")
Back to Journals » OncoTargets and Therapy » Volume 7
Exome sequencing reveals a potential mutational trajectory and treatments for a specific pancreatic cancer patient
Authors Cotterell J
Received 15 March 2013
Accepted for publication 4 October 2013
Published 3 May 2014 Volume 2014:7 Pages 655—662
DOI https://doi.org/10.2147/OTT.S45232
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
James Cotterell1,2
1Center for Genomic Regulation, Barcelona, Spain; 2Garvan Institute for Medical Research, Sydney, NSW, Australia
Abstract: Pancreatic cancer is the fourth biggest killer, and has one of the worst prognoses, of any cancer type. Approximately 95% of patients diagnosed with pancreatic cancer will not survive beyond 5 years post diagnosis, and these statistics have barely improved in over 40 years. Here, genomic changes in one particular patient with stage IV metastatic pancreatic cancer were explored to suggest a potential personalized treatment. In particular, exome sequencing of genomic DNA extracted from blood and the cancer biopsy was utilized with the aim of identifying mutational drivers of the cancer. This analysis revealed a splice site mutation in RBCK1 as the most promising driver of the cancer and a therapy based on a pan-cyclin-dependent kinase (pan-CDK) inhibitor, flavopiridol. This study suggests that drugs whose effectiveness is unclear for general populations of cancer sufferers should possibly be reconsidered for specific patients where the drug could be rationally argued to improve outcome.
Keyword: personalized medicine, driver mutation identification, next generation sequencing
Introduction
Although pancreatic cancer is the tenth most common cancer by incidence, it is also the fourth biggest killer. Pancreatic cancer is notoriously difficult to diagnose and there are no diagnostic tests currently for the early detection of this cancer, which explains a major part of the poor prognosis. Most patients with pancreatic cancer (80%) are not amenable to surgery at the time of diagnosis and, thus, chemo- or radiotherapy is the only option.2 Gemcitabine with capecitabine (GemCap) is the current first line of treatment given to most pancreatic cancer patients. A Phase III trial has demonstrated that, typically, GemCap offers a median 2.6 month survival increase for patients with good performance status (with a Karnofsky performance status score of 90–100) when compared to gemcitabine alone.3 In all, even with GemCap, the median overall survival of a pancreatic cancer patient has been shown to be only 7.1–8.4 months in Phase III trials.3,4
The major drive to improve these statistics focuses on improved early detection of the cancer such as screening high risk patients.5 Furthermore, personalized medicine promises to improve the survival of patients that are beyond surgery. Personalized medicine aims to tailor a treatment to that specific patient. General strategies that have been suggested for personalized cancer treatment include synthetic lethality and oncogene addiction.6,7 Implementing such strategies typically requires knowing the patients’ underlying somatic and germ line mutations. These molecular phenotypes are then used to guide a therapeutic strategy.8
Recently, the predominant effort for determining the molecular phenotypes of cancer patients has focused on exome sequencing. For example, exome sequencing has recently been used to identify other potential pancreatic cancer drivers, highlighting the axon guidance pathway as a common pathway mutated in pancreatic cancer.9 In recent years, the technology to sequence cancer genomes in this way has exploded, becoming commonplace for many projects. However, the uptake of such technologies by national health services for the use of diagnosis and improved outcome is, for various reasons, understandably slow. For example, many oncologists would require extensive additional training in order to interpret the results of such sequencing to guide choices in patient care.
Citizen science can be loosely defined as scientific research conducted, in whole or in part, by amateur or nonprofessional scientists. The lines of citizen science become blurred when the person performing the science is indeed a professional scientist but performing the science in a different field to their normal field of study or outside of the normal remit. Faced with a relative with pancreatic cancer, knowing that sequencing techniques exist with the power to discern molecular phenotypes for which improved therapies exist, yet none of which are available to that patient with that particular cancer type, the motivation for such professional citizen science is clear.
