")
Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 10
Evaluation of the single-dose pharmacokinetics and pharmacodynamics of apixaban in healthy Japanese and Caucasian subjects
Authors Frost C, Shenker A, Jhee S , Yu Z, Wang J, Bragat A , Pursley J, LaCreta F
Received 28 March 2018
Accepted for publication 25 August 2018
Published 24 October 2018 Volume 2018:10 Pages 153—163
DOI https://doi.org/10.2147/CPAA.S169505
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Arthur E. Frankel
Charles Frost,1 Andrew Shenker,1 Stanford Jhee,2 Zhigang Yu,1 Jessie Wang,3 Alexander Bragat,1 Janice Pursley,4 Frank LaCreta1
1Exploratory Clinical and Translational Research, Bristol-Myers Squibb, Princeton, NJ, USA; 2PAREXEL International Early Phase, Glendale, CA, USA; 3Exploratory Development Global Biometric Sciences, Bristol-Myers Squibb, Princeton, NJ, USA; 4Analytical and Bioanalytical Development, Bristol Myers Squibb, Princeton, NJ, USA
Purpose: This double-blind, placebo-controlled, intra-subject, dose-escalation study assessed single-dose safety, pharmacokinetics, and pharmacodynamics of apixaban in healthy Japanese and Caucasian subjects.
Subjects and methods: Sixteen healthy male Japanese and sixteen healthy male Caucasian subjects, matched for age, weight, and smoking status were randomized to receive four sequential single oral doses of either apixaban (2.5, 10, 25, and 50 mg) or matched placebo. Doses were separated by a ≥5-day washout. Blood samples were collected for the determination of apixaban plasma concentration, clotting times (international normalized ratio [INR], activated partial thromboplastin time, and modified prothrombin time [mPT]), and ex vivo thrombin generation (TG). Urine samples were collected for the analysis of apixaban concentration.
Results: Ascending single doses of apixaban 2.5–50 mg were safe and well tolerated by all subjects. Apixaban exposure increased the dose proportionally up to 10 mg. Apixaban reached maximum concentrations (Cmax) 3–4 h postdose, with mean Cmax ranging from 52.5–485.0 to 44.8–494.3 ng/mL in Japanese and Caucasian subjects. The mean half-life was ~8 and ~13 h and the renal clearance was 1.1 and 0.8 L/h in Japanese and Caucasian subjects, respectively. Pharmacodynamic assessments were similar between ethnic groups, with comparable dose-related prolongation of INR and mPT and inhibition of TG.
Conclusion: Ascending single doses of apixaban over a 20-fold dose range were safe and well tolerated in Japanese and Caucasian subjects in this study. The consistency between pharmacokinetic and pharmacodynamic measures in Japanese and Caucasian subjects indicates that apixaban may be administered as a fixed dose with no need for adjustment in Japanese patients.
Keywords: safety, pharmacokinetics, pharmacodynamics, Japanese, apixaban, anticoagulation, Asian, novel, Eliquis
Introduction
Apixaban (BMS-562247) is an orally bioavailable, selective inhibitor of coagulation factor Xa (FXa) that directly and reversibly binds to the active site of FXa, exerting anticoagulant and antithrombotic effects by diminishing the conversion of prothrombin to thrombin.1–3 Apixaban has been shown to have predictable pharmacokinetics and a limited potential for drug or food interactions.4–6 This oral anticoagulant has been approved for use in several countries to reduce the risk of stroke and systemic embolism in atrial fibrillation and for thromboprophylaxis after knee or hip replacement surgery, for the treatment of venous thromboembolism, and reducing the risk of its recurrence.7,8
Patients who may be treated with apixaban represent a broad demographic; therefore, it is important to understand the factors within a diverse demographic that may influence apixaban pharmacokinetics and pharmacodynamics. Race and ethnicity are among the factors considered to have the potential to influence pharmacokinetics and/or pharmacodynamics, thereby influencing a therapy’s safety and efficacy within a given population.9,10 Evaluations of these parameters in different ethnic populations are thus part of International Council on Harmonization guidelines.11 Whether a compound is more or less likely to be susceptible to ethnic influences appears to be in part related to its pharmacokinetic properties. Compounds with characteristics such as nonlinear pharmacokinetics, poor bioavailability, limited routes of elimination, and elimination via a pathway subject to genetic polymorphism appear to be more susceptible to ethnic factors, whereas compounds with linear pharmacokinetics, multiple routes of elimination, and a low potential for protein binding or drug interactions appear to be less susceptible to ethnic influence.11,12
Apixaban pharmacokinetics exhibit several characteristics suggesting that it may be less susceptible to racial or ethnic influences. These characteristics include good oral bioavailability, linear pharmacokinetics, multiple routes of elimination, and modest protein binding.4,5,13 Previous studies have suggested that Asian race does not significantly influence apixaban pharmacokinetics or pharmacodynamics.14–18 This study provides the first direct comparison of apixaban pharmacokinetics and pharmacodynamics in healthy Japanese and Caucasian subjects matched for age, weight, and smoking status at multiple dose levels, in order to better understand the potential for ethnic influences on apixaban pharmacokinetics and pharmacodynamics.
Methods
Study design and treatments
A four-period, four-treatment, randomized, double-blind, placebo-controlled, sequential, ascending single-dose study was conducted in healthy Japanese and Caucasian men. Japanese subjects were randomly assigned to receive either four sequential single oral doses of apixaban (2.5, 10, 25, and 50 mg) or matched placebo in a 3:1 ratio according to a computer-generated randomization scheme. Japanese subjects were matched to Caucasian subjects for age (±10 years), weight (±10%), and current smoking status (smoker vs nonsmoker) and received the same treatment assignment as their matched counterpart. Subjects remained on either apixaban or placebo for the duration of the study. Subjects received each dose sequentially in an ascending order, beginning with the 2.5 mg dose; each dose was separated by a washout of ≥5 days. Study medication was administered with 240 mL of water in the morning after a fast of ≥10 h.
The study was conducted at a single-center, PAREXEL International Early Phase (Glendale, CA, USA), between December 24, 2004, and April 25, 2005, in accordance with the ethical principles of the Declaration of Helsinki. All subjects were required to provide written informed consent prior to the initiation of any study-specific procedures. The protocol and informed consent were approved by the institutional review board (IRB) of the California IRB, Inc. (Beverly Hills, CA, USA).
Subjects
Healthy Japanese and Caucasian men were eligible for participation. More specifically, those eligible were able to prove both maternal and paternal Japanese ancestries, born in Japan, not living outside Japan for >10 years, and Caucasian men of European descent living in the USA for ≥10 years. Health status was determined by routine examination including medical history, physical examination, vital signs and electrocardiogram (ECG) assessment, and laboratory testing. Subjects were excluded if they did not meet the requirements for ethnicity or if they had any evidence of acute or chronic illness, risk factors for bleeding, or recent use of concomitant medication (prescription or over-the-counter) that could increase the risk of bleeding, influence apixaban pharmacokinetics, or interfere with clinical laboratory assessments.
