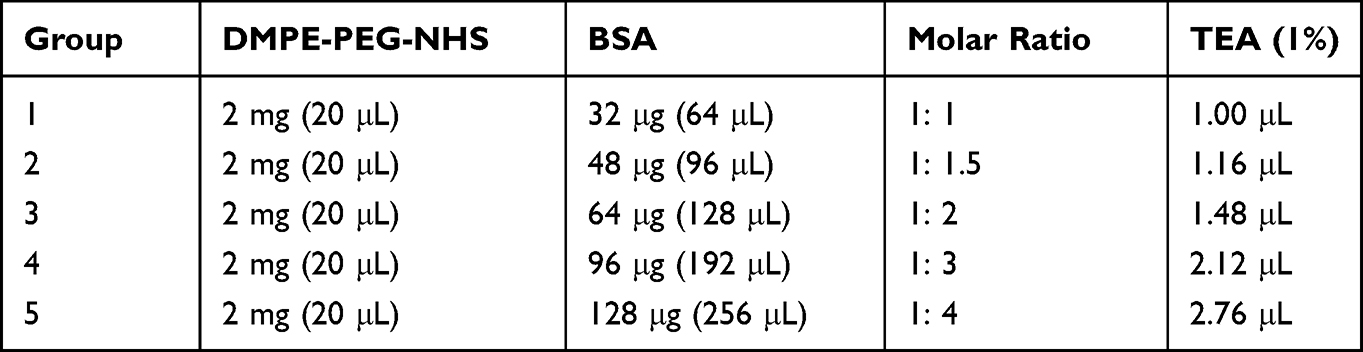 |
Table 1 Optimization of BSA-PEG-DMPE Generation |
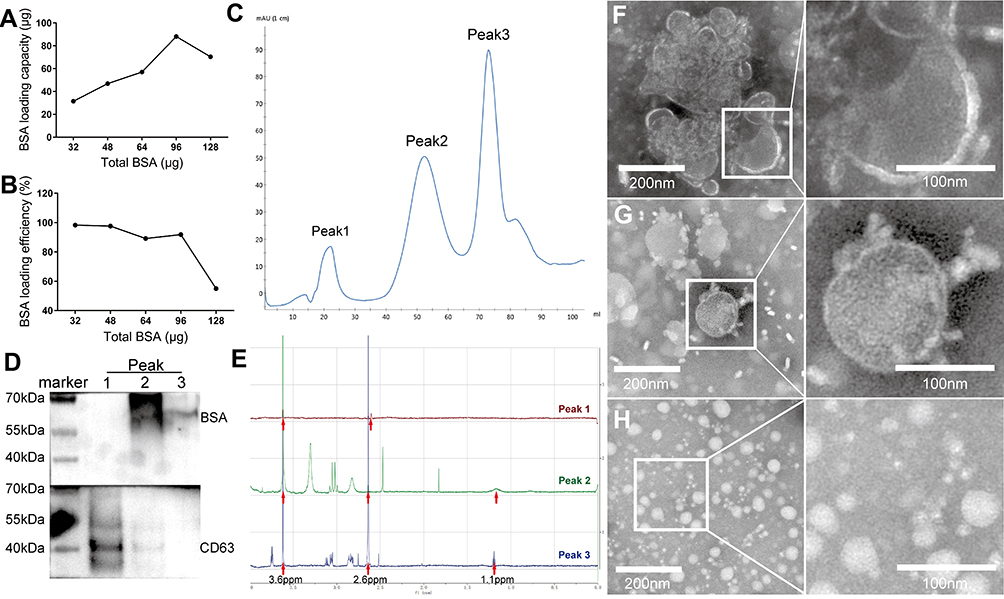 |
Figure 3 Purification and identification of BSA-PEG-lipid-exosome. A series of amount of BSA was conjugated with 2 mg of DMPE-PEG-NHS first and then mixed with DSPE-PEG to form the BSA-PEG micelles. The free BSA was quantified and BSA loading capacity (A) and loading efficacy (B) in the BSA-PEG micelles was calculated, respectively. Furthermore, the BSA-PEG micelles were regenerated at 1:3 molar ratio of DMPE-PEG-NHS with BSA and incubated with RBC-exosomes for 2 h at 40℃ to generate BSA-PEG-lipid-exosomes, and instantly purified using size-exclusion chromatography with the FPLC profile displaying three major peaks (C). The fractions of three peaks were subjected to Western blotting with anti-BSA and anti-CD63 for the identification of BSA and exosomes in each peak (D), and also subjected to liquid NMR for the identification of PEG, hydrolytic NHS and DMPE elements in each peak (E). Meanwhile, the fractions were measured using TEM to inspect the morphology of exosomes in peak 1 (F), peak 2 (G) and peak 3 (H). |
The FPLC profile of the BSA-PEG-lipid-exosomes showed three major peaks (Figure 3C). Western blot analysis showed that most of the BSA at peak 2 and low BSA levels at peak 3, whereas, exosome marker CD63 was found at higher concentrations at peak 1 and in smaller quantities at peak 2 (Figure 3D). The liquid NMR analysis showed PEG (fl = 3.6 ppm), hydrolytic NHS (fl = 2.6 ppm), and DMPE (fl = 1.17 ppm) in peaks 1, 2, and 3, with the highest levels in peak 2 (Figure 3E). The morphology of exosomes in peaks 1 and 2 was measured by TEM. Peak 1 exosomes showed large piles of stacked vesicle structure (Figure 3F). Peak 2 exosomes were regular-sized exosomes with protein or micelle-like structures attached to the surface (Figure 3G). Exosome vesicle structures (100–200 nm in diameter) were not found at peak 3 (Figure 3H). Taken together, these data demonstrated that the second peak represented the BSA-PEG-lipid-exosome complex because this mixture contained high-levels of BSA, DMPE-PEG, hydrolytic NHS, and exosome (CD63inter), and displayed typical exosome morphology.
