")
Back to Journals » Cancer Management and Research » Volume 12
Exosomal Transfer of lncRNA H19 Promotes Erlotinib Resistance in Non-Small Cell Lung Cancer via miR-615-3p/ATG7 Axis
Received 4 December 2019
Accepted for publication 28 April 2020
Published 8 June 2020 Volume 2020:12 Pages 4283—4297
DOI https://doi.org/10.2147/CMAR.S241095
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Seema Singh
Rongtao Pan,1 Haiyan Zhou2
1Department of Oncology, Taishan Hospital of Shandong Province, Taian 271000, Shandong, People’s Republic of China; 2Department of Medical Oncology, Shandong Cancer Hospital and Institute, Shandong First Medical University and Shandong Academy of Medical Sciences, Jinan 250117, Shandong, People’s Republic of China
Correspondence: Haiyan Zhou
Department of Medical Oncology, Shandong Cancer Hospital and Institute, Shandong First Medical University and Shandong Academy of Medical Sciences, No. 440 Jiyan Road, Jinan City, Shandong Province, People’s Republic of China
Tel +86-15098935816
Email [email protected]
Background: Drug resistance restrains the effect of drug therapy in non-small cell lung cancer (NSCLC). However, the mechanism of the acquisition of drug resistance remains largely unknown. This study aims to investigate the effect of exosomal lncRNA H19 on erlotinib resistance in NSCLC and the underlying mechanism.
Methods: HCC827 and A549 cells were continuously grafted into erlotinib-containing culture medium to establish erlotinib-resistant cell lines. The expression of H19 and miR-615-3p was detected by qRT-PCR. The protein levels of MMP2, MMP9, CD9, CD63 and ATG7 were measured by Western blot. Cell viability and proliferation were determined by Cell Counting Kit-8 (CCK-8) and 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay, respectively. Migration and invasion were assessed by transwell assay. Xenograft tumor models were used to investigate the effect of H19 on erlotinib resistance in vivo. Online software and dual-luciferase reporter assay were used to predicate the downstream targets and confirm the targeted relationships.
Results: H19 was upregulated in erlotinib-resistant cells, and knockdown of H19 inhibited cell proliferation, migration and invasion in erlotinib-resistant cells. Extracellular H19 can be packaged into exosomes. Exosomes containing H19 induced erlotinib resistance of sensitive cells, while knockdown of H19 abolished this effect. miR-615-3p was a target of H19 and can bind to ATG7. Exosomal H19 affected erlotinib resistance of erlotinib-resistant NSCLC cells via targeting miR-615-3p to regulate ATG7 expression. In addition, the serum exosomal H19 was upregulated in patients with erlotinib resistance. Furthermore, downregulated H19 decreased the resistance of tumor cells to erlotinib in vivo.
Conclusion: Our study demonstrated that exosomal H19 facilitated erlotinib resistance in NSCLC via miR-615-3p/ATG7 axis, which might provide a potential target for the diagnosis and treatment of NSCLC.
Keywords: lncRNA H19, exosome, erlotinib resistance, miR-615-3p, ATG7, non-small cell lung cancer
Introduction
Lung cancer is a prevalent malignancy around the world, which accounts for 18.4% of the total cancer deaths.1 Among all the cases of lung cancer, non-small-cell lung cancer (NSCLC) account for about 85%.2 Radiotherapy, chemotherapy and surgery are the common treatment strategies of NSCLC. Despite many efforts and advances have been made, the prognosis of NSCLC patients is still poor. Drug resistance is a vital factor that affects the 5-year survival rates of NSCLC patients. Therefore, it is of great significance to investigate the regulatory mechanism of drug resistance in NSCLC.
Exosomes are a kind of membrane vesicles with a diameter of 40–100 nm, which can be generated and secreted by multiple types of cells. Exosomes contain various biomolecules, such as microRNAs (miRNAs), long non-coding RNAs (lncRNAs), circular RNAs (circRNAs), DNAs and proteins, and mediate the transfer of these biomolecules, thereby participating in many physiological processes.3,4 In recent years, exosomal lncRNAs have been elucidated to exert important functions in cancer development.5 For example, exosomal lncRNA lymph node metastasis-associated transcript 2 (LNMAT2) facilitates lymph node metastasis in bladder cancer.6 Exosomal lncRNA growth arrest-specific transcript 5 (GAS5) is downregulated in NSCLC and considered as a potential biomarker for the diagnosis of early-stage NSCLC.7 In addition, exosomal lncRNAs are also related to drug resistance during chemotherapy. It has been reported that exosomal lncRNA SBF2-AS1 enhances temozolomide resistance in glioblastoma.8 A previous study revealed that exosomal lncRNA H19 facilitated gefitinib resistance in NSCLC.9 However, the effect of H19 on erlotinib resistance in NSCLC still remains unknown.
MiRNAs are crucial regulators on gene expression through degradation or transcriptional inhibition of target mRNA.10,11 Increasing evidence indicates that miRNAs participate in the development and progression of cancers, including NSCLC. For instance, miR‑138‑5p represses proliferation and induced apoptosis via regulating CDK8 in NSCLC.12 miR-605-5p contributes to proliferation and invasion of NSCLC cells by targeting tumor necrosis factor α-induced protein 3 (TNFAIP3).13 Moreover, studies have shown that miRNAs can serve as biomarkers for the diagnosis and prognosis of NSCLC.14,15
Autophagy-related proteins (ATGs) are highly conserved, which function as regulators of cell autophagy.16 ATG7, a member of the ATG family, has been manifested to regulate cancer progression and drug resistance.17 Tripartite motif (TRIM) 65 promotes autophagy and cisplatin resistance of NSCLC cells through targeting miR-138-5p to regulate ATG7 expression.18 Knockdown of urothelial carcinoma-associated 1 (UCA1) impedes bladder cancer development and drug resistance via miR-582-5p/ATG7 axis.19 Interestingly, a previous study suggested that ATG7 might be not necessary for tumor growth and chemotherapy efficacy in lung cancer.20 Therefore the precise function of ATG7 in NSCLC needs further study.
In this study, we elucidated the role of exosomal H19 on erlotinib resistance in NSCLC, and the molecular mechanism was also investigated. Our results might provide a potential target for the therapy of erlotinib-resistant NSCLC.
Materials and Methods
Serum Samples
The present study has obtained approval from the Ethics Committee of Taishan Hospital of Shandong Province. Serum samples were collected from 58 patients with NSCLC. All patients signed informed consents and received erlotinib treatment at Taishan Hospital of Shandong Province. 5 mL of venous blood from each patient was collected by venipuncture before chemotherapy was started. Serum was separated from blood and stored at −80°C.
Cell Culture
The human NSCLC cell lines HCC827 and A549 (erlotinib-sensitive cells) were purchased from the Chinese Academy of Sciences (Shanghai, China). Cells were cultured in Dulbecco’s modified Eagle medium (DMEM; Invitrogen, Carlsbad, CA, USA) with 10% fetal bovine serum (Invitrogen), maintaining the conditions of 37°C and 5% CO2. Erlotinib (S7786; Selleckchem, Houston, TX, USA) was dissolved in dimethyl sulfoxide (DMSO; Sigma, St. Louis, MO, USA). To establish erlotinib-resistant cell lines, HCC827 and A549 cells were cultured in growth medium with erlotinib and gradually increasing concentrations of erlotinib from 0.002 to 0.2 µM within 3–4 months to generate the resistant cell lines (HCC827/ER and A549/ER).
Cell Viability Assay
Cell viability was determined by the Cell Counting Kit-8 (CCK-8; Beyotime, Shanghai, China) following the instructions. Cells were placed into a 96-well plate. After erlotinib treatment or transfection, CCK-8 reagent was added into each well to incubate the cells for 2 h. A microplate reader (Thermo Fisher Scientific, Waltham, MA, USA) was used for detection of the absorbance at 450 nm to assess cell viability.
Cell Proliferation Assay
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay kit (Sigma) was used for detection of cell proliferation. Cells were placed in a 96-well plate. After treatment, cells were added with 20 µL of MTT (5 mg/mL) and incubated for 4 h. Then, cells were collected and added 150 µL of dimethyl sulfoxide to dissolve the formazan crystals. The absorbance at 490 nm wavelength was detected using a microplate reader (Thermo Fisher Scientific).
Transwell Assay
Transwell assay was carried out for the detection of cell migration and invasion. For migration detection, the upper chamber was added with serum-free medium containing cells. While for invasion, the upper chamber was additionally covered with Matrigel (BD Biosciences, San Diego, CA, USA). The basolateral chamber was added cell medium with 10% serum. After cultured for 24 h, the cells passed through the membrane were fixed by 4% paraformaldehyde and then dyed with 0.5% crystal violet solution. The stained cells were counted under a microscope (Thermo Fisher Scientific).
