")
Back to Journals » OncoTargets and Therapy » Volume 13
Evaluation of Tumor Specificity and Immunity of Thymidine Kinase-Deleted Vaccinia Virus Guang9 Strain
Authors Ding Y , Fan J, Deng L, Peng Y, Zhou B, Huang B
Received 28 April 2020
Accepted for publication 17 July 2020
Published 5 August 2020 Volume 2020:13 Pages 7683—7697
DOI https://doi.org/10.2147/OTT.S260288
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr William C. Cho
Yuedi Ding ,1 Jun Fan ,1 Lili Deng ,1 Ying Peng ,1 Bin Zhou ,1 Biao Huang 1
NHC Key Laboratory of Nuclear Medicine, Jiangsu Key Laboratory of Molecular Nuclear Medicine, Jiangsu Institute of Nuclear Medicine, Wuxi, Jiangsu 214063, People’s Republic of China
Correspondence: Biao Huang; Yuedi Ding
NHC Key Laboratory of Nuclear Medicine, Jiangsu Key Laboratory of Molecular Nuclear Medicine,Jiangsu Institute of Nuclear Medicine, Wuxi, Jiangsu 214063, People’s Republic of China
Email [email protected]; [email protected]
Purpose: Oncolytic viruses are emerging as promising options for clinical cancer treatment due to their inherent ability of tumor tropism and oncolytic property. Aside from tumor lysis, oncolytic viruses can induce host immune responses against tumor cells and may thus be viewed as a form of immunotherapy.
Methods: The attenuated vaccinia VG9-Luc, which originated from Chinese vaccinia Tian Tan strain, was constructed to express firefly luciferase for bioluminescence imaging and to disrupt the thymidine kinase gene for promoting tumor specificity. An in vivo bioluminescence imaging was performed to observe the virus distribution in live mice. The titers of neutralizing antiviral and antitumor antibodies in plasma were determined by time-resolved fluoroimmunoassay.
Results: Except BALB/c mice treated with intravenous virus injection, all immunocompromised and immunocompetent mice showed obvious tumor targeting ability of vaccinia VG9-Luc. Besides, host immune response activated by vaccinia VG9-Luc showed the production of antiviral and antitumor antibodies, the process of which was similar between intravenous and intratumoral viral delivery systems. The results indicated that virus infection promoted tumor-specific immunity by increasing the production of antitumor antibodies. Moreover, virus reinjection was performed and a more rapid viral clearance was observed in immunocompetent mice compared with first virus infection.
Conclusion: The thymidine kinase-deleted vaccinia Guang9 strain, which has the properties of tumor specificity and antitumor immunity, is a promising candidate vector for cancer therapy.
Keywords: oncolytic virotherapy, vaccinia virus, tumor specificity, bioluminescence imaging, antitumor immunity
Introduction
Oncolytic virotherapy is being developed as a promising platform for cancer therapy due to its ability to lyse cancer cells in a tumor-specific manner.1,2 Vaccinia virus Tian Tan strain (VTT) was historically used for the vaccination of millions of Chinese people during the worldwide smallpox prevention campaign, and such programs led to the eradication of Variola in China prior to 1980.3,5 Now, vaccinia viruses have emerged as attractive therapeutic candidates for cancer treatment due to their inherent ability of tumor tropism and oncolytic property, which appears to preferentially infect and lyse cancer cells without causing excessive damage to surrounding normal tissue.6
A series of vaccinia virus worldwide have been applied as both an oncolytic agent and vector for therapeutic gene delivery in clinical cancer treatment. JX-594, a Wyeth strain vaccinia-vaccine-derived oncolytic virus with disruption of the viral thymidine kinase (TK) gene for cancer selectivity and insertion of human granulocyte-macrophage colony-stimulating factor (hGM-CSF) and β-galactosidase transgenes for immune stimulation and replication assessment, is used to treat patients with hepatocellular carcinoma or metastatic melanoma or other solid tumors.7,9 JX-963, a Western Reserve strain of vaccinia (WR) with deletions in the viral TK and vaccinia growth factor (VGF) genes and expressing human GM-CSF, was also generated and tested in preclinical tumor models.10,11 GLV-1h153, an oncolytic Lister strain vaccinia virus attenuated by gene deletion and carrying a human sodium iodide symporter (hNIS), was used as a viable imaging method. In addition, combination of GLV-1h153 and radiation therapy (131I) was effective in treatment of prostate cancer.12,13 Otherwise, modified vaccinia virus Ankara (MVA), which was obtained by extensive serial passage of vaccinia virus Ankara strain, or NYVAC, which was derived from the Copenhagen strain of vaccinia virus, were both highly attenuated by genome deletions and used as candidates for cancer therapy.14,15
Recently, the attenuated vaccinia Guang 9 strain (VG9), which was derived from Chinese VTT by successive plaque-cloning purification, has become a promising replicative viral vector for cancer therapy.16,17 Deng et al2 constructed the VG9 strain expressing murine GM-CSF and found strong antitumor activity in a murine melanoma model. Besides the attenuated pathogenicity, tumor selectivity is another important index for safety evaluation of VG9. TK, an enzyme needed for nucleic acid metabolism, is high in proliferating cancer cells but not in normal cells. Deletion of the TK gene results in dependence of vaccinia virus on cellular TK expression, and leads to further selectivity of vaccinia virus in cancers.18,19 To evaluate the tumor selectivity of VG9 with TK deficiency, a non-invasive image was applied to monitor the localization of viral replication in tumor or healthy tissue.
In addition to direct tumor lysis, oncolytic viruses can induce host immune responses against tumor cells and may thus be viewed as a form of immunotherapy.1,20,23 Following oncolytic cell death, tumor cells release tumor-associated antigens that can serve to promote an adaptive immune response and the antitumor antibodies mediating complement-dependent cytotoxicity were induced after virus treatment with liver tumors.24,25 However, the natural ability of virus to induce a host antiviral immune response may result in clearance of the virus through neutralizing antiviral antibodies and/or cytotoxic T-cell-mediated immune responses.21 Although immune stimulation is critical to the antitumor activity of oncolytic viruses, this effect is balanced by the potentially rapid clearance of the virus by antiviral immunity. Therefore, the identification of immune-mediated viral clearance and induction of antitumor immunity that are activated by TK-deleted VG9 strain is important to direct its application in virotherapy.
As live viral particles, the TK-deleted VG9 strain must consider its characteristics of antitumor efficacy, tumor selectivity, and immune system activation. For this purpose, VG9 with TK deficiency and firefly luciferase (Luc) expression was used to monitor the replication and dissemination of the virus using an in vivo bioluminescence imaging method. Meanwhile, activation of systemic immune response after virus infection was monitored by detection of antitumor and antiviral neutralizing antibodies in plasma. In addition, intravenous and intratumoral routes, which are major delivery systems in clinical application of virotherapy, were both evaluated.
Materials and Methods
Cell Lines
The mammary carcinoma cell lines MDA-MB-231 (human) and 4T1 (murine), the murine melanoma cell line B16 and the colon adenocarcinoma cell line CT26.WT were obtained from the Cell Library of Biochemistry and Cell Biology, CAS, and cultured in RPMI 1640 medium (Life Technologies, catalog #23,400) supplemented with 10% FBS. The MC38 murine colon adenocarcinoma cancer cells were originally obtained from Cell Resource Center, Peking Union Medical College, NSTI. MC38 cells were cultured in minimum essential medium (Life Technologies, catalog #41,500) containing 10% FBS. Cells were maintained in a humidified atmosphere with 5% CO2 at 37°C.
Animals
BALB/c nu, BALB/c, and C57BL/6 mice were acquired from Cavens Laboratory Animal (Changzhou, China). All animals were maintained under specific pathogen-free conditions; all procedures were performed in accordance with the Laboratory Animal-Guideline of welfare ethical review of Chinese Institutional Animal Care and Use Committee (IACUC).
