")
Back to Archived Journals » Advances in Genomics and Genetics » Volume 4
Exonic deletion of OPHN1 resulting in seizures, intellectual disability, and brain malformations
Authors Larson A, LeRoux J, Elias E
Received 17 March 2014
Accepted for publication 24 April 2014
Published 14 July 2014 Volume 2014:4 Pages 107—113
DOI https://doi.org/10.2147/AGG.S63848
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Austin Larson,1 Jamie LeRoux,2 Ellen Roy Elias1
1Department of Pediatrics, University of Colorado Denver Anschutz Medical Campus, Aurora, CO, USA; 2Colorado Genetics Laboratory, Denver, CO, USA
Abstract: We report the case of a 9-year-old boy with autism, intellectual disability, and complex partial seizures as well as cerebellar vermian hypoplasia, caudate nucleus hypoplasia, and ventriculomegaly. He was found to have a deletion within the oligophrenin 1 gene (OPHN1), affecting exons 2–5. OPHN1 mutations result in a rare but well-characterized syndrome of neuroanatomical anomalies, epilepsy, and intellectual disability. This is a novel mutation in OPHN1 that adds to the spectrum of pathogenic variants of the gene. Additionally, the case illustrates the significant benefit that patients and families can derive from a definitive genetic diagnosis, even in the absence of direct therapeutic interventions.
Keywords: X-linked intellectual disability, autism, cerebellar hypoplasia, chromosomal microarray, oligophrenin 1
Introduction
The number of diagnostic tests available to the clinical geneticist has exploded in recent years. Many patients who were initially evaluated in genetics, neurology, or developmental pediatric clinics with the available cytogenetic studies prior to the advent of chromosomal microarray (CMA) and next-generation sequencing panels did not receive genetic diagnoses and were diagnosed with idiopathic autism or epilepsy. Although the majority of children with epilepsy or autism remain without genetic diagnoses, those who are diagnosed may derive significant benefit from determination of the genetic etiology of their symptoms.1 CMA is considered a first-line diagnostic test for children with developmental disabilities or congenital anomalies. CMA has a diagnostic yield of 5%–35%, depending on the indication for the test, the clinical setting, the resolution of the CMA, and the criteria used to define pathogenic variants.2 As large databases of CMA data are compiled for affected patients and unaffected controls, the ability of the clinician to interpret the detected variants improves.3 In some cases, such as the 1q21.1 deletion syndrome, there is a broad array of resultant phenotypes, and prognostic information for families can be vague.4 In other cases, such as the one reported here, a variant may be detected that results in a narrower range of phenotypes and provides the ability for the clinician to make a more specific prognosis. In this case, a novel deletion was found within oligophrenin 1 (OPHN1), a gene that encodes a Rho guanosine triphosphatase (Rho GTPase) activating protein that is necessary for maintenance of neuronal synapses.5
Case report
The patient is a 9-year-old male who was born full-term via uncomplicated vaginal delivery to a 30-year-old woman with one prior unremarkable pregnancy and delivery. He was macrosomic at birth, weighing 4.65 kilograms. His initial clinical presentation was for strabismus and nystagmus at 6 months of age. Magnetic resonance imaging (MRI) of the brain obtained by his ophthalmologist showed inferior vermian hypoplasia and retrocerebellar cyst consistent with a Dandy–Walker spectrum malformation. In addition, there was mild diffuse ventriculomegaly and distinctive hypoplasia of the caudate nuclei, as shown in Figure 1. The corpus callosum was intact. He underwent surgery for strabismus at 18 months of age.
