")
Back to Journals » International Journal of Nanomedicine » Volume 13
Evaluation of the potential of a simplified co-delivery system with oligodeoxynucleotides as a drug carrier for enhanced antitumor effect
Authors Liu CX, Liu TX, Liu YJ, Zhang N
Received 25 October 2017
Accepted for publication 17 February 2018
Published 20 April 2018 Volume 2018:13 Pages 2435—2445
DOI https://doi.org/10.2147/IJN.S155135
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Linlin Sun
Chunxi Liu,1,* Tingxian Liu,2,* Yongjun Liu,2 Na Zhang2
1Department of Pharmacy, Qilu Hospital, Shandong University, Ji’nan, China; 2School of Pharmaceutical Science, Shandong University, Ji’nan, China
*These authors contributed equally to this work
Background: We previously developed a simple effective system based on oligodeoxynucleotides with CGA repeating units (CGA-ODNs) for Dox and siRNA intracellular co-delivery.
Methods: In the present study, the in vitro cytotoxicity, gene transfection and in vivo safety of the co-delivery system were further characterized and discussed.
Results: Compared with poly(ethyleneimine) (PEI), both CGA-ODNs and the pH-sensitive targeted coating, o-carboxymethyl-chitosan (CMCS)-poly(ethylene glycol) (PEG)-aspargine-glycine-arginine (NGR) (CMCS-PEG-NGR, CPN) showed no obvious cytotoxicity in 72 h. The excellent transfection capability of CPN coated Dox and siRNA co-loaded nanoparticles (CPN-PDR) was confirmed by real-time PCR and Western blot analysis. It was calculated that there was no significant difference in silencing efficiency among Lipo/siRNA, CPN-modified siRNA-loaded nanoparticles (CPN-PR) and CPN-PDR. Furthermore, CPN-PDR was observed to be significantly much more toxic than Dox- and CPN-modified Dox-loaded nanoparticles (CPN-PD), implying their higher antitumor potential. Both hemolysis tests and histological assessment implied that CPN-PDR was safe for intravenous injection with nontoxicity and good biocompatibility in vitro and in vivo.
Conclusion: The results indicated that CPN-PDR could be a potentially promising co-delivery carrier for enhanced antitumor therapy.
Keywords: co-delivery, doxorubicin, VEGF, cytotoxicity, transfection
Introduction
Cancer has been one of the leading causes of death worldwide, and mortality and morbidity continue to increase.1 Chemotherapy is one of the major strategies for cancer therapy. However, mono-chemotherapy may encounter problems including drug resistance and unspecific toxicity. To overcome the high mortality rate of cancer, new therapeutic strategies, such as combinational therapy, have been developed.2,3 It has been reported that a combination of chemotherapy and gene therapy could potentially achieve synergistic effects and improve target selectivity, and deter the development of cancer drug resistance.4 For example, a double-modulating strategy based on the combination of chemotherapeutic agent 5-fluorouracil (5-FU) and siRNA has been developed, in which chemotherapy enhances intratumoral siRNA delivery and the delivered siRNA enhances the chemosensitivity of tumors. Furthermore, combination therapy may compensate for the limited delivery of siRNA to tumor tissue and probably manage the 5-FU-resistant tumors.5
RNA interference (RNAi) is a natural cellular process that regulates gene expression level. Small interfering RNAs (siRNAs), which are small double-stranded RNAs, 20–24 nucleotides (nts) in length with sequences complementary to a gene’s coding sequence, can induce degradation of corresponding messenger RNAs (mRNAs), thus blocking the translation of the mRNA into protein.6,7 The key therapeutic advantage of using RNAi lies in its ability to specifically and potently knock down the expression of disease-causing genes of known sequence. The high specificity and potency makes it widely used in treating various cancers such as breast, liver, and lung cancers.8–10 In recent years, combination of chemotherapy and siRNA-induced RNAi has become a hot topic in cancer treatment. For example, co-delivery of siRNA targeting multidrug resistance protein 1 (MDR1) or anti-survivin siRNA and chemotherapeutic drugs was demonstrated to be a promising strategy to overcome drug resistance.11–13 Anti-apoptotic gene Bcl-2 is a potential combinational therapy target owing to its important role in cell apoptosis, and combination of Bcl-2 siRNA and 5-FU could induce a remarkable increase of cell apoptosis both in vitro and in vivo.14
