")
Back to Journals » Journal of Pain Research » Volume 11
Evaluation of the safety and efficacy of an intravenous nanocrystal formulation of meloxicam in the management of moderate-to-severe pain after bunionectomy
Authors Gottlieb IJ, Tunick DR, Mack RJ, McCallum SW , Howard CP , Freyer A , Du W
Received 31 August 2017
Accepted for publication 10 November 2017
Published 16 February 2018 Volume 2018:11 Pages 383—393
DOI https://doi.org/10.2147/JPR.S149879
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Katherine Hanlon
Video abstract presented by Ira J Gottlieb
Views: 714
Ira J Gottlieb,1 Deborah R Tunick,1 Randall J Mack,2 Stewart W McCallum,2 Campbell P Howard,3 Alex Freyer,2 Wei Du4
1Chesapeake Research Group, Pasadena, MD, USA; 2Recro Pharma, Inc., Malvern, PA, USA; 3Howard Medical Consulting for the Pharmaceutical Industry, Yardley, PA, USA; 4Clinical Statistics Consulting, Blue Bell, PA, USA
Objective: This randomized, double-blind, placebo-controlled study evaluated the safety and efficacy of an intravenous (IV) nanocrystal formulation of meloxicam in subjects with moderate-to-severe pain following a standardized unilateral bunionectomy.
Methods: Fifty-nine subjects aged 18–72 years were randomized to receive doses of either 30 mg (n=20) or 60 mg (n=20) meloxicam IV or placebo (n=19), administered once daily as bolus IV injections over 15–30 seconds (two or three doses). Safety, the primary objective, was assessed by physical examination, clinical laboratory tests, and the incidence of adverse events (AEs). Efficacy was evaluated by examining summed pain intensity differences over the first 48 hours (SPID48) using analysis of covariance models. Use of opioid rescue analgesic agents was evaluated.
Results: Generally, AEs were mild-to-moderate in intensity, and their incidence was similar across the three treatment groups. No serious AEs were reported; there were no withdrawals due to AEs, including injection-related AEs. The estimated effect size for SPID48 versus placebo was 1.15 and 1.01 for meloxicam IV doses 30 mg and 60 mg, respectively (P≤0.01). Both doses produced significantly greater pain reductions versus placebo (P≤0.05) at all evaluated times/intervals during the 48-hour period. The proportions of subjects with ≥30% and ≥50% overall reduction in pain from baseline after 6 and 24 hours were significantly higher with meloxicam IV 30 mg doses versus placebo, but not with meloxicam IV 60 mg doses. The time to first use of rescue medication was significantly longer versus placebo with meloxicam IV 60 mg (P<0.05), but not with meloxicam IV 30 mg doses.
Conclusion: Meloxicam IV was generally safe and well tolerated in subjects with moderate-to-severe post-bunionectomy pain. Once-daily administration of meloxicam IV 30 mg and 60 mg exhibited rapid onset of analgesia (as early as 15 minutes) with maintenance of analgesic effect for two consecutive 24-hour periods.
Keywords: bunionectomy, postoperative pain, meloxicam IV, COX-2 inhibitor, safety, efficacy
Introduction
Intense pain is common after bunionectomy,1 particularly in the first few days after surgery. Post-bunionectomy pain is generally classified as a hard-tissue pain model,2 and its evaluation is useful for assessing the effectiveness of analgesic agents that have a rapid onset of action.3 As with other postoperative pain settings, effective management of pain is an important component of patient care and affects the patient’s ability to resume normal activities after surgery.4 Although opioids traditionally have been the mainstay of peri- and postoperative pain management, opioid-related adverse events (AEs) such as respiratory depression, sedation, nausea/vomiting, and constipation can potentially limit the benefit of these medications. Thus, there is a need for non-opioid analgesics that provide pain control while reducing the risk of AEs.5,6
Selection of pain control strategies for bunion surgery – which include local infiltration of anesthetics and/or peripheral nerve block procedures,1,7,8 opioids,9–11 nonsteroidal anti-inflammatory drugs (NSAIDs),12 and various other analgesics (eg, acetaminophen) – is generally guided by the intensity of the post-bunionectomy pain, the duration of the analgesia provided, and the tolerability and patient acceptance of the chosen strategy. Patient acceptance of opioid analgesics may be influenced by AEs such as respiratory depression, nausea/vomiting, urinary retention, dysphoria, and possible long-term dependence. Potential AEs of nonselective (ie, COX-1 and COX-2 inhibitors) NSAIDs include impaired platelet and renal function, and gastrointestinal intolerance, which could limit patient acceptance of these agents. Patient tolerability of the chosen medication is crucial for attaining rapid and adequate pain relief.
Meloxicam, a preferential COX-2 inhibitor with analgesic, antipyretic, and anti-inflammatory properties, with better gastrointestinal tolerability compared with nonselective NSAIDs, has been proven effective when administered orally for ameliorating the signs and symptoms of rheumatoid arthritis and osteoarthritis.13–16 However, largely due to its poor solubility, orally administered meloxicam is not rapidly absorbed; peak plasma concentrations after a dose of 30 mg are not reached until 9–11 hours after administration.14,17,18 Consequently, this slow onset of action is a reason why oral meloxicam is not currently approved for the management of acute pain.
A novel nanocrystal colloidal dispersion formulation of meloxicam (N1539; Recro Pharma, Inc., Malvern, PA, USA) has recently been developed for bolus intravenous (IV) administration, providing faster onset of analgesia than can be achieved with oral administration.19 In a randomized, double-blind, placebo-controlled study in females who underwent abdominal hysterectomy, the IV nanocrystal formulation of meloxicam was effective in relieving moderate-to-severe postoperative pain.20 All single doses of meloxicam IV evaluated (5 mg to 60 mg) resulted in significantly lower pain intensity (PI) scores and better global pain-control scores than placebo; doses >5 mg also achieved significantly better pain-relief scores than morphine (10 mg to 15 mg), and the use of rescue medication was lower in all meloxicam IV dose groups than in the morphine and placebo groups.20
The present Phase 2 study (NCT02675907) was designed to evaluate the safety and efficacy of the IV nanocrystal formulation of meloxicam administered in doses of 30 mg and 60 mg compared to placebo in subjects with moderate-to-severe pain following a standardized bunionectomy procedure. The primary objective was to evaluate the safety of meloxicam IV administered as a bolus injection over 15–30 seconds (rather than 1–2 minutes, as in earlier studies) by assessing vital signs, clinical laboratory findings, electrocardiography (ECG) changes, wound healing, and the occurrence of AEs. The principal efficacy objective was the estimated effect size of the two doses of meloxicam IV, determined via time-weighted summed PI differences (PID) over the first 48 hours (SPID48), relative to placebo.
Methods
Study design and subjects
This Phase 2, randomized, double-blind, placebo-controlled trial was performed at a single center in the United States (Chesapeake Research Group, Pasadena, MD) in males and females aged 18–75 years in good health (American Society of Anesthesiology class 1 or 2) who were scheduled to undergo a primary, unilateral, first metatarsal osteotomy and internal fixation (without collateral procedures) during the period August 10 to November 20, 2015.
On the first postoperative day, subjects eligible for study participation were required to have moderate-to-severe pain on a 4-point Likert scale and a score of ≥4 on the 11-point numeric pain rating scale (NPRS) after cessation of a popliteal sciatic nerve block and discontinuation of other pain management measures.