Western Blot
A protein extraction kit (Beyotime) was used to extract the proteins, and then proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA, USA). The membrane was blocked with 5% nonfat milk which was dissolved with TBST. After that the membrane was incubated with the primary antibodies against MMP2 (1:1000; ab97779; Abcam, Cambridge, UK), MMP9 (1:1000; ab38898 ab92742; Abcam), CD9 (1:2000; ab92726; Abcam), CD63 (1:2000; ab134045; Abcam), ATG7 (1:1000; ab53255; Abcam) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1:5000; ab9485; Abcam) overnight at 4°C. Subsequently, the secondary antibody (1:5000; ab205718; Abcam) was used to incubate the membrane for 1.5 h at room temperature. Protein blot was detected by enhanced chemiluminescence reagents (Millipore).
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
TRIzol Reagent (Invitrogen) was used to isolate the total RNA following the instructions. Then RNA was reversely transcription into cDNA by PrimeScript RT Reagent Kit (Takara, Dalian, China) and microRNA First-Strand cDNA Synthesis Kit (Sangon Biotech, China). SYBR Green Master Mix (Takara) was used for gene quantification. Finally, gene expressions were normalized by GAPDH and U6 and calculated with 2−∆∆Ct method. The primers were listed as follows. H19: 5ʹ-ATCGGTGCCTCAGCGTTCGG-3ʹ (forward), 5ʹ-CTGTCCTCGCCGTCACACCG-3ʹ (reverse); miR-615-3p: 5ʹ-ACACTCCAGCTGGGTCCGAGCCTGGGTCTC-3ʹ (forward), 5ʹ-TGGTGTCGTGGAGTCG-3ʹ (reverse); GAPDH: 5ʹ-AAGCTGGTCATCAATGGGAAAC-3ʹ (forward), 5ʹ-ACCCCATTTGATGTTAGCGG-3ʹ (reverse); U6: 5ʹ-ATGACGTCTGCCTTGGAGAAC-3ʹ (forward), 5ʹ-TCAGTGTGCTACGGAGTTCAG-3ʹ (reverse).
Cell Transfection
Small interfering RNA (siRNA) of H19 (si-H19), miR-615-3p mimic (miR-615-3p), anti-miR-615-3p and their controls (si-NC, miR-NC and anti-miR-NC) were obtained from GenePharma (Shanghai, China). ATG7 cDNA was cloned and inserted into the pcDNA3.1 vector (Invitrogen) for the overexpression of ATG7. Cell transfection was conducted with Lipofectamine 2000 reagent (Invitrogen).
Exosome Isolation
ExoQuick precipitation kit (System Biosciences, Mountain View, CA, USA) was used to extract exosomes from cell culture medium or serum samples. In brief, Cells were collected when reached 80% confluency and centrifuged at 3000×g for 10 minutes to eliminate cell debris. Then, 63 μL ExoQuick precipitation kit was mixed with 250 μL supernatant and chilled at 4°C for 30 min. The Mixtures were centrifuged at 1500×g for 30 min. After the supernatant was removed, the pellet was re-centrifuged at 1500×g for 5 min to eliminate the residual liquid. The exosome was re-suspended with 200 μL phosphate-buffered saline (PBS). All centrifugations were performed at 4°C.
Transmission Electron Microscopy (TEM)
A drop of isolated exosome samples was placed on Parafilm. A carbon-coated copper grid was floated on the drop for 5 min. After that, the grid was removed and a piece of clean filter paper was used to touch the grid edge to blot up the redundant liquid. Next, the grid was placed onto a drop of 2% phosphotungstic acid with pH 7.0 for 5 s, and the redundant liquid was removed. After drying for 5 min, the grid was observed by a JEM-1200 EX microscope (JEOL, Ltd., Tokyo, Japan) at 80 keV.
Dual-Luciferase Reporter Assay
The putative binding sequences between H19 and miR-615-3p, miR-615-3p and ATG7 were predicted by starBase 3.0. The wild type sequences of H19 and the 3ʹ-untranslated region (3ʹ-UTR) of ATG7 (H19 WT and ATG7 3ʹUTR WT) containing the putative binding sites of miR-615-3p and their mutant sequences (H19 MUT and ATG7 3ʹUTR MUT) were cloned into pGL3 reporter vectors (Promega, Madison, WI, USA), respectively. Luciferase reporter vectors and miR-615-3p or miR-NC were co-transfected into HCC827 or A549 cells. The luciferase activities were detected by a Dual-Luciferase reporter system (Promega).
In vivo Tumor Formation Assay
Short hairpin H19 (sh-H19) and the control sh-NC were synthesized by GenePharma and inserted into pLKO.1-puro Lentiviral vector (Sigma). HCC827 cells were transfected with sh-H19 or sh-NC vector and selected with 1 µg/mL puromycin (Invitrogen) for 4 weeks. The animal experiment was approved by the Animal Care Committee of Taishan Hospital of Shandong Province. Animal studies were performed in compliance with the ARRIVE guidelines and the Basel Declaration. All animals received humane care according to the National Institutes of Health (USA) guidelines. 4–5-week-old nude mice were randomly divided into two groups (sh-H19 and sh-NC) and subcutaneously injected with HCC827 cells (1×106 cells/mouse) transfected with sh-H19 or sh-NC, respectively. After a week, the mice were treated with once-daily 25 mg/kg erlotinib, and tumor length and width were measured weekly. Tumor volume was computed as the formula length × width2/2. After 5 weeks, the tumor tissues were resected and weighed.
Statistical Analysis
Data were presented as mean ± standard deviation and analyzed by SPSS 22.0 with at least three repeats for each experiment. Differences were compared by Student’s t-test or One-Way Analysis of Variance (ANOVA) with LSD post hoc test. Statistically significant was determined at P <0.05.
Results
H19 Was Upregulated in Erlotinib-Resistant NSCLC Cells
To investigate the regulatory mechanism of erlotinib resistance, erlotinib-resistant NSCLC cell lines (HCC827/ER and A549/ER) were established. The cell viability was determined after erlotinib treatment. Compared with the parental cells, the cell viability and IC50 values of HCC827/ER and A549/ER cells were significantly elevated, indicating that HCC827/ER and A549/ER cells have high resistance to erlotinib (Figure 1A and B). Also, the proliferation of HCC827/ER and A549/ER cells was enhanced compared with HCC827 and A549 cells (Figure 1C and D). Transwell assay displayed that the abilities of migration and invasion of HCC827/ER and A549/ER cells were markedly higher than that of parental cells (Figure 1E and F). Besides, the levels of migration-related proteins MMP2 and MMP9 were also increased in HCC827/ER and A549/ER cells (Figure 1G and H). In addition, the expression of H19 was measured, and the qRT-PCR result showed that H19 was upregulated in HCC827/ER and A549/ER cells (Figure 1I and J). These results suggested that H19 was associated with the erlotinib resistance in NSCLC cells.