Vaccinia
VG9-Luc is an oncolytic vaccinia virus strain TianTan modified by insertion of Luc gene into the TK locus. For TK inactivation, we used the pCB-Luc shuttle plasmid containing the Luc gene under the control of the vaccinia synthetic early/late P-se/I promoter surrounded by the flanking sequences of the vaccinia J2R (TK) gene, which facilitated homologous recombination into the whole TK gene. VG9-Luc was generated and produced by methods described previously.26
In vitro Infection Ability of VG9-Luc on Tumor Cells
Tumor cells MDA-MB-231, B16, 4T1, and MC38 were plated in 24-well plates at the density of 1×105 cells/well, while CT26 cells were plated at 0.4×105 cells/well into 24-well plates. After overnight growth, the cells were infected with VG9-Luc at different MOIs (0, 0.02, 0.05, 0.1, 0.2, and 0.4). Twenty-four hours after infection, bioluminescence was quantified by adding 150 μg/mL D-luciferin (Promega) to culture medium 5 minutes before luminescent image with the IVIS system (PerkinElmer, CA, USA).
Tumor Models
To establish human mammary carcinoma tumor models, the immunocompromised nude mice were subcutaneously implanted with 2×106 MDA-MB-231 cells on the dorsal surface or left oxter or right hind leg. Similarly, the immunocompetent C57BL/6 mice or BALB/c mice were inoculated with B16, CT26, 4T1, or MC38 cells by subcutaneous implantation of 2×106 cells on the dorsal surface or left oxter or right hind leg of the mouse. Mice were 4–6 weeks old for all studies. The numbers of male and female mice in various experimental groups were matched, although we did not observe a difference between the sexes in susceptibility to vaccinia infection. After 10 days of tumor growth, the tumors reached a volume of approximately 100 mm3. Then, 1×107 PFU of purified VG9-Luc, which suspended in 100 μL of PBS, were injected intravenously via the tail vein or intratumorally into mice. Beginning the day after vaccinia virus inoculation, animals were imaged daily for bioluminescence detection.
Bioluminescence Imaging Analysis
Bioluminescence imaging was performed using IVIS Spectrum Imaging System (PerkinElmer). Before imaging, D-luciferin (potassium salt, Promega) in normal saline was intraperitoneally injected at a dose of 150 mg/kg body weight. Mice were placed in the light-tight chamber of the CCD camera system and 2% isoflurane anesthesia in oxygen was delivered via a nose cone system. Mice were imaged in the dorsal or ventral positions, and the acquisition time was set at 1 minute. The signal intensity was quantified as the flux of all detected photon counts within a region of interest (ROI) prescribed over the ventral or dorsal torso using the LivingImage software package.
Virus Titration from Infected Organs
Mice were sacrificed each day from day 1 to day 5 after intravenous injection of vaccinia VG9-Luc via the tail vein. Normal organs including heart, liver, spleen, lung, kidney, and brain, were removed and homogenized in 1 mL PBS, then stored at −80°C immediately. Whole blood was obtained by cardiac puncture, and diluted to one tenth with 1 mL PBS. Tissue homogenates and diluted blood were lysed by three freeze-thaw cycles, and centrifuged for 5 minutes at 3000 ×g at 4°C. The viral titers of supernatants were determined by plaque assays on BSC-40 cells. Meanwhile, the luciferase activity of the organ solutions was quantified with the Firefly Luciferase Reporter Gene Assay Kit (Beyotime).
Additionally, tumors were harvested and homogenized in 1 mL PBS on the last day of luminescence imaging. After three cycles of freezing and thawing procedure, the viral titer and luciferase activity in tumor supernatants were determined by plaque assay and luciferase assay kit, respectively.
Virus Re-Infection
2 weeks after infection of vaccinia VG9-Luc, the immunocompetent C57BL/6 or BALB/c mice were rechallenged with 1×107 PFU of purified VG9-Luc intratumorally or intravenously. Then, bioluminescence imaging was performed daily again to detect the biodistribution of the virus.
Blood Samples Collection and Immunological Factors Assay
Blood samples were taken from the tail vein at different time points. Approximately 200 μL blood from the tail vein of mice was collected into anti-coagulant K2-EDTA vacutainer tubes, and kept stable for 2 hours at 4°C. Plasma was separated by centrifugation at 1500 ×g for 15 minutes and stored at <−70°C until testing. The immunological factors including IL-4, IL-6, and IFNγ were determined by ELISA kits, respectively (Invitrogen 88–7044, 88–7064, 88–7314, CA, USA).
Neutralizing Antibodies Measurement
The titers of neutralizing antibodies against vaccinia virus or tumor cells in plasma were determined by time-resolved fluoroimmunoassay (TRIFA).27 Briefly, vaccinia VG9-Luc (15 μg/mL) were coated into 96-well plates overnight at 4°C. Then, the wells were blocked with 1% BSA, followed by diluted plasma samples incubated for 2 hours at 37°C. After washing 3-times, Eu3+-labeled anti-mouse IgG was incubated for 1 hour at 37°C. With the addition of enhancement solution, fluorescence emission spectra for Eu3+ were obtained using a Perkin–Elmer LS-55 fluorescence spectrometer, and time-resolved fluorescence measurements were carried out with an AutoDELFIA-1235 automatic analyzer (PerkinElmer, CA, USA).
For measurement of antitumor neutralizing antibody, CT26 cells were plated into 96-well plates at the density of 2×104 cells/well. After overnight growth, the cells were immobilized with methanol for 30 minutes at 4°C. Then, diluted plasma samples were incubated for 2 hours at 37°C, followed by Eu3+-labeled anti-mouse IgG incubation and Eu3+ fluorescence emission spectra detection.
Statistical Analysis
All graphs were generated using Prism 5.0 software (GraphPad, San Diego, CA). Student’s t-tests were used in all analysis and P-values less than 0.05 were considered statistically significant.
Results
Vaccinia VG9-Luc is Capable of Infecting Tumor Cells in vitro
Before bioluminescence imaging is performed on tumor-bearing mouse models, it is necessary to determine whether vaccinia VG9-Luc could infect tumor cells as expected. Thus, tumor cells MDA-MB-231, B16, CT26, 4T1, and MC38, all of which were used to construct tumor models, were infected with increasing titers of vaccinia VG9-Luc and the luciferase activity was detected 24 hours after infection. As shown in Figure 1A–J, all the cells could be infected by vaccinia VG9-Luc, and the luminescence intensity increased with the enhanced infection titers. The positive correlation between luciferase activity and infection titer fully indicated that vaccinia VG9-Luc could effectively infect tumor cells in vitro.
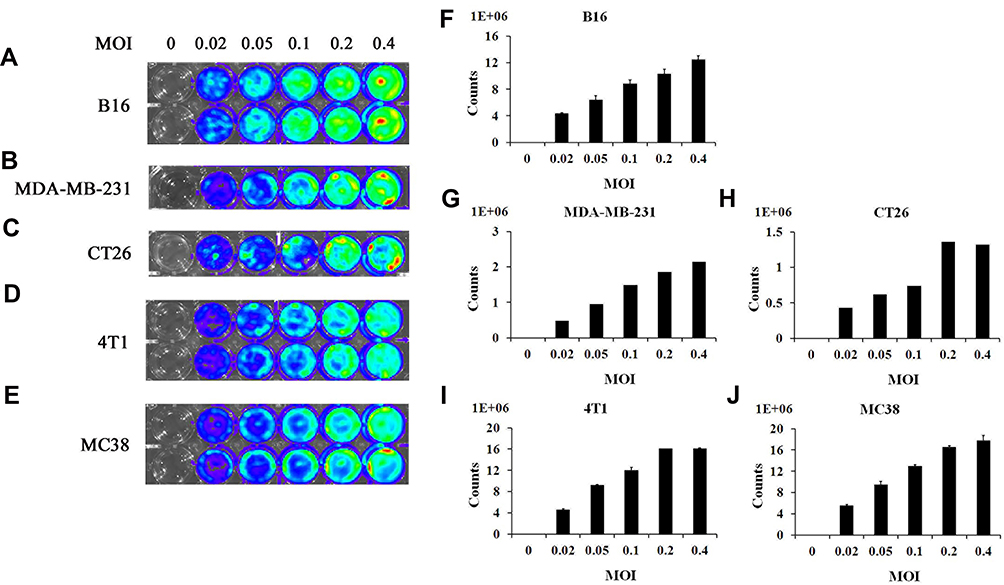 |
Figure 1 In vitro infection ability of vaccinia VG9-Luc on tumor cells. (A–E), Bioluminescence image of B16 (A), MDA-MB-231 (B), CT26 (C), 4T1 (D), and MC38 (E) cells infected by vaccinia VG9-Luc in vitro for 24 hours. (F–J) Bar graphs represent the total photon counts of bioluminescence accumulated in each well (means±SD), corresponding to A and E, respectively. Tumor cells were plated into 24-well plates and infected with increasing titers of VG9-Luc (0, 0.02, 0.05, 0.1, 0.2, or 0.4 MOI/well) for 24 hours. Bioluminescence image was applied by addition of 150 μg/mL D-luciferin and 5 minutes of incubation. |
Vaccinia VG9-Luc is Capable of Targeting Tumor Sites in Nude Mice
To evaluate tumor specificity of vaccinia VG9-Luc, immunocompromised nude mice challenged with human mammary carcinoma MDA-MB-231 cells were firstly investigated. The day after intravenous inoculation of VG9-Luc via the tail vein, bioluminescence imaging was performed each day until day 7. As shown in Figure 2A and B, the day after intravenous infection of VG9-Luc, the luciferase activity on the ventral and dorsal surface was high, both in non- and tumor-bearing nude mice. The bioluminescence of non-specific background gradually decreased over the course of infection and luciferase activity was clearly observed in tumor site from day 3 to day 7, including tumors located on the dorsal surface or oxter. However, the non-specific luminescence intensity which occurred in both hind legs of nude mice failed to prove the specificity of VG9-Luc for tumors located on hind legs, though it was confirmed by viral titer assay. These data indicated that vaccinia VG9-Luc had strong ability of tumor targeting after intravenous infection of nude mice, no matter where the tumor was located.