Ineligible participants included those with known hypersensitivity to aspirin, NSAIDs, or any of the peri- or postoperative medications used in the study; active gastrointestinal bleeding or a history of peptic ulcer disease; known bleeding disorders affecting coagulation; evidence of respiratory insufficiency, hypotension, or bradycardia; a history of migraine, frequent headaches, seizures, or significant renal, hepatic, cardiovascular, metabolic, neurologic, or psychiatric disease; or a history of alcohol or drug abuse, hepatitis B or C, or human immunodeficiency virus infection. Further ineligibility criteria included subjects with other painful physical conditions that could interfere with the assessment of postoperative pain; those with a body mass index >35 kg/m2; and pregnant or lactating females. Subjects receiving various other medications were also excluded, including those taking opioids long term (ie, for >30 consecutive days in the past year) and those taking antiepileptic and antidepressant drugs, neuroleptics, systemic or intra-articular corticosteroids (within 6 weeks prior to the study), sedative/hypnotic drugs, warfarin, lithium, and/or combinations of furosemide with either an angiotensin-converting enzyme inhibitor or angiotensin receptor blocker. Also prohibited were other analgesics (except prespecified drugs for postoperative rescue use), prophylactic antiemetics, epidural or spinal anesthetics, and pre-, intra-, or postoperative corticosteroids.
The study was approved by an independent institutional review board (Sterling IRB, Atlanta, GA, USA). All subjects enrolled provided written informed consent prior to their participation. All clinical work was conducted in compliance with Good Clinical Practices (as referenced in the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use guideline E6), local regulatory requirements, and the principles of the Declaration of Helsinki.
Study procedures
All subjects underwent the Austin bunion procedure21 involving osteotomy of the first metatarsal with internal fixation, which was required to be completed within 120 minutes and no later than approximately 6 pm on Day –1 of the study. The surgery was performed while the subject was under a standardized anesthesia regimen consisting of a continuous popliteal sciatic nerve block and local anesthesia, with IV midazolam and propofol for sedation. On postoperative Day 1, the continuous popliteal sciatic nerve block was maintained until 3 am, and other analgesic measures (eg, topical ice and ketorolac and/or morphine) were discontinued prior to this time.
After cessation of the popliteal sciatic nerve block and discontinuation of all other analgesic measures, subjects with an NPRS score of ≥4 and a rating of moderate or severe pain on a 4-point categorical pain rating scale (ie, none, mild, moderate, severe) prior to 12 pm on postoperative Day 1 were deemed eligible for study participation (n=59) and were randomized (1:1:1 via a computer-generated method) within 15 minutes to one of three treatment groups: meloxicam IV 30 mg, meloxicam IV 60 mg, or placebo IV (5% dextrose in water). The study medication, meloxicam IV or placebo, was administered as an IV bolus over 15–30 seconds under double-blind conditions, such that the subjects, investigators, and site staff involved with collecting safety and efficacy data were unaware of the assigned treatment. A second IV dose of study medication was administered 24 hours after the first dose, with the option of a third dose prior to discharge from the study center (after the initial 48 hours), at the discretion of the investigator and the subject.
Subjects were assessed for AEs and PI at various time points after administration of the first dose of study medication (Hour 0). PI was evaluated using the 11-point NPRS (0= no pain, 10= worst pain imaginable) prior to and at 0.25, 0.5, 0.75, 1, and 2 hours after the first dose. Thereafter, pain assessments were made every 2 hours until Hour 48. PI also was assessed within 5 minutes before administration of each dose of rescue medication (oral oxycodone 5 mg administered up to every 2 hours as needed).
Subjects were discharged following the 48-hour evaluations. Follow-up assessments were performed on Day 7 (at a clinic visit) and Day 30 (via a telephone call).
Outcome measures
Safety endpoints
These included the occurrence of AEs and serious AEs; wound-healing status; changes in vital signs from baseline and the incidence of clinically significant changes in vital signs; changes from baseline in clinical laboratory test parameters and the incidence of abnormal values; and the incidence of clinically significant abnormal ECG findings. Vital signs including resting blood pressure, resting pulse, respiratory rate, and peripheral oxygen saturation were measured and recorded 15 minutes before dosing and 0.5, 1, 2, and 6 hours after dosing (±15 minutes). Vital signs were also recorded at Hour 48 and at Day 7. A 12-lead ECG was completed at screening, at check-in on Day -1 (if screening ECG was done >7 days prior to Day -1), at Hour 48 prior to discharge, at the Day 7 visit, and at time of early discontinuation. Urine and blood samples were collected for routine clinical laboratory testing during the screening visit, on Day -1 during check-in, at the Day 7 visit, and in the event of subject early discontinuation.
Efficacy endpoints
The PID at each time point was calculated, and SPID values were determined by multiplying a weight factor to each score prior to summation; the weight factor at each time point was the time (in minutes) elapsed since the previous observation. The primary efficacy endpoint, the effect size (point estimates with 95% CIs), was determined based on the difference in SPID48 values for each dose of meloxicam IV as compared to placebo.
Secondary efficacy endpoints were as follows:
- SPID values at other time points, specifically 6, 12, and 24 hours (SPID6, SPID12, and SPID24, respectively) and for the intervals 12–24, 12–48, and 24–48 hours (SPID12–24, SPID12–48, and SPID24–48, respectively), all of which were determined by the same method used for SPID48 values.
- The proportions of subjects with ≥30% and ≥50% overall reductions in pain from baseline within the first 6 and 24 hours after administration of the first dose of study medication.
- Patients’ global assessment (PGA) of their pain-control method at 24 and 48 hours after the first dose, scored on a 5-point scale (0= poor, 1= fair, 2= good, 3= very good, 4= excellent).
- The time to administration of the first dose of rescue analgesia (oral oxycodone 5 mg administered up to every 2 hours, if necessary), and the number of times rescue analgesia was required during each of the following periods: 0–24, 24–48, and 0–48 hours.
Statistical analysis
All randomized subjects (ie, the intention-to-treat population) were included in the safety and efficacy analyses. For the safety analysis, AEs were summarized by treatment group. The Medical Dictionary for Regulatory Activities (version 18.1) was used to classify all AEs by system organ class and preferred term.
Changes in vital signs at each assessment point were summarized by treatment group using descriptive statistics. The numbers and proportions of subjects with abnormal ECG findings at each assessment point were summarized by treatment group.
For the efficacy analysis, differences in SPID values between the study groups were assessed by analysis of covariance, with the baseline pain score as a covariate.
Least-squares (LS) mean and standard error (SE) SPID values for each treatment group were determined, and the effect size was calculated from differences in LS mean values versus placebo and the pooled SD derived from the analysis of covariance model. Between-group differences in LS mean SPID values were also tested using paired-sample Student’s t-tests. Differences in the proportions of subjects achieving ≥30% and ≥50% overall reductions in pain from baseline were tested using Fisher’s exact test. Exact CIs of the differences and common odds ratios for the proportions of subjects meeting the criteria were calculated.
Differences in PGA scores between treatment groups were evaluated by the exact Mantel-Haenszel chi-square test. Time to first use of rescue analgesia was analyzed by Kaplan-Meier methodology, and the differences in these times were evaluated by a log-rank test. All tests were two-sided, and P-values of ≤0.05 were deemed significant. Nominal P-values were reported without adjustment for multiple comparisons.