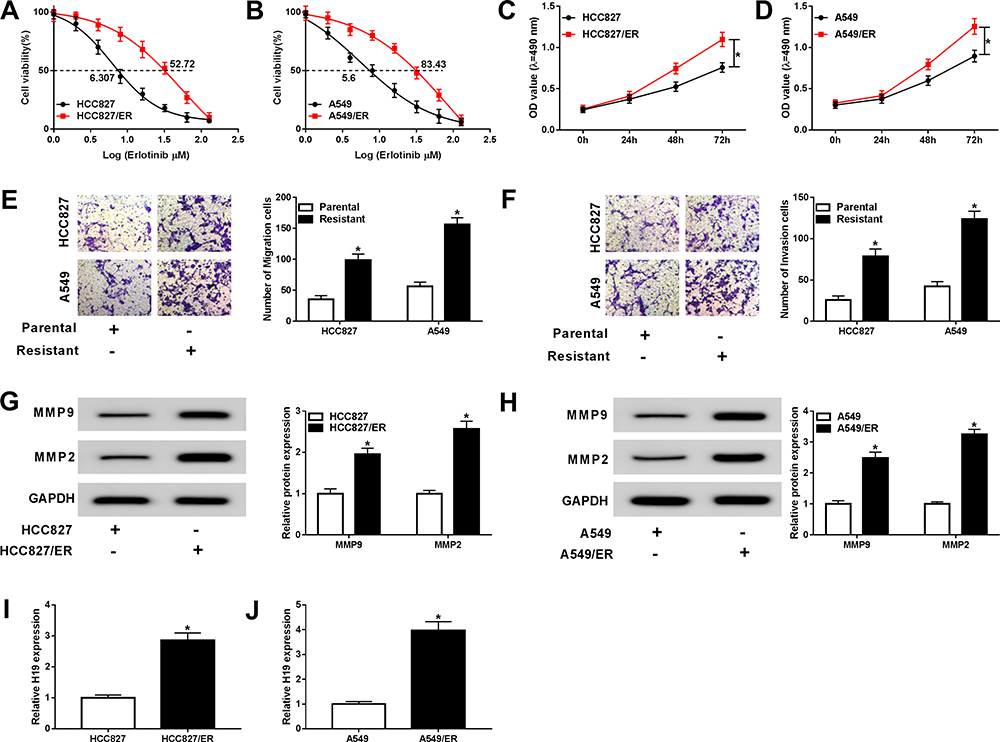 |
Figure 1 H19 was upregulated in erlotinib-resistant NSCLC cells. (A and B) The IC50 value of erlotinib was detected for both parental cells and erlotinib-sensitive cells by cell viability assay. (C and D) Proliferation of parental and erlotinib-sensitive NSCLC cells was determined by MTT assay. (E and F) Migration and invasion of parental and erlotinib-sensitive NSCLC cells were assessed by transwell assay. (G and H) The levels of migration-related proteins MMP2 and MMP9 were detected in parental and erlotinib-sensitive NSCLC cells by Western blot. (I and J) The expression of H19 was detected in parental and erlotinib-sensitive NSCLC cells by qRT-PCR. *P<0.05. |
Knockdown of H19 Decreased the Resistance of Erlotinib-Resistant NSCLC Cells to Erlotinib
To explore the role of H19 in erlotinib resistance of NSCLC cells, si-H19 was used to silence H19. The expression of H19 was evidently downregulated by si-H19 in both HCC827/ER and A549/ER cells (Figure 2A and B, Fig S1). When treated with erlotinib, HCC827/ER and A549/ER cells transfected with si-H19 had lower cell viability and IC50 compared with the si-NC group (Figure 2C and D). MTT assay revealed that knockdown of H19 inhibited the proliferation of HCC827/ER and A549/ER cells (Figure 2E and F). Moreover, migration and invasion were remarkably suppressed in HCC827/ER and A549/ER cells transfected with si-H19 (Figure 2G and H). And the protein levels of MMP2 and MMP9 were also downregulated by knockdown of H19 in HCC827/ER and A549/ER cells (Figure 2I and J). These results indicated that H19 was essential for erlotinib resistance of erlotinib-resistant NSCLC cells.
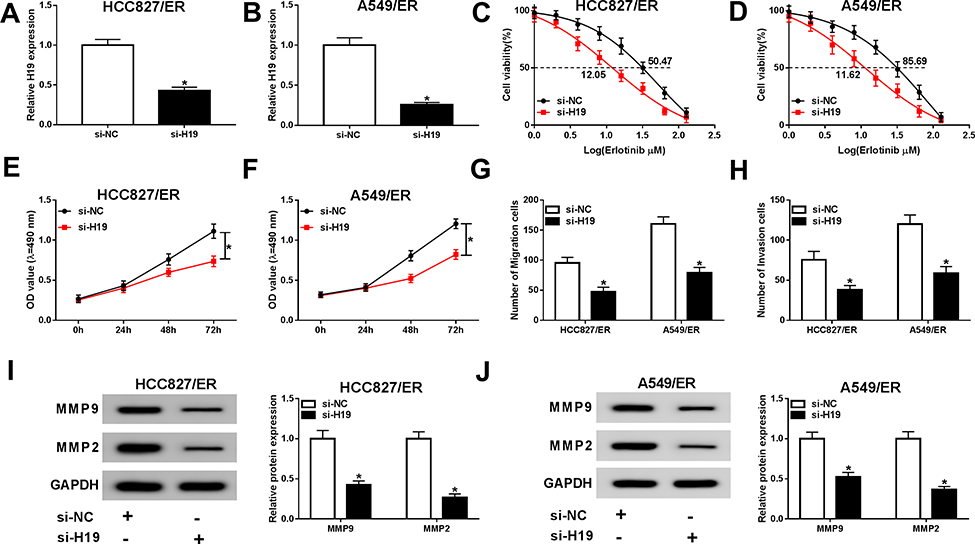 |
Figure 2 H19 was essential for erlotinib resistance of NSCLC cells. HCC827/ER and A549/ER cells were transfected with si-H19 for 48 h. (A and B) The silencing efficacy was evaluated by qRT-PCR. (C and D) The IC50 value of erlotinib was detected for HCC827/ER and A549/ER cells by cell viability assay. (E and F) Proliferation of HCC827/ER and A549/ER cells were determined by MTT assay. (G and H) Migration and invasion of HCC827/ER and A549/ER cells were assessed by transwell assay. (I and J) The protein levels of MMP2 and MMP9 were detected by Western blot in HCC827/ER and A549/ER cells. *P<0.05. |
Extracellular H19 Was Transferred Through Incorporation in Exosomes in Erlotinib-Resistant NSCLC Cells
Many types of cells can secrete exosomes into the culture medium, which affect various cellular processes. To investigate whether H19 was packaged into exosomes and transferred via exosomes, the culture medium of erlotinib-resistant NSCLC cells was collected. Then the expression of H19 was detected in culture medium following treated with RNase A or RNase A+Triton X-100. As shown in Figure 3A, the level of H19 was unacted on RNase A treatment but strikingly decreased upon RNase A and Triton X-100 treatment simultaneously, implying that extracellular H19 was packaged in membranes. Subsequently, we isolated exosomes from the culture medium and observed the characteristics by TEM, and a typical lipid bilayer membrane was observed (Figure 3B). Then the levels of exosomal marker proteins CD9 and CD63 were measured by Western blot, and the result displayed that CD9 and CD63 were enriched in exosomes (Figure 3C). In addition, the qRT-PCR result revealed that the level of H19 was notably higher in exosomes extracted from erlotinib-resistant NSCLC cells than that of parental cells (Figure 3D). These findings indicated that extracellular H19 was released via incorporation in exosomes in erlotinib-resistant NSCLC cells.
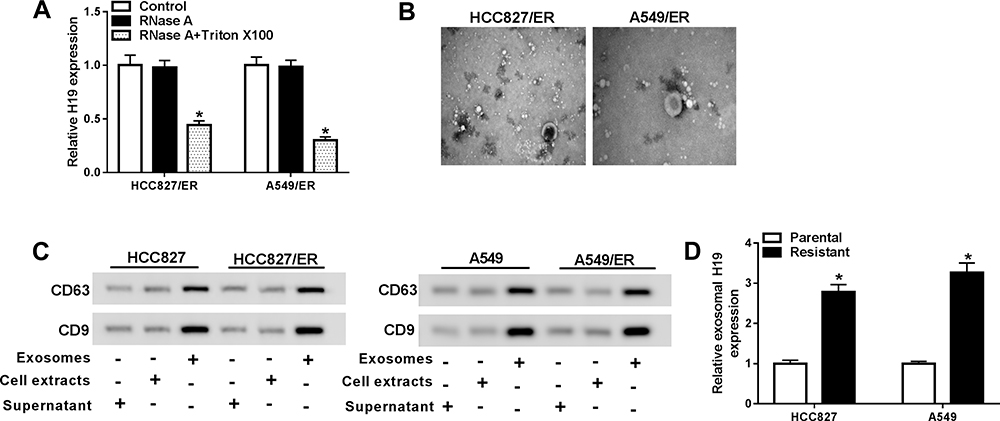 |
Figure 3 Extracellular H19 was packaged into exosomes in NSCLC cells. (A) The expression of H19 was detected by qRT-PCR after cells were treated with 1 μg/mL RNase A or 1 μg/mL RNase A+ 0.1% Triton X100 for 30 min. (B) The exosomes images secreted by HCC827/ER and A549/ER cells were showed by TEM scanning. (C) The levels of exosomal marker proteins CD9 and CD63 were measured by Western blot in HCC827/ER and A549/ER cells. (D) The expression of H19 in exosomes of parental and erlotinib-sensitive NSCLC cells was detected by qRT-PCR. *P<0.05. |
Knockdown of H19 Attenuated Exosome-Mediated Erlotinib Resistance
In order to explore the role of exosome and the molecular mechanism, HCC827 and A549 cells were treated with exosomes which were extracted from HCC827/ER and A549/ER cells culture medium or additionally transfected with si-H19. The expression of H19 was upregulated by exosome treatment and abated by si-H19 in HCC827 and A549 cells (Figure 4A and B). The CCK-8 assay manifested that cell viability and IC50 value were elevated in HCC827 and A549 cells treated with exosome and reversed by knockdown of H19 (Figure 4C and D). The proliferation of HCC827 and A549 cells was significantly induced by exosome and restored when transfected with si-H19 (Figure 4E and F). Moreover, exosome treatment promoted migration and invasion of HCC827 and A549 cells, and H19 knockdown abolished this effect (Figure 4G and H). Besides, the protein levels of MMP2 and MMP9 were increased after exosome treatment but reduced by si-H19 in both HCC827 and A549 cells (Figure 4I and J). Therefore, these data suggested that exosome conferred erlotinib resistance through elevating the level of H19 in NSCLC cells.