Under this backdrop, here, genomic sequence data of one patient related to the author with stage IV metastatic exocrine adenocarcinoma of the pancreas is explored to see whether it could be used to identify the mutational drivers of the cancer and hence direct a more suitable therapy than currently on offer in the National Health Service (UK). Genomic DNA was extracted from blood (normal) and a cancer biopsy and exome sequencing performed (a single sample of each). Exome sequencing revealed a germ line mutation in BRCA2 and six somatic mutations between the cancer and normal sequence.
Methods
Ethics statement
Informed consent from patient and next of kin was obtained at all steps throughout this work. This work was performed outside of the normal protocol of a scientific study since ethical approval from an ethical review board was not sought beforehand. Originally, the sequencing was performed with the intention of diagnosis and improved patient outcome. However, during this process it became obvious that the results would be helpful for cancer research in general and thus a decision was made to publish the paper after patient/next of kin approval. Hence, this work can be considered a form of citizen science. However, for the purposes of clarity, the paper has been written as much as ethically possible in the form of a typical research paper.
DNA extraction and quality
Cancer tissue was obtained via a percutaneous liver biopsy of a secondary lesion. The cancer was confirmed by histological analysis to be derived from exocrine tissue. DNA extraction was performed at the John Radcliffe hospital. The Qiagen blood/tissue kit (Qiagen, Venlo, the Netherlands) was used to extract genomic DNA from the cancer biopsy sample and a blood sample. Quality and quantity of the DNA was measured using a NanoDrop. Quality control metrics of the cancer DNA sample were 260/230 = 1.91 and 260/280 = 2.00 (total DNA 1.4 μg) and of the blood DNA sample were 260/230 = 2.10 and 260/280 = 1.90 (total DNA 38.2 μg). Further quality checks were performed at Oxford Gene Technology using NanoDrop, Qubit, and agarose gel electrophoresis. Importantly, as the cancer biopsy was taken from secondary liver lesions, the sequenced DNA should have the early metastasis driving mutations.
Exome selection and sequencing
Exome selection and sequencing was performed at Oxford Gene Technology using the Sureselect exome selection kit (Agilent Technologies, Santa Clara, CA, USA). Samples were prepared according to Agilent’s SureSelect protocol version 1.2 (Agilent Technologies). Concentration of each library was determined using Agilent’s QPCR NGS Library Quantification Kit (G4880A; Agilent Technologies). Quality assessment of indexed samples was performed using a bioanalyzer and quantitative PCR. Samples were pooled prior to sequencing with each sample at a final concentration of 10 nmol. The Illumina HiSeq 2000 (Illumina, Inc, San Diego, CA, USA) for sequencing using TruSeq v3 (Illumina, Inc) chemistry. 30× coverage was used. Over 80% of base reads of both the cancer and normal sample had greater than 30× coverage and over 90% had a greater than 20× coverage.
Bioinformatical analysis
Bioinformatical analysis of the samples was performed at Oxford Gene Technology. Briefly, the pipeline of analysis was as follows: sequences were aligned to the human reference genome 19 using BWA (Burrows-Wheeler Aligner).39 Picard was used to mask PCR duplicates. GATK was used for local realignment around indels and score quality recalibration. Base quality (Phred scale) scores were recalibrated using GATK’s covariance recalibration. Single-nucleotide polymorphism and indel variants were called using the GATK Unified Genotyper for each sample. Single-nucleotide polymorphism novelty was determined against dbSNP (Release 135). Variants were annotated with gene and gene-function data from Ensembl. Known variants from dbSNP (Release 135) are annotated within the dataset so that novel variants with serious predicted consequences may be rapidly identified. The SomaticSniper software was utilized to identify differing genomic sequences between the tumor and normal samples.20 The SIFT (Sorting Intolerant From Tolerant),40 Condel,41 and polyphen (Polymorphism Phenotyping v2)42 algorithms were utilized when a mutation was predicted to make an amino acid change to assess whether the amino acid change was predicted to be functionally neutral or result in a functional change. Data is visualized using OGTs interactive report software.