Restrictions during the study included the following: subjects were to remain at the center for ≥72 h after dosing; alcohol-containing beverages and grapefruit-containing products were not permitted from 3 days before dosing through Day 4; and caffeine-containing products were not permitted from 24 h before admission for each period through Day 4. Subjects were required to fast from 10 h before study drug administration until 4 h after; all were required to consume 240 mL of water 1 h after dosing to promote urine production; lunch, dinner, and a light snack were served 4, 8, and 12 h after dosing, respectively. Substances that interfere with fecal occult testing (including >250 mg vitamin C from any source and red meat or raw broccoli, cauliflower, horseradish, parsnips, radishes, turnips, and melons) were not permitted from 3 days before admission through Day 4, and any red meat consumed during the study was required to be cooked well done.
Pharmacokinetic and pharmacodynamic variables
Blood samples for the determination of apixaban plasma concentrations were collected in 3.2% sodium citrate tubes each period prior to apixaban administration (0 h) and then 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12, 18, 24, 36, 48, and 72 h after study drug administration. Following collection, blood samples were centrifuged for 15 min at ~1,500¥ g and room temperature to separate plasma. The plasma was transferred to a cryogenic vial and stored immediately at or below –20°C. Spot urine samples were collected at predose, with pooled urine samples collected in 12 h increments for 36 h after study drug administration and in a final 36–72 h period after study drug administration.
Plasma and urine samples were analyzed for apixaban with a validated liquid chromatography–tandem mass spectrometry assay within the known period of stability (Intertek Pharmaceutical Services, El Dorado Hills, CA, USA) as previously described.19 Briefly, the lower limit of quantification was 1 ng/mL for apixaban. The between-run and within-run variabilities (each expressed as coefficient of variation [CV] for apixaban) in plasma quality control (QC) samples were ≤5.42% and ≤10.2%, respectively, with deviations from the nominal concentration of no more than ±16.0%. The between-run and within-run CVs for apixaban in urine QC samples were ≤7.80% and ≤22.7%, respectively, with deviations from nominal concentration of no more than ±13.5%.
Single-dose pharmacokinetic parameters (maximum observed plasma concentration [Cmax], time to maximum plasma concentration [Tmax], area under the concentration–time curve to the last observation [AUC(0–T)], area under the concentration–time curve to infinity [AUC(INF)], terminal half-life [T1/2], and renal clearance [CLR]) were derived from plasma concentration versus time and urinary excretion data. Pharmacokinetic parameters were derived by validated noncompartmental methods using Kinetica (Thermo Fisher Scientific, Waltham, MA, USA).
Blood samples for pharmacodynamics analysis were drawn into 3.2% sodium citrate Vacutainer® tubes (Becton Dickinson, Franklin Lakes, NJ, USA) predose and serially at 0.5, 1.5, 3, 6, 12, 24, 48, and 72 h postdose for the determination of international normalized ratio (INR), activated partial thromboplastin time (aPTT), and modified prothrombin time (mPT) and at 3, 6, 12, and 24 h postdose for thrombin generation (TG). Immediately after collection, the samples were gently inverted six to eight times without shaking and processed within 30 min. Pharmacodynamic samples were first centrifuged for 10 min at 2,500¥ g at 4°C. The resulting supernatant was then transferred to a second tube and centrifuged a second time for 10 min at 2,500¥ g at 4°C. The resulting platelet-poor plasma supernatant was transferred into three cryovials and stored at or below –20°C until analysis.
Analysis of INR and aPTT was performed on an STA Compact coagulation analyzer (Diagnostica Stago, Parsippany, NJ, USA) and Diagnostica Stago reagents (STA-Neoplastin CI with an international sensitivity index [ISI] of 1.76–1.80 and STA-PTT Automate 5 reagent). The mPT was measured with a validated method at Covance Laboratories, Inc. (Chantilly, VA, USA) using an MLA Electra 1600C automatic analyzer and Dade Thromboplastin C+ (ISI =1.79). The reportable range of the assay was 7–600 s.20 The TG assay was performed on platelet-poor plasma samples by Synapse B.V. (Maastricht, the Netherlands). The thrombin concentration in each sample was calculated over time by measuring the cleavage of a slow-reacting fluorogenic substrate in the context of a paired calibration sample. A dedicated software program (Thrombinoscope®; Thrombinoscope B.V., Maastricht, the Netherlands) was used to derive TG parameters. TG parameters were derived from the TG curve and included time to peak thrombin concentration (TTP), peak observed thrombin concentration (PEAK), endogenous thrombin potential (ETP), and lag time (LAG).21
Safety variables
Safety assessments were based on medical review of adverse event (AE) reports and the results of vital sign measurements, ECGs, physical examinations, and routine clinical laboratory tests including serum chemistries, liver function, complete blood count, fecal occult blood assessment, bleeding time, and urinalyses. Bleeding time assessment was performed using the Surgicutt® device (ITC, Edison, NJ, USA) according to the manufacturer’s instructions.
All recorded AEs were listed and tabulated by MedDRA (v8.1) system organ class, preferred term, ethnic group, and dose. Severity of AEs was classified as mild (awareness of event but easily tolerated), moderate (discomfort enough to cause some interference with usual activity), severe (inability to carry out usual activity), or very severe (debilitating, significantly incapacitates subject despite symptomatic therapy). Causality of AEs was classified as certain (a reasonable causal relationship exists between study drug and AE; the event responds to the withdrawal of study drug [dechallenge] and recurs with rechallenge when clinically feasible), probable (a reasonable causal relationship exists; the event responds to dechallenge, but rechallenge is not required), possible (reasonable causal relationship; dechallenge information is lacking or unclear), not likely (temporal relationship to study drug administration but no reasonable causal relationship), or unrelated (no temporal relationship, or there is a reasonable causal relationship with another drug, concurrent disease, or circumstance).
Statistical methods
The number of subjects enrolled in this study was not based on statistical power considerations. However, administration of apixaban to 12 subjects in each subject group (Japanese or Caucasian) would provide 83% confidence that the estimated Japanese:Caucasian ratios of Cmax and AUC(INF) geometric means at each dose level would be within 20% of the true population ratios.
All statistical analyses were carried out using SAS/STAT Version 8.2 (SAS Institute Inc., Cary, NC, USA). Summary statistics were tabulated for the pharmacokinetic parameters (Cmax, AUC[INF], AUC[0–T], Tmax, T1/2, and CLR) of apixaban by group and dose. Geometric means and CVs were provided for Cmax, AUC(INF), and AUC(0–T). Medians and ranges were presented for Tmax. Means and SD were provided for T1/2 and CLR.