Generation of Peptides-PEG-Lipid-Exosomes
Next, a mixture of 31 T cell epitope peptides validated from SARS-CoV-2 proteins and presented by the HLA-A0201 molecule was conjugated with DMPE-PEG-NHS and fused with the RBC-derived exosomes to form the peptides-PEG-lipid-exosome complex according to the procedure established previously for the BSA-PEG-lipid-exosome complex. The size-exclusion chromatography showed three major peaks (Figure 4A) with a similar FPLC profile as observed during the purification of the BSA-PEG-lipid-exosome complex. The peak 2 fractions were collected and concentrated by ultrafiltration. Then, the peptide content in the peptides-PEG-lipid-exosome complex was calculated by subtracting the protein concentration in the peptide-PEG-lipid-micelle by the protein concentration in the peak 3 fraction.
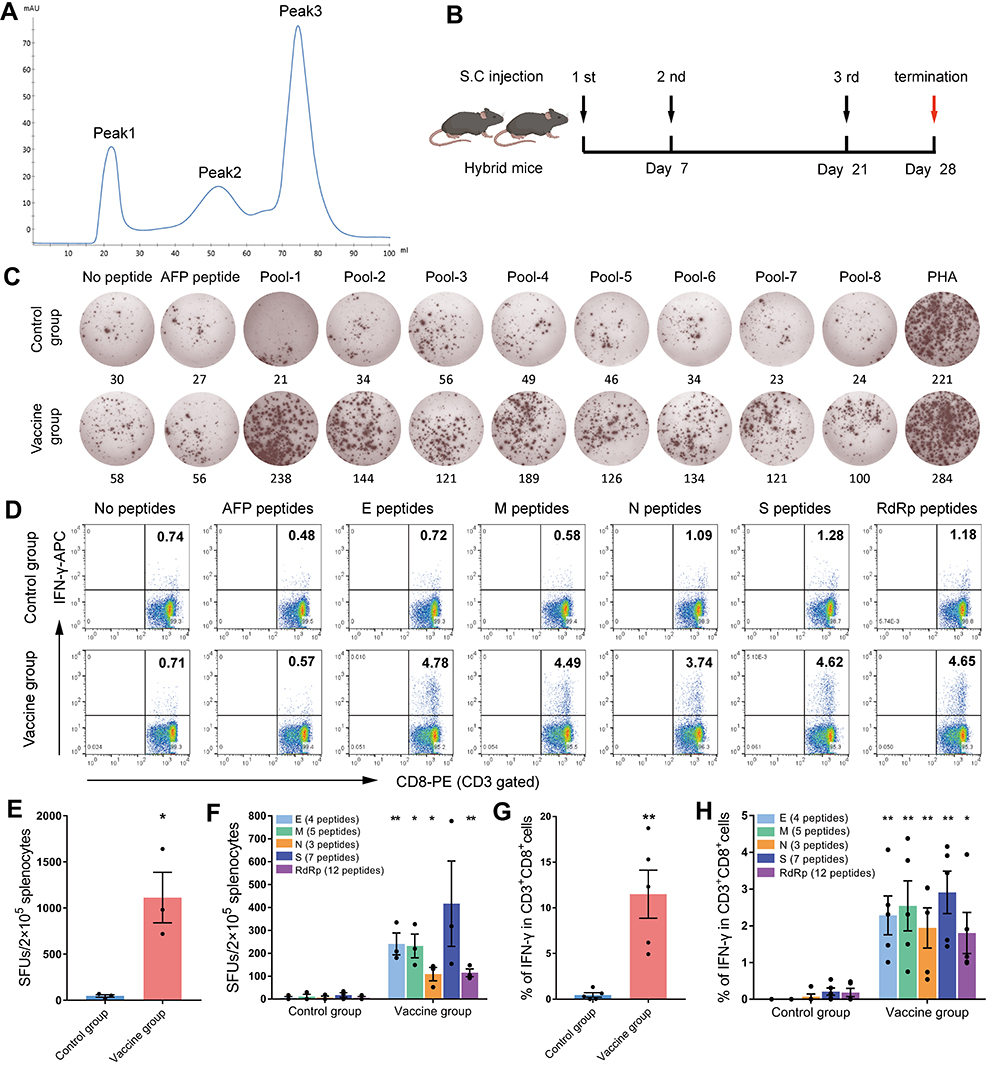 |
Figure 4 Peptides-PEG-lipid-exosomes elicited robust CD8+ T cell responses in hybrid mice. The cocktail of 31 VEPs was used to generate peptides-PEG-lipid-exosomes, and followed by three rounds of immunizations to the hybrid mice of HLA-A0201/DR1 transgenic C57BL/6 mice with wild-type mice. Then, splenocytes were collected 7 days after the final booster and ex vivo stimulated with each peptide pool, irrelevant peptide (HLA-A24 restricted AFP158-166, AFP424-432 epitope), no peptide (negative control), or PHA (positive control) overnight followed by IFN-γ ELISPOT and ICS. (A) The FPLC profile of peptides-PEG-lipid-exosomes purification. (B) The timeline of immunization. (C) Representative SFUs spot plots and SFUs numbers of splenocytes from each group in the IFN-γ ELISPOT assay under stimulation with each peptide pool. (D) Representative flow cytometric dot plots of IFN-γ+/CD8+ T cells in splenocytes from each group in the intracellular IFN-γ staining under stimulation with each peptide pool. (E) The IFN-γ SFUs number responding to all peptide pools (31 VEPs) in each group. Data in the histograms were presented as mean ± SEM, n = 3 per group. (F) Deconvolution of the SFUs numbers from (E) into the single SARS-CoV-2 protein. (G) The frequency of IFN-γ+ T cells reacting to all peptide pools (31 VEPs) in CD3+CD8+ T cell population in each group. Data in the histograms were presented as mean ± SEM, n = 5 per group. (H) Deconvolution of the frequency from (G) into the single SARS-CoV-2 protein. Control group: poly(I:C) plus normal saline; Vaccine group: peptides-PEG-lipid-exosomes plus poly(I:C). *p < 0.05; **p < 0.01. |
Exosomal Vaccine Elicits Robust in vivo Epitope Specific CD8+ T Cell Response
The hybrid mice of HLA-A0201/DR1 transgenic C57BL/6 mice with wild-type C57BL/6 mice were bred in house and the expression levels of HLA-A0201 and DR1 molecules on the peripheral white blood cells were confirmed by Flow cytometry analysis (data not shown). The hybrid mice were randomly divided into two groups and vaccinated with either peptides-PEG-lipid-exosome/Poly (I:C) (Vaccine group) or Poly (I:C)/normal saline (Control group) three times on days 0, 7, and 21 (Figure 4B). Seven days after the final injection (day 28), splenocytes of the primed mice were tested for epitope-specific T cell responses by IFN-γ ELSPOT and IFN-γ ICS.