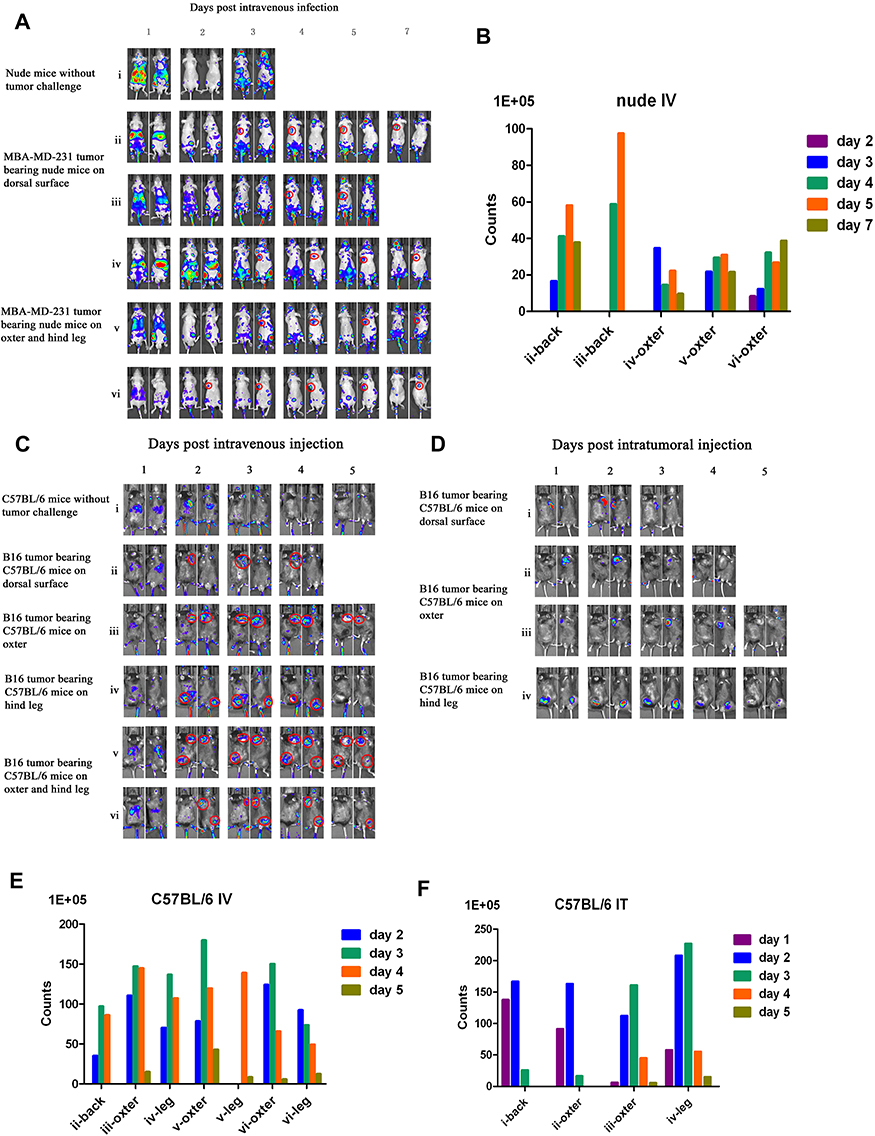 |
Figure 2 Characterization of luciferase activity in tumor-bearing mice infected with VG9-Luc intravenously or intratumorally. (A) Nude mice were either challenged with MDA-MB-231 or without tumor challenge. Ten days after tumor cells inoculation, mice were intravenously injected with 1×107 PFU of VG9-Luc. Luciferase activity was characterized by bioluminescence imaging on days 1, 2, 3, 4, 5, and 7 after vaccinia infection. (B) Corresponding to A, the bar graph represents the total photon counts of bioluminescence accumulated in tumor sites. (C and D) C57BL/6 mice were either challenged with B16 cells or without tumor challenge. Ten days after tumor inoculation, mice were intravenously (C) or intratumorally (D) injected with 1×107 PFU of VG9-Luc. Luciferase activity was characterized by bioluminescence imaging on days 1, 2, 3, 4, and 5 after vaccinia infection. (E and F) Corresponding to (C and D), respectively, bar graphs represent the total photon counts of bioluminescence accumulated in tumor sites. |
Biodistribution of vaccinia VG9-Luc in nude mice were performed by plaque assays on normal organs, including heart, liver, spleen, lung, kidney, brain, and blood. The viral titers showed there was little or no plaques formed in excised organs (Table 1), and luciferase activity was not detected in tissue homogenates (data not shown). In tumor tissues that were harvested on the last day of luminescence imaging, both the luciferase activity and viral titers were significantly high (Table 2), suggesting effective tumor targeting ability of VG9-Luc after intravenous injection. In addition, there was a positive relationship between luciferase activity, viral titers, and luminescence intensity, which demonstrated the tumor specificity of vaccinia VG9-Luc from three aspects.