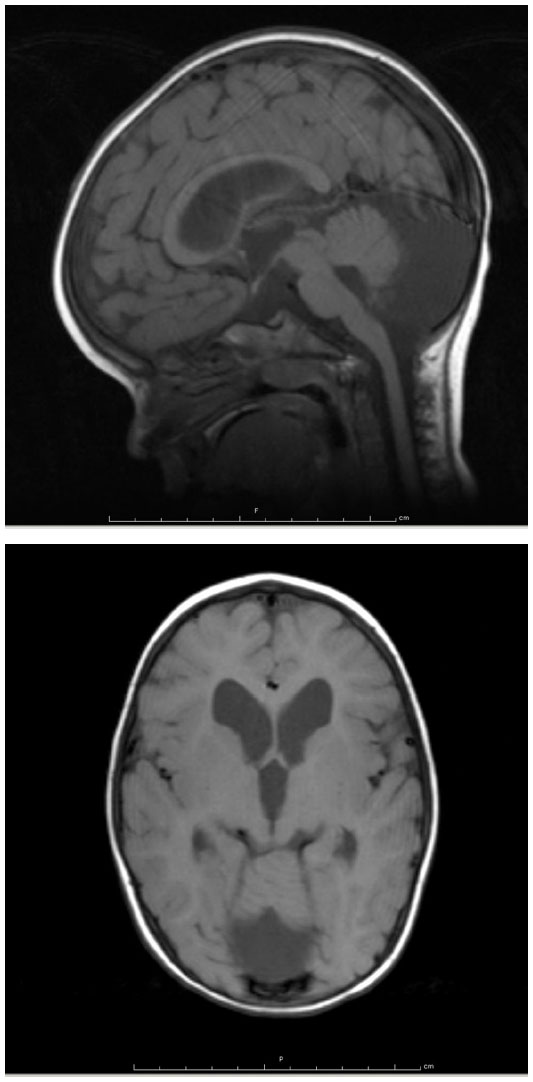 | Figure 1 Sagittal and axial T1-weighted magnetic resonance images showing cerebellar hypoplasia, ventriculomegaly, and caudate atrophy. |
Global developmental delays were apparent from an early age. The patient did not walk until 26 months of age, and currently cannot pedal a tricycle (a typical 3-year-old skill). At age 9 years, he is unable to feed or dress himself. Verbal communication skills are at the level of a 2-year-old. Behaviors consistent with autism are present, including echolalia, hand-flapping, self-stimulatory behaviors, poor eye contact, and lack of shared attention. He was diagnosed with autism at the age of 3 years. He did not have episodes of developmental regression. Anxious behaviors and poor sleep were particularly difficult to manage for the patient’s family.
At 17 months of age the patient had his first apparent seizures, characterized as complex partial seizures with secondary generalization. He was found to have an abnormal electroencephalogram (EEG) with left temporal spike-and-wave discharges. He has been treated with multiple anticonvulsant medications with only fair control of his seizures. With diagnoses of idiopathic autism and epilepsy but no definitive underlying genetic diagnosis, he was being given large doses of a nutritional supplement containing over 500% of daily value for many vitamins as empiric therapy by his family with hope for improvement in autistic symptoms.
Karyotype and testing for fragile X syndrome were carried out in the patient’s second year of life and were normal. Apart from EEG and MRI, no other diagnostic testing was performed until the age of 9 years. Evaluation in a genetics clinic revealed normal lactate, pyruvate, carnitine, and acylcarnitine profiles. Physical exam was pertinent for mildly dysmorphic facial features with downslanting palpebral fissures, epicanthal folds, and large prominent pinnae, as shown in Figure 2. Strabismus was not apparent on exam after surgical correction. There was symmetrical fifth finger clinodactyly, truncal hypotonia, and a mildly ataxic gait. Behavior was generally anxious and agitated, using occasional single words for communication. His height and head circumference were at about the 50th percentile, with weight at the 75th percentile. Family history was significant for the absence of epilepsy or intellectual disability (ID). CMA obtained at that visit revealed a deletion on chromosome X that included part of the gene OPHN1.
 | Figure 2 Photographs of the patient showing epicanthal folds, downslanting palpebral fissures, and large pinnae. |
The deletion was determined to be a de novo variant and his family was counseled about their low recurrence risk and the patient’s prognosis. Alternative therapies for autism (vitamin supplements) were stopped by the family at that time.