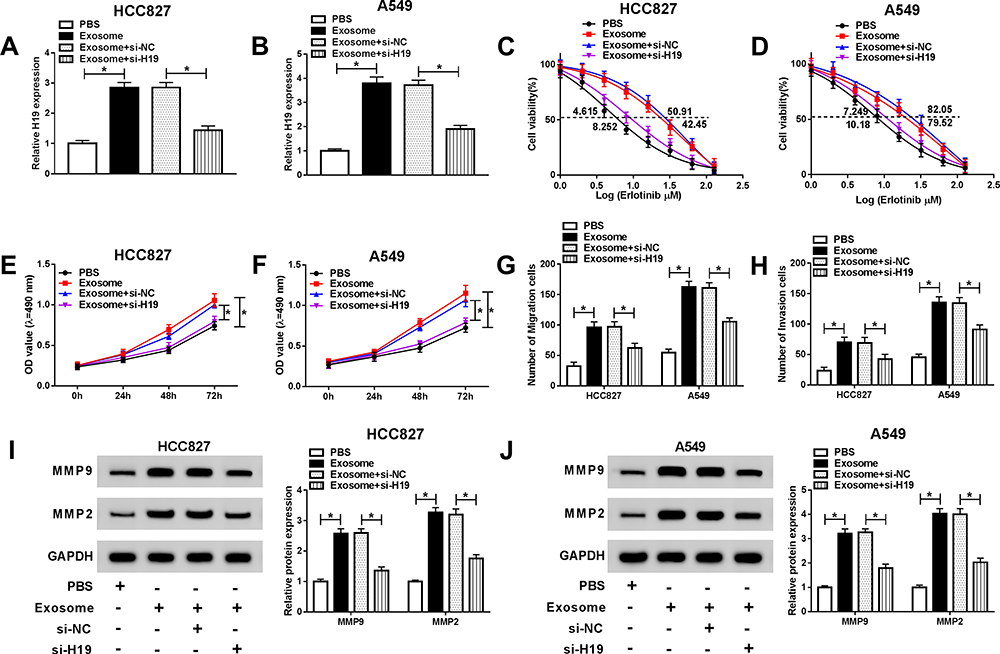 |
Figure 4 Exosomal transfer of H19 induced the resistance of NSCLC cells to erlotinib. Exosomes were isolated from erlotinib-sensitive NSCLC cells, and HCC827 and A549 cells were treated with exosome or exosome+si-H19. (A and B) The expression of H19 was detected by qRT-PCR in HCC827 and A549 cells. (C and D) The IC50 value of erlotinib was detected for HCC827 and A549 cells by cell viability assay. (E and F) Proliferation of HCC827 and A549 cells was determined by MTT assay. (G and H) Migration and invasion of HCC827 and A549 cells were assessed by transwell assay. (I and J) The protein levels of MMP2 and MMP9 were detected by Western blot in HCC827 and A549 cells. *P<0.05. |
miR-615-3p Was a Direct Target of H19
LncRNAs usually play their functions by sponging miRNAs. StarBase 3.0 predicated that H19 had putative binding sites of miR-615-3p (Figure 5A). Therefore, H19 WT and H19 MUT were constructed to confirm the relationship between H19 and miR-615-3p by dual-luciferase reporter assay. The luciferases activities were greatly reduced by miR-615-3p in HCC827 and A549 cells transfected with H19 WT, while there was no change when transfected with H19 WUT (Figure 5B and C). Subsequently, the expression of miR-615-3p was detected by qRT-PCR. Compared with the parental cells, miR-615-3p was downregulated in HCC827/ER and A549/ER cells (Figure 5D and E). In addition, knockdown of H19 enhanced the expression of miR-615-3p in HCC827 and A549 cells (Figure 5F and G), indicating that H19 regulated miR-615-3p expression by directly targeting miR-615-3p.
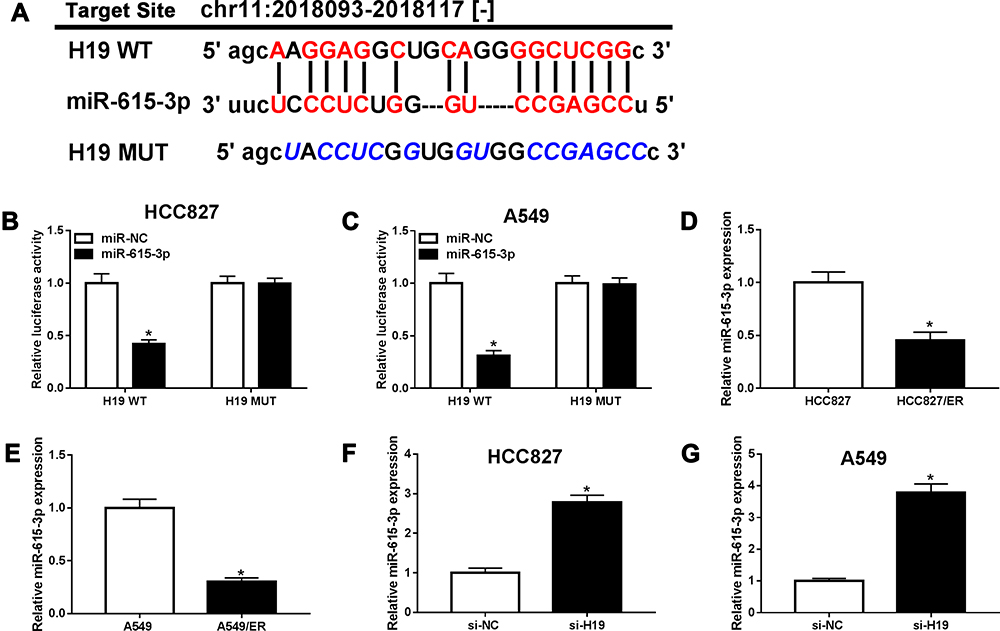 |
Figure 5 miR-615-3p was a target of H19. (A) The putative binding sites between H19 and miR-615-3p were predicated by starBase 3.0. (B and C) The luciferase activities of H19 WT and H19 MUT were measured in HCC827 and A549 cells transfected with miR-NC or miR-615-3p. (D and E) The expression of miR-615-3p was detected in parental and erlotinib-sensitive NSCLC cells by qRT-PCR. (F and G) The expression of miR-615-3p was detected by qRT-PCR in HCC827 and A549 cells transfected with si-H19. *P<0.05. |
Inhibition of miR-615-3p Reversed the Effects of H19 Knockdown on Erlotinib Resistance in NSCLC Cells
To further explore the interaction between H19 and miR-615-3p, we first measured the expression of miR-615-3p in HCC827 and A549 cells treated with exosome or exosome+si-H19. The resulted showed that the expression of miR-615-3p was downregulated by exosome treatment and reversed when knocked down H19 simultaneously (Figure 6A and B). Then, HCC827/ER and A549/ER cells were transfected with si-H19 or si-H19+anti-miR-615-3p, and the upregulated miR-615-3p level resulted by si-H19 was decreased by anti-miR-615-3p (Figure 6C and D). The cell viability and IC50 of HCC827/ER and A549/ER cells were markedly reduced by knockdown of H19 and reverted by inhibition of miR-615-3p upon erlotinib treatment (Figure 6E and F). And the cell proliferation was consistent with this result, which was repressed by si-H19 but rescued when co-transfected with anti-miR-615-3p into HCC827/ER and A549/ER cells (Figure 6G and H). Besides, knockdown of H19 suppressed migration and invasion and inhibition of miR-615-3p weakened this effect (Figure 6I and J). Moreover, the downregulated MMP2 and MMP9 levels in HCC827/ER and A549/ER cells transfected with si-H19 were also reversed by anti-miR-615-3p (Figure 6K and L). These data revealed that exosomal H19 regulated erlotinib resistance of NSCLC cells through inhibiting miR-615-3p expression.