To address the impact of rescue analgesia on the response to study medication, two methods were used: a last observation carried forward (LOCF) method and a 2-hour windowed LOCF (W2LOCF) method. For the LOCF analysis, the PI score obtained before the first dose of rescue medication was carried forward to replace all PI scores obtained after the rescue dose. For the W2LOCF analysis, the PI score obtained before each dose of rescue medication was carried forward to replace the PI scores collected in the following 2-hour window. Although LOCF initially was considered the primary analysis method for this study, it was later determined, from consultations with the regulatory agency, that W2LOCF was a more appropriate method for imputation in this setting. Consequently, the W2LOCF method was used for all results, and the LOCF method became a supportive analysis tool. All analyses were performed using SAS versions 9.1 and 9.3 (SAS Institute Inc., Cary, NC, USA).
Results
Of the 93 subjects screened for suitability for participation, 59 who satisfactorily completed the surgery and met all study criteria (including the required level of postoperative pain) were randomized to one of the three study groups. All randomized subjects received study medication and were included in the safety and efficacy analyses.
The demographic and clinical characteristics of the study groups were comparable (Table 1). The age range of the study population was 18–72 years. Most subjects (81.4%) were female; 50.8% were black or African American, and 47.5% were white. The study site did not target recruitment to any demographic subgroup and the predominance of females is consistent with the epidemiology of this condition.
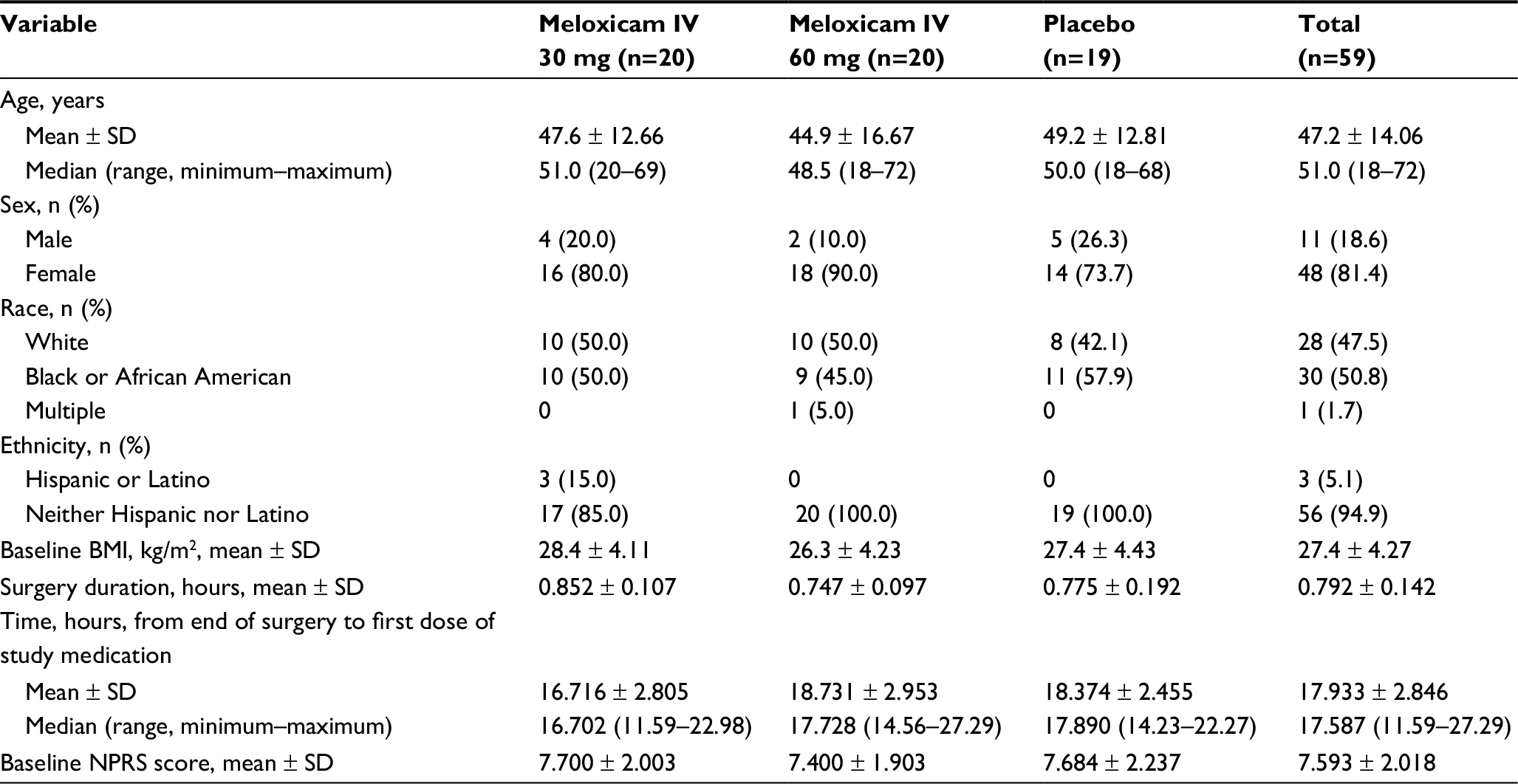 | Table 1 Demographic and clinical characteristics of enrolled subjects Abbreviations: BMI, body mass index; IV, intravenous; NPRS, numeric pain rating scale (score range, 0–10). |
During the study, treatment was discontinued in two subjects. One subject in the meloxicam IV 60 mg group was withdrawn at her request. The other subject, a placebo recipient, was withdrawn by the investigator. Therefore, 57 subjects completed the study.
Safety findings
All randomized subjects received at least one dose of study medication; 57 subjects (96.6%) received two doses, and 30 (50.8%) received a third dose after the 48-hour assessment. The findings indicated that both doses of meloxicam IV (30 and 60 mg) were generally well tolerated. Although most subjects experienced at least one treatment-emergent AE (Table 2), the majority of AEs were rated as mild and there were no meaningful differences between the study groups. No deaths or other serious AEs were reported, and no subject was discontinued from the study due to an AE. Moreover, there were no injection-related events and no apparent trends in clinically meaningful abnormal laboratory results between the study groups.