Methods
CMA was performed on deoxyribonucleic acid (DNA) extracted from leukocytes in peripheral blood. Testing was performed in a licensed clinical laboratory using standard techniques. CMA was performed using the CytoChip oligonucleotide 180K platform (BlueGnome, Cambridge, UK). Patient and pooled same-sex reference DNA (Promega Corporation, Fitchburg, WI, USA) were labeled with Cy3-dUTP and Cy5-dUTP, respectively, and hybridized to the array platform, as specified by the manufacturer’s protocol. The array was scanned using the G2505C microarray scanner (Agilent Technologies, Santa Clara, CA, USA). Data analysis was performed using BlueFuse Multi v2.6 with the ADM-2 algorithm set at a threshold of 5.0 and a minimum of four continuous probes (BlueGnome). Findings were mapped to Genome Reference Consortium Human Build 37 (GRCh37).
The deletion was confirmed using a bacterial artificial chromosome clone corresponding to the sequence RP3-360E18 for fluorescent in situ hybridization (FISH). The same FISH probe was used to evaluate the patient’s mother for the deletion. The FISH probe spanned linear positions 67,342,256–67,474,160 and largely overlapped with the detected deletion from CMA (see Results section).
Results
CMA revealed a deletion within chromosome Xq12 spanning linear positions 67,362,279–67,552,882, as shown in Figure 3. This is an intragenic deletion affecting exons 2–5 of the gene OPHN1. Given that the patient is male and hence hemizygous for OPHN1, an exonic deletion would be expected to be pathogenic. His mother did not carry the deletion in leukocyte DNA. Figure 4 demonstrates confirmation of the presence of the deletion in the proband via FISH and the absence of the deletion in the mother of the proband.
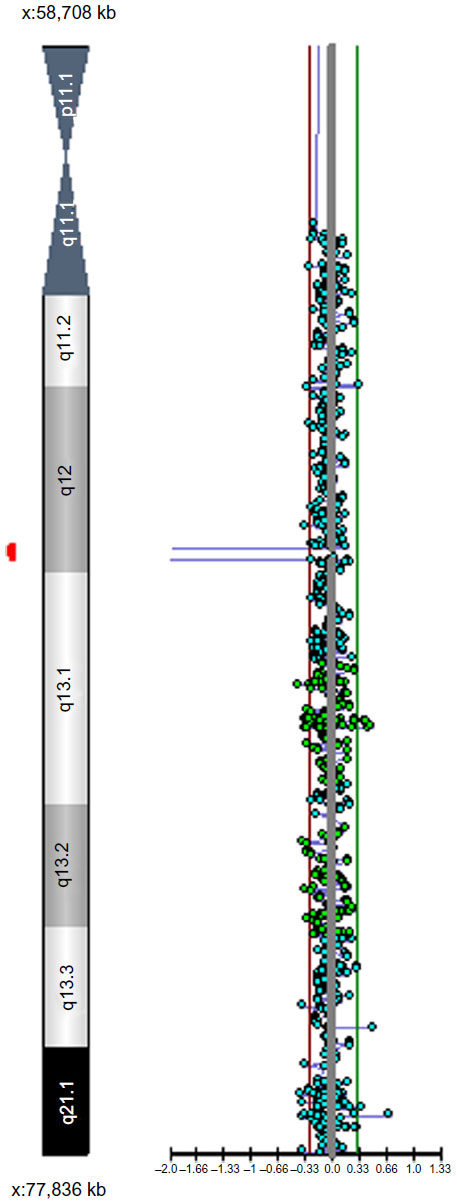 | Figure 3 Chromosomal microarray representation showing copy number loss for probes corresponding to linear positions 67,362,279–67,552,882 in Genome Reference Consortium Human Build 37. |