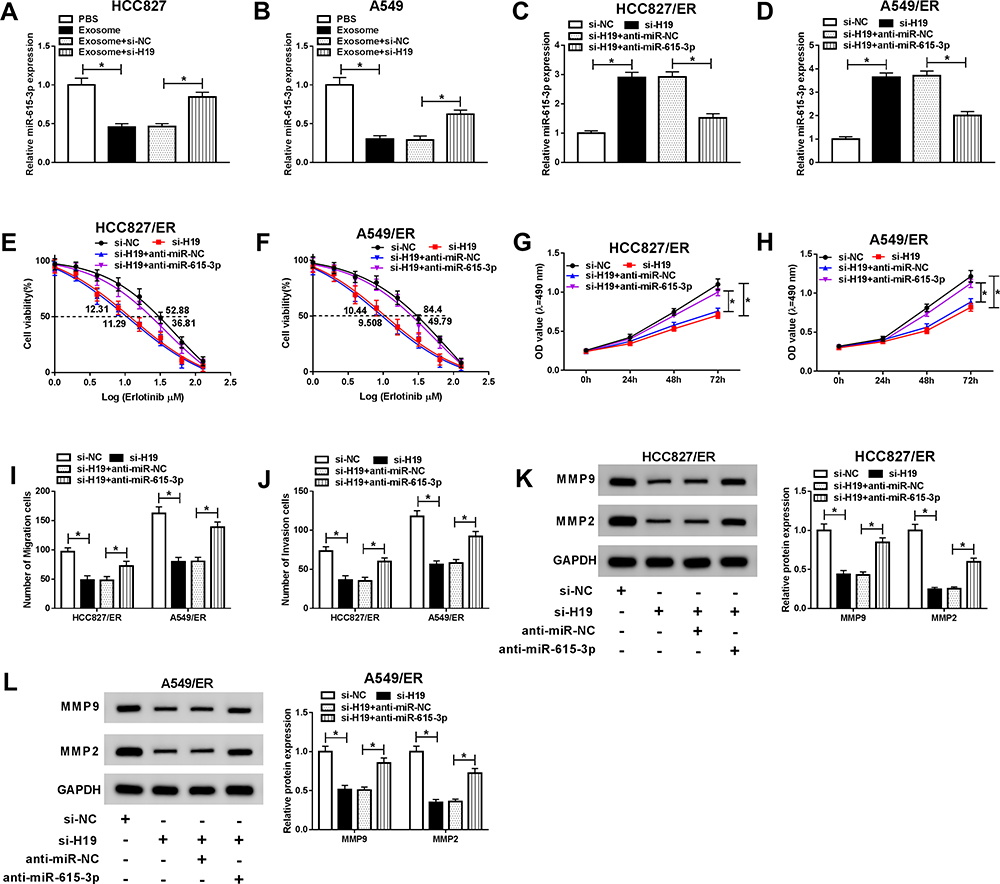 |
Figure 6 Inhibition of miR-615-3p restored the effects of exosomal H19 knockdown on erlotinib resistance in NSCLC cells. HCC827 and A549 cells were treated with exosome or exosome+si-H19, and HCC827/ER and A549/ER cells were transfected with si-H19 or si-19+anti-miR-615-3p. (A and B) The expression of miR-615-3p was detected by qRT-PCR in HCC827 and A549 cells treated with exosome or exosome+si-H19. (C and D) The expression of miR-615-3p was detected by qRT-PCR in HCC827/ER and A549/ER cells transfected with si-H19 or si-19+anti-miR-615-3p. (E and F) The IC50 value of erlotinib was detected for HCC827/ER and A549/ER cells by cell viability assay. (G and H) Proliferation of HCC827/ER and A549/ER cells were determined by MTT assay. (I and J) Migration and invasion of HCC827/ER and A549/ER cells were assessed by transwell assay. (K and L) The protein levels of MMP2 and MMP9 were detected by Western blot in HCC827/ER and A549/ER cells. *P<0.05. |
H19 Regulated ATG7 Expression via miR-615-3p
To further investigate the regulatory mechanism of H19 in NSCLC, StarBase 3.0 further predicated ATG7 as a potential target of miR-615-3p (Figure 7A). Then ATG7 3ʹUTR WT and ATG7 3ʹUTR MUT were used for dual-luciferase reporter assay to confirm whether miR-615-3p could bind to the 3ʹUTR of ATG7. The result showed that miR-615-3p weakened the luciferases activities in HCC827 and A549 cells transfected with ATG7 3ʹUTR WT rather than ATG7 3ʹUTR MUT (Figure 7B and C). Subsequently, the expression of ATG7 was detected using Western blot. ATG7 was evidently upregulated in HCC827/ER and A549/ER cells compared with the parental cells (Figure 7D and E). Overexpression of miR-615-3p decreased the level of ATG7 in HCC827 and A549 cells (Figure 7F and G). Furthermore, the protein level of ATG7 was downregulated in HCC827 and A549 cells transfected with si-H19 and rescued by anti-miR-615-3p (Figure 7H and I), suggesting that H19 regulated ATG7 expression via miR-615-3p.
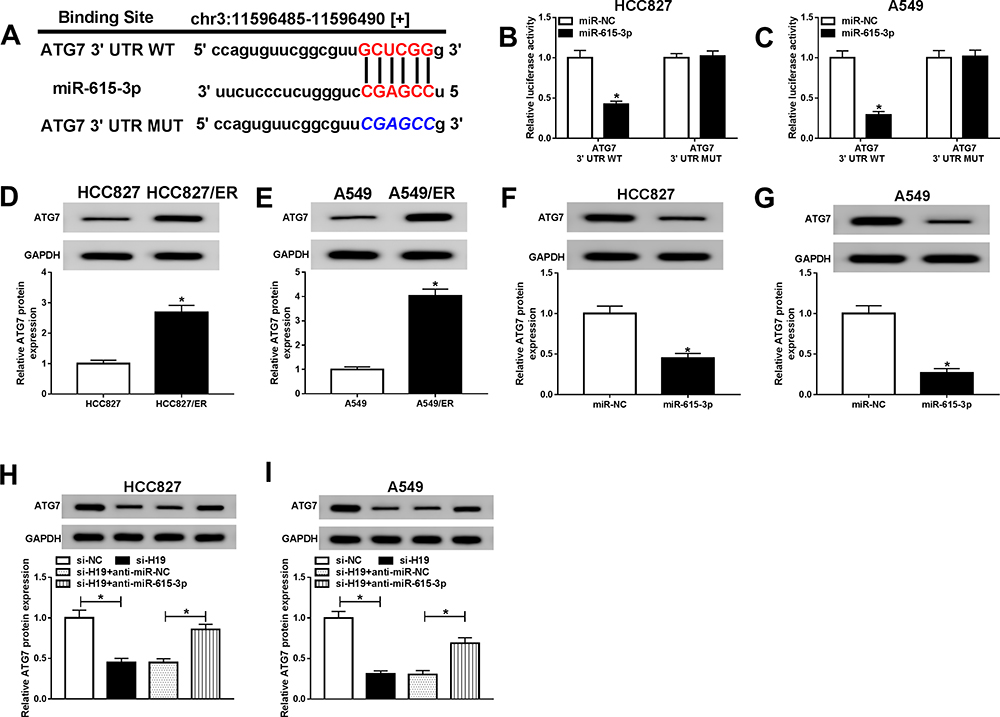 |
Figure 7 H19 regulated ATG7 expression through miR-615-3p. (A) The putative binding sites between miR-615-3p and ATG7 were predicated by starBase 3.0. (B and C) The luciferase activities of ATG7 3ʹUTR WT and ATG7 3ʹUTR MUT were measured in HCC827 and A549 cells transfected with miR-NC or miR-615-3p. (D and E) The level of ATG7 was detected in parental and erlotinib-sensitive NSCLC cells by Western blot. (F and G) The expression of miR-615-3p was detected by Western blot in HCC827 and A549 cells transfected with miR-615-3p. (H and I) The expression of miR-615-3p was detected by Western blot in HCC827 and A549 cells transfected with si-19 or si-19+anti-miR-615-3p. *P<0.05. |
Overexpression of ATG7 Restored the Effects of H19 Knockdown on Erlotinib Resistance in NSCLC Cells
To further explore the interaction between H19 and ATG7, the protein level of ATG7 was determined in HCC827 and A549 cells treated with exosome or exosome+si-H19. ATG7 was upregulated when treated with exosome and recovered by knockdown of H19, indicating that exosomal transfer of H19 promoted ATG7 expression (Figure 8A and B). Then HCC827/ER and A549/ER cells were transfected with si-H19 or si-H19+ATG7, and the level of ATG7 was significantly downregulated by knockdown of H19 and rescued by overexpression of ATG7 (Figure 8C and D). The inhibited cell viability and reduced IC50 of HCC827/ER and A549/ER cells transfected with si-H19 were elevated when co-transfected with ATG7 (Figure 8E and F). Also, overexpression of ATG7 reversed si-H19-mediated inhibitory effect on cell proliferation of HCC827/ER and A549/ER cells (Figure 8G and H). As expect, the repressed migration and invasion resulted by si-H19 were also regained by overexpression of ATG7 (Figure 8I and J). In addition, knockdown of H19 restrained the expression of MMP2 and MMP9, while overexpression of ATG7 relieved this effect in HCC827/ER and A549/ER cells (Figure 8K and L). These results suggested that exosomal transfer of H19 affected erlotinib resistance of NSCLC cells through regulating ATG7 expression.