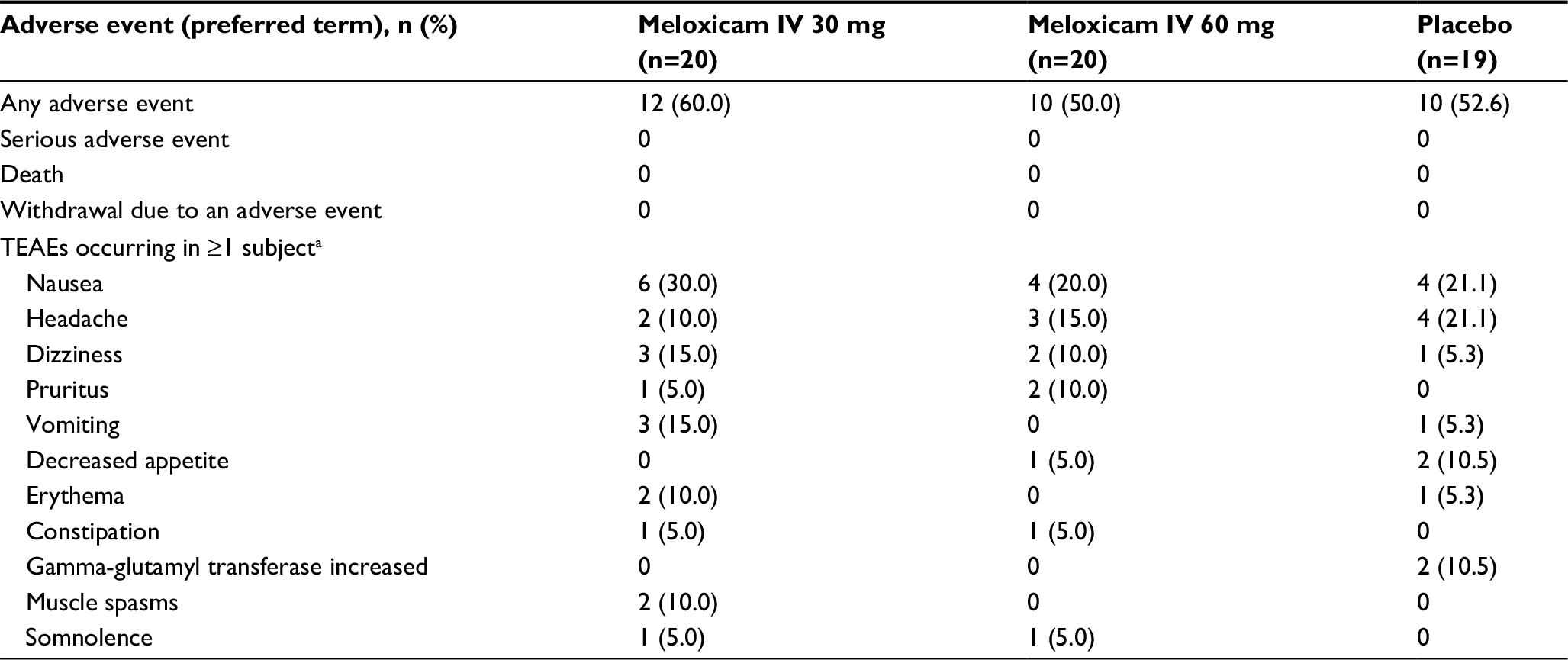 | Table 2 Treatment-emergent adverse events (TEAEs) in the three study groups Note: aThe events listed are those reported by ≥1 subject in any study group. Abbreviation: IV, intravenous. |
The results of liver function tests were normal in subjects who received meloxicam IV, as were all findings of renal function tests and urinalyses. The only clinically meaningful laboratory abnormality among recipients of meloxicam IV was a decreased white blood cell count in one subject. Because the subject had a history of this blood disorder, the abnormality was not considered related to the study treatment. Three subjects in the placebo group had clinically meaningful laboratory abnormalities (including two with increased gamma-glutamyl transferase levels), all of which resolved subsequently. There were no clinically meaningful changes in blood pressure or heart rate in subjects who received meloxicam IV or placebo.
Two subjects, one who received meloxicam IV 60 mg and one who received placebo, had abnormal wound healing. The subject who received meloxicam IV had local cellulitis (reported as an AE) associated with erythema and edema, none of which were considered treatment related. The placebo subject’s wound was described as slightly macerated; this finding was not considered clinically significant or an AE. No clinically significant abnormal ECG findings were detected during the study, and there were no injection-related AEs reported.
Efficacy findings
SPID48 and other post-dose SPID values
The effect sizes for doses of 30 mg and 60 mg meloxicam IV during the first 48 hours after administration, using the W2LOCF and LOCF analysis methods, are shown in Figure 1 and Table 3. Subjects treated with either dose of meloxicam IV had significantly greater reductions in SPID48 than subjects who received placebo (P≤0.01) according to both methods of analysis (Table 3). Both doses of meloxicam IV also produced significantly greater pain reductions than placebo at other post-dose intervals (ie, in SPID6, SPID12, SPID24, SPID12–24, SPID12–48, and SPID24–48 values) according to W2LOCF and LOCF analysis methods (P≤0.05; Table 4).