To assess the differences in apixaban pharmacokinetics between Japanese and Caucasian subjects and to assess the dose proportionality within each ethnic group, mixed-effects repeated-measures linear models were fitted on logarithmic scales for Cmax, AUC(INF), and AUC(0–T). To compare the two groups, mixed-effect models with dose and group were fitted. Point estimates and 90% CIs were provided in the original scale for the Japanese:Caucasian ratios of apixaban Cmax, AUC(INF), and AUC(0–T). Mixed-effect log-linear models were fitted to relate apixaban pharmacokinetic parameters Cmax, AUC(INF), and AUC(0–T) with apixaban dose.
To further assess the dose proportionality of apixaban, the following relationship was estimated for each group (Japanese or Caucasian) and each of the parameters Y=Cmax, AUC(INF), and AUC(0–T) by regressing log(Y) on log(dose):
|
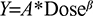
Point estimates, standard error, and 95% confidence limits were generated for the dose proportionality parameter b. A value near 1 for b would support the conclusion of dose proportionality.
Summary statistics were tabulated by group (Japanese or Caucasian), dose, and time point for INR, aPTT, mPT, and TG parameters, and the corresponding percent change from baseline. Plots of mean profiles over time for INR, aPTT, mPT, and TG parameters by dose were provided for each group. Scatter plots of INR, aPTT, and mPT values vs apixaban plasma concentration were provided by dose and group.
All available data from all subjects who received study medication were included in the summaries of physical examination findings, clinical laboratory data, vital signs, and AEs. Subject demographics, physical examinations, laboratory data, and vital signs were summarized. Incidence of AEs was summarized by system organ class, preferred term, relationship to study drug, and severity.
Results
Demographics
Thirty-two healthy male subjects (16 Japanese and 16 Caucasian subjects) participated in the study and were well matched for age, body weight, body mass index (BMI), and smoking status (Table 1). One Japanese subject withdrew informed consent after receiving a single dose of placebo, and all other subjects completed the study.
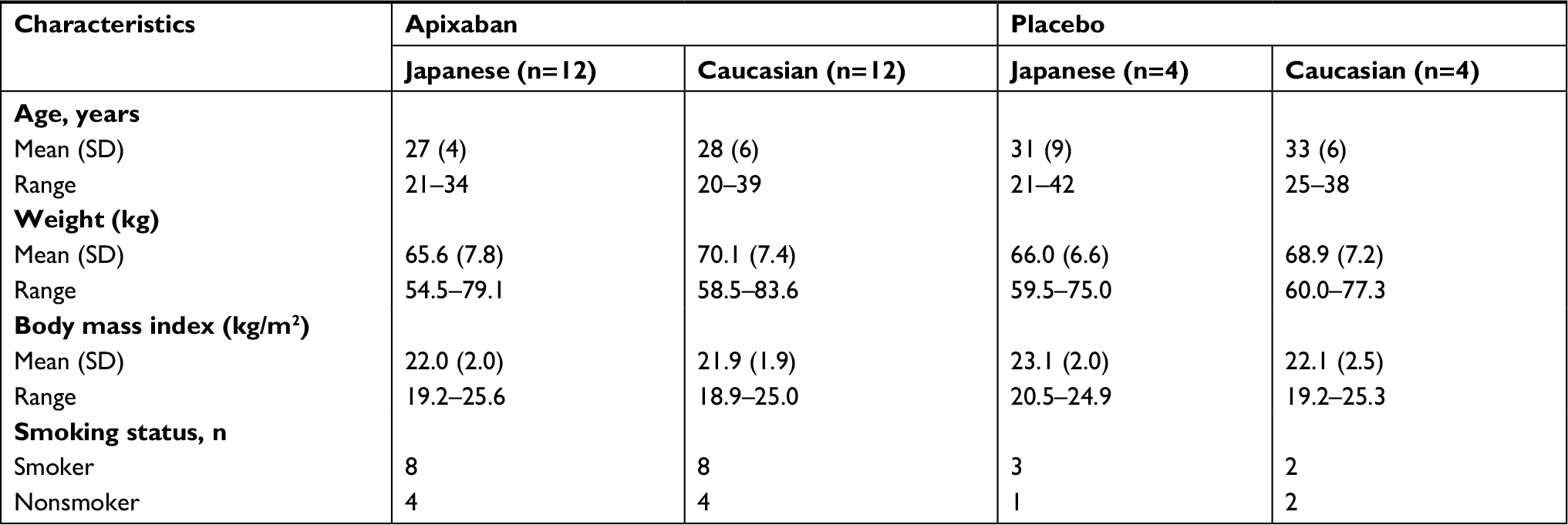 | Table 1 Demographic characteristics and physical measurements |
Pharmacokinetics
Figure 1 shows the mean (SD) plasma concentration–time profiles of apixaban in Japanese and Caucasian subjects. Apixaban pharmacokinetic parameters for Japanese and Caucasian subjects are summarized in Table 2. Apixaban Tmax values ranged from 3 to 4 h across doses and were comparable between Japanese and Caucasian subjects. Apixaban geometric mean Cmax, AUC(0–T), and AUC(INF) for Japanese subjects were within 84%–117% of Caucasian values across all doses except at the 50 mg dose, where AUC values were ~20%–22% lower in Japanese subjects (Table 3). In the dose proportionality analysis, the point estimates and 95% CIs for b were <1 for Cmax and AUC in both Japanese and Caucasian subjects (Table 4) indicating that increases in apixaban exposure were less than proportional to dose across the 2.5–50 mg dose range. This appears to be driven by less than proportional increases in exposure following administration of the two highest doses, 25 and 50 mg (Figure 2). Increases in Cmax and AUC were in proportion to dose following the administration of 2.5 and 10 mg apixaban in Caucasians and nearly proportional in Japanese subjects based on inspection of the dose and exposure ratios.