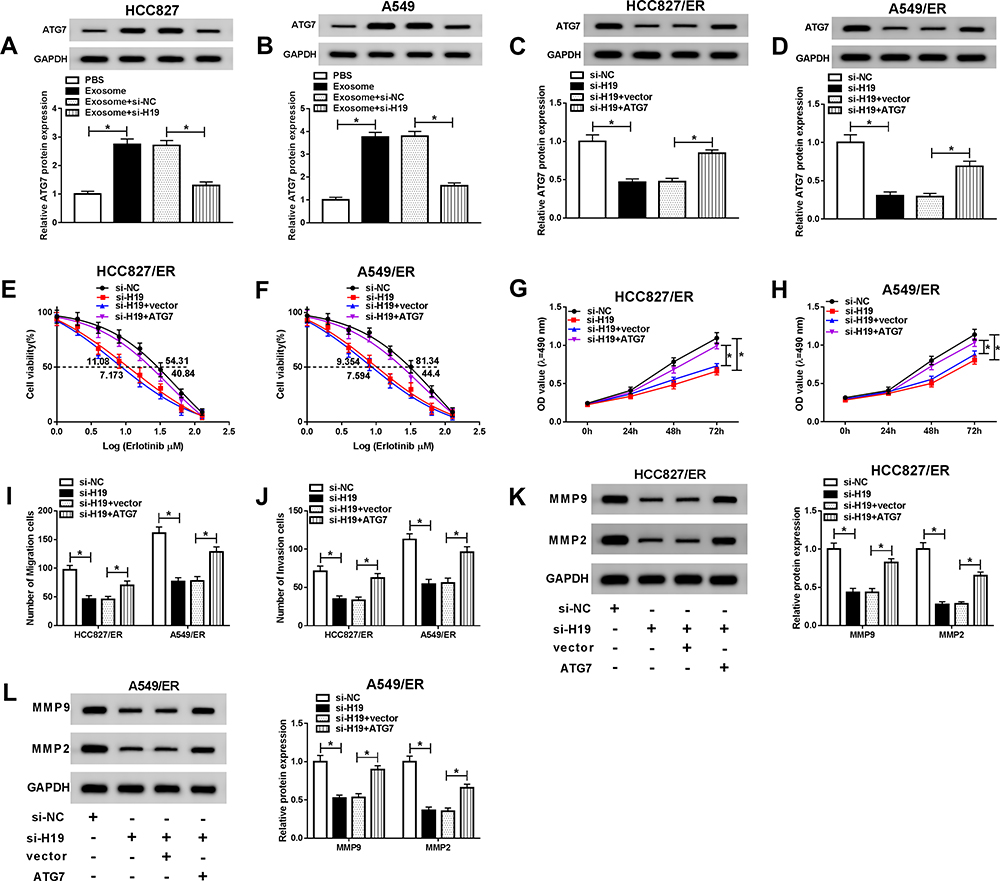 |
Figure 8 Overexpression of ATG7 reversed the effects of exosomal H19 knockdown on erlotinib resistance in NSCLC cells. HCC827 and A549 cells were treated with exosome or exosome+si-H19, and HCC827/ER and A549/ER cells were transfected with si-H19 or si-19+ATG7. (A and B) The level of ATG7 was detected by Western blot in HCC827 and A549 cells treated with exosome or exosome+si-H19. (C and D) The level of ATG7 was detected by Western blot in HCC827/ER and A549/ER cells transfected with si-H19 or or si-19+ATG7. (E and F) The IC50 value of erlotinib was detected for HCC827/ER and A549/ER cells by cell viability assay. (G and H) Proliferation of HCC827/ER and A549/ER cells were determined by MTT assay. (I and J) Migration and invasion of HCC827/ER and A549/ER cells were assessed by transwell assay. (K and L) The protein levels of MMP2 and MMP9 were detected by Western blot in HCC827/ER and A549/ER cells. *P<0.05. |
Serum Exosomal H19 May Serve as a Potential Diagnostic Biomarker for Erlotinib-Resistant NSCLC Patients
Exosomes were isolated from the serum of 28 responding and 30 non-responding patients with advanced NSCLC receiving erlotinib treatment. Next, the expression of serum exosomal H19 was detected. The result exhibited that serum exosomal H19 was upregulated in patients who did not respond to erlotinib (Figure 9A). Then the stability of serum exosomal H19 was determined, and the qRT-PCR results showed that the level of H19 had no change after the serum exosome was incubated at room temperature for 0, 3, 6, 12 and 24 h as well as treated with RNaseA, NaOH or HCl solution for 3 h at room temperature (Figure 9B–D). In addition, ROC analysis was performed to assess the diagnostic potential of H19, and the AUC, diagnostic sensitivity and specificity reached 0.799, 70 and 85.71% (95% CI =0.6794 to 0.9182) (Figure 9E). Based on the cut-offs (1.967) established by ROC, patients were divided into the low and high H19 expression groups, and the proportion of patients who did not respond to erlotinib treatment was significantly higher in the high exosomal H19 expression group than in the low expression group (Figure 9F). Altogether, exosomal H19 in serum was stable and might serve as a potential biomarker for the erlotinib treatment of NSCLC patients.
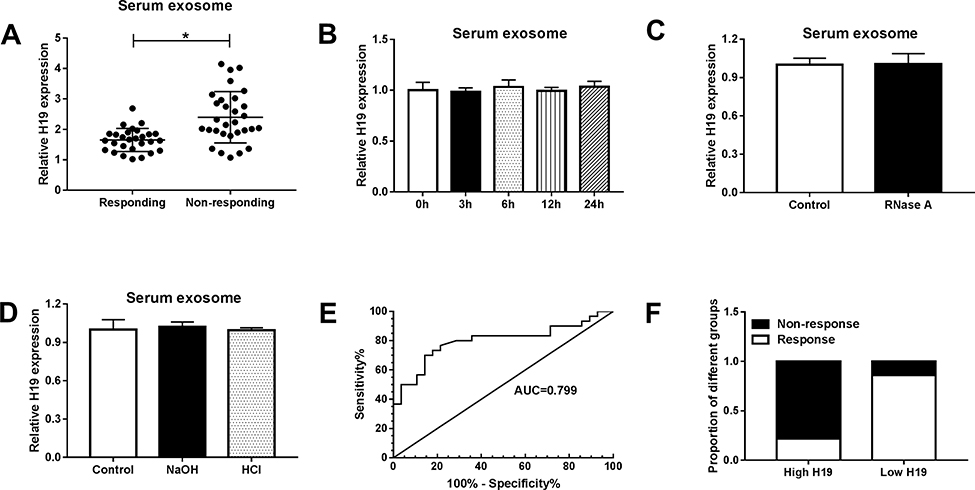 |
Figure 9 Expression of serum exosomal H19 was associated with erlotinib resistance in NSCLC patients. (A) The expression of serum exosomal H19 was detected by qRT-PCR in NSCLC patients who received erlotinib treatment. (B) The expression of H19 was detected by qRT-PCR after the serum exosomes were incubated at room temperature for 0, 3, 6, 12 and 24 h. (C) The expression of H19 was detected by qRT-PCR after the serum exosomes were treated with RNase A. (D) The expression of H19 was detected by qRT-PCR after the serum exosomes were treated with NaOH or HCl solution. (E) The diagnostic value of exosomal H19 in NSCLC patients who received erlotinib treatment receiving erlotinib treatment was assessed by ROC curve analysis. (F) The proportion of patients with or without response to erlotinib treatment in high H19 and low H19 group was analyzed. *P<0.05. |
Knockdown of H19 Decreased Erlotinib Resistance in NSCLC in vivo
To explore the effect of H19 on erlotinib resistance in NSCLC in vivo, a model of nude mice xenografts was established. HCC827 cells transfected with sh-NC or sh-H19 lentiviral vector were established successfully (Fig S2). Then the cells were injected into nude mice following treated with erlotinib, and the mice were divided into two group: sh-NC+erlotinib group (control group) and sh-H19+erlotinib group. Tumor volume and weight were measured to evaluate the tumor growth, and the results indicated that knockdown of H19 repressed tumor growth compared with the control group upon erlotinib treatment (Figure 10A and B). The expression level of H19 was lower in sh-H19+erlotinib group, while miR-615-3p was upregulated by knockdown of H19 (Figure 10C and D). And a Western blot analysis showed that the level of ATG7 was decreased in sh-H19+erlotinib group (Figure 10E). These findings uncovered that knockdown of H19 attenuated erlotinib resistance via regulating miR-615-3p and ATG7 expression in vivo.