The 31 VEPs were grouped into eight pools according to their original proteins and their acidic and alkaline features (Table 2). Then, splenocytes from each of the primed mice were co-incubated with each peptide pool, irrelevant CD8+ T cell epitope peptides (AFP158-166 and AFP424-432), PHA or PBS for 20 h in a 96-well PVDF membrane plate followed by the IFN-γ ELSPOT assay. The total number of SFUs per 2 × 105 splenocytes from each primed mouse was approximately 10–82 times higher in the vaccine group compared to the control group (1113 ± 274 vs 46.67 ± 14.31; Figure 4C, E and F). Two irrelevant CD8+ T cell epitope peptides (AFP158-166 and AFP424-432) were used as non-specific antigen control in ELISPOT assay and displayed results similar to the negative control. Figure S1 shows positive spots in the ELISPOT assay from 3 mice per group.
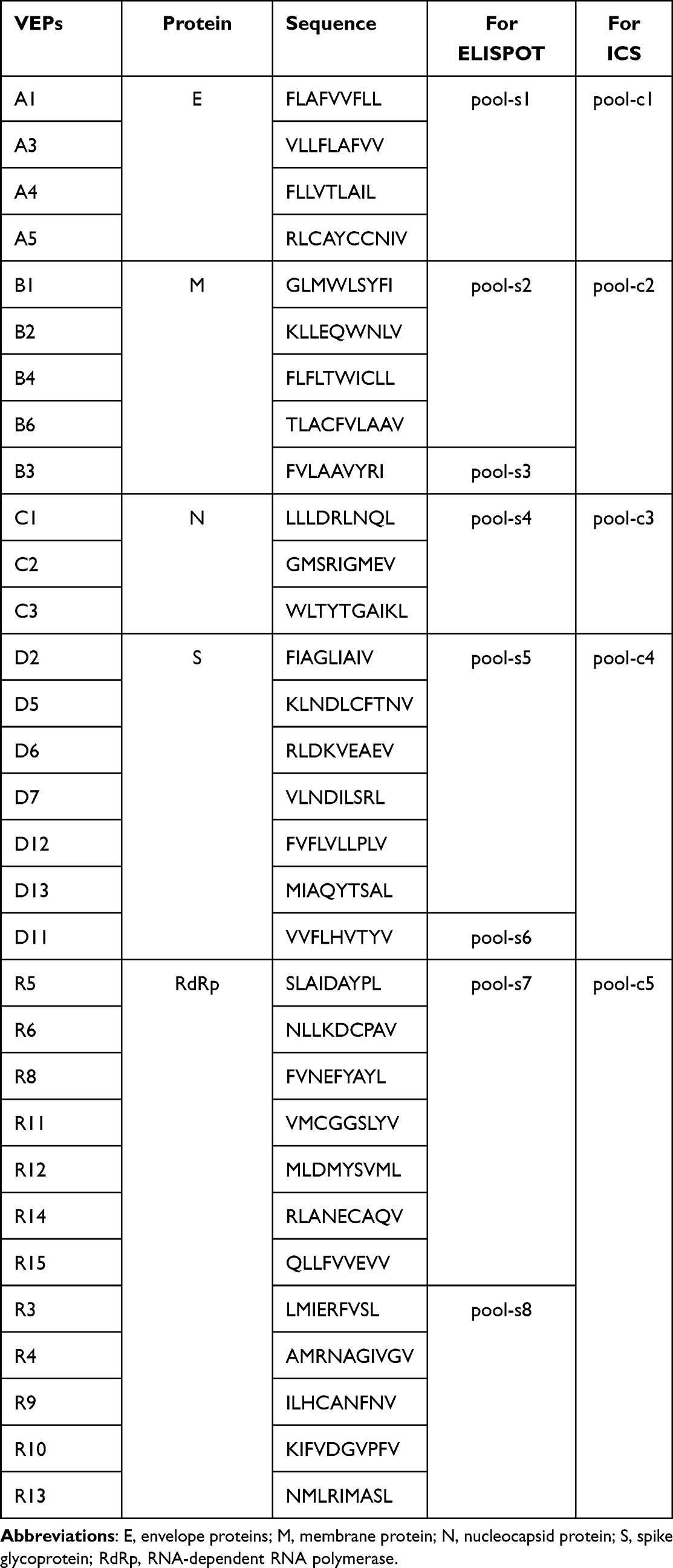 |
Table 2 31 VEPs Binding to the HLA-A0201 Molecule Were Grouped into Different Peptide Pools for IFN-γ ELISPOT and IFN-γ ICS Assay |
Furthermore, an intracellular cytokine staining (ICS) assay was performed for IFN-γ to confirm the specific CD8+ T cell responses. The 31 VEPs were grouped into five pools according to their original proteins (Table 2). Each peptide pool was co-incubated with the splenocytes from each of the primed mice for 16 h in a 48-well plate. Then, the splenocytes were co-incubated for 6 h with a BFA/monensin mixture. The frequency of IFN-γ+ cells in the CD3+CD8+ T cell populations of the vaccine group was 13–65 folds higher than in the Poly (I:C)/normal saline control group (11.5 ± 2.634% vs 0.462 ± 0.246%; Figure 4D, G and H). The groups with two non-specific CD8+ T cell epitope peptides showed results similar to negative control. Figure S2 shows the flow cytometry plots of IFN-γ ICS for each mouse. As shown, peptides-PEG-lipid-exosome/poly (I:C) elicited robust epitope-specific CD8+ T cell response, whereas poly (I:C)/normal saline did not activate CD8+ T cells.