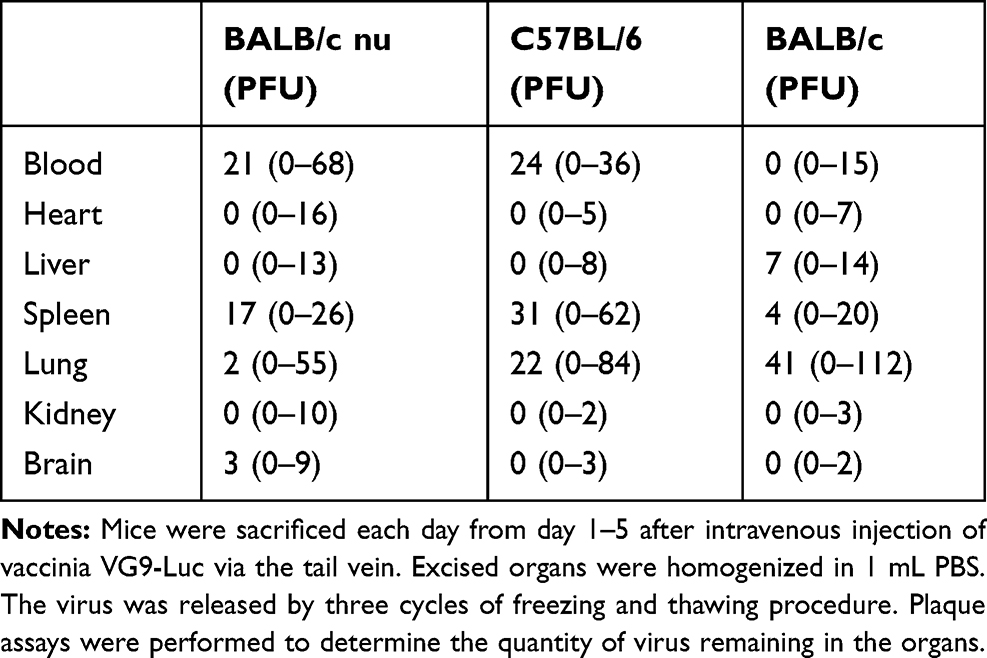 |
Table 1 Biodistribution of Vaccinia VG9-Luc in Normal Tissues |
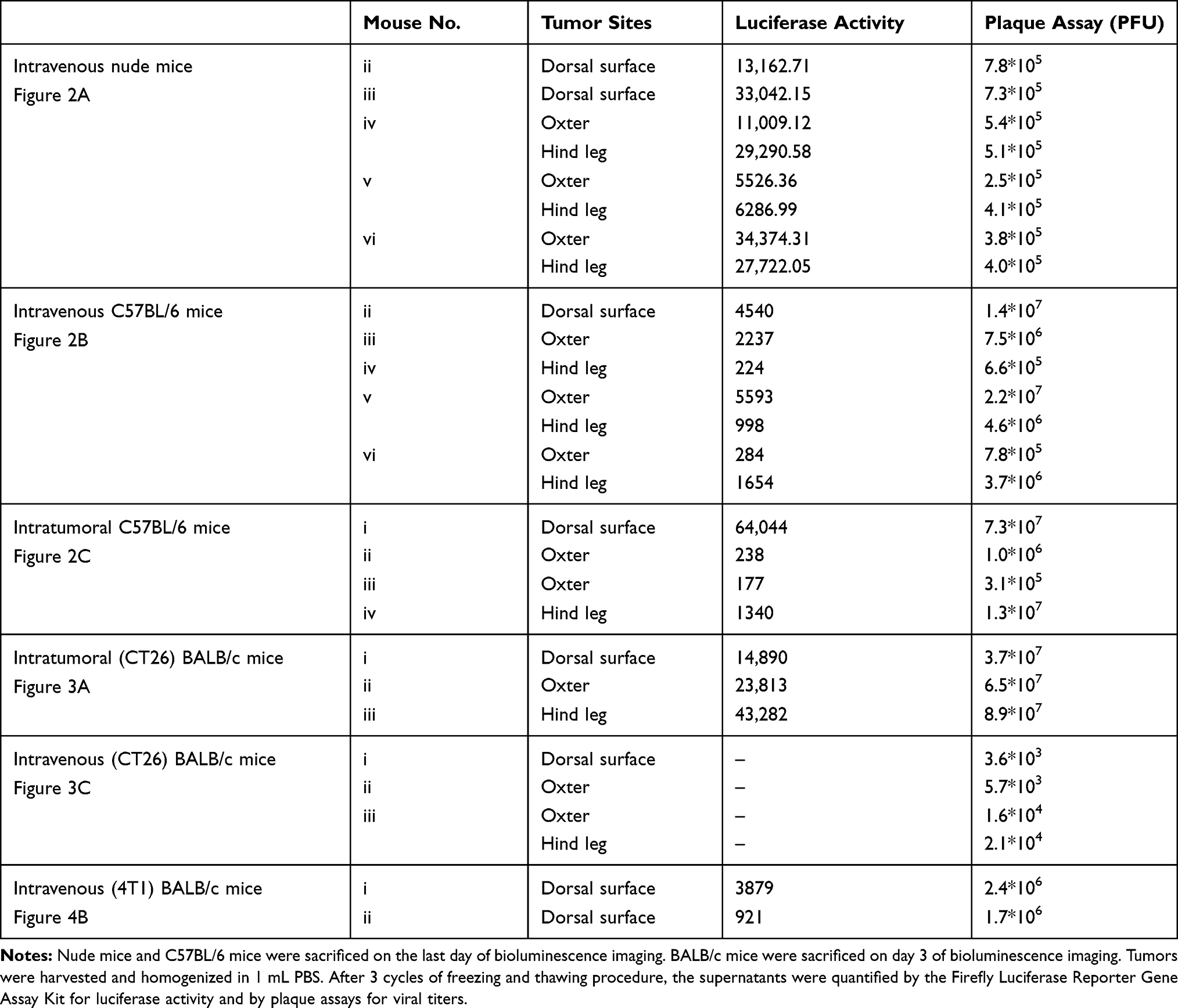 |
Table 2 Luciferase Activity and Plaque Assays of Vaccinia VG9-Luc Located in Tumor Sites of Mice |
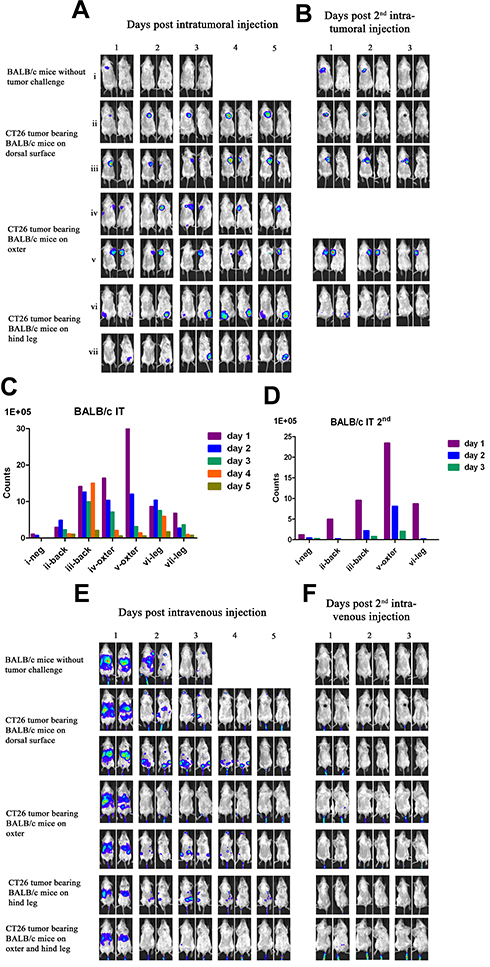 |
Figure 3 Characterization of luciferase activity in CT26 tumor-bearing BALB/c mice infected with VG9-Luc intratumorally or intravenously. (A) BALB/c mice were either challenged with CT26 cells or without tumor challenge. Six days after tumor cells inoculation, BALB/c mice were intratumorally injected with 1×107 PFU of VG9-Luc. Luciferase activity was characterized by bioluminescence imaging on days 1, 2, 3, 4, and 5 after vaccinia infection. (B) Two weeks after first virus injection, intratumoral re-injection of VG9-Luc was performed and luciferase activity was characterized by bioluminescence imaging on days 1, 2, and 3 after vaccinia re-infection. (C and D) Corresponding to (A and B), respectively, bar graphs represent the total photon counts of bioluminescence accumulated in tumor sites. (E) Six days after CT26 tumor cells inoculation, BALB/c mice were intravenously injected with 1×107 PFU of VG9-Luc. Luciferase activity was characterized by bioluminescence imaging on days 1, 2, 3, 4, and 5 after vaccinia infection. (F) Two weeks after first virus injection, intravenous re-injection of VG9-Luc was performed and luciferase activity was characterized by bioluminescence imaging on days 1, 2, and 3 after vaccinia re-infection. |
 |
Figure 4 Repeated characterization of luciferase activity in tumor-bearing C57BL/6 and BALB/c mice. (A) C57BL/6 mice challenged with MC38 or B16 cells were intravenously injected with 1×107 PFU of VG9-Luc. (B) BALB/c mice challenged with 4T1 or CT26 cells were intravenously injected with 1×107 PFU of VG9-Luc. Luciferase activity was characterized by bioluminescence imaging on days 1, 2, 3, 4, and 5 after vaccinia infection. (C and D) Two weeks after first virus injection, intravenous (C) or intratumoral (D) re-injection of VG9-Luc in C57BL/6 mice was performed. Luciferase activity was characterized by bioluminescence imaging on days 1, 2, and 3 after vaccinia re-infection. |
Vaccinia VG9-Luc is Capable of Targeting Tumor Sites in C57BL/6 Mice
Immunocompetent C57BL/6 mice challenged with B16 murine melanoma cells were investigated to further demonstrate the tumor specificity of vaccinia VG9-Luc. Mice were infected with 1*107 PFU of VG9-Luc by intratumoral or intravenous inoculation 10 days after tumor cells implantation. From the next day, bioluminescence imaging was performed daily and continued for 5 days. As shown in Figure 2C and E, the day after intravenous infection, C57BL/6 mice without tumor challenge showed high luminescence intensity in the dorsal and ventral surfaces. The luminescence dramatically decreased on day 2, and little luciferase expression was observed in the dorsal and ventral surfaces from day 3. C57BL/6 mice challenged with B16 cells on the dorsal surface, oxter, or hind leg, showed significant tumor specificity of VG9-Luc. High luciferase activity was observed in all tumor sites, no matter where it is located. The luminescence intensity was extremely high from day 2, and gradually decreased with the days passed. The data indicated that vaccinia VG9-Luc had the ability to target tumor cells in immunocompetent C57BL/6 mice.