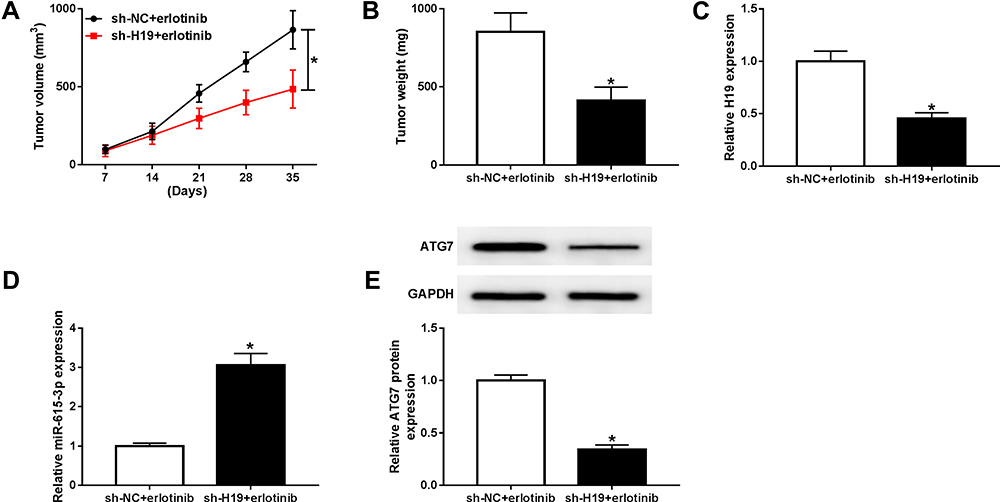 |
Figure 10 Knockdown of H19 restrained erlotinib resistance in NSCLC in vivo. Nude mice were injected HCC827 cells transfected with sh-NC or sh-H19 lentiviral vector and treated with erlotinib. (A) The tumor volume was measured weekly after implantation for 7 days. (B) tumor weight was measured at 35 days after implantation. (C and D) The levels of H19 and miR-615-3p were detected by qRT-PCR. (E) The level of ATG7 was detected by Western blot. *P<0.05. |
Discussion
Chemotherapy is a common therapy for cancer, while the acquirement of drug resistance severely restricts the curative effect. Erlotinib, an epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI), is a therapeutic drug of NSCLC. It is very significant to understand the molecular mechanism of erlotinib resistance for the therapy of NSCLC. In our study, lncRNA H19 was upregulated in erlotinib-resistant NSCLC cells compared with the parental cells, implying the potential effect of H19 on erlotinib resistance.
Numerous studies have proved that lncRNA H19 plays a regulatory role in many diseases, including cancer. LncRNA H19 involved in the development of esophageal squamous cell cancer and predicated a poor prognosis.21 Bioinformatics analysis identified H19 as a critical factor of oxaliplatin or irinotecan resistance in colorectal cancer.22 Meanwhile, H19 may act as a promising diagnostic target for human cancers.23 However, there is no study to elucidate the role of H19 in erlotinib resistance. Our data showed that knockdown of H19 suppressed the resistance of NSCLC cells to erlotinib, as evidence by the inhibited cell viability, proliferation migration and invasion in erlotinib-resistant cells.
Exosome has been revealed to participate in cellular communication and regulate various physiological processes. It is also indicated that exosome-mediated transfer of lncRNAs or miRNAs was associated with drug resistance in the chemotherapy of cancers.24,25 Exosomal lncARSR was reported to enhance the resistance of renal cancer to sunitinib.26 Exosomal miR-501 enhanced doxorubicin resistance in gastric cancer.27 TEM analysis proved that extracellular H19 was packaged into exosomes. Moreover, the exosomes isolated from erlotinib-resistant cells conferred erlotinib resistance to sensitive cells, while knockdown of H19 abolished this effect. A previous study manifested that exosomal H19 induced gefitinib resistance in NSCLC.9 Also, exosomal H19 was indicated to reinforce chemoresistance in colorectal cancer.28 Furthermore, serum exosomal H19 can serve as a biomarker for the diagnosis and prognosis of bladder cancer.29 In consistent with these studies, our results revealed that exosomal H19 facilitate erlotinib resistance in NSCLC in vitro and in vivo. H19 was highly expressed in the serum exosome of NSCLC patients who had no response to erlotinib treatment. Besides, ROC analysis proved the diagnostic potential of H19, suggesting that serum exosomal H19 might act as a promising diagnostic target for NSCLC patients.
LncRNAs function as competing endogenous RNAs (ceRNAs) to regulate gene expression. Bioinformatics analysis and dual-luciferase reporter assay verified miR-615-3p as a direct target of H19. miR-615-3p was manifested to promote tumor progression in prostate cancer and gastric cancer.30,31 On the contrary, in esophageal squamous cell carcinoma and NSCLC, miR-615-3p played an antitumor effect.32,33 In addition, miR-615 was proved to suppress glioblastoma development via targeting EGFR,34 and this finding implicated that miR-615 might be associated with the resistance to EGFR TKIs. However, the effect of miR-615 in drug resistance has not been reported. In our study, restoration experiments exhibited that downregulation of miR-615-3p restored the inhibiting effect of H19 knockdown on erlotinib resistance in NSCLC cells, which uncovered that miR-615-3p played an inhibiting effect on erlotinib resistance.
Autophagy is a highly conserved process that can degrade dysfunctional proteins and organelles by the lysosome. Previous studies indicated that autophagy was closely related to chemoresistance.35,36 As an autophagy-related protein, ATG7 regulated cancer development and chemoresistance through promoting autophagy. Knockdown of ATG7 overcame the 5-FU resistance in esophageal cancer.37 MiR-17 targeted ATG7 to inhibit autophagosome formation and temozolomide resistance in glioblastoma.38 Moreover, Huang et al reported that ATG7 induced autophagy and facilitated chemoresistance in NSCLC.39 However, Eng et al suggested that ATG7 was not necessary for the growth and drug resistance in Kirsten rat sarcoma (KRAS) mutant tumors, which relied on autophagy for growth and survival.40 Besides, Sun et al demonstrated that ATG7 was essential for the tumorigenesis of lung cancer but had no influence on tumor growth and chemoresistance.20 These contradictory results may due to the different mouse models and tumor microenvironments. We identified ATG7 as the downstream target of miR-615-3p, and exosomal H19 regulating ATG7 expression via miR-615-3p. Overexpression of ATG7 rescued the inhibited erlotinib resistance which was resulted by H19 knockdown, indicating that ATG7 played a promotor effect on erlotinib resistance, which was consistent with the previous study.39 Consequently, our results elucidated that exosomal H19 enhanced erlotinib resistance of NSCLC cells by upregulating ATG7.
In conclusion, we found that H19 was upregulated in erlotinib-resistant NSCL cells, and knockdown of H19 restrained erlotinib resistance in vitro and in vivo. Moreover, our data revealed that H19 could be transferred through exosome and conferred erlotinib resistance to sensitive cells. Based on dual-luciferase reporter assay and restoration experiments, we demonstrated that exosomal transfer of H19 regulated erlotinib resistance via miR-615-3p/ATG7 in NSCLC. Our findings provide a new molecular mechanism of drug resistance, which may contribute to the development of diagnosis and treatment strategies of NSCLC.