To achieve the desired combinational and synergistic effects of chemotherapy and RNAi, selection of siRNA is important, and the selection of siRNA is also the section of silencing target. The mechanism for combinational anticancer effect varies with different silencing targets. For example, siRNA targeting MDR1 or anti-apoptotic gene Bcl-2 were used to prevent drug resistance response and sensitize chemotherapeutic drugs, or enhance the apoptosis of cancer cells, both offering enhanced anticancer effect.15,16 Tumor tissues have abundant new blood vessels that pump sufficient nutrition and oxygen for tumor growth and metastasis. The dependency of tumor tissues on these new blood vessels is important in anti-angiogenesis therapy for cancer treatment.17 Recently, there have been many efforts in achieving anti-angiogenesis, including delivering siRNAs to silence specific pro-angiogenic genes, such as vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF) and interleukin-8 (IL-8).18 Of these targets, VEGF has received a lot of attention owing to its key role in tumor angiogenesis. With the development of RNAi technology, siRNA with VEGF target has been widely explored and referred to as anti-VEGF treatment.19–21 In 2004, the success of bevacizumab showed VEGF to be a potential target for angiogenesis therapy. There are a number of novel anti-VEGF agents in phase III clinical trials that may come to market in the next few years.22,23 In our design, siRNA with VEGF target was selected to co-deliver with chemotherapeutic drugs, resulting in a combinational anticancer effect. The mechanism for this could be that anti-VEGF treatments sensitize the cells to chemotherapy agents by blocking blood supply, improve drug penetration by disruption or normalization of tumor vasculature and in addition directly kill cancer cells by gene therapy.24,25
However, naked siRNAs may not induce efficient transfection by themselves because they are unstable and vulnerable, especially to nuclease-mediated degradation; also, the negatively charged surface blocks them from cellular endocytosis. Moreover, successful transfection of siRNA into cells is based on siRNA with complete structure, which could further initiate the RNAi process for targeted gene silencing.26,27 Thus, nanoscale delivery systems, which could co-load drug and siRNA and protect siRNA from degradation, were widely used and demonstrated to be excellent candidates. These include nanoparticles,28 liposomes,29 polyplexes30 and dendrimers.31 The basic standard involves employing cationic polymers or lipids to condense negatively charged siRNA and protect them from degradation.32,33 In our previous study,25 oligodeoxynucleotides with CGA repeating units (CGA-ODNs) were introduced to load Dox to obtain Dox-loaded CGA-ODNs (CGA-ODNs-Dox). Poly(ethyleneimine) (PEI) was then used to condense siRNA and CGA ODNs-Dox to obtain Dox and siRNA co-loaded nanoparticles (PEI/CGA-ODNs-Dox and siRNA[PDR]). Finally, the pH-sensitive targeted material, o-carboxymethyl-chitosan (CMCS)-poly(ethylene glycol) (PEG)-aspargine-glycine-arginine (NGR) (CMCS-PEG-NGR[CPN]), was used to modify PDR to obtain multifunctional CPN-PDR. CPN-PDRs were demonstrated to be multifunctional, being able to co-deliver Dox and siRNA into cells, induce pH-responsive disassembly and realize endosomal escape of gene and drug. Thus, in the present study, the cytotoxicity of the materials and the delivery system are further evaluated. Then, the transfection efficiency of siRNA is monitored by semiquantitative real-time polymerase chain reaction (real-time PCR) and Western blot techniques for mRNA level and protein level, respectively. Finally, the safety of the delivery system is evaluated by hemolysis test in vitro and histological assessment.
Materials and methods
Materials
Dox was purchased from Dalian Meilun Biology Technology Co. Ltd (Dalian, China). Targeting human VEGF siRNA (sense: 5′-ACAUCACCAUGCAGAUUAUdTdT-3′, anti-sense: 5′-dTdTUGUAGUGGUACGUCUAAUA-3′) was obtained from Guangzhou Ribobio Co., Ltd (Guangzhou, China). CGA oligodeoxynucleotides (sequence: 5′-CGACGACGACGACGACGACGA-3′; complementary sequence: 5′-TCGTCGTCGTCGTCGTCGTCG-3′) were purchased from BGI Co. (Shenzhen, China). Fetal bovine serum (FBS) was obtained from Sijiqing Co., Ltd, (Zhejiang, China) and 3-[4,5- dimethyl-2-thiazolyl]-2,5-diphenyl-2H-tetrazolium bromide (MTT) was from Sigma-Aldrich (St Louis, MO, USA). Trizol RNA extraction was from Thermo Fisher Scientific (Waltham, MA, USA). SYBR®Green and ReverTra Ace qPCR RT Kit were purchased from Toyobo (Osaka, Japan). All solutions were made up in Millipore ultrapure water (EMD Millipore, Billerica, MA, USA) and sterilized for cell culture. All other chemicals and reagents were of analytical grade and used as received. All the primers used in real-time PCR were synthesized and purified by BGI Co. with sequences: VEGF – forward: 5′-CTGGAGTGTGTGCCCACTGA-3′; VEGF – reverse: 5′-TCCTATGTGCTGGCCTTGGT-3′; actin – forward: GAGCTACGAGCTGCCTGACG; actin – reverse: CCTAGAAGCATTTGCGGTGG.
Cells and animals
The MCF-7 cells were purchased from the Chinese Academy of Sciences Cells Bank (Shanghai, China), and the cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% fetal bovine serum (FBS), streptomycin at 100 μg/mL and penicillin at 100 U/mL. All cells were cultured in a 37°C incubator with 5% CO2.
Kunming mice (20±2 g) were obtained from Experimental Animal Center of Shandong University. Animal experiments were carried out according to the requirements of the Animal Management Rules of the Ministry of Health (China) and approved by the Laboratory Animal Ethics Review Committee of Qilu Medical College of Shandong University.