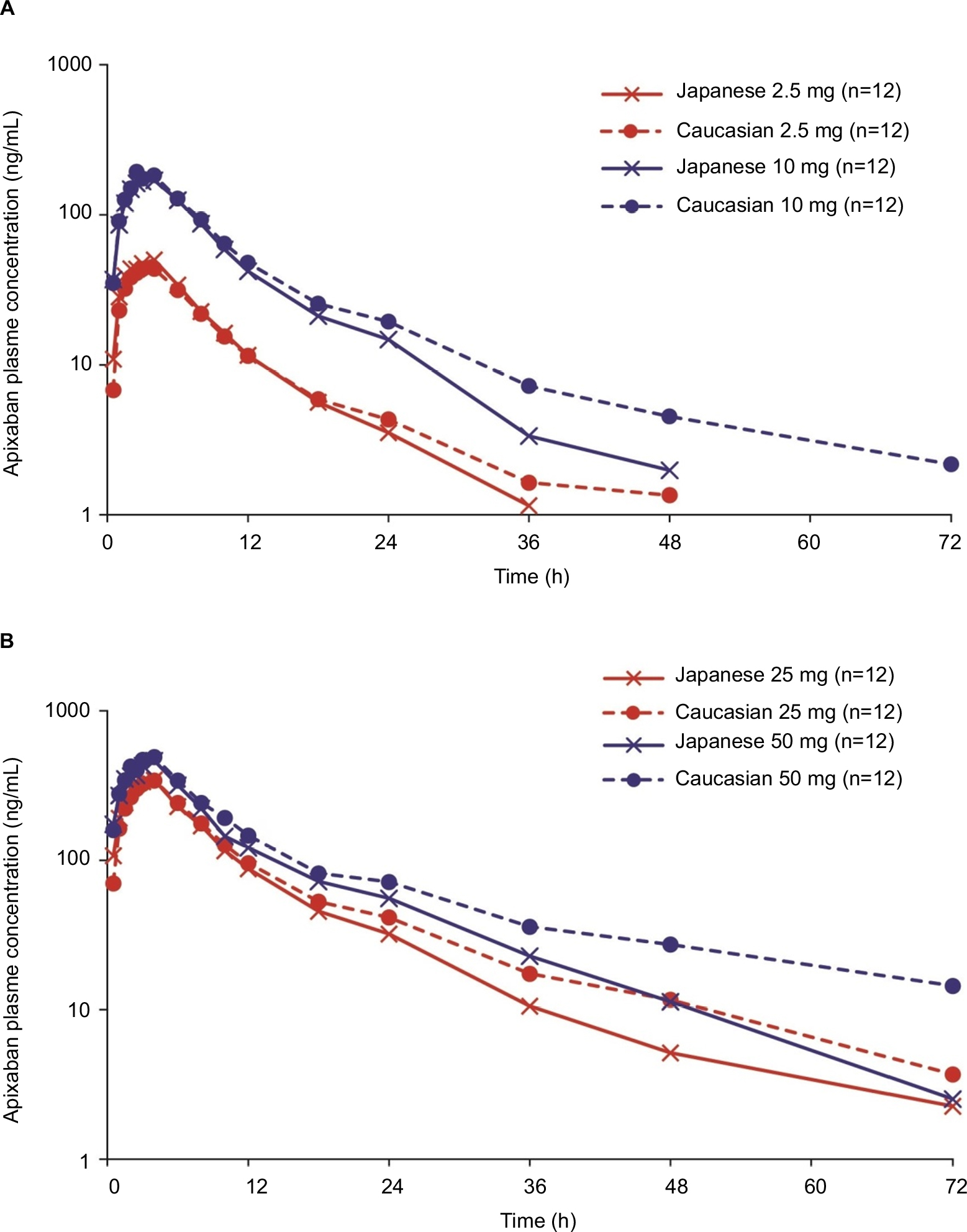 | Figure 1 Mean apixaban plasma concentration versus time profiles after single oral doses of (A) apixaban 2.5 and 10 mg and (B) apixaban 25 and 50 mg. |
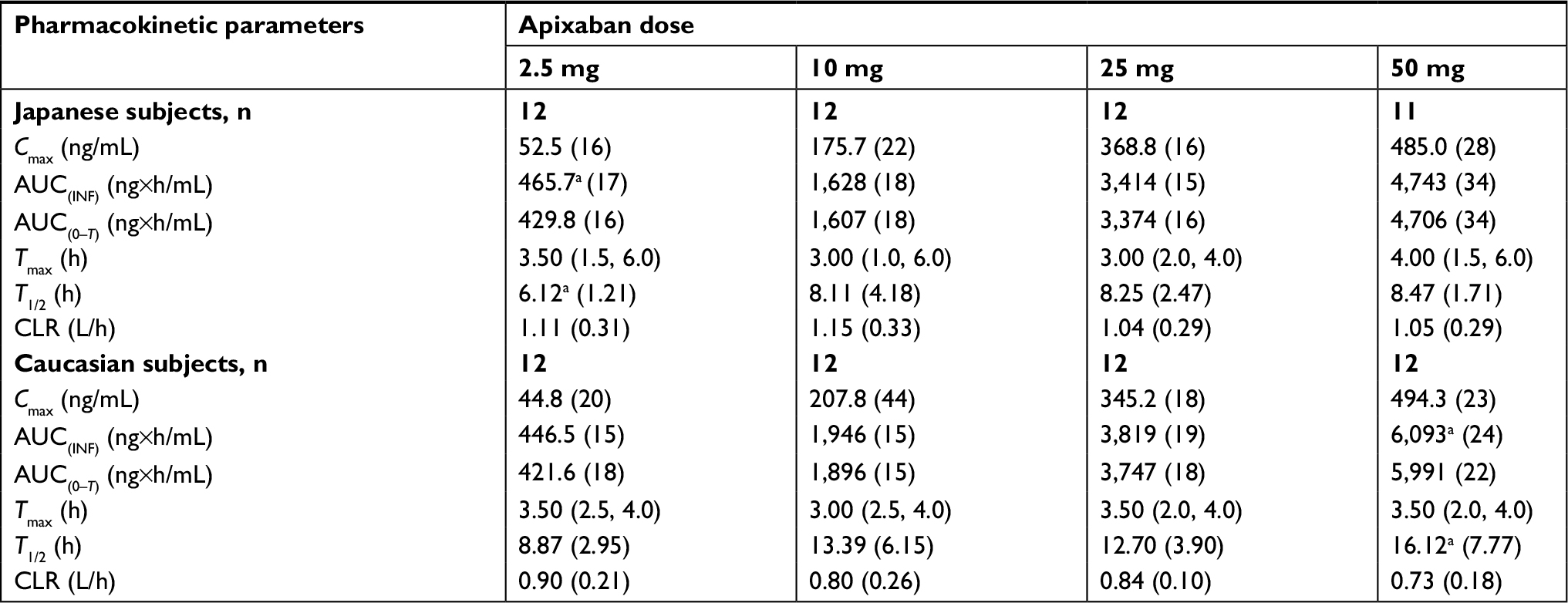 | Table 2 Apixaban pharmacokinetic parameter summary statistics in Japanese and Caucasian subjects Notes: an=10. Values for Cmax, AUC(INF), and AUC(0–T) are reported as geometric mean (CV%). Values for Tmax are reported as median (minimum, maximum). Values for T1/2 and CLR are reported as mean (SD). Abbreviations: AUC(0–T), area under the concentration–time curve to the last observation; AUC(INF), area under the concentration–time curve to infinity; CLR, renal clearance; CV, coefficient of variation; Cmax, maximum observed plasma concentration; T1/2, terminal half-life; Tmax, time to maximum plasma concentration. |
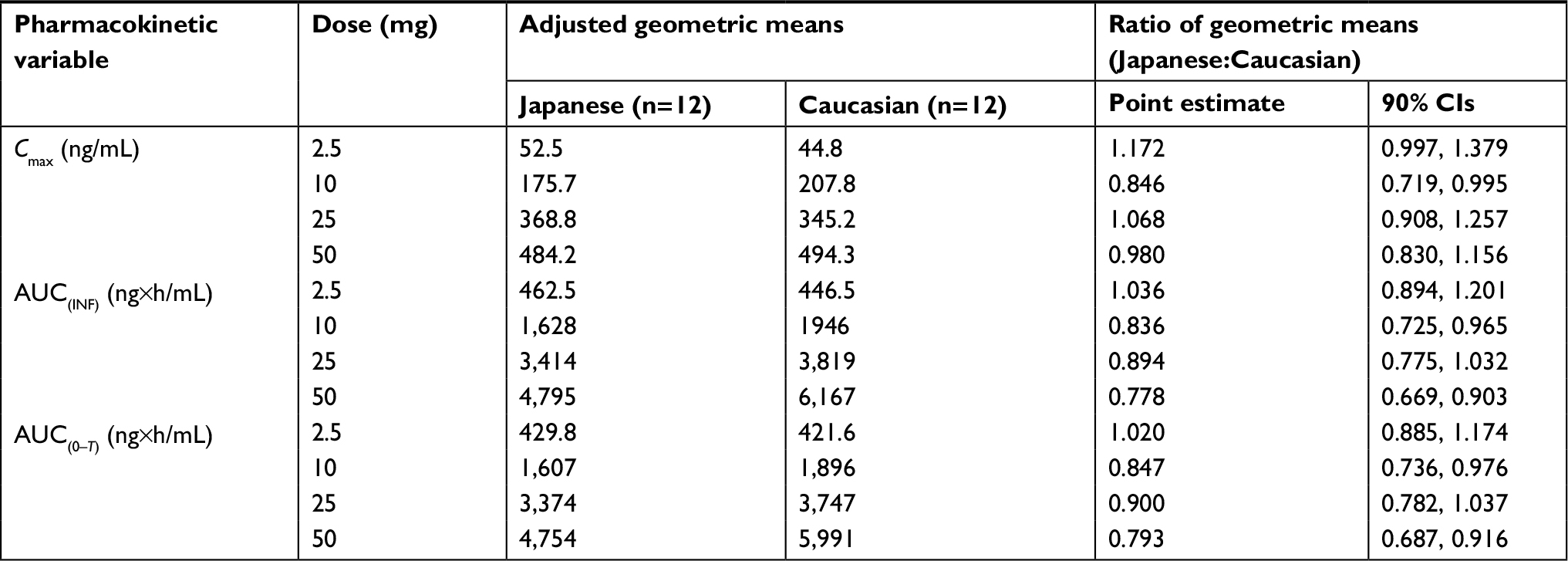 | Table 3 Results of statistical analyses comparing apixaban Cmax, AUC(INF), and AUC(0–T) between Japanese and Caucasian subjects Note: General linear models were fitted on logarithmic scales and 90% CIs were calculated separately for ratios of geometric means. Abbreviations: AUC(0–T), area under the concentration–time curve to the last observation; AUC(INF), area under the concentration–time curve to infinity; Cmax, maximum observed plasma concentration. |