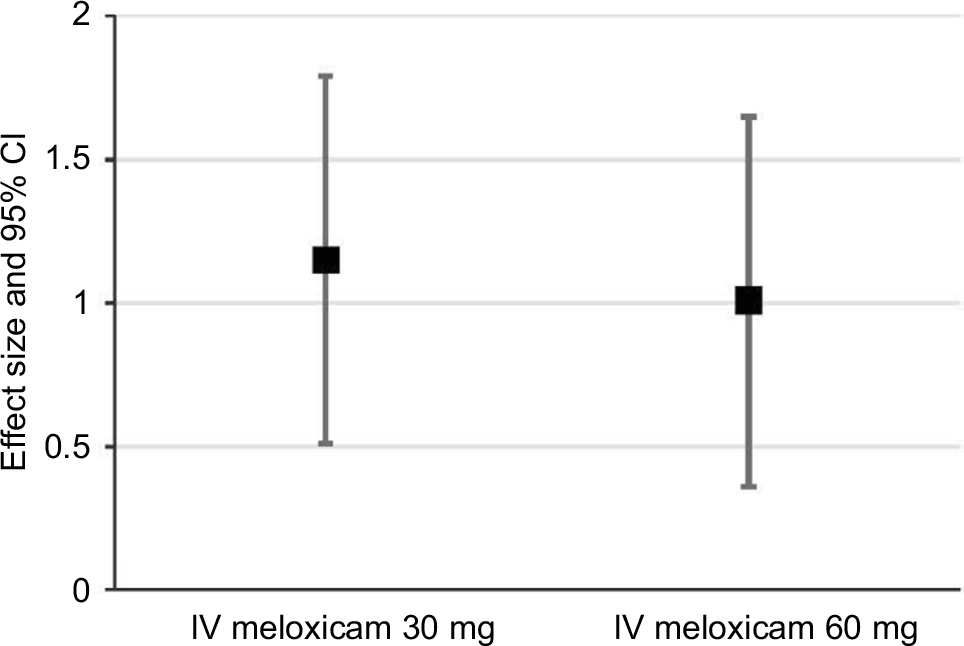 | Figure 1 SPID48 effect sizes and 95% CIs for the two doses of meloxicam IV, according to W2LOCF analysis. Abbreviations: IV, intravenous; SPID48, summed pain intensity differences over the first 48 hours; W2LOCF, 2-hour windowed last observation carried forward. |
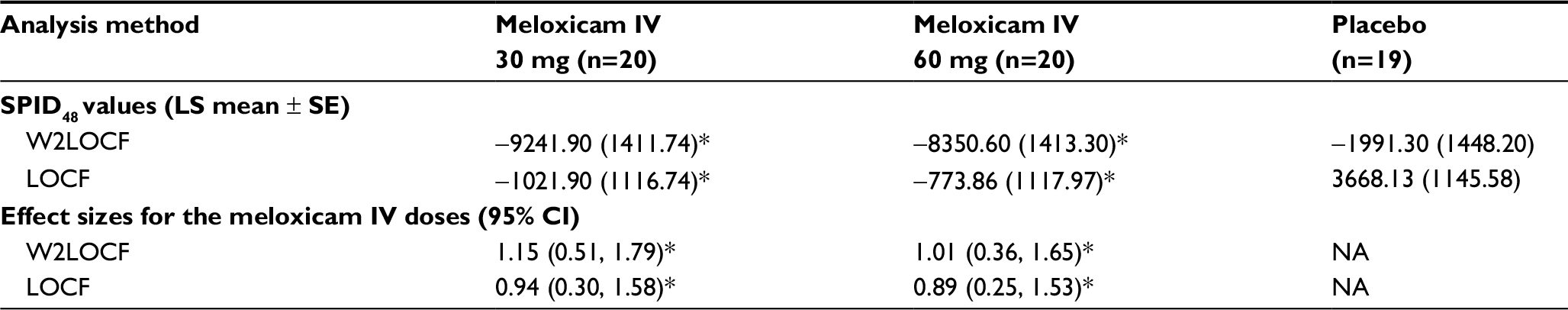 | Table 3 SPID48 values (LS mean and SE) and effect sizes (95% CI) for the two doses of meloxicam IV, by analysis method Note: *P≤0.01 versus placebo. Abbreviations: IV, intravenous; LOCF, last observation carried forward; LS, least-squares; NA, not applicable; SE, standard error; SPID48, summed pain intensity differences over the first 48 hours; W2LOCF, 2-hour windowed last observation carried forward. |
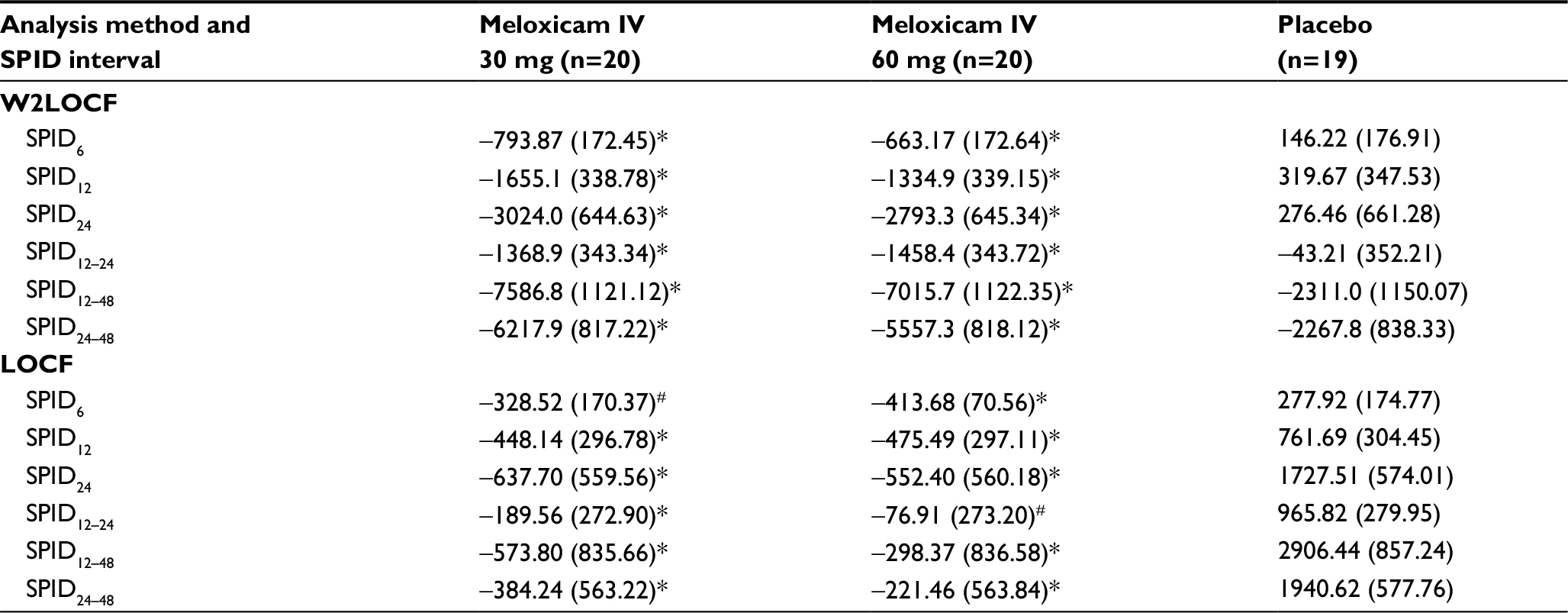 | Table 4 SPID values at other times/intervals, by analysis method (LS mean and SE) Notes: *P≤0.01 versus placebo; #P≤0.05 versus placebo. Abbreviations: LOCF, last observation carried forward; LS, least-squares; SE, standard error; SPID, summed pain intensity differences; W2LOCF, 2-hour windowed last observation carried forward. |
PID at each time point
Mean PID values versus baseline for the three treatment groups over 48 hours, using the W2LOCF assessment method, are shown in Figure 2A. Statistically significant decreases in pain from baseline (ie, negative PID values) were detected as early as 15 minutes after the first dose of both meloxicam IV 30 mg and meloxicam IV 60 mg (Figure 2B). At this time, mean PID values versus baseline with the two doses were −1.5 ± 2.44 (P<0.05) and −0.7 ± 1.22 (P<0.05), respectively.
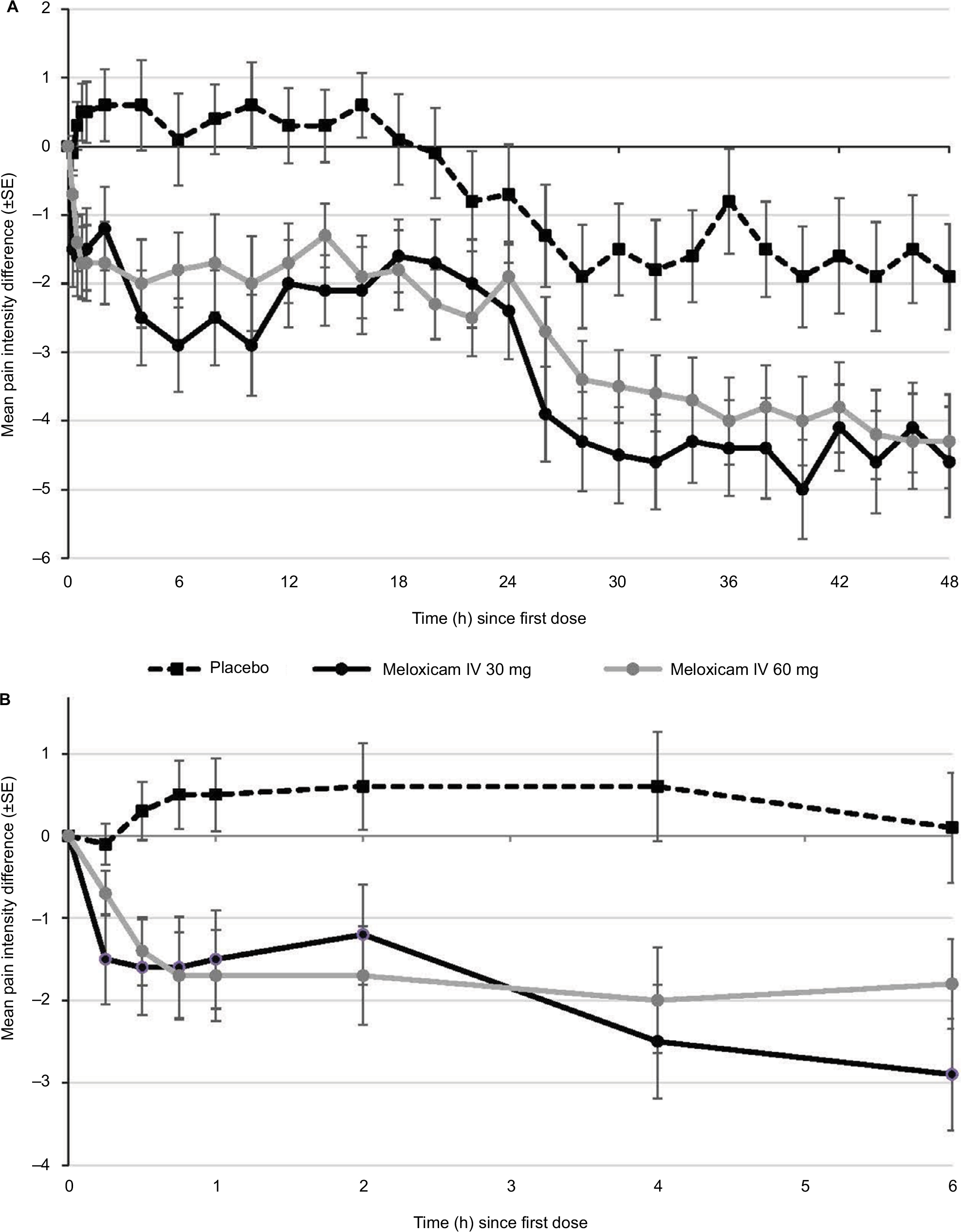 | Figure 2 Mean pain intensity differences according to W2LOCF analysis in the three study groups from (A) baseline through 48 hours, and (B) baseline through the first 6 hours. Abbreviations: IV, intravenous; SE, standard error; W2LOCF, 2-hour windowed last observation carried forward. |