Preparation of nanoparticles
PDR and CPN-PDR were prepared by the method reported in our previous study.25 Briefly, CGA-ODNs-Dox was first obtained by incubating Dox with CGA-ODNs at room temperature. Then, PEI, siRNA, ODNs-Dox and CPN were dissolved and diluted to the corresponding concentrations with deionized water. After that, siRNA and CGA-ODNs-Dox were mixed by vortexing for several seconds to obtain the mixture. The mixture was added dropwise into PEI solution and mixed by vortexing and then incubated for 30 min at room temperature to form PDR. CPN-PDR was obtained by adding PDR suspension into the CPN solution under vortexing, followed by 30-min incubation at room temperature. When preparing the blank nanoparticles, the DOX and siRNA were not involved in above method. The construction of different kinds of nanoparticles is shown in Figure 1.
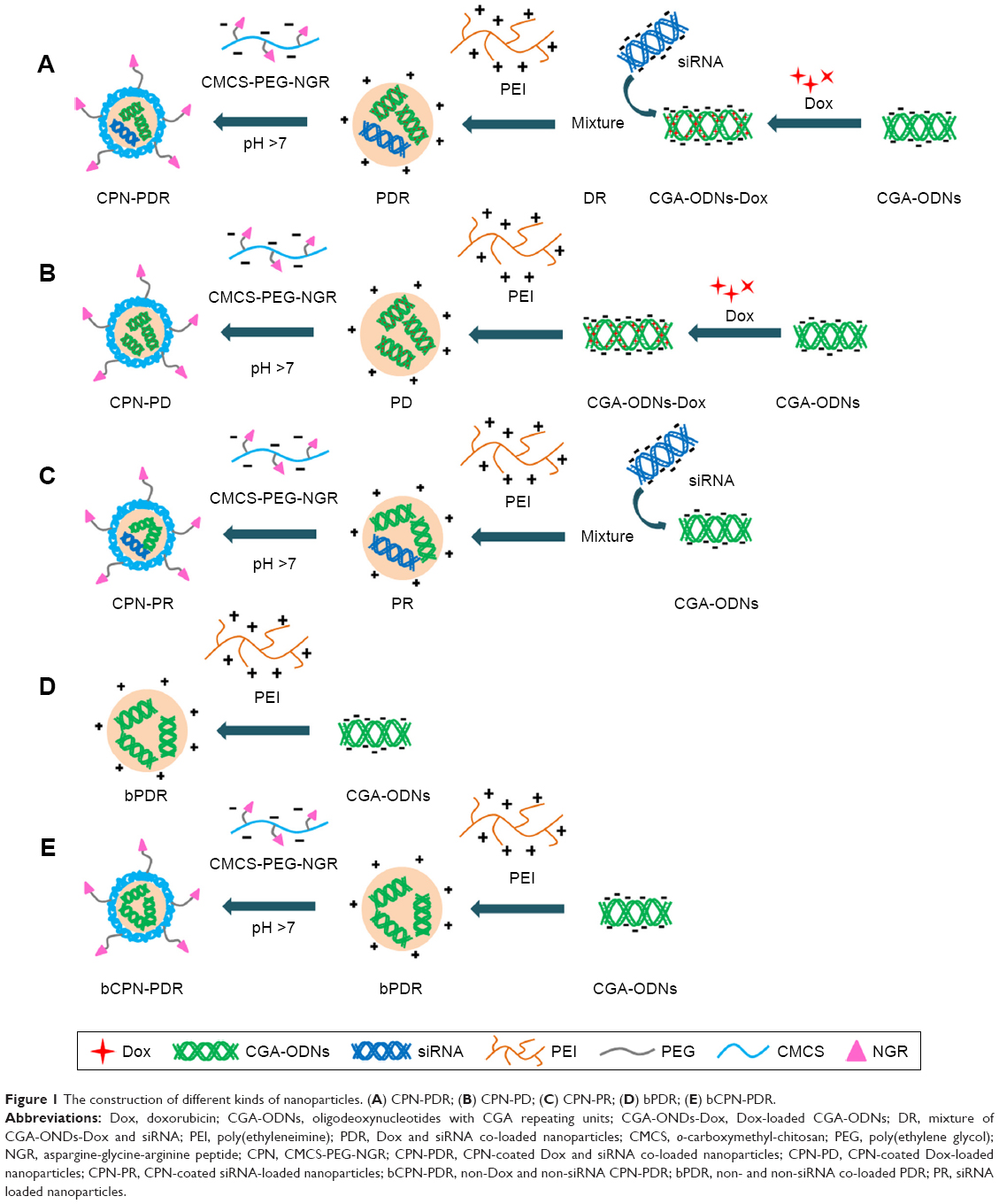 | Figure 1 The construction of different kinds of nanoparticles. (A) CPN-PDR; (B) CPN-PD; (C) CPN-PR; (D) bPDR; (E) bCPN-PDR. |
The loading content of DOX and siRNA was calculated using the following formula:34
|
|
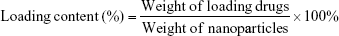
In vitro cytotoxicity of materials
MTT assays were carried out on MCF-7 cells to evaluate the in vitro cytotoxicity of CGA-ODNs, PEI and CPN, respectively.35 MCF-7 cells were seeded with density at 7,000 cells/well in the 96-well plates and allowed to adhere overnight. CGA-ODNs, PEI and CPN solutions, which were corresponding to Dox concentrations (0.25, 0.625, 1.25, 2.5 and 5 μM), were added and incubated with the cells for 24, 48 and 72 h, respectively. Five wells were set for each concentration, and cells incubated with fresh media were taken as control. At a determined time point, 20 μL MTT solutions (5 mg/mL) were added. After 4 h incubation, the plates were centrifuged (3,000 rpm, 10 min) and the supernatant was removed. Then, 200 μL DMSO solutions were added to dissolve the formazan crystals formed by the living cells. The cell viability was calculated according to the following formula (Equation 2) after recording the absorbance at 570 nm by a microplate reader (Model 680; Bio-Rad Laboratories Inc., Hercules, CA, USA). All the assays were repeated three times.