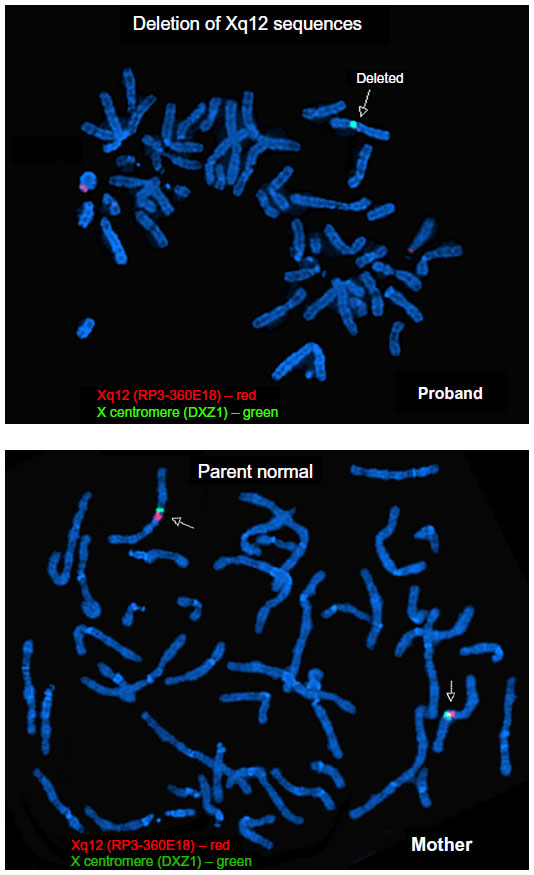 | Figure 4 Fluorescent in situ hybridization analysis showing absence of the red probe on the X chromosome in the proband and presence of the red probe on both X chromosomes of the proband’s mother. Note that the red probe hybridizes with the Y chromosome centromere in the proband and does not correspond to the sequences within OPHN1. |
Discussion
Loss of function of OPHN1 was discovered to cause X-linked ID in 1998.6 Though initially thought to result in ID without other features, additional phenotypic data found that OPHN1 mutations cause cerebellar hypoplasia.7 The clinical phenotype has been further expanded to include epilepsy, hypotonia, and strabismus.8 The facial dysmorphisms associated with OPHN1 mutations and deletions are variable and have been reported to include deep-set eyes, infraorbital creases, short philtrum, broad nasal root, prominent chin, and large pinnae.9 MRI findings in patients with OPHN1 mutations include inferior cerebellar vermian hypoplasia, malformations of the cerebellar hemispheres, enlargement of the lateral ventricles, caudate atrophy, and mild cortical thinning.10 The radiological phenotype of caudate atrophy with cerebellar vermian hypoplasia is relatively distinctive and should promote strong consideration of OPHN1 mutations, particularly in the setting of a male with epilepsy and ID.
Since its initial description, about 50 cases of OPHN1 mutations have been published.11 In an attempt to determine the prevalence of OPHN1 mutations in males with ID, mutation and deletion analysis was conducted for 196 males with at least one other affected male relative, as well as 17 males with ID and cerebellar abnormalities. In each group, two individuals were diagnosed with OPHN1 mutations for a prevalence of 1 percent (two of 196) in X-linked ID generally and 12% (two of 17) in males with ID and posterior fossa abnormalities.12 Next-generation sequencing has made possible the diagnostic use of sequencing via large panels of ID-associated genes. As OPHN1 is included in many clinically available panels, diagnoses of OPHN1 mutations are likely to increase in coming years. As clinical use of CMA continues to expand for patients with autism and epilepsy, OPHN1 deletions and duplications will increasingly be detected as well.