Meanwhile, tumor specificity of VG9-Luc infection by intratumoral inoculation was also investigated. Figure 2D and F showed significant high luciferase activity accumulated in tumor sites, no matter if tumor cells were located on the dorsal surface, oxter, or hind leg. The day after virus infection, the bioluminescence in tumor sites was obviously observed and reached a peak on day 2, then the luminescence intensity gradually decreased with the passage of time. In addition, we have previously investigated the tumor specificity of VG9-Luc infected by intratumoral injection in nude mice, which also confirmed its ability of tumor targeting.26
Plaques assays and luciferase activity were also determined in normal organs of C57BL/6 mice, which were harvested 1–5 days post intravenous injection. Similar to nude mice, there was little or no plaques formed in normal organs (Table 1), and no luciferase activity was detected in tissue homogenates (data not shown). Tumor tissues were harvested on the last day of luminescence imaging, the luciferase activity and plaque assays were performed as well. Table 2 showed the luciferase activity and viral titers in tumor homogenates, which corresponded with the results of bioluminescence imaging. The results confirmed the tumor specificity of VG9-Luc on the other hand, no matter if the virus was infected intravenously or intratumorally.
Tumor Targeting Ability of Vaccinia VG9-Luc in BALB/c Mice
Another immunocompetent mouse strain of BALB/c was further investigated to strengthen the conclusion that vaccinia VG9-Luc had the property of tumor specificity. As before, BALB/c mice challenged with CT26 colon adenocarcinoma cells were infected with 1*107 PFU of VG9-Luc by intratumoral or intravenous inoculation 6 days after tumor cells implantation. Bioluminescence imaging showed that strong bioluminescence was accumulated in tumor sites of BALB/c mice after intratumoral injection of VG9-Luc (Figure 3A and C). The luminescence intensity was higher on day 1 and 2, and progressively decreased with time passed. Otherwise, only weak luminescence was observed in the site of subcutaneous injection in BALB/c mice without tumor challenge, and the luminescence quickly disappeared 2 days after virus infection. These results illustrated that vaccinia virus could not effectively replicate in healthy tissue, however, the replication of VG9-Luc in tumor sites was high efficiency and strong luciferase activity was accumulated in tumor tissues, which demonstrated the tumor selectivity of vaccinia VG9-Luc. In addition, the luciferase activity and viral titers in tumor homogenates were consistent with in vivo bioluminescence imaging (Table 2), providing further evidence that vaccinia VG9-Luc was tumor specific.
However, when tumor-bearing BABL/c mice were injected with vaccinia VG9-Luc intravenously, the situation was quite different. As shown in Figure 3E, no luciferase activity was observed in tumor sites, no matter where the tumor was located. To confirm the conclusion, excised tumor tissues harvested 3 days after intravenous injection were used to detect their luciferase activity and viral titers. The results showed that no luciferase activity was detected in any tumor homogenates, but three in 10 mice were detected as low degree of viral titers (Table 2). Additionally, there were little or no plaques formed in normal organs (Table 1), corresponding to nude mice and C57BL/6 mice.
It was greatly disappointing that vaccinia VG9-Luc showed no tumor specificity in BALB/c mice after intravenous infection. However, we believe that different mouse strains and tumor models may cause the phenomenon. Therefore, B16 and MC38 cell lines generated from C57BL/6 mice, as well as CT26 and 4T1 cell lines generated from BALB/c mice, were used to build immunocompetent models in their matched hosts. Bioluminescence imaging was repeated after intravenous infection of VG9-Luc in tumor models. As shown in Figure 4A, obvious tumor targeting ability was shown in C57BL/6 mice, no matter whether they were challenged with MC38 or B16 cells, and whether the tumor was located on the dorsal surface or oxter. The progression of virus infection was consistent with previous results, except for the lower luminescence intensity acquired this time. Once again, no tumor specificity was observed in BALB/c mice, neither in 4T1 tumor models nor in CT26 tumor models (Figure 4B). However, there is an exception in 4T1 tumor-bearing BALB/c mice challenged on the dorsal surface, which was shown in the first line of Figure 4B. Though not significant, there was weak luminescence discovered in the tumor site after 2 or 4 days of intravenous infection. Yet, we cannot conclude whether vaccinia VG9-Luc was tumor specific in 4T1 tumor models. Therefore, bioluminescence imaging was performed again after intravenous infection of VG9-Luc on 4T1 tumor models, and the results are shown in the last two lines of Figure 4B. It was observed that the bioluminescence was detected close to the tumor, but definitely no photon flux was acquired in the exact tumor site. In order to further confirm the conclusion, tumor tissues were harvested and homogenized after 3 days of intravenous injection. Results show there was weak luciferase activity existent in 4T1 tumors and approximately 106 PFU of viral titers were detected in 4T1 tumor homogenates (Table 2). Although vaccinia VG9-Luc failed to target CT26 or 4T1 tumor tissue in the BALB/c mouse model, we suppose there may be some reason that prevents the vaccinia invasion into the tumor tissues.
Re-Infection of Vaccinia VG9-Luc in Immunocompetent Mice
To study the effect of immunity activated by vaccinia virus on virotherapy, two immunocompetent tumor-bearing BALB/c and C57BL/6 models were reinjected with vaccinia VG9-Luc 2 weeks after the first infection, and bioluminescence imaging was performed again to observe the replication of the virus. As shown in Figure 3F and Figure 4C, intravenous reinjection of vaccinia VG9-Luc failed to cause the replication of the virus in mice. No luminescence was detected in both BALB/c and C57BL/6 tumor-bearing mice the day after reinjection (day 1), neither in the next 2 days (day 2 and 3), except for the non-specific luminescence accumulated in the tails. However, if vaccinia VG9-Luc was infected intratumorally, significant virus replication was observed in tumor sites, though the luminescence intensity was much lower than the first infection, as shown in Figure 3B, D and Figure 4D. It is supposed that first systemic virus infection induced strong antiviral immune response, which rapidly cleared the second systemic injected virus. The different situation of intratumoral injection may be due to the difference in local immune response and the larger amount of virus injected into tumor sites.
Vaccinia VG9-Luc Induced Tumor-Specific Immunity
To evaluate the immune response after vaccinia VG9-Luc infection, the titers of plasma antibodies against virus or tumor cells were determined by TRIFA. As mentioned before, CT26 tumor cells were implanted in BALB/c mice 6 days before virus infection, so the antibodies against tumor cells were produced earlier and higher than antiviral antibodies. Figure 5A and B showed the production of neutralizing antibodies and immunological factors within 7 days after intravenous virus infection. As shown in Figure 5A, no antitumor antibodies were detected 6 days after tumor cells implantation (day 0). After virus infection, the antitumor antibodies were gradually produced, and a sharp rise in antibodies was observed from day 3 to day 5, and then remained stable on day 6 and day 7. Meanwhile, the antiviral antibodies were produced slowly and uniformly from day 2 to day 7 after virus infection. The immunological factors including IL-4, IL-6, and IFNγ were also determined to evaluate the immune response in BALB/c mice after tumor cells implantation and virus infection. Although IL-4 and IL-6 were not detected in plasma (data not shown), IFNγ showed the process of the immunity activated by vaccinia VG9-Luc. Before tumor cells implantation, IFNγ was not detected in plasma. Then, a low level of IFNγ was produced 6 days after tumor cells inoculation (day 0). Later, the production of IFNγ sharply increased from day 1 to day 4 and dropped quickly from day 4 to day 6 after virus infection. By day 7, the amount of IFNγ remained low and cannot be detected after that (Figure 5B).