Twenty Eight of the 31 VEPs Induced Robust in vivo T Cell Responses
Next, we identified the VEPs that elicited T cell responses in the hybrid mice. Briefly, each of the 31 VEPs were co-incubated with the splenocytes from the three mice in the vaccine group and two mice from the control group followed by the IFN-γ ELSPOT assay. The assay set up also included controls such as non-specific epitope peptides, PHA, and PBS control wells. As shown in Figure 5, 28 out of the 31 VEPs induced specific T cell responses in either three or two of the primed mice with at least 2-fold higher SFUs than the PBS control (no peptide stimulation). Furthermore, 3 VEPs (R3, R11, and R12) did not induce T cell responses in the three primed mice. The two mice in the Poly (I:C)/normal saline control group did not show any positive T cell responses specific to any of the 31 VEPs and their SFUs were similar to the PBS control. Figure S3 shows the spots from the ELISPOT assay for each of the 31 VEPs in each of the primed mice.
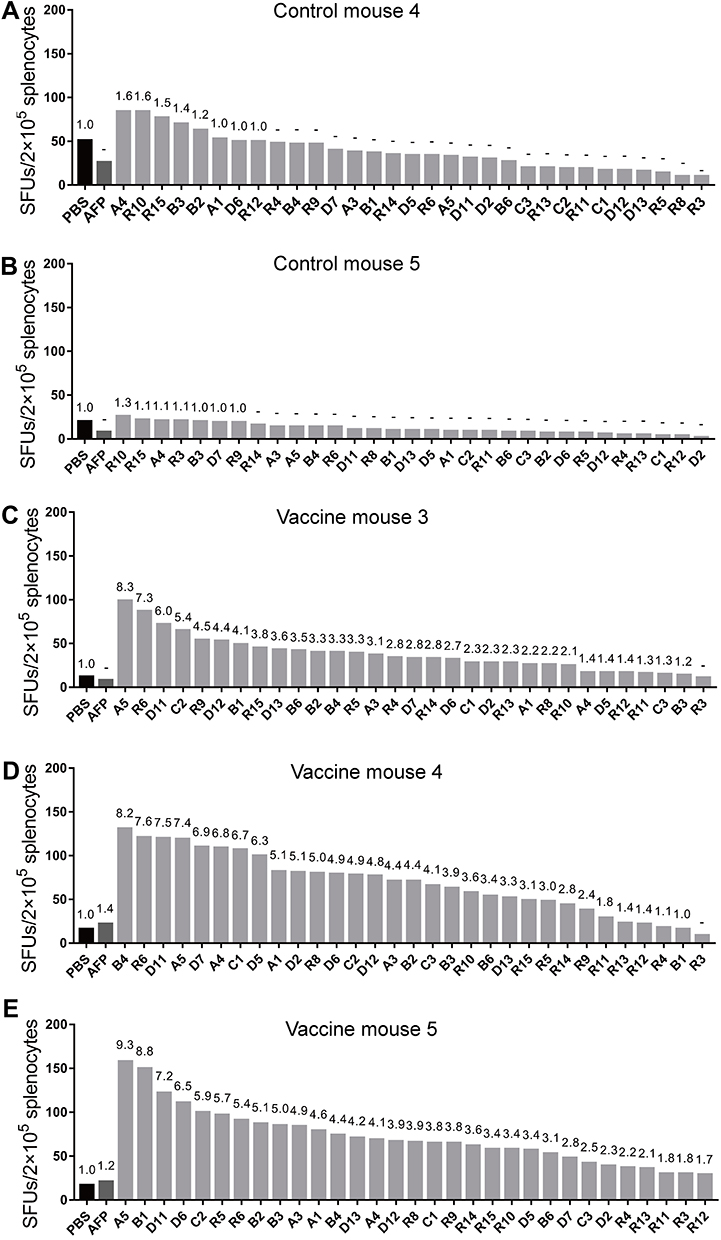 |
Figure 5 28 of 31 VEPs elicited T cell response in hybrid mice. Splenocytes from each primed mouse were harvested 7 days after the last booster and ex vivo stimulated with each of the 31 VEPs, AFP irrelevant peptides, PHA, or PBS, and followed by IFN-γ ELSPOT assay. Two mice in Control group (A and B) and three mice from Vaccine group (C–E) were tested. The IFN-γ SFUs responding to each peptide in 2×105 splenocytes from each mouse was presented as histograms. The fold of SFUs responding to each peptide compared to the SFUs responding to PBS (no peptide) was displayed on each histogram. |
Exosomal Vaccine Did Not Cause Significant Organ Toxicity
Exosomes are biocompatible and biodegradable with low biotoxicity. Therefore, we investigated if the peptide-loaded exosomes in our in vivo experiments caused organ toxicity by checking the heart, liver, lung, and kidney tissues from all the experimental mice on day 28. Hematoxylin-eosin staining assay did not show any significant organ injury in any of the mice belonging to all the experimental groups (Figure 6).