The OPHN1 clinical phenotype results from loss of protein function due to deletion of the entire gene,13 intragenic deletions,14,15 intragenic insertions (without frameshift),16 chromosomal translocations with breakpoints within the gene, and nonsense and frameshift mutations.10 One patient with severe ID has been reported with a duplication of the entire OPHN1 gene. His phenotype differed from males with null alleles due to his absence of cerebellar hypoplasia.17 The androgen receptor gene AR is immediately proximal (centromeric) to OPHN1, and there are reports of a contiguous gene deletion syndrome resulting in 46, XY disorder of sex development due to complete androgen insensitivity as well as the manifestations of OPHN1 deletion.18,19 A separate contiguous gene deletion syndrome includes OPHN1 as well as the distal gene EFNB1. Deletion of EFNB1 results in craniofrontonasal syndrome with hypertelorism, nasal clefting, and skeletal asymmetry, with the distinctive feature that the phenotype is expressed largely in females with mutations and not males.13 As would be expected in an X-linked condition, there is wide variability in the phenotype of female carriers of OPHN1 mutations, ranging from asymptomatic to isolated strabismus to moderate ID with similar brain malformations to affected males.8,20 More severely affected females have been found to have skewed X chromosome inactivation with increased expression of the mutated allele.21
OPHN1 is a large gene, consisting of 25 exons and spanning about 390 kilobases of genomic DNA.22 The gene is expressed in developing fetal brain tissue as well as in mature structures. In particular, its protein product is found on both the axonal and dendritic sides of synapses and is found in all major types of neurons in the brain.23,24 It acts as a Rho GTPase activating protein, regulating G protein signaling in the neuron as it relates to the cytoskeleton.5,25 The result of disrupted function of the cytoskeleton is shortened or immature dendritic spines, likely resulting in impaired synaptic plasticity. There are several major functional domains within the protein product of OPHN1: a GTPase activating protein (GAP) domain, a Homer-binding domain, an actin-binding domain at the C-terminus, and, at the N-terminus of the protein, a Bin/amphiphysin/Rvs (BAR) domain.23,24 The GAP domain interacts with Rho-GTPases in the neuron to inhibit RhoA and other GTPases. GTPase inhibition preserves dendrite spine length, and the Homer-binding domain mediates interaction with the glutamate receptors of the synapse.23 The BAR domain facilitates interaction of the protein with the cell membrane and may provide autoinhibition on the GAP activity of the protein.26 Additional smaller proline-rich domains near the C-terminus of the protein interact with regulators of endocytosis of synaptic vesicles.27 Analysis of messenger ribonucleic acid transcripts was not performed for our patient, so it is unknown whether the mutated allele is transcribed. If it were translated, the resultant protein would lack a functional BAR domain, given the deletion of exons 2–5. Pirozzi et al16 showed that a 16 amino acid in-frame insertion in the BAR domain resulted in phenotypes equivalent to patients with frameshift and nonsense mutations. Thus, it is highly likely that the deletion of exons 2–5 of OPHN1 results in a pathogenic allele.
An Ophn-1 knockout mouse showed hyperactivity, impaired social and procedural learning, and inappropriately decreased aggression. Brain malformations included ventriculomegaly but not cerebellar hypoplasia in the mouse model.28 An in vitro model using interfering ribonucleic acid knockdown in rat hippocampal neurons showed that excitatory glutamatergic synapses could be neither formed nor maintained normally in the absence of Ophn-1 expression.29 Ophn-1 knockdown also prevents long-term depression in the hippocampus by impairing α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor endocytosis, representing a distinct mechanism of dysfunction of synaptic plasticity.30 Using a similar assay, it was shown that neurons not expressing Ophn-1 are unable to perform normal synaptic vesicle recycling, further refining the understanding of the molecular pathogenesis of Ophn-1 dysfunction.27 Using this model, it was shown that synaptic function in Ophn-1-knockdown neurons could be rescued with application of Rho-kinase inhibitors. Thus, separate mechanisms of impaired long-term potentiation and long-term depression have been elucidated in models of OPHN1 dysfunction. Given that these processes are a fundamental mechanism of learning at a cellular and molecular level, it is not surprising that patients with OPHN1 mutations have severe ID.31
The determination of a genetic diagnosis for the patient described in this report significantly altered his care. Prior to the diagnostic finding on CMA, with diagnoses of idiopathic autism and epilepsy, his parents treated him with large doses of vitamins well in excess of recommendations. Subsequent to the diagnosis, with an improved understanding of the underlying cause of his autism and ID, his parents chose to forgo alternative therapies. As in this case, a thorough diagnostic evaluation of individuals considered to have idiopathic epilepsy, autism, or ID may provide significant benefit to families and patients via improved understanding of prognosis, therapy, and recurrence risk.