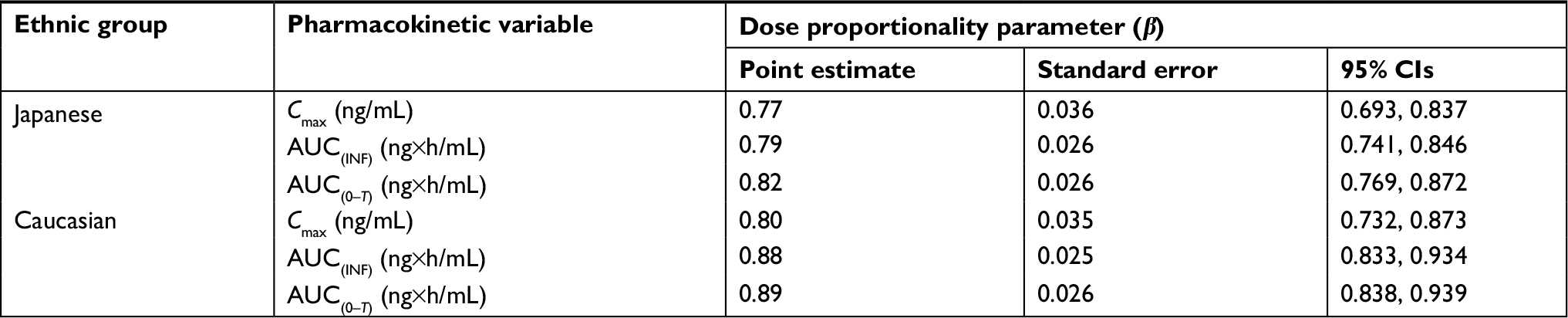 | Table 4 Results of dose-proportionality analyses of apixaban Cmax, AUC(INF), and AUC(0–T) by ethnic group Abbreviations: AUC(0–T), area under the concentration–time curve to the last observation; AUC(INF), area under the concentration–time curve to infinity; Cmax, maximum observed plasma concentration. |
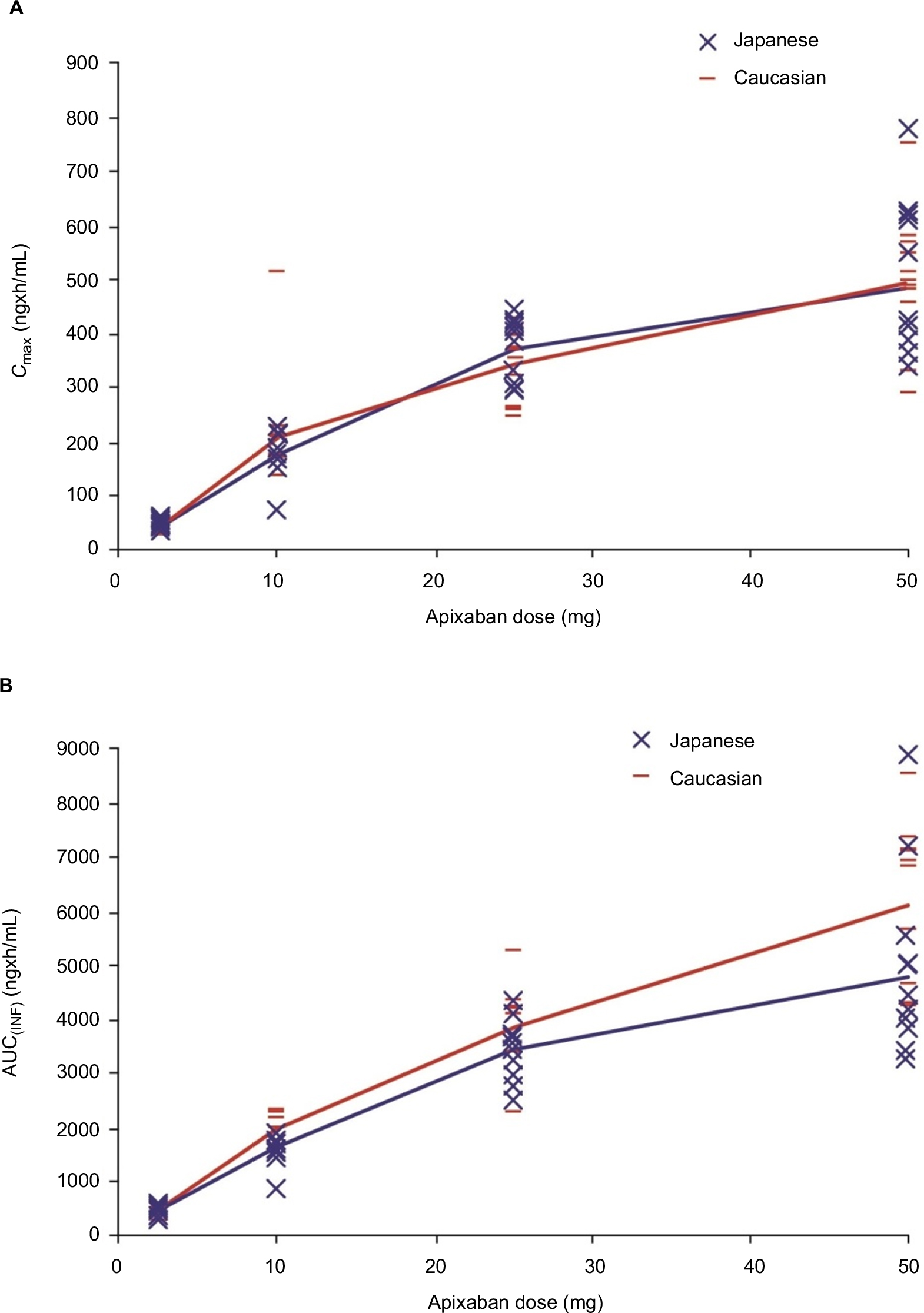 | Figure 2 Dose proportionality of apixaban (A) Cmax and (B) AUC(INF) for Caucasians and Japanese. Note: Geometric mean values from each group were connected by red (Caucasian) and blue (Japanese) lines. Abbreviations: AUC(INF), area under the concentration–time curve to infinity; Cmax, maximum observed plasma concentration. |
Mean CLR appeared to be slightly higher in Japanese subjects compared with that observed in Caucasian subjects, and mean T1/2 values across doses in Japanese subjects (6.1–8.5 h) appeared to be shorter than in Caucasian subjects (8.9–16.1 h; Table 2).
Pharmacodynamics
Clotting time measures (INR, aPTT, and mPT) and TG parameters were comparable between Japanese and Caucasian subjects prior to the first dose of apixaban or placebo. Following the administration of apixaban, changes in clotting time measures and TG parameters closely followed the apixaban plasma concentration–time profile, resulting in comparable dose-related changes in both Japanese and Caucasian subjects (Figure 3). Generally, changes in pharmacodynamic measures were evident within 30 min of apixaban