Disclosure
The authors declare that they have no financial conflicts of interest.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong KK. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer. 2014;14(8):535–546. doi:10.1038/nrc3775
3. ELA S, Mager I, Breakefield XO, Wood MJ. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov. 2013;12(5):347–357. doi:10.1038/nrd3978
4. Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2(8):569–579. doi:10.1038/nri855
5. Luo J, Xiong Y, Fu PF, et al. Exosomal long non-coding RNAs: biological properties and therapeutic potential in cancer treatment. J Zhejiang Univ Sci B. 2019;20(6):488–495. doi:10.1631/jzus.B1900039
6. Chen C, Luo Y, He W, et al. Exosomal long noncoding RNA LNMAT2 promotes lymphatic metastasis in bladder cancer. J Clin Invest. 2019. doi:doi:10.1172/JCI130892
7. Li C, Lv Y, Shao C, et al. Tumor-derived exosomal lncRNA GAS5 as a biomarker for early-stage non-small-cell lung cancer diagnosis. J Cell Physiol. 2019;234(11):20721–20727. doi:10.1002/jcp.28678
8. Zhang Z, Yin J, Lu C, Wei Y, Zeng A, You Y. Exosomal transfer of long non-coding RNA SBF2-AS1 enhances chemoresistance to temozolomide in glioblastoma. J Exp Clin Cancer Res. 2019;38(1):166. doi:10.1186/s13046-019-1139-6
9. Lei Y, Guo W, Chen B, Chen L, Gong J, Li W. Tumorreleased lncRNA H19 promotes gefitinib resistance via packaging into exosomes in nonsmall cell lung cancer. Oncol Rep. 2018;40(6):3438–3446. doi:10.3892/or.2018.6762
10. Ambros V. The functions of animal microRNAs. Nature. 2004;431(7006):350–355. doi:10.1038/nature02871
11. Singh SK, Pal Bhadra M, Girschick HJ, Bhadra U. MicroRNAs–micro in size but macro in function. FEBS J. 2008;275(20):4929–4944. doi:10.1111/j.1742-4658.2008.06624.x
12. Xing S, Xu Q, Fan X, Wu S, Tian F. Downregulation of miR1385p promotes nonsmall cell lung cancer progression by regulating CDK8. Mol Med Rep. 2019. doi:doi:10.3892/mmr.2019.10741
13. Liao Y, Cao L, Wang F, Pang R. miR-605-5p promotes invasion and proliferation by targeting TNFAIP3 in non-small-cell lung cancer. J Cell Biochem. 2019. doi:doi:10.1002/jcb.29323
14. Jin X, Chen Y, Chen H, et al. Evaluation of tumor-derived exosomal miRNA as potential diagnostic biomarkers for early-stage non-small cell lung cancer using next-generation sequencing. Clin Cancer Res. 2017;23(17):5311–5319. doi:10.1158/1078-0432.CCR-17-0577
15. Tian F, Wang J, Ouyang T, et al. miR-486-5p serves as a good biomarker in nonsmall cell lung cancer and suppresses cell growth with the involvement of a target PIK3R1. Front Genet. 2019;10:
16. Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:
17. Galluzzi L, Pietrocola F, Bravo-San Pedro JM, et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015;34(7):856–880. doi:10.15252/embj.201490784
18. Pan X, Chen Y, Shen Y, Tantai J. Knockdown of TRIM65 inhibits autophagy and cisplatin resistance in A549/DDP cells by regulating miR-138-5p/ATG7. Cell Death Dis. 2019;10(6):429. doi:10.1038/s41419-019-1660-8
19. Wu J, Li W, Ning J, Yu W, Rao T, Cheng F. Long noncoding RNA UCA1 targets miR-582-5p and contributes to the progression and drug resistance of bladder cancer cells through ATG7-mediated autophagy inhibition. Onco Targets Ther. 2019;12:
20. Sun S, Wang Z, Tang F, et al. ATG7 promotes the tumorigenesis of lung cancer but might be dispensable for prognosis predication: a clinicopathologic study. Onco Targets Ther. 2016;9:
21. Li X, Yang H, Wang J, et al. High level of lncRNA H19 expression is associated with shorter survival in esophageal squamous cell cancer patients. Pathol Res Pract. 2019;215(11):152638. doi:10.1016/j.prp.2019.152638
22. Sun F, Liang W, Qian J. The identification of CRNDE, H19, UCA1 and HOTAIR as the key lncRNAs involved in oxaliplatin or irinotecan resistance in the chemotherapy of colorectal cancer based on integrative bioinformatics analysis. Mol Med Rep. 2019;20(4):3583–3596. doi:10.3892/mmr.2019.10588
23. Liu Y, He A, Liu B, Huang Z, Mei H. Potential role of lncRNA H19 as a cancer biomarker in human cancers detection and diagnosis: a pooled analysis based on 1585 subjects. Biomed Res Int. 2019;2019:
24. Yousefi H, Maheronnaghsh M, Molaei F, et al. Long noncoding RNAs and exosomal lncRNAs: classification, and mechanisms in breast cancer metastasis and drug resistance. Oncogene. 2019. doi:doi:10.1038/s41388-019-1040-y
25. Kulkarni B, Kirave P, Gondaliya P, et al. Exosomal miRNA in chemoresistance, immune evasion, metastasis and progression of cancer. Drug Discov Today. 2019. doi:doi:10.1016/j.drudis.2019.06.010
26. Qu L, Ding J, Chen C, et al. Exosome-transmitted lncARSR promotes sunitinib resistance in renal cancer by acting as a competing endogenous RNA. Cancer Cell. 2016;29(5):653–668. doi:10.1016/j.ccell.2016.03.004
27. Liu X, Lu Y, Xu Y, et al. Exosomal transfer of miR-501 confers doxorubicin resistance and tumorigenesis via targeting of BLID in gastric cancer. Cancer Lett. 2019;459:
28. Ren J, Ding L, Zhang D, et al. Carcinoma-associated fibroblasts promote the stemness and chemoresistance of colorectal cancer by transferring exosomal lncRNA H19. Theranostics. 2018;8(14):3932–3948. doi:10.7150/thno.25541
29. Wang J, Yang K, Yuan W, Gao Z. Determination of serum exosomal H19 as a noninvasive biomarker for bladder cancer diagnosis and prognosis. Med Sci Monit. 2018;24:
30. Laursen EB, Fredsoe J, Schmidt L, et al. Elevated miR-615-3p expression predicts adverse clinical outcome and promotes proliferation and migration of prostate cancer cells. Am J Pathol. 2019. doi:10.1016/j.ajpath.2019.08.007
31. Wang J, Liu L, Sun Y, et al. miR-615-3p promotes proliferation and migration and inhibits apoptosis through its potential target CELF2 in gastric cancer. Biomed Pharmacother. 2018;101:
32. Liu J, Jia Y, Jia L, Li T, Yang L, Zhang G. MicroRNA 615-3P inhibits the tumor growth and metastasis of NSCLC via Inhibiting IGF2. Oncol Res. 2019;27(2):269–279. doi:10.3727/096504018X15215019227688
33. Sun Y, Wang J, Pan S, et al. LINC00657 played oncogenic roles in esophageal squamous cell carcinoma by targeting miR-615-3p and JunB. Biomed Pharmacother. 2018;108:
34. Ji Y, Sun Q, Zhang J, Hu H. miR-615 inhibits cell proliferation, migration and invasion by targeting EGFR in human glioblastoma. Biochem Biophys Res Commun. 2018;499(3):719–726. doi:10.1016/j.bbrc.2018.03.217
35. Wu WK, Coffelt SB, Cho CH, et al. The autophagic paradox in cancer therapy. Oncogene. 2012;31(8):939–953. doi:10.1038/onc.2011.295
36. Sui X, Chen R, Wang Z, et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 2013;4:
37. O’Donovan TR, O’Sullivan GC, McKenna SL. Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy. 2011;7(5):509–524. doi:10.4161/auto.7.6.15066
38. Comincini S, Allavena G, Palumbo S, et al. microRNA-17 regulates the expression of ATG7 and modulates the autophagy process, improving the sensitivity to temozolomide and low-dose ionizing radiation treatments in human glioblastoma cells. Cancer Biol Ther. 2013;14(7):574–586. doi:10.4161/cbt.24597
39. Huang FX, Chen HJ, Zheng FX, et al. LncRNA BLACAT1 is involved in chemoresistance of nonsmall cell lung cancer cells by regulating autophagy. Int J Oncol. 2019;54(1):339–347. doi:10.3892/ijo.2018.4614
40. Eng CH, Wang Z, Tkach D, et al. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc Natl Acad Sci U S A. 2016;113(1):182–187. doi:10.1073/pnas.1515617113
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.