Acknowledgments
The authors wish to thank the family of the patient for their consent to participate in a published case report, including consent for publication of photographs. In addition, we would like to thank Billie Carstens of Colorado Genetics Laboratory for assistance with figures relating to microarray and FISH.
Disclosure
The authors report no conflicts of interest in this work.
References
Lenhard W, Breitenbach E, Ebert H, Schindelhauer-Deutscher H, Henn W. Psychological benefit of diagnostic certainty for mothers of children with disabilities: lessons from Down syndrome. Am J Med Genet A. 2005;133(2):170–175. | |
Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86(5):749–764. | |
Cooper GM, Coe BP, Girirajan S, et al. A copy number variation morbidity map of developmental delay. Nat Genet. 2011;43(9):838–846. | |
Mefford HC, Sharp AJ, Baker C, et al. Recurrent rearrangements of chromosome 1q21. 1 and variable pediatric phenotypes. N Engl J Med. 2008;359(16):1685–1699. | |
Nakayama AY, Harms MB, Luo L. Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J Neurosci. 2000;20(14):5329–5338. | |
Billuart P, Bienvenu T, Ronce N, et al. Oligophrenin-1 encodes a rhoGAP protein involved in X-linked mental retardation. Nature. 1998;392(6679):923–926. | |
Philip N, Chabrol B, Lossi A, et al. Mutations in the oligophrenin-1 gene (OPHN1) cause X linked congenital cerebellar hypoplasia. J Med Genet. 2003;40(6):441–446. | |
Bergmann C, Zerres K, Senderek J, et al. Oligophrenin 1 (OPHN1) gene mutation causes syndromic X-linked mental retardation with epilepsy, rostral ventricular enlargement and cerebellar hypoplasia. Brain. 2003;126(7):1537–1544. | |
Chabrol B, Girard N, N’Guyen K, et al. Delineation of the clinical phenotype associated with OPHN1 mutations based on the clinical and neuropsychological evaluation of three families. Am J Med Genet A. 2005;138(4):314–317. | |
Portes Vd, Boddaert N, Sacco S, et al. Specific clinical and brain MRI features in mentally retarded patients with mutations in the oligophrenin-1 gene. Am J Med Genet A. 2004;124(4):364–371. | |
Al-Owain M, Kaya N, Al-Zaidan H, et al. Novel intragenic deletion in OPHN1 in a family causing XLMR with cerebellar hypoplasia and distinctive facial appearance. Clin Genet. 2011;79(4):363–370. | |
Zanni G, Saillour Y, Nagara M, et al. Oligophrenin 1 mutations frequently cause X-linked mental retardation with cerebellar hypoplasia. Neurology. 2005;65(9):1364–1369. | |
Wieland I, Weidner C, Ciccone R, et al. Contiguous gene deletions involving EFNB1, OPHN1, PJA1 and EDA in patients with craniofrontonasal syndrome. Clin Genet. 2007;72(6):506–516. | |
Madrigal I, Rodríguez-Revenga L, Badenas C, Sánchez A, Milà M. Deletion of the OPHN1 gene detected by aCGH. J Intellect Disabil Res. 2008;52(3):190–194. | |
Froyen G, Van Esch H, Bauters M, et al. Detection of genomic copy number changes in patients with idiopathic mental retardation by high-resolution X-array-CGH: important role for increased gene dosage of XLMR genes. Hum Mutat. 2007;28(10):1034–1042. | |