|
|

In vitro cytotoxicity of blank nanoparticles
The cytotoxicity of non-DOX- and siRNA-loaded nanoparticles (blank PDR[bPDR]) and CPN-coated blank PDR (bCPN-PDR) were also evaluated in MCF-7 cells. bPDR and bCPN-PDR solutions, corresponding to Dox concentrations (0.25, 0.625, 1.25, 2.5 and 5 μM), were added and incubated with the cells for 24, 48 and 72 h, respectively. Five wells were set for each concentration and cells incubated with fresh media were taken as control. After the indicated time periods, cell viability was evaluated according to the procedure described above.
siRNA transfection with nanoparticles
MCF-7 cells were seeded into 24-well plates at densities of 8×104, 6×104 and 5×104 cells/well for 24, 48 and 72 h transfection, respectively. After culturing at 37°C overnight, MCF-7 cells were treated with siRNA specific for VEGF. The siRNA was delivered either by Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s instructions or by CPN-modified siRNA-loaded nanoparticles (CPN-PR) or by CPN-modified Dox- and siRNA-loaded nanoparticles (CPN-PDR). The final concentrations of Dox and siRNA were 1 μM and 55 nM, respectively. After siRNA transfection, cells were harvested for RNA extraction and Western blot PCR analysis or subjected to Western blot assay.
Real-time PCR
The expression level of VEGF mRNA was monitored by real-time PCR technique with the standard procedure. Total RNA was extracted using Trizol agent under the standard procedure (Thermo Fisher Scientific), and cDNA synthesis was carried out using Rever Tra Ace qPCR RT Kit (Toyobo) according to the manufacturer’s instructions. The final concentration of RNA was measured by Nano Drop 2000 (Thermo Fisher Scientific), and the final product cDNA was stored at −20°C until use.
Each real-time PCR system contained 20 μL solutions including cDNA (2 μL), SYBR®Green mix (10 μL), primer (1.6 μL) and double distilled water (6.4 μL). The reaction system was placed into PCR instrument (Roche LightCycler™; Hoffman-La Roche Ltd, Basel, Switzerland) for analysis. β-Actin served as a reference gene. Three wells were set for each group. The expression level of VEGF mRNA was normalized to the β-actin expression level and calculated by recording the Ct value at the end of the reaction.
Western blot assay
A Western blot assay was employed to investigate the expression level of total VEGF protein after transfection. At a determined time, cells were trypsinized and pelleted by centrifugation. The protein collecting process was carried out based on the standard procedure of Total Protein Extraction Kit (BestBio, Shanghai, China). The total protein was stored at −80°C until use. Protein was quantified by BCA protein assay kit (Beyotime Biotechnology, Shanghai, China) based on the standard carve calculation (A=0.006 C+0.0919, r=0.9987). After quantification, all the samples were diluted to 2 μg/μL with sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) electrophoresis, denatured in boiling water for 5 min and stored at −80°C until use. Equal amounts of protein (50 μg) were subjected to 12% SDS-PAGE and transferred to nitrocellulose membranes using standard procedures. The membrane was blocked in 2% skimmed milk in phosphate buffered saline for 2 h and washed by Tris-buffered saline and Tween-20 (TBST) solution three times. Then the membranes were probed with primary antibodies against VEGF and glyceraldehyde 3-phosphate dehydrogenase (GAPDN) overnight at 4°C. After 3×10 min washing in TBST on shaker, membranes were incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit IgG for 1 h at room temperature. With another 3×10 min washes in TBST, the membranes were developed with electrochemiluminescence (ECL) using a gel-imaging system and analyzed using Image Analysis software.
Cell proliferation assay
The anti-proliferation activity of Dox, CPN-PD and CPN-PDR against MCF-7 cells was tested via MTT method. MCF-7 cells were seeded into 96-well plates at a density of 7,000 cells/well. After overnight incubation, the cells were treated with various concentrations (0.002, 0.01, 0.05, 0.25 and 0.5 μM) of Dox, CPN-PD and CPN-PDR solutions and incubated for another 48 h. Five wells were set for each concentration, and cells incubated with fresh media were taken as control. After indicated time periods, cell viability was evaluated according to the procedure described in the “In vitro cytotoxicity of materials” section.