administration, reaching peak values at approximately 3 h postdose and nearing baseline values within 12–24 h after the administration of doses ≤10 mg. The pharmacodynamic effects of apixaban extended beyond 24 h for the 25 and 50 mg dose panels. There were no significant changes in clotting times or TG parameters in the subjects who received placebo (Figure 3 and Tables S1–S4).
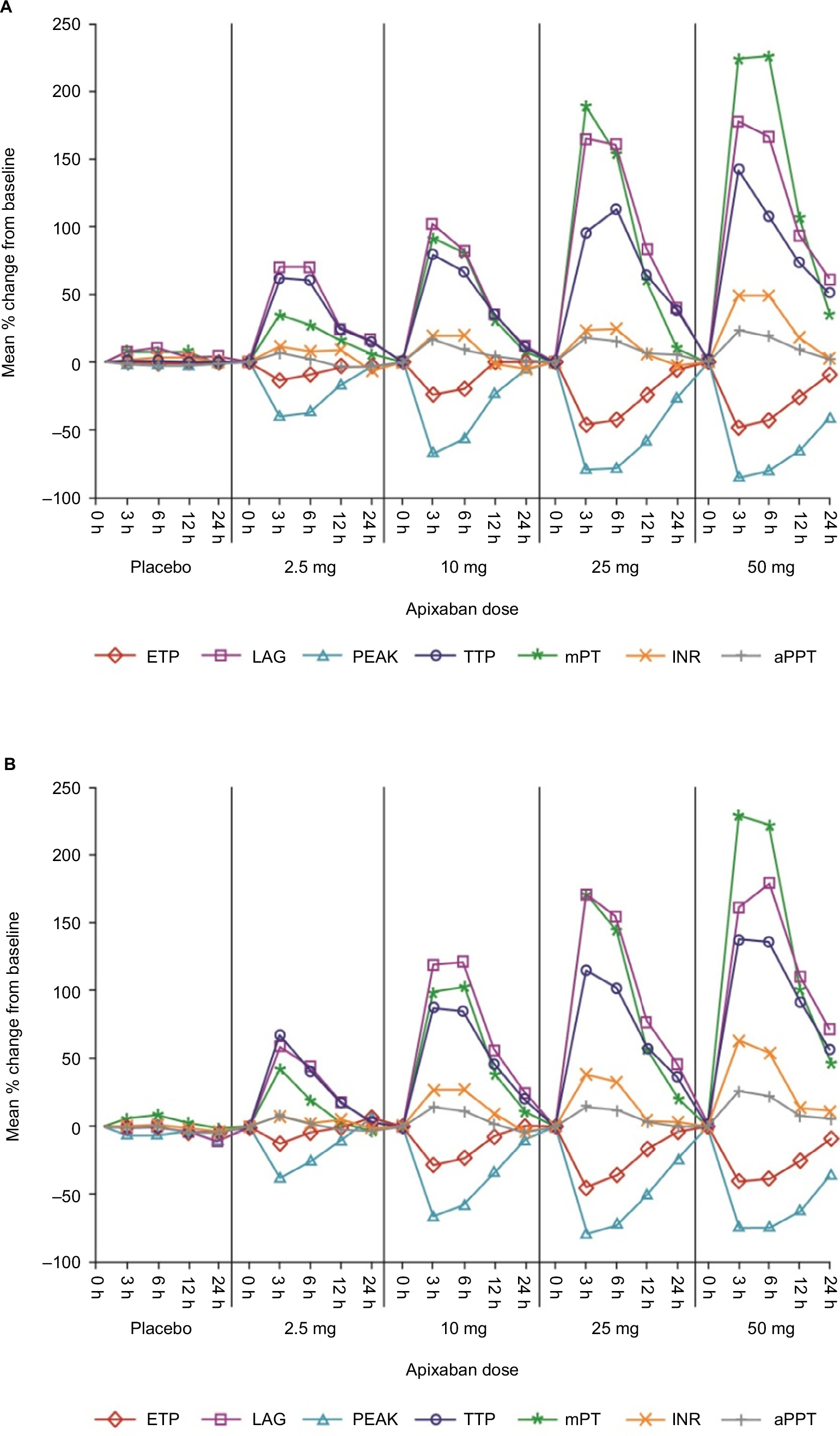 | Figure 3 Mean percentage change from baseline profiles for pharmacodynamic variables for (A) Japanese and (B) Caucasian subjects. Abbreviations: aPTT, activated partial thromboplastin time; ETP, endogenous thrombin potential; INR, international normalized ratio; LAG, lag time; mPT, modified prothrombin time; PEAK, peak observed thrombin concentration; TTP, time to peak thrombin concentration. |
Among the clotting time measures, mPT was the most sensitive to the effects of apixaban with a mean maximum increase from the baseline of approximately 226%–229%, following administration of a 50 mg single dose, and exhibited a steeper concentration–response relationship than that for INR, which exhibited approximately a 50%–64% increase at the 50 mg dose (Figures 3, S1, and S2 and Tables S1 and S2). The effect of apixaban on aPTT was relatively limited compared with the other clotting time measures with a mean maximum increase from the baseline of ~25% at the 50 mg dose (Figures 3 and S3 and Table S3).