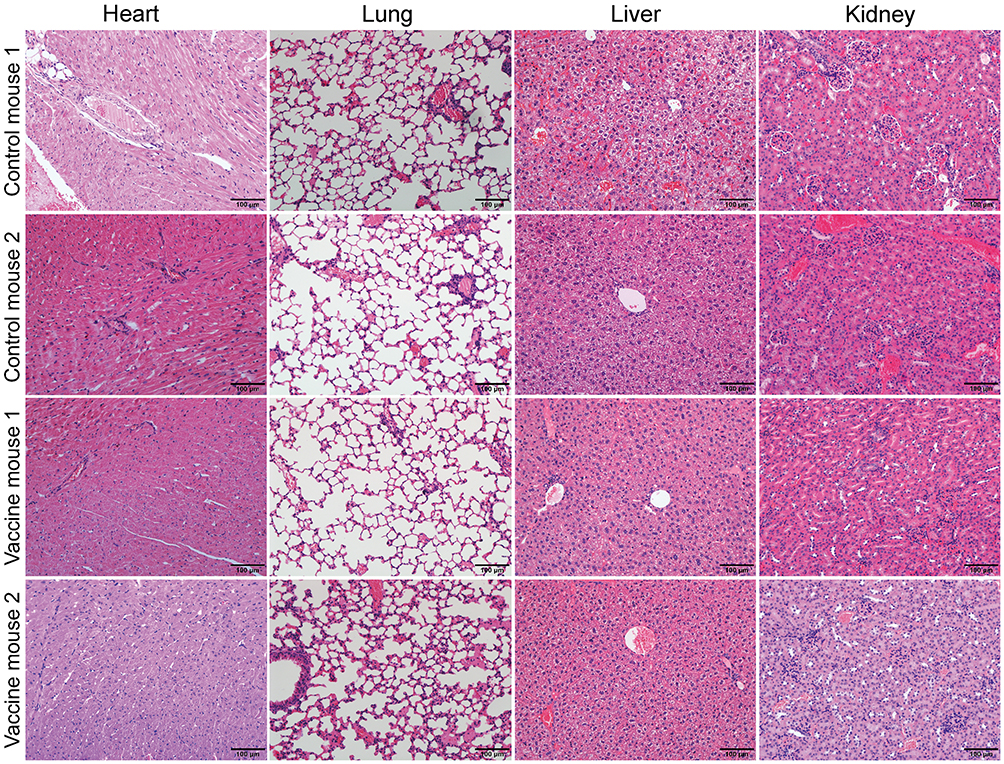 |
Figure 6 The peptide cocktail vaccine have no visible toxicity on the organs. Seven days after the last booster, all mice were executed. Heart, liver, lung and kidney were taken out, immersed and were finally stained with Hematoxylin-Eosin. No obvious pathological damage was found in all organs in each group. The representative HE staining of heart, liver, lung and kidney in two mice from each group were exhibited. |
Discussion
In this study, we generated an exosomal vaccine carrying 31 T cell epitope peptides of SARS-CoV-2 and confirmed its potential to elicit robust CD8+ T cell responses in HLA-A transgenic mice. Exosomes can be collected from a variety of sources including human biological fluids and cells. RBCs are a valuable natural source of large-scale exosomes because of their abundance and lack nucleus and other organelles.27 Therefore, in this study, we used RBC-derived exosomes to generate the exosomal vaccine. However, how to load multiple peptides onto the exosomes remains a challenge. The key points of this study were that: firstly, we established a new procedure to load peptides into exosome membrane without peptide and EVs pre-modification. Secondly, many T cell epitope peptides were loaded into exosome membrane rather than one peptide at a time for the spatial and temporal co-delivery. Thirdly, the exosomal vaccine carrying T cell epitope peptides of SARS-CoV-2 was established for the first time, and may have a much stronger potential to elicit epitope-specific CD8+ T cell responses in vivo than the peptides cocktail vaccine generated in our recent works.24
Exosomes transfer multiple biological cargos between cells and organs through blood circulation. Therefore, they are a good vehicle for the clinical delivery of therapeutic agents, thus numerous exosome engineering methods have been proposed, including endogenous and exogenous loading approaches. Endogenous loading refers to a system that involves deposition of the biological cargo in the engineered cell directly into the EVs prior to its shedding.25,28 Exogenous loading includes passive loading that requires the incubation of EVs with cargos and active loading that requires disruption to the EV membrane through electroporation, saponin permeabilization, or sonication which may cause cargo and EVs aggregation and damage EVs phospholipid bilayer and cargos. Proteins or peptides can be loaded into EVs or displayed on the surface of EVs by various passive exogeneous techniques, such as liposome fusing with exosome, click chemistry conjugation, and aptamer coating.25 However, these commonly used passive cargo-loading techniques require pre-modifications of the cargo proteins/peptides and EVs. In click chemistry, alkynes are grafted onto the EV membranes and the cargos are tagged by azide.29,30 Freeze-thaw-induced or PEG-induced hybrid EV/liposomes require embedding the peptides or proteins into synthetic liposomes.31,32 Aptamer coating mediates the connection between proteins to the EV membrane, but requires pre-modification of the EVs.33
We used the amino group-conjugated lipid-PEG molecules (DMPE-PEG-NHS) to load a mixture of peptides into exosomes without pre-modification of the peptides or the EVs. This simplified the process and enabled conjugation of all peptides of interest with the lipid. This modified self-assembly approach involved passive exogeneous loading without any risk of damage to the EV membrane. In this process, the peptides were first covalently conjugated with the lipid micelles (DSPE-PEG-NHS/DMPE-PEG) through peptide bonds, which stably connected the peptides with the lipids. Then, the peptide conjugated lipid micelles were assembled into exosome membranes through simple co-incubation for 2 h at 40℃ to allow DSPE and DMPE phospholipids to fuse with the exosome membrane. Then, size-exclusion chromatography was used to isolate and purify the peptides-PEG-lipid-exosome complexes. These showed a specific FPLC profile with three apparent peaks, which related to the large pile of exosomal aggregates, peptides-PEG-lipid-exosomes, and free peptide-PEG-micelles, respectively. These were confirmed by Western blotting, liquid NMR, and TEM. A previous study used size-exclusion chromatography to isolate peptide-nanobody-PEG-lipid-EV; the required product appeared in the first peak, while the second and third peaks included EVs and peptides-PEG-micelle, respectively.22 This difference in our FPLC profile is mainly attributed to the smaller molecular size of peptide conjugated micelles compared to the peptide conjugated nanobody.