Results
First, the genes that have been shown to be associated with hereditary, familial, and increased risk forms of pancreatic cancer, which include trypsin and genes involved in DNA repair, cell cycle control, and cell polarity, were examined.10 Specifically, BRCA1/BRCA2, ATM, P53, CDKN2A, PALB2, APC, MLH1, MSH2, MSH6, PMS2, PRSS1, and STK1 were analyzed. For this analysis, the exome sequence from the blood sample was compared to that of reference human genome 19 and only those variations that were detected with at least a 20-fold coverage were explored in greater detail. These variations that are predicted to have serious consequences are summarized in Table 1. The most striking of these variations is that the patient has a C > G mutation at genomic position 32,913,771 of the BRCA2 gene. This mutation would change a serine to a termination codon in exon 10 of the BRCA2 gene.
 | Table 1 |
Consequences of the BRCA2 mutation
The BRCA2 mutation that leads to a termination codon in the middle of the mRNA is possibly leading to the production of a dominant negative protein product, though most likely it is leading to haploinsufficiency due to nonsense mediated decay (NMD) of mutated BRCA2 mRNA. NMD is a cellular mechanism for detecting the potential production of erroneous or truncated proteins, acting to degrade mRNA coding for such proteins.11 Nonsense mediated decay works by detecting nonsense stop codons in the middle of an mRNA molecule. The typical large average intron size of eukaryotic cells makes it likely that the inclusion of an intron will by chance contain one of the nonsense stop codons (UAA, UAG, UGA).
Next, the somatic mutations were explored by comparing the exome sequences of the genomic DNA extracted from the blood to that of the tumor. Six somatic mutational changes were apparent between the normal and cancer samples, which are summarized in Table 2. Of the six mutations, four resulted in nonsynonymous changes in the resulting protein amino acid that were predicted to be neutral. Two mutations, however, showed possible deleterious effects; a nonsynonymous amino acid change in FAT3 and a splice site mutation in RBCK1. Both mutations were detected with statistically significant read depths so the predicted effects of these mutations were explored in further detail.
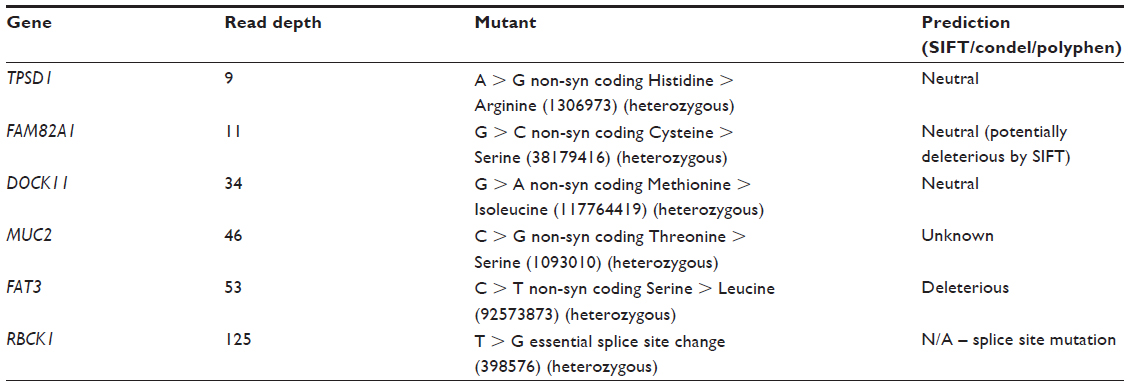 | Table 2 |
Consequences of the Rbck1 mutation
RBCK1 is a ring finger containing E3 ubiquitin ligase (Figure 1A).12 RBCK1 was originally identified as an interactor of protein kinase C.12 RBCK1 contains both a nuclear export signal and a nuclear localization signal.13 It has been shown to shuttle between the nucleus and the cytoplasm though is more often present in the cytoplasm. The RBCK1 mutation detected here manifests itself as a heterozygote that affects a splice site of intron 4 between exons 4 and 5 (Figure 1B). Specifically, at the five prime donor site of intron 4 the GT genomic sequence is mutated to GG meaning it is not available for splicing. There are several potential splicing consequences of such a mutation and these are summarized in Figure 1C. Intriguingly, only one combination of exon skipping would result in a viable mRNA as all other combinations would result in a frame shift (and, thus, degradation of the resultant mRNA by NMD). While the predicted translation of intron 4 of RBCK1 would maintain the protein product in frame, it nevertheless codes for 18 stop codons, most probably marking such a transcript for NMD. Hence, the unique splicing event that results in a viable transcript is the skipping of exons 2, 3, and 4 such that exon 1 joins directly to exon 5.