TG parameters exhibited a curvilinear response with increases from baseline up to approximately 178% and 142% for LAG and TTP, respectively, representing the greatest changes after apixaban administration followed by decreases in PEAK and ETP (86% and 49% decreases from baseline, respectively). Unlike INR and aPTT, changes in TG parameters and mPT were evident for 12 h or more after apixaban administration (Figures 3 and S2 and Tables S2 and S4).
Safety and tolerability
There were no serious AEs or discontinuations due to AEs in this study. A total of 32 AEs were reported from five (41.7%) Japanese and 11 (91.7%) Caucasian subjects who received apixaban, and a total of 25 events were reported from two (50%) Japanese and four (100%) Caucasian subjects who received placebo. All reported AEs were mild or moderate in intensity, and all but two (back pain and ear congestion) were resolved by study completion. Although AEs were reported more frequently in Caucasian subjects than in Japanese subjects, the overall frequencies of AEs were similar for apixaban- and placebo-treated subjects within each ethnic group and there were no observed dose-related AEs trends in either group.
The most frequently reported treatment-emergent AE in this study was headache, reported by five Caucasian subjects and one Japanese subject who received apixaban, as well as one Caucasian subject and one Japanese subject who received placebo. Dizziness was reported by four Caucasian subjects: two subjects received apixaban and two subjects received placebo. Fatigue was reported by three Caucasian subjects, of whom one subject received apixaban and two subjects received placebo. Infusion site (ie, venipuncture site for blood sampling) pain was reported by one Caucasian subject and two Japanese subjects, of whom all received apixaban. All other AEs in this study were reported by two or fewer subjects during the study.
One bleeding-related AE of mild intensity, a prolonged bleeding time of 20 min, occurred in a Caucasian subject 1 week after the administration of apixaban 25 mg. The AE was observed during predose procedures prior to administration of the 50 mg dose. Following administration of the 50 mg dose, this subject’s bleeding time was 30 min at 3.5 h after dosing and decreased to 9 min by 12 h after dosing. The subject was questioned and denied taking aspirin or aspirin-containing medication during his clinic furlough. The investigator considered the event to be unrelated to study drug. The subject had no other prolonged bleeding times during the study.
Two subjects tested positive for fecal occult blood after the administration of study drug: one subject 7 days after the administration of apixaban 25 mg and one subject after placebo administration. There were no clinically relevant changes in vital signs or ECGs in either group.
Discussion
This study directly compared apixaban pharmacokinetics and pharmacodynamics between Japanese and Caucasian subjects for the first time and evaluated apixaban following the administration of ascending doses across the approved dose range within the same individual, extending the available information with important, unique intra-subject variability data. In this study, oral, single-dose administration of 2.5–50 mg apixaban was well tolerated and resulted in consistent pharmacokinetic and pharmacodynamic profiles among these healthy male Japanese and Caucasian subjects. The matching for baseline age, body weight, and smoking status suggests little or no potential confounding by differences in these characteristics.
In both Japanese and Caucasian subjects, apixaban exposure increased in proportion to dose across the approved dose range (2.5–10 mg) while less than proportional increases were observed for dose greater than 10 mg (ie, 25 and 50 mg), and similar biphasic elimination was evident for both groups. Apixaban Cmax and AUC estimates were largely consistent between Japanese and Caucasian subjects across the 2.5–50 mg dose range given that the 90% CIs included 1 in most instances. In those instances where the 90% CI excluded 1, the point estimates and the CI were less than l suggesting lower exposure, particularly with respect to AUC, in the Japanese subjects. Nominally, these Japanese subjects also appeared to exhibit a shorter mean T1/2 and a higher mean CLR than that observed in the Caucasian subjects.
The dose- and concentration-dependent increases in coagulation tests (aPTT, INR, and mPT) observed in this study were consistent with previous reports demonstrating a similar range of sensitivity to the effects of apixaban, with aPTT being the least sensitive and mPT being the most sensitive among these tests.4,5,20 The observed changes in TG parameters were also consistent with the expected effects of a direct FXa inhibitor in prolonging the initiation phase and inhibiting the propagation phase of TG. Changes in LAG, TTP, and PEAK remained evident at 12 h postdose for all dose panels in both Japanese and Caucasian subjects. ETP appeared to be the least sensitive TG parameter. Overall, changes in these pharmacodynamic parameters were consistent with pharmacokinetics within each group and did not reveal any significant differences in the pharmacodynamic response to apixaban between Japanese and Caucasian subjects.
Following multiple-dose administration, apixaban pharmacokinetics and pharmacodynamics appear to be consistent among Japanese, Chinese, and non-Asian healthy subjects.5,14-18 While these reports are generally informative, there are limitations to comparing data across small studies such as these. A larger population-based pharmacokinetic analysis including data from Phase II and Phase III clinical trials in patients with atrial fibrillation suggests that apixaban clearance may be 15% lower in Japanese patients compared to the overall population, which would correspond to an 18% higher exposure.16,17 A similar small effect of Asian race was found in patients treated with apixaban for venous thromboembolism.18 The effect of apixaban on safety and efficacy endpoints in Phase II and III clinical trials appears to be consistent between Japanese patients and the broader population, despite potential small differences in apixaban clearance.22–25 The present study’s findings of comparable pharmacokinetics and pharmacodynamics between Japanese and Caucasian subjects are in line with such results; however, both Japanese and Caucasian groups in this study comprised a limited number of healthy young males, which may be a constraint in the generalization of these results to the patient population.
Conclusion
In this study, single oral doses of apixaban over a 20-fold dose range were safe and well tolerated by both Japanese and Caucasian healthy subjects. Consistent dose-related increases in apixaban exposure and corresponding changes in pharmacodynamic measures were observed in both Japanese and Caucasian subjects indicating that apixaban may be administered as a fixed dose with no need for adjustment in Japanese patients.