Our study demonstrates that many peptides of interest could be co-loaded into the exosomes through this modified protocol. This will pave the way to generate vaccines inducing T cell immune responses against multiple viral variants. The current vaccines are mostly designed to induce neutralizing antibodies that bind to the conformational B cell epitopes on the receptor-binding domain (RBD) of the S protein, thereby blocking the binding of SARS-CoV-2 to ACE2 surface receptor protein on the host cells. However, since the first outbreak, many variants have emerged with mutations in the RBD, which have resulted in conformational changes of the B cell epitopes and significant decline in the neutralizing activity of the vaccine-induced antibodies.4–6 However, T cell epitopes are 9–10-mer or 15–19-mer short peptides, and distributed along the entire viral proteome with a much larger amount than the B cell epitopes in the RBD. Therefore, the mutations of viral variants have less impact on these T cell epitopes than B cell epitopes. The more T cell epitope peptides are co-delivered by exosomal vaccine, the more virus-specific T cell clones will be activated in vivo. Therefore, the peptide cocktail-loaded exosomal vaccine will possess much stronger potential to protect against infection by the viral variants than the conventional B cell epitope vaccines. In addition, the peptide cocktail-loaded exosomal vaccine will be more effective in individuals with distinct HLA allotypes because a cocktail of peptides can be presented by many HLA subtypes, which will cover a larger population. More importantly, several studies have demonstrated that memory T cells persist in vivo for 6–17 years after natural infections by viruses, such as SARS-CoV and MARS-CoV.14,34,35 In contrast, the life-span of memory B cells is significantly shorter in the host.34,36 Therefore, T cell epitope-based vaccines are expected to provide long-lasting preventive and therapeutic effects against SARS-CoV-2 infection by inducing specific T cell responses.
Currently, the SARS-CoV-2 vaccine candidates designed to prime T cell responses are very limited. In a self-experimentation study, 13 peptides (15-mer) derived from the SARS CoV-2 S protein and N protein and predicted to cross-bind to several HLA-DR allotypes were emulsified with montanide and the TLR 1/2 ligand XS15 and vaccinated in a healthy donor as a single subcutaneous injection. The results showed that 9 out of 13 peptides induced robust CD4+ T cell responses, but five 9-mer peptides restricted by HLA-A01 or B08 molecules did not elicit CD8+ T cell responses after vaccination with the same adjuvants.37 Meanwhile, an ongoing Phase I clinical trial (NCT04546841) is testing the safety and immunogenicity of a peptide vaccine containing SARS-CoV-2 HLA-DR binding peptides in volunteers without prior or current SARS-CoV-2 infections using XS15 adjuvant.37 Emergex Vaccines Holding Ltd is evaluating the safety and immunogenicity of a T-cell priming synthetic SARS-CoV-2 peptide vaccine mounted on gold nanoparticles in a clinical trial (NCT05113862). This vaccine is expected to be effective against all currently sequenced viral mutations. In the animal model, Venezuelan equine encephalitis replicon particles expressing single CD8+ or CD4+ T cell epitopes of the SARS-CoV-2 protein induced robust T cell responses in the Ad5-hACE2-transduced and SARS-CoV-2-infected mice and led to faster viral clearance than the neutralizing antibodies and attenuated lung pathological damage.38 Furthermore, 54 peptides that derived from the nonstructural polyprotein 1a of SARS-CoV-2 and showed high or medium binding affinities to HLA-A0201 molecule were encapsulated into liposomes, and further administered into HLA-A0201 transgenic (HHD) mice. Eighteen of 54 peptides elicited peptide-specific CD8+ T cell activation and cytotoxicity, and 10 peptides induced high responses.39 To the best of our knowledge, this is the first study to provide experimental data regarding the efficacy of an exosomal vaccine candidate carrying multiple T cell epitope peptides of SARS-CoV-2 in a mouse model.
Whether the exosomal vaccine has advantages over the nanoparticle vaccine or liposome vaccine remains controversial. However, the lipid and protein composition of exosomes can affect the pharmacokinetic properties of peptides and act as an adjuvant to enhance the immune response, while their natural constituents may play a role in enhanced bioavailability and in minimizing adverse reactions.15,21–23 Notably, this exosomal vaccine candidate may have a stronger potential to elicit specific CD8+ T cell responses than the classical peptide cocktail vaccine with an adjuvant. In a recent study,24 we used the 31 T cell epitope peptides of SARS-CoV-2 and restricted by HLA-A0201 molecule as peptide cocktail vaccine with R848 or Poly (I:C) to inject into the hybrid mice of HLA-A0201/DR1 transgenic C57BL/6 mice with wild-type C57BL/6 mic. The vaccination regimen was identical to the current exosomal vaccine, including the same 31 T cell epitope peptides, the same usage dose of each epitope peptide (10μg/peptide), the same kind of hybrid mice, the same poly(I:C) control group, the same injection time line, the same time point to collect spleen cells of immunized mice, and the same IFN-γ ELISPOT and intracellular cytokine staining protocols. Additionally, both vaccine immunization experiments and later ELISPOT/ICS analysis were mainly carried out by the same person during around half year. IFN-γ ELISPOT assay showed 9-fold higher SFUs in the 2 × 105 splenocytes tested from each of the primed mice in the Vaccine B group (peptides/R848) and Vaccine C group (peptides/poly I:C) compared to the control group with adjuvants and normal saline without the peptides. The frequencies of IFN-γ+ cells in CD3+CD8+ T cell populations from each of the primed mice from the Vaccine B and C groups were approximately 5 folds higher than the control mice, as detected by IFN-γ ICS and flow cytometry experiments.24 However, in the current study, the SFUs were approximately 10–82 times higher in the vaccine group than in the control group (Figure 4C and E), while the frequencies of IFN-γ+ cells in CD3+CD8+ T cell populations in the vaccine group were 13–65 folds higher than in the control group (Figure 4D and G). Of note is that we did not directly compare the indicated IFN-γ SFUs number and IFN-γ+/CD8+ T cells frequency between the current vaccine group and the early vaccine group, so the deviation between the two experiments should be decreased to some extent. These results suggested that the exosomal vaccine may have a much stronger ability to induce specific T cell responses in vivo than the peptide cocktail vaccine, but more immunization experiments are needed to confirm this issue.