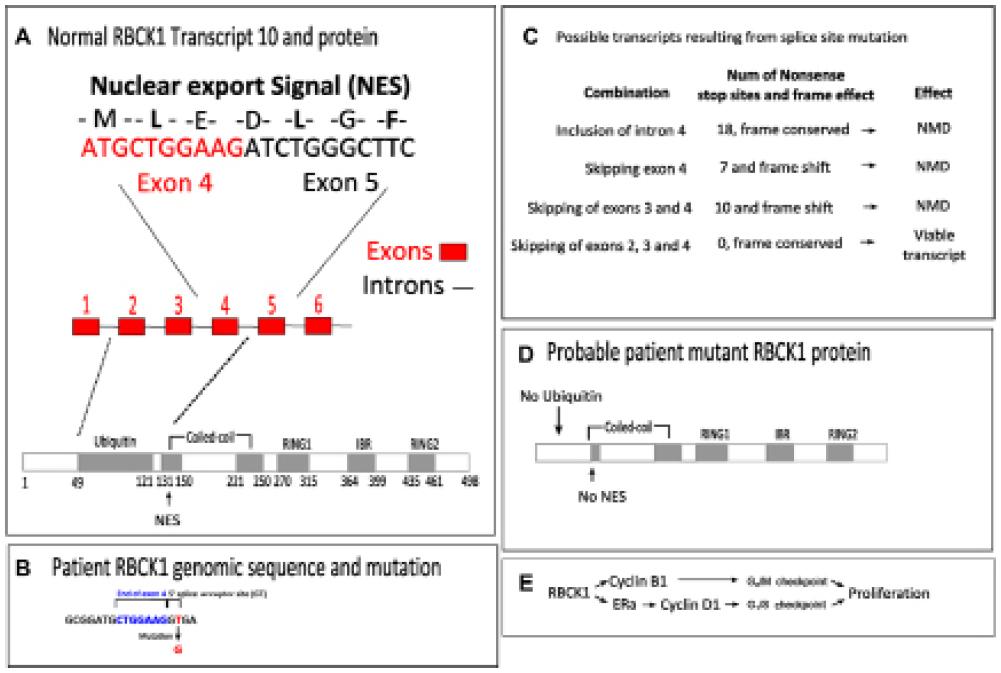 | Figure 1 A splice site mutation in RBCK1 would be expected to lead to loss of the nuclear export signal and the ubiquitin domain. |
If exons 2, 3, and 4 were skipped, the resulting protein would be identical to the normal protein but lacking its entire ubiquitin domain and nuclear export signal (Figure 1D). Ubiquitin is an important small regulatory peptide that tags protein for degradation. Typically, long chains of ubiquitin are added to a protein making it more likely to be degraded. The lack of an ubiquitin domain in RBCK1 most likely means that it cannot be degraded. Furthermore, the nuclear export signal of RBCK1 intriguingly bridges exons 4 and 5 of the normal protein. The skipping of exon 4 most likely completely destroys this nuclear export signal leading to RBCK1 being retained in the nucleus (Figure 1D). Overall the combination of the effects on the ubiquitin domain and the NES most likely result in RBCK1 lacking a mark for degradation and being retained in the nucleus.
E3 ubiquitin ligases have been demonstrated to be important proteins in breast cancer often showing elevated expression levels.14 Indeed, RBCK1 has been shown to drive proliferation in breast cancers (Figure 1E).15 Knockdown of RBCK1 in breast cancer cell lines results in substantially reduced proliferation, with cells blocked in G1/S1. Breast cancer cell lines with higher levels of RBCK1 activity also demonstrate higher expression of cyclin B1 and estrogen receptor expression, which is an activator of cyclin D1 expression. The failure of nuclear export and degradation of RBCK1 would thus be expected to increase its nuclear transcriptional activity, leading to an increase in cyclin B1 and D1 and thus driving cellular proliferation.