Pirozzi F, Di Raimo FR, Zanni G, et al. Insertion of 16 amino acids in the BAR domain of the oligophrenin 1 protein causes mental retardation and cerebellar hypoplasia in an Italian family. Hum Mutat. 2011;32(11):E2294–E2307. | |
Bedeschi MF, Novelli A, Bernardini L, et al. Association of syndromic mental retardation with an Xq12q13. 1 duplication encompassing the oligophrenin 1 gene. Am J Med Genet A. 2008;146(13):1718–1724. | |
Tentler D, Gustavsson P, Leisti J, et al. Deletion including the oligophrenin-1 gene associated with enlarged cerebral ventricles, cerebellar hypoplasia, seizures and ataxia. Eur J Hum Genet. 1999;7(5):541–548. | |
Davies H, Hughes I, Savage M, et al. Androgen insensitivity with mental retardation: a contiguous gene syndrome? J Med Genet. 1997;34(2):158–160. | |
Menten B, Buysse K, Vermeulen S, et al. Report of a female patient with mental retardation and tall stature due to a chromosomal rearrangement disrupting the OPHN1 gene on Xq12. Eur J Med Genet. 2007;50(6):446–454. | |
Rocas D, Alix E, Michel J, et al. Neuropathological features in a female fetus with OPHN1 deletion and cerebellar hypoplasia. Eur J Med Genet. 2013;56(5):270–273. | |
Billuart P, Chelly J, Carrié A, et al. Determination of the gene structure of human oligophrenin-1 and identification of three novel polymorphisms by screening of DNA from 164 patients with non-specific X-linked mental retardation. Ann Genet. 2000;43(1):5–9. | |
Govek E-E, Newey SE, Akerman CJ, Cross JR, Van der Veken L, Van Aelst L. The X-linked mental retardation protein oligophrenin-1 is required for dendritic spine morphogenesis. Nat Neurosci. 2004;7(4):364–372. | |
Fauchereau F, Herbrand U, Chafey P, et al. The RhoGAP activity of OPHN1, a new F-actin-binding protein, is negatively controlled by its amino-terminal domain. Mol Cell Neurosci. 2003;23(4):574–586. | |
Ng J, Luo L. Rho GTPases regulate axon growth through convergent and divergent signaling pathways. Neuron. 2004;44(5):779–793. | |
Eberth A, Lundmark R, Gremer L, et al. A BAR domain-mediated autoinhibitory mechanism for RhoGAPs of the GRAF family. Biochem J. 2009;417:371–377. | |
Nakano-Kobayashi A, Kasri NN, Newey SE, Van Aelst L. The Rho-linked mental retardation protein OPHN1 controls synaptic vesicle endocytosis via endophilin A1. Curr Biol. 2009;19(13):1133–1139. | |
Khelfaoui M, Denis C, Van Galen E, et al. Loss of X-linked mental retardation gene oligophrenin1 in mice impairs spatial memory and leads to ventricular enlargement and dendritic spine immaturity. J Neurosci. 2007;27(35):9439–9450. | |
Kasri NN, Nakano-Kobayashi A, Malinow R, Li B, Van Aelst L. The Rho-linked mental retardation protein oligophrenin-1 controls synapse maturation and plasticity by stabilizing AMPA receptors. Genes Dev. 2009;23(11):1289–1302. | |
Nadif Kasri N, Nakano-Kobayashi A, Van Aelst L. Rapid synthesis of the X-linked mental retardation protein OPHN1 mediates mGluR-dependent LTD through interaction with the endocytic machinery. Neuron. 2011;72(2):300–315. | |
Ba W, van der Raadt J, Nadif Kasri N. Rho GTPase signaling at the synapse: implications for intellectual disability. Exp Cell Res. 2013;319(15):2368–2374. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.