The mean PI changes from baseline at all assessment points were less than zero with both doses of meloxicam IV (ie, the pain level was lower than at baseline); at the 48-hour assessment, the changes were −4.6 ± 3.61 and −4.3 ± 3.06 with meloxicam IV doses of 30 mg and 60 mg, respectively (both P<0.001 versus baseline). With placebo, the mean PID at 0.25 hours was –0.1. Thereafter, PI changes from baseline were positive until the 20-hour assessment, and a significant difference versus baseline was not detected until Hour 28.
Response analysis
The proportions of subjects with pain reduction of ≥30% and ≥50% from baseline during the first 6 and 24 hours were determined by the W2LOCF and LOCF analysis methods. According to W2LOCF analysis, significantly more subjects who received meloxicam IV 30 mg doses had pain reductions of ≥30% and ≥50% over 6 hours and 24 hours after receiving the first dose in comparison with those who received placebo (P≤0.05; Figure 3). However, the LOCF analysis method did not show a statistically significant difference versus placebo with doses of 30 mg, and neither analysis method demonstrated a significant difference with meloxicam IV doses of 60 mg versus placebo.
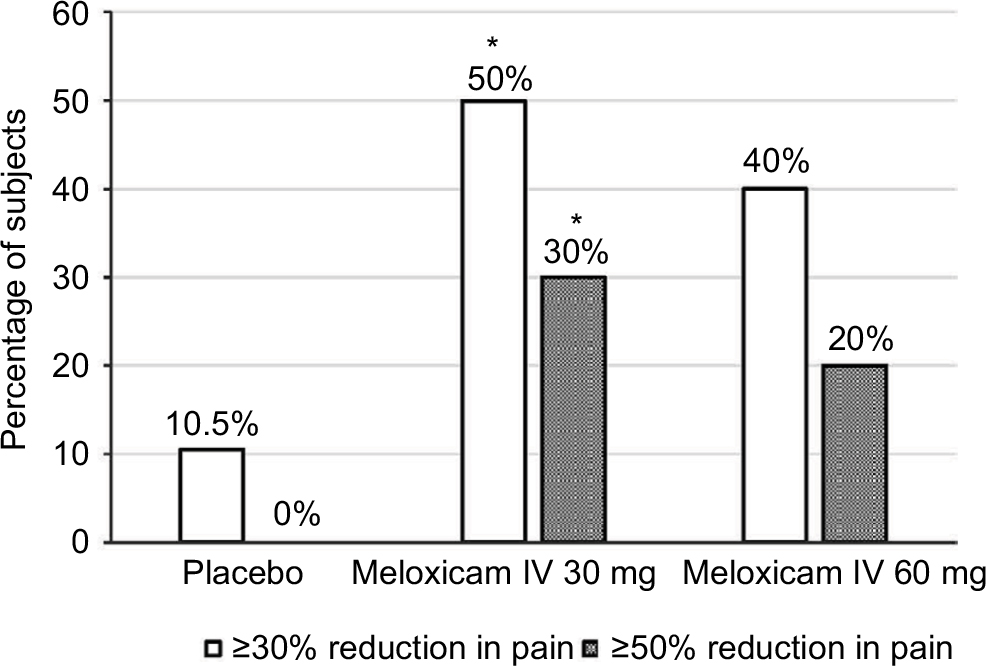 | Figure 3 Percentages of subjects with pain reductions of ≥30% and ≥50% from baseline to 24 hours, according to W2LOCF analysis (*P≤0.05 vs placebo). Abbreviations: IV, intravenous; W2LOCF, 2-hour windowed last observation carried forward. |
PGA of the pain-control method
No statistically significant differences in PGA scores between the three study groups were observed at 24 or 48 hours after administration of the first dose of study medication. However, compared with placebo recipients, more subjects in both meloxicam IV groups reported “good” or better pain control (ie, a rating of ≥2 on the 5-point scale) at these times (Figure 4).