Consequences of the Fat3 mutation
The FAT3 protein is a cadherin; a family of calcium-dependent cell adhesion molecules that can mediate cell aggregation and cell sorting in vitro in a homophilic fashion (for example, cells expressing N-cadherin tend to segregate with other cells expressing the same cadherin). The Fat3 gene encodes a large protein of 4,555 amino acids with 34 cadherin domains, four epidermal growth factor-like motifs, a laminin A–G motif, and a cytoplasmic domain. The FAT3 protein is similar to that encoded by the Drosophila tumor suppressor gene FAT.16 Recessive lethal mutations in the fat locus of Drosophila cause hyperplastic, tumor-like overgrowth of larval imaginal discs.17
The heterozygous FAT3 mutation detected here would cause a change from a serine residue to a leucine residue in the resulting protein in three of the four transcripts of this gene. The mutation occurs in the last of 34 extracellular cadherin repeats similar to the position of the Gull allele of the Drosophila fat gene that causes similar defects.17 Cadherins are suspected to have important roles in cancer invasion and metastasis due to their roles in cell-cell adhesion (switching from E to N cadherin has been identified as a feature of aggressive tumors, for example).18 Hence, the FAT3 mutation may be a candidate for driving the early stages of metastasis if it has somehow caused an epithelial to mesenchymal type transition in the cancerous cells. However, FAT3 is not normally expressed in the pancreas, hence bringing into question its role as a driver of the pancreatic cancer of this patient.
Discussion
Here it has been demonstrated how exome sequencing of genomic DNA extracted from a pancreatic cancer biopsy and from a blood sample can be utilized to identify potential mutational drivers of a cancer. A single mutation in BRCA2 in the germ line that may well have influenced subsequent mutations has been identified (Figure 2). Six somatic mutations have been identified, with a mutation in RBCK1 most likely to be the main driver. Intriguingly, there was no observed somatic mutation in KRAS, in which approximately 95% of pancreatic cancer patients typically have a mutation.9,19 This finding suggests that this particular patient may have a different mutational profile from typical pancreatic cancer patients. Alternatively, it is possible that a mutation in KRAS has been missed by sampling inherent in the analysis.
 | Figure 2 Stages of a potential mutational trajectory for this cancer and potential points of treatment. |
Typical pancreatic cancer biopsies are known to contain a substantial amount of normal tissue (in particular, desmoplastic stroma), which could potentially lead to false negatives in somatic mutation calling. It has been demonstrated that the SomaticSniper software can detect 90% of mutations at 30× sequencing depth if the tumor allele frequency in the tumor sample is greater than 30% (assuming a pure normal sample).20 The normal sample can be considered pure as circulating tumor cells from solid tumors are predicted to only constitute approximately 0.01% of nucleated blood cells even in metastatic disease such as the case described here (on average 10–20 circulating tumor cells compared to several million nucleated white blood cells per mL of blood).21 Furthermore, it has also been demonstrated that, on average, biopsies from pancreatic cancer have 38% tumor cellularity.9 Hence, at least 90% of somatic mutations should be successfully called from an average pancreatic cancer biopsy at a sequencing depth of 30×. Therefore, although there is a possibility that somatic mutations have been missed, the vast majority should be detected in this analysis (at least in the exome).
As mentioned, a key issue in cancer genomics is discerning driver mutations from passenger mutations as genomic instability is a typical feature of many cancers.22 As has been shown, functional inference can come from exploring the effect of the exact mutation on the resulting RNA and protein. Furthermore, functional inference can come from the COSMIC database that catalogues known somatic mutations in cancer.23 Indeed, multiple entries can be found in the COSMIC database for exon 4 and a breakpoint can be found in intron 4 of RBCK1 that results in an intrachromosomal tandem duplication (COST29521). Multiple mutations are also found in the cadherin repeats of FAT3 and there are over 5,000 entries for exon 10 of BRCA2 alone, though in this study that BRCA2 mutation was detected as germline rather than somatic. These entries further strengthen the hypothesis that the mutations in BRCA2, RBCK1 and FAT3 are driver mutations and not passenger mutations.