Conclusions
This study established a novel approach for passively conjugating many T cell epitope peptides of SARS-CoV-2 to the RBC-derived exosomes without pre-modification to both peptides and exosomes. Furthermore, this exosomal vaccine elicited robust peptide-specific CD8+ T cell responses in the HLA-A transgenic mice without any visible organ toxicity. This approach provides an alternative way to generate SARS-CoV-2 vaccines inducing T cell responses that may provide long-lasting protection from SARS-CoV-2 and its emerging variants.
Abbreviations
COVID-19, coronavirus disease 2019; SARS-CoV-2, severe acute respiratory syndrome coronavirus-2; HLA, Human leukocyte antigens; RBC, Red blood cells; EVs, extracellular vesicles; PBS, Phosphate-buffered saline; TEM, Transmission electron microscopy; DMPE, 1,2-Dimyristoyl-sn-glycero-3-phosphoethanolamine; DSPE, 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine; TEA, triethylamine; FPLC, fast protein liquid chromatography; NMR, nuclear magnetic resonance; VEPs, validated epitope peptides; ELISPOT, enzyme-linked immunosorbent spot; PHA, phytohemagglutinin; SFU, spot forming units; ICS, intracellular cytokine staining; RBD, receptor-binding domain.
Acknowledgment
This work was supported by the National Nature Science Foundation of China (82041006) and COVID-19 Emergency Research Fund of Zhejiang University of China (2020XGZX021). The sponsors had no role in study design, data collection and analysis, preparation of the manuscript, or decision to submit the article for publication.
Disclosure
The authors declare no competing financial interests related to this study.
References
1. Chen Y, Yin S, Tong X, et al. Dynamic SARS-CoV-2-specific B-cell and T-cell responses following immunization with an inactivated COVID-19 vaccine. Clin Microbiol Infect. 2021;28(3):410–418. doi: 10.1016/j.cmi.2021.10.006
2. Anand SP, Prévost J, Nayrac M, et al. Longitudinal analysis of humoral immunity against SARS-CoV-2 Spike in convalescent individuals up to 8 months post-symptom onset. Cell Rep Med. 2021;2(6):100290. doi: 10.1016/j.xcrm.2021.100290
3. Koerber N, Priller A, Yazici S, et al. Dynamics of spike-and nucleocapsid specific immunity during long-term follow-up and vaccination of SARS-CoV-2 convalescents. Nat Commun. 2022;13(1):153. doi: 10.1038/s41467-021-27649-y
4. Cele S, Gazy I, Jackson L, et al. Escape of SARS-CoV-2 501Y.V2 from neutralization by convalescent plasma. Nature. 2021;593(7857):142–146. doi: 10.1038/s41586-021-03471-w
5. Liu C, Ginn HM, Dejnirattisai W, et al. Reduced neutralization of SARS-CoV-2 B.1.617 by vaccine and convalescent serum. Cell. 2021;184(16):4220–4236 e4213. doi: 10.1016/j.cell.2021.0
6. Murano K, Guo Y, Siomi H. The emergence of SARS-CoV-2 variants threatens to decrease the efficacy of neutralizing antibodies and vaccines. Biochem Soc Trans. 2021;49(6):2879–2890. doi: 10.1042/BST20210859
7. Ni L, Ye F, Cheng ML, et al. Detection of SARS-CoV-2-specific humoral and cellular immunity in COVID-19 convalescent individuals. Immunity. 2020;52(6):971–977 e973. doi: 10.1016/j.immuni.2020.04.023
8. Melenotte C, Silvin A, Goubet AG, et al. Immune responses during COVID-19 infection. Oncoimmunology. 2020;9(1):1807836. doi: 10.1080/2162402X.2020.1807836
9. Tay MZ, Poh CM, Renia L, MacAry PA, Ng LFP. The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol. 2020;20(6):363–374. doi: 10.1038/s41577-020-0311-8
10. Goletti D, Petrone L, Manissero D, et al. The potential clinical utility of measuring severe acute respiratory syndrome coronavirus 2-specific T-cell responses. Clin Microbiol Infect. 2021;27(12):1784–1789. doi:10.1016/j.cmi.2021.07.005
11. Sekine T, Perez-Potti A, Rivera-Ballesteros O, et al. Robust T cell immunity in convalescent individuals with asymptomatic or mild COVID-19. Cell. 2020;183(1):158–168 e114. doi:10.1016/j.cell.2020.08.017
12. Mathew D, Giles JR, Baxter AE, et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science. 2020;369(6508):eabc8511. doi:10.1126/science.abc8511
13. Sauer K, Harris T. An effective COVID-19 vaccine needs to engage T cells. Front Immunol. 2020;11:581807. doi:10.3389/fimmu.2020.581807
14. Sariol A, Perlman S. Lessons for COVID-19 immunity from other coronavirus infections. Immunity. 2020;53(2):248–263. doi:10.1016/j.immuni.2020.07.005
15. Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020;367(6478). doi:10.1126/science.aau6977
16. Lv LL, Feng Y, Wu M, et al. Exosomal miRNA-19b-3p of tubular epithelial cells promotes M1 macrophage activation in kidney injury. Cell Death Differ. 2020;27(1):210–226. doi:10.1038/s41418-019-0349-y
17. Tang TT, Lv LL, Wang B, et al. Employing macrophage-derived microvesicle for kidney-targeted delivery of dexamethasone: an efficient therapeutic strategy against renal inflammation and fibrosis. Theranostics. 2019;9(16):4740–4755. doi:10.7150/thno.33520