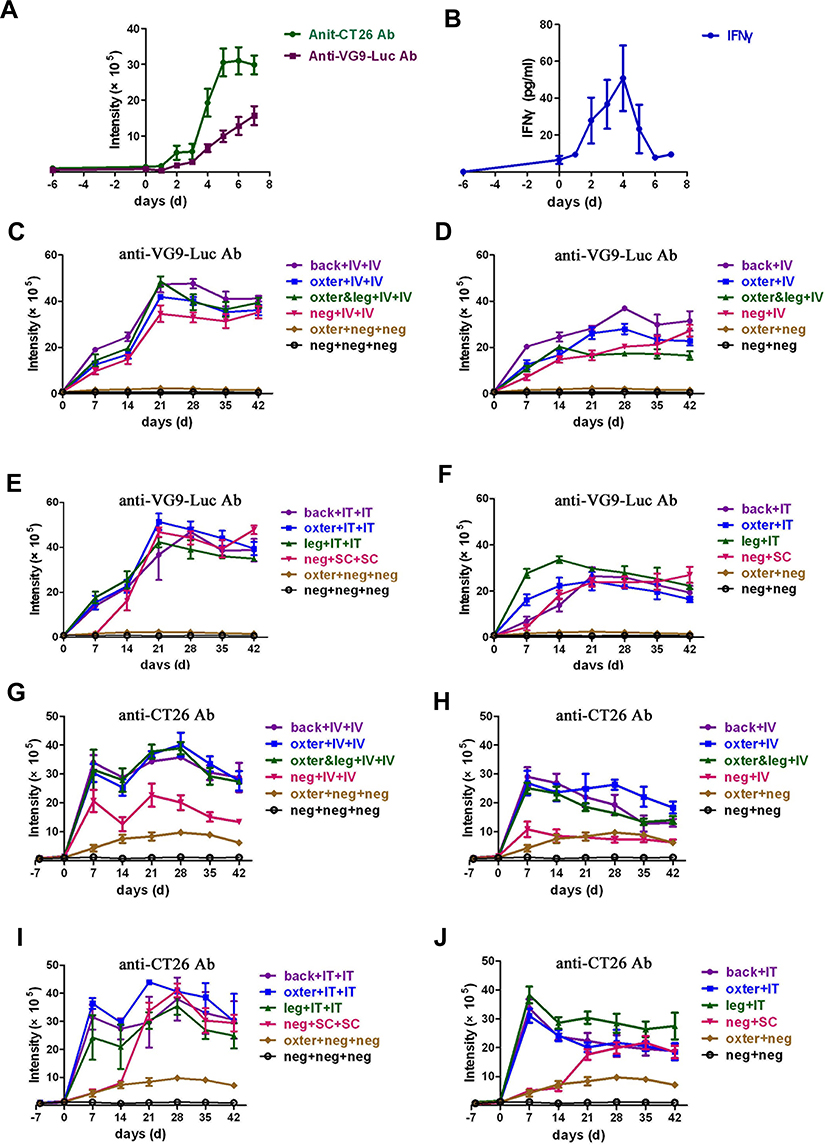 |
Figure 5 Neutralizing antiviral and antitumor antibodies produced in tumor-bearing BALB/c mice activated by virus infection. (A) Neutralizing antiviral and antitumor antibodies produced within 7 days after intravenous virus infection. Six days after CT26 tumor cells inoculation, BALB/c mice were intravenously injected with 1×107 PFU of VG9-Luc. Blood samples were taken from tail vein each day until 7 days after virus infection. The neutralizing antiviral and antitumor antibodies in plasma were determined by TRIFA. (B) Immunological factor IFNγ detected by an ELISA Kit. (C–F) Neutralizing antiviral antibodies determined by TRIFA. CT26 tumor-bearing BALB/c mice were intravenously (D) or intratumorally (F) injected with 1×107 PFU of VG9-Luc. Two weeks after first virus injection, intravenous (C) or intratumoral (E) re-injection of VG9-Luc was performed. Blood samples were taken from tail vein each week after virus infection. (G–J) Neutralizing antitumor antibodies determined by TRIFA. CT26 tumor-bearing BALB/c mice were intravenously (H) or intratumorally (J) injected with 1×107 PFU of VG9-Luc. Two weeks after first virus injection, intravenous (G) or intratumoral (I) re-injection of VG9-Luc was performed. Back or oxter or leg represents the site where the tumor was located; IV or IT or SC represent the mode of virus delivery, as intravenous or intratumoral or subcutaneous, respectively; neg represents the absence of the corresponding operation. Data are expressed as means±SEM of three independent mice. |
Long-term neutralizing antibodies were also detected to evaluate the systemic immune response process activated by the first and second viral infections. As shown in Figure 5C–F, the antiviral antibodies were quickly produced and rapidly increased within 14 days after viral infection in tumor-bearing mice. Then, the production of antiviral antibodies kept a slight rise in vein-infected mice (Figure 5D) or a slight drop in tumor-infected mice (Figure 5F) in the following month. However, when the virus re-infected tumor-bearing mice 14 days after the first viral infection, antiviral antibodies sharply increased on day 21 and decreased slightly over the next 3 weeks, no matter if the tumor was located on the dorsal surface, oxter, or hind leg (Figure 5C and E). In addition, mice without tumor challenge showed the same performance as tumor-bearing mice after the first and second viral infection. Meanwhile, tumor-bearing and normal mice without viral infection did not produce any antiviral antibodies, as expected. Taken together, these data indicated that viral infection induced a significant antiviral immune response in mice, no matter where the tumor is located or whether there is a tumor. Moreover, re-infection of the virus induced a stronger secondary immune response, both in intravenous or intratumor routes.
The situation of antibodies against CT26 tumor cells was a bit similar. As shown in Figure 5G–J, no antitumor antibodies were produced 6 days after tumor cells implantation. However, when vaccinia VG9-Luc was injected, the antitumor antibodies were sharply produced and reached a peak on day 7. Then, a marked decline was observed on day 14 in all groups (Figure 5G–J). In the following month, the production of antitumor antibodies kept stable or showed a slight drop without any operation (Figure 5H and J). However, when the virus was re-infected on day 14, the antitumor antibodies increased again and reached a peak on day 28. In the following 2 weeks, the production of antibodies gradually decreased (Figure 5G and I), but the baseline of the antibodies was higher than that in mice infected only once. Notably, tumor-bearing mice without viral infection produced extremely lower antitumor antibodies and kept stable in the whole process. While viral infection enhanced the production of antitumor antibodies by 5-times, it is suggested that vaccinia VG9-Luc strongly induced tumor-specific immunity by increasing the production of antitumor antibodies.
It is noticeable that there was no significant difference in antibody production between intravenous or intratumoral delivery systems, except for normal mice infected with virus. As shown in Figure 5G and H, normal mice produced lower antibodies than tumor-bearing mice, who received intravenous viral infection once or twice, though their production processes of antitumor antibodies were the same. Nevertheless, if virus was intratumorally infected in normal mice, the production of antitumor antibodies was extremely low within the first 2 weeks and then showed a sharp rise on day 21, whether or not the virus was re-infected. Then, the antibody production was the same level as tumor-bearing mice in the following 3 weeks. In theory, normal mice could not produce antibodies against CT26 tumor cells, and normal mice without any operation showed no antitumor (Figure 5G–J) or antiviral (Figure 5C–F) antibodies produced. So, why did normal mice infected with viruses produce significantly high antitumor antibodies? As we know, immune stimulation is an essential part of virotherapy, represented by the production of neutralizing antibodies. However, the antibodies could recognize antigens expressed by viruses, which limited the extent of oncolysis; antigens expressed by tumor cells, which increased the antitumor efficacy; as well as antigens expressed by normal cells, which induced autoimmunity and weakened the body. The production of antitumor antibodies in normal mice injected with virus is considered to be caused by the activation of an autoimmune response.