Potential therapies
The strategy of synthetic lethality for combating cancer has received much attention in recent times. The basic concept of synthetic lethality states that either of two mutations alone are nonlethal but in combination are lethal.24 The strategy of synthetic lethality can be applied in this context when a patient is known to have a mutation (typically familial) in a DNA repair gene such as BRCA1/BRCA2.25 The concept in this context works on the fact that a cell only has two different pathways in which to fix double stranded breaks (DSBs) in DNA; nonhomologous end joining and homologous recombination ([HR] of which there are 2 distinct branches). BRCA2 is thought to play a direct role in HR repair, specifically in the error-free repair of DSBs via the gene conversion branch.26 When BRCA2 is mutated, either the nonhomologous end joining pathway or the single-strand annealing branch of HR is used, both of which are error prone. Hence, mutations in BRCA2 are thought to sensitize cells to DSB DNA damage. Inhibition of Poly ADP ribose polymerase (PARP) 1 is also thought to sensitize cells to double stranded breaks because the failure of base excision repair (PARP1’s function) leads to the replication fork stalling and potential formation of double stranded breaks.25,27 Hence, it is thought that PARP1 inhibition in combination with a BRCA2 mutation would increase the frequency of formation of replication associated DNA lesions leading to cell cycle arrest and/or death.25 Indeed, PARP inhibitors such as olaparib have been shown to be effective in more than half of the pancreatic cancer patients in a Phase I trial.28 However, any strategy, when based on a germ line mutation, would be expected to also target normal healthy cells as they too have the mutation. This feature would be expected to reduce the size of the therapeutic window and, thus, the effectiveness of the treatment.29
Hence, the most effective choices of anticancer therapy are likely to be based on somatic mutations such as those identified in Table 2. In this case, the most likely success will come from a therapy based on the mutation in RBCK1 as this appears to be the major driver of the cancer. Directly or indirectly inhibiting the function of RBCK1 could be considered a form of oncogene addiction strategy as RBCK1 appears to be acting as the predominant oncogene in this case. No known direct specific inhibitor for RBCK1 function exits; however, one can take advantage of the manner by which RBCK1 appears to be driving the cell cycle (through driving cyclin expression) to inhibit this function indirectly.
Cyclins are a family of proteins that control the progression of cells through the cell cycle by activating cyclin-dependent kinase (CDK) enzymes. CDKs are relatively small proteins containing little more than a kinase domain that bind to cyclins to activate their kinase domain regulating progression through the cell cycle.30 In cancer, the misregulation of CDKs is a common event and, therefore, much attention has been focused on inhibiting these enzymes as cancer therapy. Adenosine triphosphate competitive inhibitors and the last generation of non-adenosine triphosphate competitive inhibitors are now emerging as one of the most potentially powerful target therapies.31 Hence, inhibition of CDKs is an indirect and promising way of inhibiting the Rbck1 proliferation driving function.
Flavopiridol is a pan-cyclin-dependent kinase (pan-CDK) inhibitor that has been used in clinical trials for various different cancer types. In vitro flavopiridol has been shown to inhibit tumor cell growth through the blockade of cell cycle progression at G1 or G2.32,33 As a single agent in a 150–300 nM range, flavopiridol also inhibits the colony formation of a series of human cancer cell lines.34 Flavopiridol inhibits CDK 1, 2, 4, and 6 in the nanomolar range. Cyclin B1 and D1, which are expected to be overexpressed in this patient’s cancer, perform their function with CDK 1 and 4/6, respectively. Biologically active plasma concentrations of flavopiridol (approximately 300–500 nM) are easily achievable in patients receiving flavopiridol via infusion. Hence, flavopiridol has the potential to be effective against this patient’s specific cancer. Indeed, the combination of flavopiridol and docetaxel has shown some promise in pancreatic cancer patients.35 Half of those patients showed a response or stable disease in the aforementioned Phase I trial and one had a complete response. Furthermore, a Phase I trial testing flavopiridol in combination with FOLFOX for advanced solid tumors demonstrated a complete response and three stable diseases for six patients with pancreatic cancer.36 However, a Phase II study in ten refractory metastatic pancreatic cancer patients demonstrated minimal activity and significant toxicity.37 Hence, it is an open question as to whether flavopiridol is simply not effective or it is only effective in a specific subpopulation as none of the patients in the aforementioned trials were genetically tested.