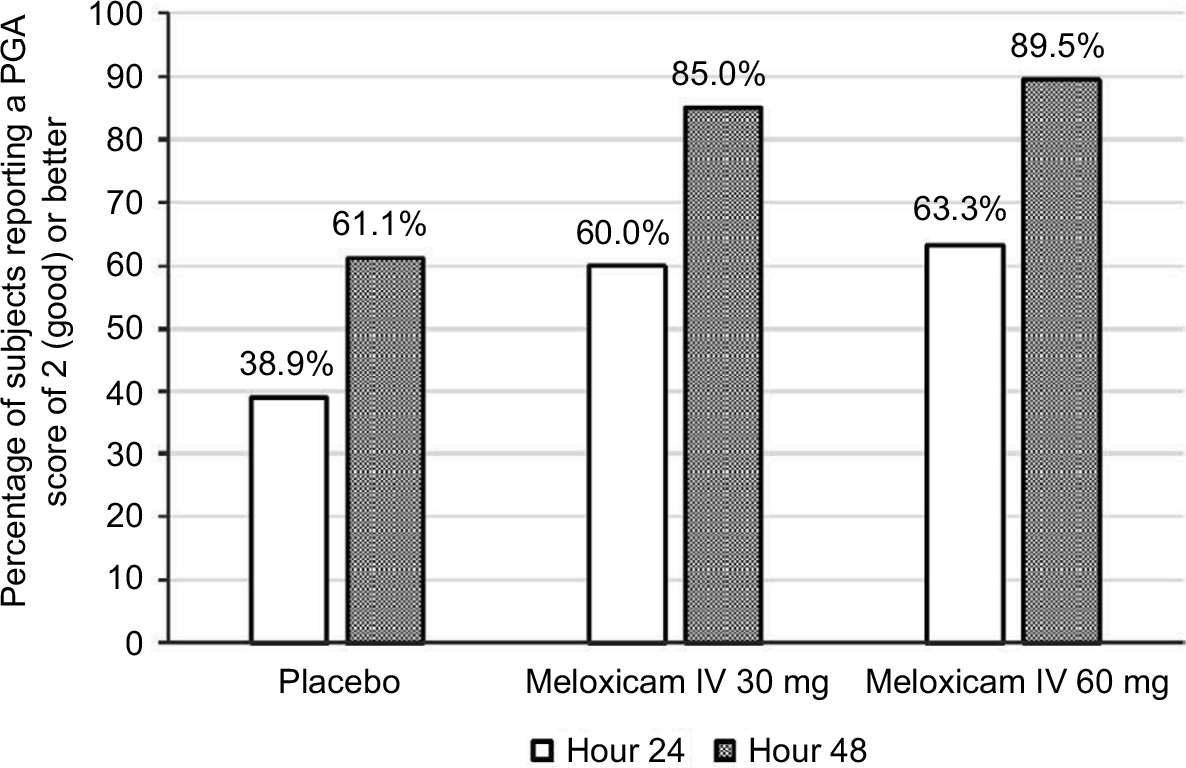 | Figure 4 Percentages of subjects who reported “good” or better pain control (score ≥2 on the 5-point patient global assessment [PGA] scale) at 24 and 48 hours after the first dose of study medication. Abbreviation: IV, intravenous. |
Rescue analgesia
Oral oxycodone 5 mg, the rescue medication used in this study, was required by ≥90% of subjects in all three study groups within the first 24 hours of receiving the initial dose of study medication. However, the percentage of subjects who required rescue analgesia during the second 24 hours (Hours 24–48) was lower with both doses of meloxicam IV than with placebo (55.0%, 52.6%, and 77.8%, respectively). Although the time to first use of rescue medication was significantly longer with meloxicam IV doses of 60 mg than with placebo (median, 3.10 hours versus 1.57 hours; P<0.05), there was no significant difference between the meloxicam IV 30 mg dose and placebo.
Subjects treated with either dose of meloxicam IV required fewer doses of rescue analgesia than those who received placebo (average number of rescue doses: 8.2, 6.9, and 11.1 with meloxicam IV 30 mg and 60 mg doses and placebo, respectively), but the differences were not statistically significant.
Discussion
This randomized, double-blind, placebo-controlled study was conducted in subjects who experienced moderate-to-severe pain after bunionectomy, a postoperative pain model that has proven useful for assessing the analgesic efficacy of NSAIDs, COX-2 inhibitors, and other drugs.3 The study’s design was consistent with current Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials (IMMPACT) recommendations for short-duration, acute-pain trials.2 This included using standardized surgical, anesthetic, and postoperative care regimens.
The primary objective of the study was to evaluate the safety of the IV nanocrystal formulation of meloxicam administered as a bolus injection over 15–30 seconds in subjects who had undergone bunionectomy. At doses of both 30 mg and 60 mg, meloxicam IV was generally well tolerated when administered as a bolus injection once daily. Most AEs that occurred with meloxicam IV were rated mild in intensity, and their incidence was similar to that in the placebo group – including adverse gastrointestinal events such as nausea, constipation, gastric or peptic ulcers, and gastroesophageal reflux disease. There was no increase in the incidence or severity of AEs over time. No injection-related AEs were reported with either dose of meloxicam IV, and there were no clinically meaningful changes as determined by the primary investigator, in vital signs or laboratory parameters (including hepatic and renal function) and no clinically significant abnormal ECG changes. There was no evidence of abnormal wound healing attributable to meloxicam IV as evaluated by the PI.
Although the study originally was not statistically powered to demonstrate efficacy, results numerically favored both doses of meloxicam IV push (30 mg and 60 mg, given once daily) over placebo in relieving moderate-to-severe post-bunionectomy pain at all assessment times during the 48-hour study period, regardless of the analysis method (LOCF or W2LOCF). A statistically significant difference versus placebo was observed with both doses of meloxicam IV for SPID48 values, and also for SPID6, SPID12, SPID24, SPID12–24, SPID12–48, and SPID24–48 values. The significant differences in SPID12–24 values versus placebo with the two doses indicate that the analgesic effect of meloxicam IV remains significant throughout the second half of the dosing interval.
Meloxicam IV had a rapid onset of action; significant decreases in PID values from baseline were detected with both doses as early as 15 minutes after administration. Moreover, compared with placebo, meloxicam IV 30 mg resulted in significantly more subjects with PI reductions of ≥30% and ≥50% from baseline within the first 6 and 24 hours after the initiation of treatment, according to W2LOCF analysis. The time to first use of rescue medication was significantly longer with the meloxicam IV 60 mg doses than with placebo and, overall, fewer doses of rescue medication were used by subjects who received meloxicam IV. Although differences in rescue medication usage between the meloxicam IV and placebo groups were not statistically significant (likely due to the small sample size), the rescue analgesia findings from this study indicate that, as with other NSAIDs administered intravenously in postoperative pain settings,22–26 meloxicam IV may have an opioid-sparing effect. This may be beneficial in reducing the occurrence of opioid-induced AEs that, in some patients, may slow postoperative recovery. Determining whether the opioid-sparing effect of meloxicam IV is clinically significant will require investigation in larger studies.
To address the impact of rescue medication on the treatment response in this study, two methods of analysis were used: LOCF and W2LOCF. Although LOCF was initially stated to be the primary method of analysis, the W2LOCF method was ultimately used for the primary analysis, following its identification as a more appropriate method for imputation. The LOCF method served as a supportive analysis. For most efficacy evaluations, PI results determined by the two methods were consistent. The main difference pertained to the proportions of subjects with pain reductions of ≥30% and ≥50% from baseline in the first 6 hours and 24 hours after the initial dose of meloxicam IV 30 mg. The proportions of subjects with these overall pain reductions were significantly greater with the 30 mg dose than with placebo according to the W2LOCF analysis method but not the LOCF method.
Limitations of this study include the relatively small sample size and the possibility of some intersubject variability. Although the surgical procedure, anesthesia protocol, and initial postoperative management regimens were standardized as much as possible to minimize intersubject variability in this study, it is recognized that hemodynamic fluctuations and other intraoperative events may have necessitated some deviation from standard regimens, which may have resulted in intersubject variability. Further, while the extended duration of hospital stay (48 hours) was required for clinical assessment, this may not reflect the current standard of care.
Conclusion
Meloxicam IV was generally safe and well tolerated in subjects with moderate-to-severe post-bunionectomy pain, yielding a low incidence of AEs and no injection-related events. Most AEs were rated mild in intensity, and their incidence was similar to that in the placebo group and did not increase over time.
In terms of efficacy, doses of 30 mg and 60 mg meloxicam IV administered once daily by bolus injection over 15–30 seconds produced meaningful effect sizes according to W2LOCF and LOCF analysis methods, and achieved significantly better LS mean SPID48 values than placebo. Moreover, once-daily dosing of meloxicam IV produced a rapid onset of analgesic activity (within 15 minutes after administration) and maintained analgesia for the 48-hour study period. Consequently, findings from this study support further (Phase 3) investigations of the efficacy and safety of once-daily administration of meloxicam IV to subjects with moderate-to-severe postoperative pain.
Acknowledgments
Funding for this research was provided by Recro Pharma, Inc., Malvern, PA, USA.