Discussion
Engineered oncolytic viruses have been emerging as promising options for clinical cancer treatment since the 1990s.28,29 More than 1000 patients have now been treated with oncolytic viruses by intratumoral injection and/or intravenous infusion during Phase I–III clinical trials. Vaccinia Guang9 (VG9) strain, which originates from Chinese vaccinia Tian Tan strain (VTT), has attenuated pathogenicity and is becoming a promising engineered viral vector for expression of therapeutic anticancer genes. Usually, Thymidine kinase (TK) gene was deleted to promote tumor tropism at the same time of exogenous therapeutic gene insertion. Therefore, understanding the characteristic of the TK-deleted VG9 strain is critical to virus construction and application. In this study, we evaluate the tumor specificity and immune system activation of VG9-Luc in different tumor-bearing mice models. As intratumoral injection and intravenous infusion are the most popular delivery systems in clinical virotherapy, both routes of administration were conducted and their respective performance was compared.
For tumor targeting evaluation, an in vivo bioluminescence imaging was performed to observe the virus distribution in live mice each day. Luminescence intensity in live mice as well as luciferase activity and viral titers in excised organs showed the dramatic tumor targeting ability of VG9-luc in immunocompromised nude mice and immunocompetent C57BL/6 and BALB/c mice, and showed tumor specificity in both intravenous and intratumoral injection. The only exception occurred in immunocompetent BALB/c mice with intravenous virus infection. Repeated trials showed no tumor specificity of VG9-luc was observed in both CT26 and 4T1 tumor-bearing BALB/c mice infected intravenously. However, weak bioluminescence was detected close to 4T1 tumor tissue, and low luciferase activity was detected in excised tumor homogenates. Thereby, we suppose the stiffness of the tumor matrix may contribute to the phenomenon.
Tumor stiffness is mainly determined by the amount of extracellular matrix (ECM), particularly collagen and hyaluronan contained in the tumor. Increasing ECM stiffness is shown to induce a malignant phenotype and promote cancer cell invasion to surrounding tissues.30,32 Here, we found tumor stiffness also affected the tumor targeting ability of vaccinia VG9-Luc. The softer the tumor matrix, the easier vaccinia virus invaded and duplicated into the tumor. For example, B16 and MC38 tumor cells, which were soft in the tumor matrix, showed obvious tumor targeting ability of VG9-Luc in C57BL/6 mice. From previous experiments, we know that both 4T1 and CT26 tumors had a stiff matrix, and their tissues were hard to homogenize. Furthermore, the matrix of CT26 was stiffer than that of 4T1. Thus, we assumed both CT26 and 4T1 tumor models were difficult for virus invasion and replication. Sometimes the virus accumulated near the tumor site, such as 4T1; but sometimes the virus could not replicate in the tumor nearby, and no luminescence was detected in and around the tumor site, such as CT26. Thus, we conclude that the stiffness of the tumor matrix is responsible for the non-tumor specificity observed in BALB/c mice, which can prevent the vaccinia from invading into the tumor tissues.
It is reported some patients undergoing oncolytic virotherapy seem to spontaneously undergo a dramatic and therapeutic transition from induced antiviral response against oncolytic vector and then progress onto more effective acquired antitumor immunity.20 Besides directly killing tumor cells by oncolytic effect, immunotherapy has a different mechanism for eliminating tumors, that eradicates tumor cells by enhancing the antitumor immune response.33 Therefore, the balance of antiviral immunity and antitumor immunity activated by virotherapy is critical to the efficacy of oncolytic virus. Determination of neutralizing antibodies proved that viral infection really promoted tumor-specific immunity by increasing the antibodies against tumor cells. Results also showed the production process of antiviral antibodies was similar between the intravenous and intratumoral delivery systems, however, the bioluminescence images of the two were quite different. As mentioned before, re-infection of virus intravenously induced a stronger secondary immunization, which caused a rapid clearance of the second infected virus, and no luminescence was detected in mice. Nevertheless, re-infection of virus intratumorally induced a similar secondary immune response, but there was significant bioluminescence detected in tumor location, though the luminescence intensity was much lower than the first infection. These differences may be contributed by the difference in viral clearance between the two routes of administration. It is reported that intravenous delivery of virus caused an immediate innate humoral immune response, which may lead to viral inactivation in the bloodstream, prior to infection of tumor cells. Even without preexisting immunity, repeated intravenous viral administration results in production of antiviral antibodies that quickly render vascular delivery ineffective.34 Early viral inactivation by the innate immune system is also an issue with direct intratumoral injection, but to a lesser extent than intravenous therapy. With the immune system able to provide a systemic response after local administration, intratumoral injection also induces distant tumor responses while ensuring local tumor delivery.35,38
Besides the negative results of bioluminescence imaging, repeated intravenous delivery of virus was not recommended for its high autoimmune response. At first, repeated intravenous administration of virus was performed 7 days after first injection, however, most C57BL/6 mice died on the second day. Then, virus re-injection was performed after a 14-day interval. Although all mice were very weak, only one or two mice died among C57BL/6 mice. In brief, either intravenous or intratumoral viral administration could be chosen according to the condition of tumors. Direct intratumoral infection, the preferred method of administration for most viral platforms, is suitable for patients with easily accessible solid tumors. Otherwise, metastatic disease or hard-to-reach location tumors should be treated with intravenous injection, which has the potential for rapid viral infection at all locations of tumors.39,41 Furthermore, if it is necessary to perform a secondary virotherapy, intratumoral injection of more viruses is the only choice.
Conclusion
In this study, we constructed a vaccinia VG9-Luc strain, which expressed firefly luciferase for bioluminescence imaging and disrupted the TK gene for promoting tumor specificity. Except BALB/c mice treated with intravenous virus injection, all immunocompromised and immunocompetent mice with intravenous or intratumoral virus injection showed obvious tumor targeting ability of vaccinia VG9-Luc. For evaluation of host immune response activated by vaccinia VG9-Luc, we found no significant difference in production of neutralizing antiviral and antitumor antibodies between mice injected intravenously or intratumorally. Moreover, secondary immunization was studied and intravenous re-injection was not recommended because of a more rapid viral clearance and safety evaluation.
Abbreviations
VTT, vaccinia virus Tian Tan strain; TK, thymidine kinase; GM-CSF, granulocyte-macrophage colony-stimulating factor; WR, Western Reserve strain of vaccinia; VGF, vaccinia growth factor; hNIS, human sodium iodide symporter; MVA, modified vaccinia virus Ankara; VG9, vaccinia Guang 9 strain; Luc, firefly luciferase; FBS, fetal bovine serum; PBS, phosphate-buffered saline; ROI, region of interest; IL-4, interleukin 4; IL-6, interleukin 6; IFNγ, interferon γ; TRIFA, time-resolved fluoroimmunoassay; BSA, bovine serum albumin; ECM, extracellular matrix.
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding authors on reasonable request.
Ethics Approval
The animal experiment was approved by the Institutional Animal Care and Use Committees (IACUC) of Jiangsu Institute of Nuclear Medicine (JSINM2010007). All animal procedures were performed in accordance with the Laboratory Animal-Guideline of welfare ethical review of Chinese Institutional Animal Care and Use Committee (IACUC).
Acknowledgments
The authors sincerely thank Professor XinYuan Liu (Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, The Graduate School, Chinese Academy of Sciences) for providing the vaccinia shuttle plasmid (pCB). They thank the National Institutes for Food and Drug Control (NIFDC) for providing the vaccinia virus Tian Tan strain VG9.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Chen CY, Hutzen B, Wedekind MF, Cripe TP. Oncolytic virus and PD-1/PD-L1 blockade combination therapy. Oncolytic Virother. 2018;7:65–77. doi:10.2147/OV.S145532
2. Deng L, Fan J, Guo M, Huang B. Oncolytic and immunologic cancer therapy with GM-CSF-armed vaccinia virus of Tian Tan strain Guang9. Cancer Lett. 2016;372:251–257. doi:10.1016/j.canlet.2016.01.025
3. Fenner FH, Arita I, Jezedk Z, Ladnyi ID. Smallpox and Its Eradication. Geneva: World Health Organization; 1988:1464pp.