This study demonstrates that there is a grey area in medical research and its corresponding ethics that needs to be addressed. This gray area arises when the rate of innovation of a new technology far outstrips the uptake of such technology into medical practice. Nowhere is this expected to be more evident than in the treatment of cancers such as pancreatic cancer that have a dismal prognosis that has barely improved in the last 40 years, even with the best of modern medicine.
Combined with the explosion in the power of DNA sequencing technology, the gap between what can be done theoretically with regards to possible treatments and what can be done in a standard hospital is enormous. When such a large gap appears, professional citizen science is likely to come to the fore. When these new sequencing technologies eventually become common practice in a standard hospital setting, such professional citizen science situations are unlikely to occur.
Finally, this work demonstrates a specific example of how sequencing can suggest therapies specific to a particular patient. These forms of bench to bedside translational research are gaining popularity and are likely to change the landscape of pancreatic cancer research and treatment.38 Indeed, there have been many translational cancer science centers opening of late with the exact aim of personalized medicine based on genomic sequencing, and a continued trend in this direction is to be expected. Furthermore, sequencing the genomes of cancer patients and their cancer is starting to become so commonplace that it suggests that the way patients are enrolled on clinical trials in the future may have to change. One possibility is that genetic screening of a pool of patients willing to undergo a trial could be used to assign patients to particular trials. Rational assignment based on solid functional evidence of mutational drivers is likely to improve overall outcome.
Acknowledgment
The patient analyzed in this study is related to the author.
Disclosure
The author reports no other conflicts of interest in this work.
References
Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. | |
Butturini G, Stocken DD, Wente MN, et al. Influence of resection margins and treatment on survival inpatients with pancreatic cancer: meta-analysis of randomized controlled trials. Arch Surg. 2008;143:75–83. | |
Herrmann R, Bodoky G, Ruhstaller T, et al. Gemcitabine plus capecitabine compared with gemcitabine alone in advanced pancreatic cancer: a randomized, multicenter, phase III trial of the Swiss Group for Clinical Cancer Research and the Central European Cooperative Oncology Group. J Clin Oncol. 2007;25:2212–2217. | |
Cunningham D, Chau I, Stocken DD, et al. Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancer. J Clin Oncol. 2009;27:5513–5518. | |
Lo Menzo E, Di Vita M, Berretta M, et al. Molecular diagnosis of pancreatic cancer: where do we stand? Front Biosci (Schol Ed). 2010;2:578–590. | |
Weinstein IB, Joe AK. Mechanisms of disease: Oncogene addiction – a rationale for molecular targeting in cancer therapy. Nat Clin Pract Oncol. 2006;3:448–457. | |
Rehman FL, Lord CJ, Ashworth A. Synthetic lethal approaches to breast cancer therapy. Nat Rev Clin Oncol. 2010;7:718–724. | |
Cao Y, DePinho RA, Ernst M, Vousden K. Cancer research: past, present and future. Nature Rev Cancer. 2011;11:749–754. | |
Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. | |
Solomon S, Das S, Brand R, Whitcomb DC. Inherited pancreatic cancer syndromes. Cancer J. 2012;18:485–491. | |
Frischmeyer PA, Dietz HC. Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet. 1999;8:1893–1900. | |
Tokunaga C, Tatematsu K, Kuroda S, Nakagawa N, Kikkawa U. Molecular cloning and characterization of RBCK2, a splicing variant of a RBCC family protein, RBCK1. FEBS Lett. 1998;435:11–15. | |
Tatematsu K, Yoshimoto N, Koyanagi T, et al. Nuclear-cytoplasmic shuttling of a RING-IBR protein RBCK1 and its functional interaction with nuclear body proteins. J Biol Chem. 2005;280:22937–22944. | |