Hemolysis test
Hemolysis test was carried out to investigate the safety of CPN-PDR for intravenous injection. A 2% red blood cell suspension was collected by centrifugation and resuspension. The test was performed under the design described in Table 1. After incubated at 37°C for 3 h, all the groups were centrifuged at 3,000 rpm for 15 min and visualized by the naked eye. To measure the hemolysis ratios of each group, UV-vis spectrophotometry was carried out to record the absorbance of supernatant in each group. The quantitation of hemolysis ratios was calculated according to the following formula:
|
|
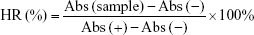
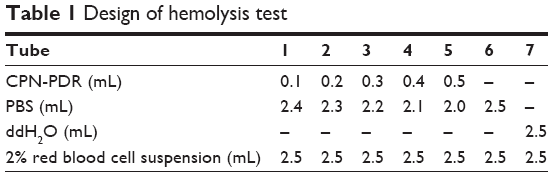 | Table 1 Design of hemolysis test |
where Abs (sample), Abs (−) and Abs (+) refer to the absorbances of the samples, negative control and positive control, respectively.
Histological assessment
In order to evaluate the compatibility and tissue toxicity of CPN-PDR in vivo, a histological observation was performed.36 Five female Kunming mice were injected with CPN-PDR at a dose of 232 μg/kg through the tail vein. In the meantime, normal saline (NS) was taken as a control reagent. After 1 week, all the mice were sacrificed, and the heart, liver, spleen, lung and kidney were separated. After washing twice with PBS, all the organs were fixed in 4% formaldehyde, dehydrated in gradient alcohol, placed in xylene, embedded in paraffin and made into sections, followed by hematoxylin-eosin (HE) staining for histological examination with microscope.
Statistical analysis
All studies were repeated a minimum of three times and measured at least in triplicate. The results are reported as the means ± SD. Statistical significance was analyzed using Student’s t-test. Differences between experimental groups were considered significant if the P-value was <0.05.
Results and discussion
Characteristics of nanoparticles
In the optimal formulation of CPN-PDR, the weight ratio of DOX to siRNA was 1:1; therefore, the siRNA loading content was equal to the DOX loading content. Based on the facts that the molar ratio of CGA-ODNs to Dox was 1:5, the weight ratio of CPN/PEI/ODNs and siRNA was 4:1:1, and consequently, siRNA and Dox loading efficiency in CPN-PDR was calculated to be 4.93%.
Cytotoxicity of materials
In the co-delivery platform, CGA-ODNs were selected as carriers of DOX, then the mixture of CGA-ODNs-Dox and siRNA (abbreviated as DR) was electrostatically interacted with PEI to obtain Dox and siRNA co-loaded nanoparticles, PEI/DR (PDR). The copolymer CPN was employed as a multifunctional material.25 In order to evaluate the safety of the carrier, MTT assays were carried out to evaluate the cytotoxicity of CGA-ODNs and CPN. In the meantime, PEI, a toxic cationic polymer, was also tested in this experiment. As shown in Figure 2A, the viability of MCF-7 cells treated with CGA-ODNs and CPN at any concentration was observed to be stable and >80% within 48 h. After 72 h incubation, the viability of MCF-7 cells treated with CPN was still >80%, but after treatment with a higher concentration of CGA-ODNs (2.5 and 5 μM), the cell viability was slightly decreased to 60%. This indicated that there is no obvious cytotoxicity of CGA-ODNs and CPN. However, the cell viability of MCF-7 cells treated with PEI was obviously dependent on the concentrations of PEI. With an increase in concentration, the MCF-7 cells treated with PEI showed lower cell viability. Moreover, this concentration-dependent trend became more obvious when incubation time increased from 24 h to 72 h.