Acknowledgments
This study was funded by Bristol-Myers Squibb, Princeton, NJ, USA, and Pfizer Inc. Medical writing and editorial assistance were provided by Andrew Shepherd at Caudex, funded by Bristol-Myers Squibb and Pfizer, Inc.
Disclosure
Charles Frost is an employee of Bristol-Myers Squibb. Frank LaCreta, Jessie Wang, Alexander Bragat, Zhigang Yu, and Andrew Shenker were employees of Bristol-Myers Squibb at the time of research. Stanford Jhee was an employee of California Clinical Trials. Andrew Shenker is currently employed at GlaxoSmithKline, King of Prussia, PA, USA. Alexander Bragat is currently employed at New York University School of Medicine, NYU Langone Health, New York, NY, USA. The authors report no other conflicts of interest in this work.
References
Cheung YW, Barco S, Hutten BA, Meijers JC, Middeldorp S, Coppens M. In vivo increase in thrombin generation by four-factor prothrombin complex concentrate in apixaban-treated healthy volunteers. J Thromb Haemost. 2015;13(10):1799–1805. | ||
Pinto DJ, Orwat MJ, Koch S, et al. Discovery of 1-(4-methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine-3-carboxamide (apixaban, BMS-562247), a highly potent, selective, efficacious, and orally bioavailable inhibitor of blood coagulation factor Xa. J Med Chem. 2007;50(22):5339–5356. | ||
Wong PC, Crain EJ, Xin B, et al. Apixaban, an oral, direct and highly selective factor Xa inhibitor: in vitro, antithrombotic and antihemostatic studies. J Thromb Haemost. 2008;6(5):820–829. | ||
Frost C, Wang J, Nepal S, et al. Apixaban, an oral, direct factor Xa inhibitor: single dose safety, pharmacokinetics, pharmacodynamics and food effect in healthy subjects. Br J Clin Pharmacol. 2013;75(2):476–487. | ||
Frost C, Nepal S, Wang J, et al. Safety, pharmacokinetics and pharmacodynamics of multiple oral doses of apixaban, a factor Xa inhibitor, in healthy subjects. Br J Clin Pharmacol. 2013;76(5):776–786. | ||
Wang L, Zhang D, Raghavan N, et al. In vitro assessment of metabolic drug-drug interaction potential of apixaban through cytochrome P450 phenotyping, inhibition, and induction studies. Drug Metab Dispos. 2010;38(3):448–458. | ||
Bristol-Myers Squibb, Pfizer EEIG. Eliquis® (apixaban tablets) Summary of product characteristics; 2018. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002148/WC500107728.pdf. | ||
Bristol-Myers Squibb Company and Pfizer Inc. Eliquis (apixaban) prescribing information. Revised 2/2018; 2016. Available from: http://packageinserts.bms.com/pi/pi_eliquis.pdf. Accessed June 25, 2018. | ||
Bjornsson TD, Wagner JA, Donahue SR, et al. A review and assessment of potential sources of ethnic differences in drug responsiveness. J Clin Pharmacol. 2003;43(9):943–967. | ||
Kim K, Johnson JA, Derendorf H. Differences in drug pharmacokinetics between East Asians and Caucasians and the role of genetic polymorphisms. J Clin Pharmacol. 2004;44(10):1083–1105. | ||
International Conference on Harmonization [homepage on the Internet]. ICH Harmonized Tripartite Guideline: ICH topic E5 ethnic factors in the acceptability of foreign clinical data; 1998. Available from: http://www.ich.org/products/guidelines/efficacy/efficacy-single/article/ethnic-factors-in-the-acceptability-of-foreign-clinical-data.html. Accessed June 25, 2018. | ||
Johnson JA. Predictability of the effects of race or ethnicity on pharmacokinetics of drugs. Int J Clin Pharmacol Ther. 2000;38(2):53–60. | ||
Frost C, Yu Z, Nepal S, et al. Apixaban, a direct factor Xa inhibitor: single-dose pharmacokinetics and pharmacodynamics of an intravenous formulation [abstract 148]. J Clin Pharmacol. 2008;48:1132. | ||
Cui Y, Song Y, Wang J, et al. Single- and multiple-dose pharmacokinetics, pharmacodynamics, and safety of apixaban in healthy Chinese subjects. Clin Pharmacol. 2013;5:177–184. | ||
Yamahira N, Frost C, Fukase H, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of multiple doses of apixaban in healthy Japanese male subjects. Int J Clin Pharmacol Ther. 2014;52(7):564–573. | ||
Kowalski K, Nielsen J, Roy A, et al. Apixaban exposure and anti-xa activity in nonvalvular atrial fibrillation patients: an application of population PK/PD analysis. Poster presented at the American Conference on Pharmacometrics, October 13, 2014. | ||
Cirincione B, Kowalski K, Nielsen J. Population pharmacokinetics of apixaban in subjects with non-valvular atrial fibrillation. CPT Pharmacometrics Syst Pharmacol. In press 2018. | ||
Byon W, Sweeney K, Frost C, Boyd RA. Population pharmacokinetics, pharmacodynamics, and exploratory exposure-response analyses of apixaban in subjects treated for venous thromboembolism. CPT Pharmacometrics Syst Pharmacol. 2017;6(5):340–349. | ||
Pursley J, Shen JX, Schuster A, et al. LC-MS/MS determination of apixaban (BMS-562247) and its major metabolite in human plasma: an application of polarity switching and monolithic HPLC column. Bioanalysis. 2014;6(15):2071–2082. | ||
Barrett YC, Wang Z, Knabb RM. A novel prothrombin time assay for assessing the anticoagulant activity of oral factor Xa inhibitors. Clin Appl Thromb Hemost. 2013;19(5):522–528. | ||
Hemker HC, Giesen P, Al Dieri R, et al. Calibrated automated thrombin generation measurement in clotting plasma. Pathophysiol Haemost Thromb. 2003;33(1):4–15. | ||
Granger CB, Alexander JH, et al. ARISTOTLE Committees and Investigators, et al. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2011;365(11):981–992. | ||
Ogawa S, Shinohara Y, Kanmuri K. Safety and efficacy of the oral direct factor Xa inhibitor apixaban in Japanese patients with non-valvular atrial fibrillation. -The ARISTOTLE-J study-. Circ J. 2011;75(8):1852–1859. | ||
Nakamura M, Nishikawa M, Komuro I, et al. Apixaban for the Treatment of Japanese Subjects With Acute Venous Thromboembolism (AMPLIFY-J Study). Circ J. 2015;79(6):1230–1236. | ||
Agnelli G, Buller HR, Cohen A, et al. Oral apixaban for the treatment of acute venous thromboembolism. N Engl J Med. 2013;369(9):799–808. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.