Burger A, Amemiya Y, Kitching R, Seth AK. Novel RING E3 ubiquitin ligases in breast cancer. Neoplasia. 2006;8:689–695. | |
Gustafsson N, Zhao C, Gustafsson JA, Dahlman-Wright K. RBCK1 drives breast cancer cell proliferation by promoting transcription of estrogen receptor alpha and cyclin B1. Cancer Res. 2010;70:1265–1274. | |
Mitsui K, Nakajima D, Ohara O, Nakayama M. Mammalian fat3: a large protein that contains multiple cadherin and EGF-like motifs. Biochem Biophys Res Commun. 2002;290:1260–1266. | |
Mahoney PA, Weber U, Onofrechuk P, Biessmann H, Bryant PJ, Goodman CS. The fat tumor suppressor gene in Drosophila encodes a novel member of the cadherin gene superfamily. Cell. 1991;67:853–868. | |
Cavallaro U, Schaffhauser B, Christofori G. Cadherins and the tumour progression: is it all in a switch? Cancer Lett. 2002;176:123–128. | |
Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–554. | |
Larson DE, Harris CC, Chen K, et al. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinforamtics. 2012;28:311–317. | |
Miller MC, Doyle GV, Terstappen LW. Significance of circulating tumor cells detected by the cellsearch system in patients with metastatic breast colorectal and prostate cancer. J Oncol. 2010;2010:617421. | |
Campbell PJ, Yachida S, Mudie LJ, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467:1109–1113. | |
Bamford S, Dawson E, Forbes S, et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer. 2004;91:355–358. | |
Dobzhansky T. Genetics of natural populations: Xiii. Recombination and variability in populations of Drosophila Pseudoobscura. Genetics. 1946;31:269–290. | |
Ashworth A. Drug resistance caused by reversion mutation. Cancer Res. 2008;68:10021–10023. | |
Gudmundsdottir K, Ashworth A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene. 2006;25:5864–5874. | |
Haber JE. DNA recombination: The replication connection. Trends Biochem Sci. 1999;24:271–275. | |
Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–134. | |
Kaelin WG Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5:689–698. | |
Morgan DO. Principles of CDK regulation. Nature. 1995;374:131–134. | |
Rizzolio F, Tuccinardi T, Caligiuri I, Lucchetti C, Giordano A. CDK inhibitors: from the bench to clinical trials. Curr Drug Targets. 2010;11:279–290. | |
Kaur G, Stetler-Stevenson M, Sebers S, et al. Growth inhibition with reversible cell cycle arrest of carcinoma cells by flavone L86-8275. J Natl Cancer Inst. 1992;84:1736–1740. | |
Carlson BA, Dubay MM, Sausville EA, Brizuela L, Worland PJ. Flavopiridol induces G1 arrest with inhibition of cyclin dependent kinase CDK2 and CDK4 in human breast carcinoma cells. Cancer Res. 1996;56:2973–2978. | |
Bible KC, Kaufmann SH. Flavopiridol: a cytotoxic flavone that induces cell death in noncycling A549 human lung carcinoma cells. Cancer Res. 1996;56:4856–4861. | |
Fornier MN, Rathkopf D, Shah M, et al. Phase I dose-finding study of weekly docetaxel followed by flavopiridol for patients with advanced solid tumors. Clin Cancer Res. 2007;13:5841–5846. | |
Rathkopf D, Dickson MA, Feldman DR, et al. Phase I study of flavopiridol with oxaliplatin and fluorouracil/leucovorin in advanced solid tumors. Clin Cancer Res. 2009;15:7405–7411. | |
Carvajal RD, Tse A, Shah MA, et al. A phase II study of flavopiridol (Alvocidib) in combination with docetaxel in refractory, metastatic pancreatic cancer. Pancreatology. 2009;9:404–409. | |
Corbo V, Tortora G, Scarpa A. Molecular pathology of pancreatic cancer: from bench-to-bedside translation. Curr Drug Targets. 2012;13:744–752. | |
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics. 2009;25:1754–1760. | |
Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. | |
González-Pérez A, López-Bigas N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet. 2011;88:440–449. | |
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.