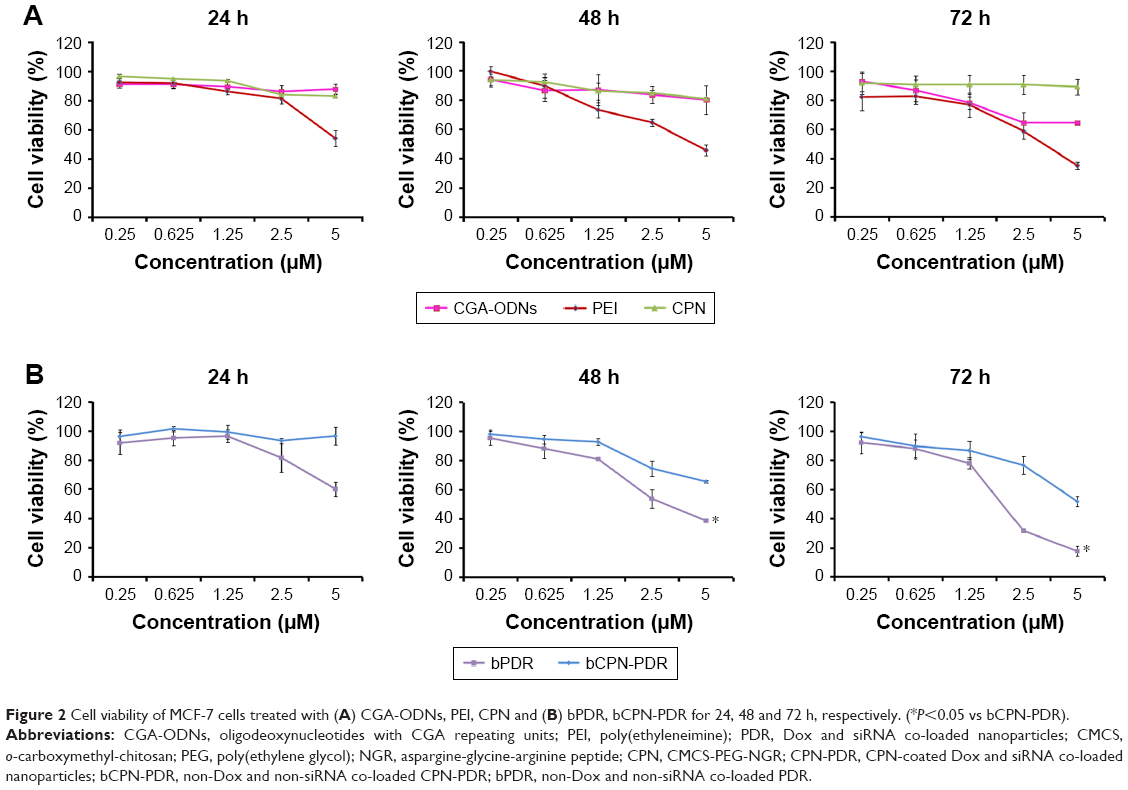 | Figure 2 Cell viability of MCF-7 cells treated with (A) CGA-ODNs, PEI, CPN and (B) bPDR, bCPN-PDR for 24, 48 and 72 h, respectively. (*P<0.05 vs bCPN-PDR). |
Cytotoxicity of blank nanoparticles
Cell viability of MCF-7 cells was also monitored to investigate the cytotoxicity of non-RNA- and non-drug-loaded carriers including bPDR and bCPN-PDR. As shown in Figure 2B, after treatment with bCPN-PDR, the cell viability was stable around 95% and shown to be concentration independent at 24 h. Meanwhile, bPDR-treated cells showed lower cell viability with time. With increased concentration, bPDR showed higher cytotoxicity, inducing 80% cell death in 72 h. Compared with the bPDR group, the cell viabilities of the bCPN-PDR group were significantly higher (P<0.05). This phenomenon could be explained by the higher positive charge of bPDR due to the positive ingredient of PEI, while the cell cytotoxicity could be significantly decreased by covering with the nontoxic negatively charged CPN (>80% cell viability in Figure 2A), which could induce the charge reversal of bPDR, implying that the negatively charged CPN coating could decrease the higher toxicity of bPDR to cells.
Expression level of VEGF mRNA
The transfection efficiency of CPN-PDR compared to Lipofectamine 2000 was demonstrated by delivering VEGF-siRNA into MCF-7 cells. The expression level of VEGF mRNA was measured using real-time PCR, and the results are shown in Figure 3. The expression level of VEGF mRNA was normalized to the β-actin expression and calculated based on the semiquantitative method. Untreated cells were selected as control, and the expression level was set as 100%. For lipo/siRNA group, the silencing efficiencies were 61.98%±6.96%, 31.93%±6.43% and 37.02%±3.17% at 24, 48 and 72 h, respectively. The decreased expression of VEGF mRNA reflected that gene silencing capability of lipo/siRNA. With incubation time extended from 24 to 48 h, the capability was enhanced (P<0.05). However, in 72 h, no further reduction was observed compared to that in 48 h, although the significantly downregulated expression could be observed (P<0.05 vs control, Dox and bCPN-PDR), implying that the silencing effect could reach the plateau period after 48 h and last for 72 h at least. The CPN-PR and CPN-PDR have shown similar behavior to lipo/siRNA. Compared with the control, a remarkable decrease of VEGF mRNA expression was observed (P<0.01) at 48 h. There was no significant difference in expression levels between CPN-PR and CPN-PDR (P>0.05) at any time point, implying their comparable gene delivery and transfection capability. More importantly, this capability was equivalent to the commercial Lipofectamine 2000.