4. Liu Q, Huang W, Nie J, et al. A novel high-throughput vaccinia virus neutralization assay and preexisting immunity in populations from different geographic regions in China. PLoS One. 2012;7:e33392. doi:10.1371/journal.pone.0033392
5. Fang Q, Yang L, Zhu W, et al. Host range, growth property, and virulence of the smallpox vaccine: vaccinia virus Tian Tan strain. Virology. 2005;335:242–251. doi:10.1016/j.virol.2005.02.014
6. Guo ZS, Lu B, Guo Z, et al. Vaccinia virus-mediated cancer immunotherapy: cancer vaccines and oncolytics. J Immunother Cancer. 2019;7. doi:10.1186/s40425-018-0495-7
7. Breitbach CJ, Burke J, Jonker D, et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature. 2011;477:99–102. doi:10.1038/nature10358
8. Breitbach CJ, Moon A, Burke J, Hwang TH, Kirn DH. A phase 2, open-label, randomized study of pexa-vec (JX-594) administered by intratumoral injection in patients with unresectable primary hepatocellular carcinoma. Methods Mol Biol. 2015;1317:343–357.
9. Hwang TH, Moon AF, Burke J, et al. A mechanistic proof-of-concept clinical trial with JX-594, a targeted multi-mechanistic oncolytic poxvirus, in patients with metastatic melanoma. Mol Ther. 2011;19:1913–1922. doi:10.1038/mt.2011.132
10. Thorne SH, Hwang TH, O’Gorman WE, et al. Rational strain selection and engineering creates a broad-spectrum, systemically effective oncolytic poxvirus, JX-963. J Clin Invest. 2007;117:3350–3358. doi:10.1172/JCI32727
11. Lee JH, Roh MS, Lee YK, et al. Oncolytic and immunostimulatory efficacy of a targeted oncolytic poxvirus expressing human GM-CSF following intravenous administration in a rabbit tumor model. Cancer Gene Ther. 2010;17:73–79. doi:10.1038/cgt.2009.50
12. Mansfield DC, Kyula JN, Rosenfelder N, et al. Oncolytic vaccinia virus as a vector for therapeutic sodium iodide symporter gene therapy in prostate cancer. Gene Ther. 2016;23:357–368. doi:10.1038/gt.2016.5
13. Haddad D, Socci N, Chen CH, et al. Molecular network, pathway, and functional analysis of time-dependent gene changes associated with pancreatic cancer susceptibility to oncolytic vaccinia virotherapy. Mol Ther Oncolytics. 2016;3:16008. doi:10.1038/mto.2016.8
14. Gomez CE, Najera JL, Domingo-Gil E, Ochoa-Callejero L, Gonzalez-Aseguinolaza G, Esteban M. Virus distribution of the attenuated MVA and NYVAC poxvirus strains in mice. J Gen Virol. 2007;88:2473–2478. doi:10.1099/vir.0.83018-0
15. Lulf AT, Freudenstein A, Marr L, Sutter G, Volz A. Non-plaque-forming virions of modified vaccinia virus ankara express viral genes. Virology. 2016;499:322–330. doi:10.1016/j.virol.2016.09.006
16. Zhu R, Liu Q, Huang W, Yu Y, Wang Y. Comparison of the replication characteristics of vaccinia virus strains Guang 9 and Tian Tan in vivo and in vitro. Arch Virol. 2014;159:2587–2596. doi:10.1007/s00705-014-2079-2
17. Zhu R, Huang W, Wang W, et al. Comparison on virulence and immunogenicity of two recombinant vaccinia vaccines, Tian Tan and Guang9 strains, expressing the HIV-1 envelope gene. PLoS One. 2012;7:e48343. doi:10.1371/journal.pone.0048343
18. Wong HH, Lemoine NR, Wang Y. Oncolytic viruses for cancer therapy: overcoming the obstacles. Viruses. 2010;2:78–106. doi:10.3390/v2010078
19. McCart JA, Ward JM, Lee J, et al. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res. 2001;61:8751–8757.
20. Bell J, McFadden G. Viruses for tumor therapy. Cell Host Microbe. 2014;15:260–265. doi:10.1016/j.chom.2014.01.002
21. Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2015;14:642–662. doi:10.1038/nrd4663
22. Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Going viral with cancer immunotherapy. Nat Rev Cancer. 2014;14:559–567. doi:10.1038/nrc3770
23. Dai P, Wang W, Yang N, et al. Intratumoral delivery of inactivated modified vaccinia virus Ankara (iMVA) induces systemic antitumor immunity via STING and Batf3-dependent dendritic cells. Sci Immunol. 2017;2:eaal1713. doi:10.1126/sciimmunol.aal1713
24. Kim MK, Breitbach CJ, Moon A, et al. Oncolytic and immunotherapeutic vaccinia induces antibody-mediated complement-dependent cancer cell lysis in humans. Sci Transl Med. 2013;5:185ra63. doi:10.1126/scitranslmed.3005361
25. Heo J, Reid T, Ruo L, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med. 2013;19:329–336. doi:10.1038/nm.3089
26. Ding Y, Fan J, Deng L, Peng Y, Zhang J, Huang B. Bioluminescence imaging of a tumor-selective, thymidine kinase-defective vaccinia virus Guang9 strain after intratumoral or intraperitoneal administration in mice. Oncotarget. 2017;8:88708–88718. doi:10.18632/oncotarget.20788
27. Huang B, Tao W, Shi J, Tang L, Jin J. Determination of ochratoxin A by polyclonal antibodies based sensitive time-resolved fluoroimmunoassay. Arch Toxicol. 2006;80:481–485. doi:10.1007/s00204-006-0112-2
28. Liu TC, Galanis E, Kirn D. Clinical trial results with oncolytic virotherapy: a century of promise, a decade of progress. Nat Clin Pract Oncol. 2007;4:101–117. doi:10.1038/ncponc0736
29. Argnani R, Marconi P, Volpi I, et al. Characterization of herpes simplex virus 1 strains as platforms for the development of oncolytic viruses against liver cancer. Liver Int. 2011;31:1542–1553. doi:10.1111/j.1478-3231.2011.02628.x
30. Wells RG. The role of matrix stiffness in regulating cell behavior. Hepatology. 2008;47:1394–1400. doi:10.1002/hep.22193
31. Wullkopf L, West AV, Leijnse N, et al. Cancer cell ability to mechanically adjust to extracellular matrix stiffness correlates with their invasive potential. Mol Biol Cell. 2018;29:2378–2385.
32. Gkretsi V, Stylianopoulos T. Cell adhesion and matrix stiffness: coordinating cancer cell invasion and metastasis. Front Oncol. 2018;8:145. doi:10.3389/fonc.2018.00145
33. Zhao X, Li L, Starr TK, Subramanian S. Tumor location impacts immune response in mouse models of colon cancer. Oncotarget. 2017;8:54775–54787. doi:10.18632/oncotarget.18423
34. Raki M, Sarkioja M, Escutenaire S, et al. Switching the fiber knob of oncolytic adenoviruses to avoid neutralizing antibodies in human cancer patients. J Gene Med. 2011;13:253–261. doi:10.1002/jgm.1565
35. Friedman A, Tian JP, Fulci G, Chiocca EA, Wang J. Glioma virotherapy: effects of innate immune suppression and increased viral replication capacity. Cancer Res. 2006;66:2314–2319. doi:10.1158/0008-5472.CAN-05-2661
36. Fulci G, Breymann L, Gianni D, et al. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc Natl Acad Sci U S A. 2006;103:12873–12878. doi:10.1073/pnas.0605496103
37. Wakimoto H, Johnson PR, Knipe DM, Chiocca EA. Effects of innate immunity on herpes simplex virus and its ability to kill tumor cells. Gene Ther. 2003;10:983–990. doi:10.1038/sj.gt.3302038
38. Dorner T, Radbruch A. Antibodies and B cell memory in viral immunity. Immunity. 2007;27:384–392. doi:10.1016/j.immuni.2007.09.002
39. Pease DF, Kratzke RA. Oncolytic Viral Therapy for Mesothelioma. Front Oncol. 2017;7:179. doi:10.3389/fonc.2017.00179
40. Woller N, Gürlevik E, Ureche CI, Schumacher A, Kühnel F. Oncolytic viruses as anticancer vaccines. Front Oncol. 2014;4:188. doi:10.3389/fonc.2014.00188
41. Ibrahim AM, Wang YH. Viro-immune therapy: a new strategy for treatment of pancreatic cancer. World J Gastroenterol. 2016;22:748–763. doi:10.3748/wjg.v22.i2.748
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.