Assistance with manuscript preparation was provided by Trevor Speight, MPS, and Susan Martin, PhD, of The Medicine Group, and was paid for by Recro Pharma, Inc., Malvern, PA, USA.
Author contributions
All authors contributed to the design of the study and its implementation. IJG and DRT managed the subjects and contributed to acquisition of the data. WD provided statistical guidance for data analysis and interpretation. All authors contributed to drafting the manuscript and/or critically reviewing it for important intellectual content. All have read and approved the final version for journal submission, and agree to be accountable for all aspects of the work.
Disclosure
Portions of this manuscript have previously been presented at the 2016 PAINWeek National Conference; September 6–10th, 2016.
Ira J Gottlieb and Deborah R Tunick are employees of Chesapeake Research Group, which conducted this trial. Wei Du and Campbell P Howard received consultancy fees from Recro Pharma, Inc., Malvern, PA, USA. Randall J Mack, Stewart W McCallum, and Alex Freyer are employees and security holders of Recro Pharma, Inc., Malvern, PA, USA.
The authors have no other conflicts of interest to declare with respect to this work.
References
Gadek A, Liszka H, Wordliczek J. Postoperative pain and preemptive local anesthetic infiltration in hallux valgus surgery. Foot Ankle Int. 2015;36(3):277–281. | ||
Cooper SA, Desjardins PJ, Turk DC, et al. Research design considerations for single-dose analgesic clinical trials in acute pain: IMMPACT recommendations. Pain. 2016;157(2):288–301. | ||
Wang H, Gargano C, Lukac S, et al. An enhanced bunionectomy model as a potential tool for early decision-making in the development of new analgesics. Adv Ther. 2010;27(12):963–980. | ||
American Society of Anesthesiologists Task Force on Acute Pain Management. Practice guidelines for acute pain management in the perioperative setting: an updated report by the American Society of Anesthesiologists Task Force on Acute Pain Management. Anesthesiology. 2004;100(6):1573–1581. | ||
White PF. What are the advantages of non-opioid analgesic techniques in the management of acute and chronic pain? Expert Opin Pharmacother. 2017;18(4):329–333. | ||
White PF, Kehlet H. Improving postoperative pain management: what are the unresolved issues? Anesthesiology. 2010;112(1):220–225. | ||
Golf M, Daniels SE, Onel E. A phase 3, randomized, placebo-controlled trial of DepoFoam(R) bupivacaine (extended-release bupivacaine local analgesic) in bunionectomy. Adv Ther. 2011;28(9):776–788. | ||
Turan I, Assareh H, Rolf C, Jakobsson J. Multi-modal-analgesia for pain management after Hallux Valgus surgery: a prospective randomised study on the effect of ankle block. J Orthop Surg Res. 2007;2:26. | ||
Singla NK, Muse DD, Evashenk MA, Palmer PP. A dose-finding study of sufentanil sublingual microtablets for the management of postoperative bunionectomy pain. J Trauma Acute Care Surg. 2014;77(3 Suppl 2):S198–203. | ||
Richards P, Riff D, Kelen R, Stern W. A phase 3, randomized, double-blind comparison of analgesic efficacy and tolerability of Q8003 vs oxycodone or morphine for moderate-to-severe postoperative pain following bunionectomy surgery. Pain Med. 2013;14(8):1230–1238. | ||
Daniels SE, Spivey RJ, Singla S, Golf M, Clark FJ. Efficacy and safety of oxycodone HCl/niacin tablets for the treatment of moderate-to-severe postoperative pain following bunionectomy surgery. Curr Med Res Opin. 2011;27(3):593–603. | ||
Willens JS, Bucior I, Bujanover S, Mehta N. Assessment of rescue opioid use in patients with post-bunionectomy pain treated with diclofenac potassium liquid-filled capsules. J Pain Res. 2015;8:53–62. | ||
Smith HS, Baird W. Meloxicam and selective COX-2 inhibitors in the management of pain in the palliative care population. Am J Hosp Palliat Care. 2003;20(4):297–306. | ||
Del Tacca M, Colucci R, Fornai M, Blandizzi C. Efficacy and tolerability of meloxicam, a COX-2 preferential nonsteroidal anti-inflammatory drug. A review. Clin Drug Invest. 2002;22(12):799–818. | ||
Chen YF, Jobanputra P, Barton P, Bryan S, Fry-Smith A, Harris G, Taylor RS. Cyclooxygenase-2 selective non-steroidal anti-inflammatory drugs (etodolac, meloxicam, celecoxib, rofecoxib, etoricoxib, valdecoxib and lumiracoxib) for osteoarthritis and rheumatoid arthritis: a systematic review and economic evaluation. Health Technol Assess. 2008;12(11):1–278, iii. | ||
Ahmed M, Khanna D, Furst DE. Meloxicam in rheumatoid arthritis. Expert Opin Drug Metab Toxicol. 2005;1(4):739–751. | ||
Busch U, Heinzel G, Narjes H. Effect of food on pharmacokinetics of meloxicam, a new non steroidal anti-inflammatory drug (NSAID). Agents Actions. 1991;32(1–2):52–53. | ||
Davies NM, Skjodt NM. Clinical pharmacokinetics of meloxicam. A cyclo-oxygenase-2 preferential nonsteroidal anti-inflammatory drug. Clin Pharmacokinet. 1999;36(2):115–126. | ||
Ochi M, Kawachi T, Toita E, Hashimoto I, Yuminoki K, Onoue S, Hashimoto N. Development of nanocrystal formulation of meloxicam with improved dissolution and pharmacokinetic behaviors. Int J Pharm. 2014;474(1–2):151–156. | ||
Mack R, Freyer A, Du W. An evaluation of the efficacy and safety of N1539, a novel intravenous formulation of nanocrystal meloxicam, in subjects with moderate to severe pain following hysterectomy (Abstr. 409). J Pain. 2016;17(4S):S77. | ||
Austin DW, Leventen EO. A new osteotomy for hallux valgus: a horizontally directed “V” displacement osteotomy of the metatarsal head for hallux valgus and primus varus. Clin Orthop Relat Res. 1981;(157):25–30. | ||
Alexander R, El-Moalem HE, Gan TJ. Comparison of the morphine-sparing effects of diclofenac sodium and ketorolac tromethamine after major orthopedic surgery. J Clin Anesth. 2002;14(3):187–192. | ||
Cepeda MS, Carr DB, Miranda N, Diaz A, Silva C, Morales O. Comparison of morphine, ketorolac, and their combination for postoperative pain: results from a large, randomized, double-blind trial. Anesthesiology. 2005;103(6):1225–1232. | ||
Southworth S, Peters J, Rock A, Pavliv L. A multicenter, randomized, double-blind, placebo-controlled trial of intravenous ibuprofen 400 and 800 mg every 6 hours in the management of postoperative pain. Clin Ther. 2009;31(9):1922–1935. | ||
Gan TJ, Singla N, Daniels SE, et al. Postoperative opioid sparing with injectable hydroxypropyl-beta-cyclodextrin-diclofenac: pooled analysis of data from two phase III clinical trials. J Pain Res. 2016;10:15–29. | ||
Apfelbaum JL, Desjardins PJ, Brown MT, Verburg KM. Multiple-day efficacy of parecoxib sodium treatment in postoperative bunionectomy pain. Clin J Pain. 2008;24(9):784–792. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.