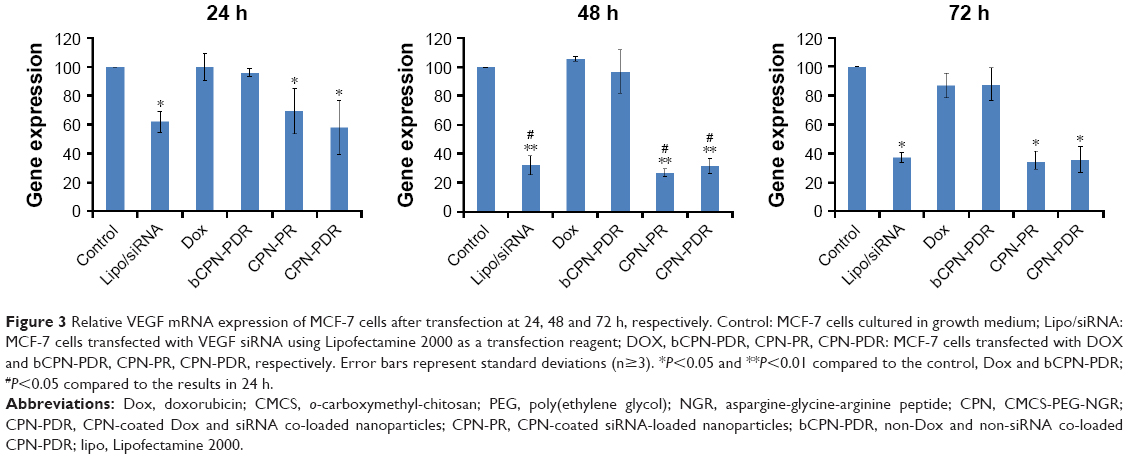 | Figure 3 Relative VEGF mRNA expression of MCF-7 cells after transfection at 24, 48 and 72 h, respectively. Control: MCF-7 cells cultured in growth medium; Lipo/siRNA: MCF-7 cells transfected with VEGF siRNA using Lipofectamine 2000 as a transfection reagent; DOX, bCPN-PDR, CPN-PR, CPN-PDR: MCF-7 cells transfected with DOX and bCPN-PDR, CPN-PR, CPN-PDR, respectively. Error bars represent standard deviations (n≥3). *P<0.05 and **P<0.01 compared to the control, Dox and bCPN-PDR; #P<0.05 compared to the results in 24 h. |
Expression level of VEGF protein
Based on the results of real-time PCR, the gene-silencing effect enhanced with time and reached a platform after 48 h. In this study, the Western blot technique was employed to observe VEGF protein expression at 48 h. The results are shown in Figure 4. GAPDH was selected as the inner control; all the bands were found to be equivalent in gray levels, and the amount of protein expression was positively correlated with the shade of gray in the imaging picture. From Figure 4, the gray levels of both control and bCPN-PDR, which were similar, were obvious. However, siRNA-loaded nanoparticles including lipo/siRNA, CPN-PR and CPN-PDR demonstrated a notably lower gray level. Furthermore, as marked with the red rectangle in Figure 4, expression of VEGF protein in CPN-PDR could hardly be identified. This suggested that co-delivery of anti-VEGF siRNA and Dox could downregulate the relevant VEGF protein level, which was in agreement with the results from semiquantitative real-time PCR. Taken together, these results indicate that nanoparticles loaded with anti-VEGF siRNA downregulated both the protein and the mRNA expression of VEGF.
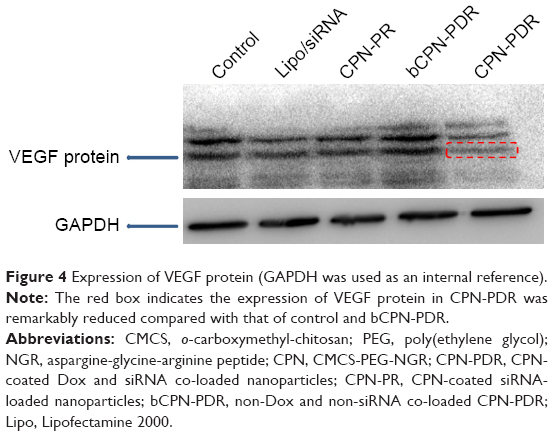 | Figure 4 Expression of VEGF protein (GAPDH was used as an internal reference). |
Inhibition of cell proliferation in vitro
The ultimate goal of siRNA intracellular delivery is to restrain the proliferation of cancer cells. Different treatments against MCF-7 cells were performed to further investigate the inhibition effect. As shown in Figure 5A, an obvious concentration dependence in terms of DOX, CPN-PD and CPN-PDR could be observed after 48 h incubation. We have calculated the differences between CPN-PDR vs CPN-PD and CPN-PDR vs DOX at each dose. The results indicated that there was a significant difference between CPN-PDR vs DOX at each dose except the dose of 0.05, and there were significant differences between CPN-PDR vs CPN-PD at a dose of 0.002. It was suggested that the in vitro antitumor activity of CPN-PDR was equivalent to CPN-PD, and both of them showed a higher antitumor activity than free drug DOX, which was mainly attributed to the design of the multifunctional nanovectors. We have also calculated the statistical differences between CPN-PDR vs CPN-PD and CPN-PDR vs DOX in the concentration range of Dox (0.002, 0.01, 0.05, 0.25 and 0.5 μM) using Student’s t-test. After statistical calculation, the significant differences were verified to exist between CPN-PDR vs CPN-PD (P<0.05) and CPN-PDR vs DOX (P<0.01). After calculation with professional software, the half maximal inhibitory concentrations (IC50) are presented in Figure 5B. They are 0.0367±0.0088 μM for Dox, 0.0266±0.0027 μM for CPN-PD and 0.0126±0.0022 μM for CPN-PDR, respectively. The IC50 of CPN-PD was 2.913-fold higher than that of CPN-PDR, and the IC50 of DOX was 2.110-fold higher than that of CPN-PDR. After statistical calculation, significant differences were found between CPN-PDR vs CPN-PD (P<0.05) and CPN-PDR vs DOX (P<0.05). With the results from real-time PCR and Western blot, the increased cytotoxicity of CPN-PDR probably resulted from siRNA delivery and successful transfection. This was because the intracellular delivery of anti-VEGF siRNA could downregulate the expression of VEGF protein. The anti-VEGF therapy may sensitize cancer cells or kill cells block the blood supply of the tumor and improve drug penetration. Taken together, the anti-VEGF siRNA and Dox co-delivery platform could induce increased cytotoxicity against cancer cells.