18. Chaput N, Schartz NE, Andre F, et al. Exosomes as potent cell-free peptide-based vaccine. II. Exosomes in CpG adjuvants efficiently prime naive Tc1 lymphocytes leading to tumor rejection. J Immunol. 2004;172(4):2137–2146. doi:10.4049/jimmunol.172.4.2137
19. Mehanny M, Lehr CM, Fuhrmann G, Extracellular vesicles as antigen carriers for novel vaccination avenues. Adv Drug Deliv Rev. 2021;173:164–180. doi:10.1016/j.addr.2021.03.016
20. Zhang W, Zhang X, Zhang L, et al. Astrocytes increase exosomal secretion of oligodendrocyte precursor cells to promote their proliferation via integrin beta 4-mediated cell adhesion. Biochem Biophys Res Commun. 2020;526(2):341–348. doi:10.1016/j.bbrc.2020.03.092
21. Kuate S, Cinatl J, Doerr HW, Uberla K. Exosomal vaccines containing the S protein of the SARS coronavirus induce high levels of neutralizing antibodies. Virology. 2007;362(1):26–37. doi:10.1016/j.virol.2006.12.011
22. Kooijmans SAA, Fliervoet LAL, van der Meel R, et al. PEGylated and targeted extracellular vesicles display enhanced cell specificity and circulation time. J Control Release. 2016;224:77–85. doi:10.1016/j.jconrel.2016.01.009
23. Chen NX, O’Neill K, Chen XM, Kiattisunthorn K, Gattone VH, Moe SM. Transglutaminase 2 accelerates vascular calcification in chronic kidney disease. Am J Nephrol. 2013;37(3):191–198. doi:10.1159/000347031
24. Jin X, Ding Y, Sun S, et al. Screening HLA-A-restricted T cell epitopes of SARS-CoV-2 and the induction of CD8+ T cell responses in HLA-A transgenic mice. Cell Mol Immunol. 2021;18(12):2588–2608. doi:10.1038/s41423-021-00784-8
25. Tang TT, Wang B, Lv LL, Liu BC. Extracellular vesicle-based Nanotherapeutics: emerging frontiers in anti-inflammatory therapy. Theranostics. 2020;10(18):8111–8129. doi:10.7150/thno.47865
26. Tang TT, Wang B, Li ZL, et al. Kim-1 targeted extracellular vesicles: a new therapeutic platform for RNAi to treat AKI. J Am Soc Nephrol. 2021;32(10):2467–2483. doi:10.1681/ASN.2020111561
27. Gangadaran P, Hong CM, Oh JM, et al. In vivo non-invasive imaging of radio-labeled exosome-mimetics derived from red blood cells in mice. Front Pharmacol. 2018;9:817. doi:10.3389/fphar.2018.00817
28. Sutaria DS, Badawi M, Phelps MA, Schmittgen TD. Achieving the promise of therapeutic extracellular vesicles: the devil is in details of therapeutic loading. Pharm Res. 2017;34(5):1053–1066. doi:10.1007/s11095-017-2123-5
29. Tian T, Zhang HX, He CP, et al. Surface functionalized exosomes as targeted drug delivery vehicles for cerebral ischemia therapy. Biomaterials. 2018;150:137–149. doi:10.1016/j.biomaterials.2017.10.012
30. Jia G, Han Y, An YL, et al. NRP-1 targeted and cargo-loaded exosomes facilitate simultaneous imaging and therapy of glioma in vitro and in vivo. Biomaterials. 2018;178:302–316. doi:10.1016/j.biomaterials.2018.06.029
31. Sato YT, Umezaki K, Sawada S, et al. Engineering hybrid exosomes by membrane fusion with liposomes. Sci Rep. 2016;6(6):21933. doi:10.1038/srep21933
32. Piffoux M, Silva AKA, Wilhelm C, Gazeau F, Tareste D. Modification of extracellular vesicles by fusion with liposomes for the design of personalized biogenic drug delivery systems. Acs Nano. 2018;12(7):6830–6842. doi:10.1021/acsnano.8b02053
33. Wan Y, Wang LX, Zhu CD, et al. Aptamer-conjugated extracellular nanovesicles for targeted drug delivery. Cancer Res. 2018;78(3):798–808. doi:10.1158/0008-5472.CAN-17-2880
34. Tang F, Quan Y, Xin ZT, et al. Lack of peripheral memory B cell responses in recovered patients with severe acute respiratory syndrome: a six-year follow-up study. J Immunol. 2011;186(12):7264–7268. doi:10.4049/jimmunol.0903490
35. Rodriguez L, Pekkarinen PT, Lakshmikanth T, et al. Systems-level immunomonitoring from acute to recovery phase of severe COVID-19. Cell Rep Med. 2020;1(5):100078. doi:10.1016/j.xcrm.2020.100078
36. Channappanavar R, Fett C, Zhao J, Meyerholz DK, Perlman S, Sandri-Goldin RM. Virus-specific memory CD8 T cells provide substantial protection from lethal severe acute respiratory syndrome coronavirus infection. J Virol. 2014;88(19):11034–11044. doi:10.1128/JVI.01505-14
37. Rammensee HG, Gouttefangeas C, Heidu S, et al. Designing a SARS-CoV-2 T-cell-inducing vaccine for high-risk patient groups. Vaccines. 2021;9(5):428. doi:10.3390/vaccines9050428
38. Zhuang Z, Lai X, Sun J, et al. Mapping and role of T cell response in SARS-CoV-2-infected mice. J Exp Med. 2021;218(4):e20202187. doi:10.1084/jem.20202187
39. Takagi A, Matsuib M, Heise MT. Identification of HLA-A*02:01-restricted candidate epitopes derived from the nonstructural polyprotein 1a of SARS-CoV-2 that may be natural targets of CD8 + T cell recognition in vivo. J Virol. 2021;95(5):e01837–20. doi:10.1128/JVI.01837-20
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.