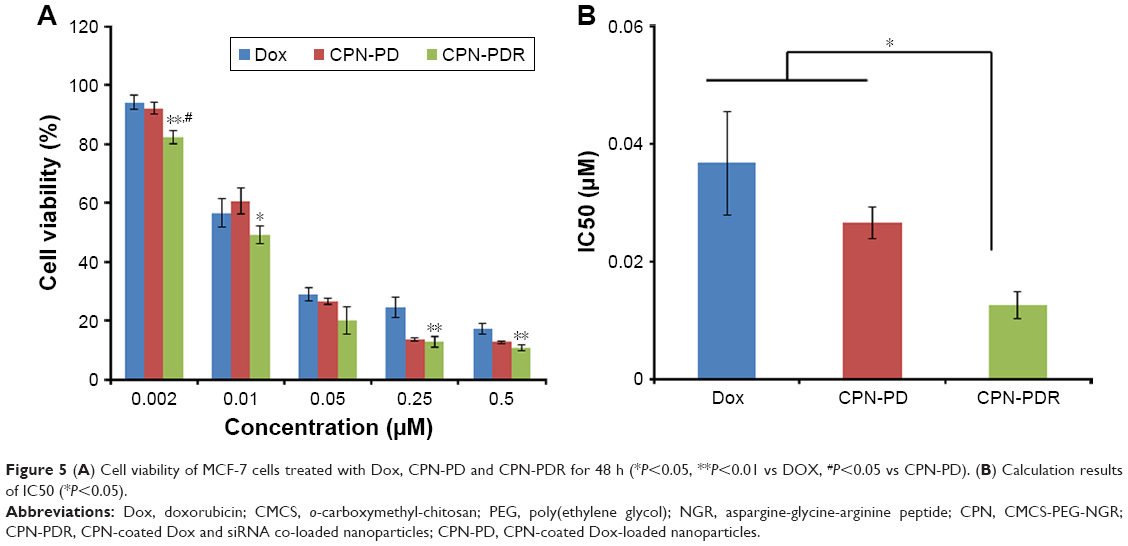 | Figure 5 (A) Cell viability of MCF-7 cells treated with Dox, CPN-PD and CPN-PDR for 48 h (*P<0.05, **P<0.01 vs DOX, #P<0.05 vs CPN-PD). (B) Calculation results of IC50 (*P<0.05). |
Hemolysis test
To investigate the safety of intravenous injection of CPN-PDR, a hemolysis test was performed. As shown in Table 1, different volume ratios of CPN-PDR to 2% red blood cell suspension were selected. After incubation and centrifugation, tubes were observed by the naked eye. As shown in Figure 6, no red blood cell hemolysis was observed in tube 6, in which 100% PBS was added serving as a negative control. For tube 7, 100% double distilled water was added and red cell hemolysis could be clearly observed. For tubes 1–5, in which different percentages of CPN-PDR were added, no red blood cell hemolysis occurred. Hemolysis of red blood cells can generate hemoprotein, which could be detected using UV-vis spectrophotometry. After scanning the absorbance wavelength of hemoprotein, 577 nm was selected as the detecting wavelength. According to Equation 3, hemolysis ratios were calculated based on the recorded absorbance and shown in Table 2. All the hemolysis ratios of tubes 1–5 were <5%, implying that no hemolysis was induced by addition of CPN-PDR.
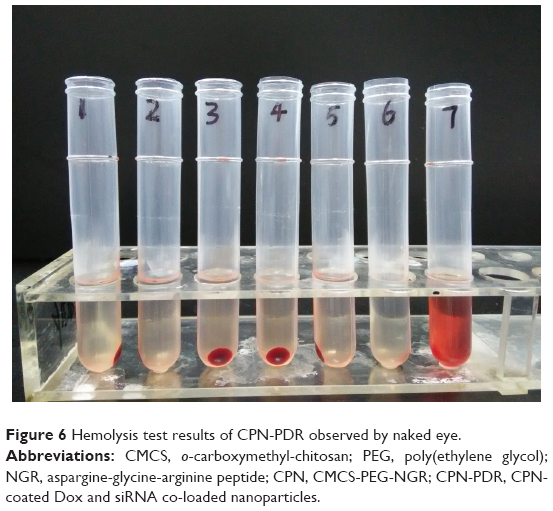 | Figure 6 Hemolysis test results of CPN-PDR observed by naked eye. |
 | Table 2 Results of hemolysis ratios of CPN-PDR |
Tissue section
In this study, a histological analysis of organs (heart, liver, spleen, lung and kidney) was performed to evaluate whether CPN-PDR could cause tissue damage, inflammation and lesions. Kunming mice were injected with CPN-PDR and normal saline (NS) by tail vein, respectively. After 1 week, all the mice were sacrificed, and the heart, liver, spleen, lung and kidney were separated. After the standard procedure of HE staining for histological examination, the organs were observed under a microscope. Mice without any treatment were chosen as a control. As shown in Figure 7, compared with the control group, there was no visible histologically difference between mice administrated with CPN-PDR group and that with NS group, implying the safety of CPN-PDR in vivo.
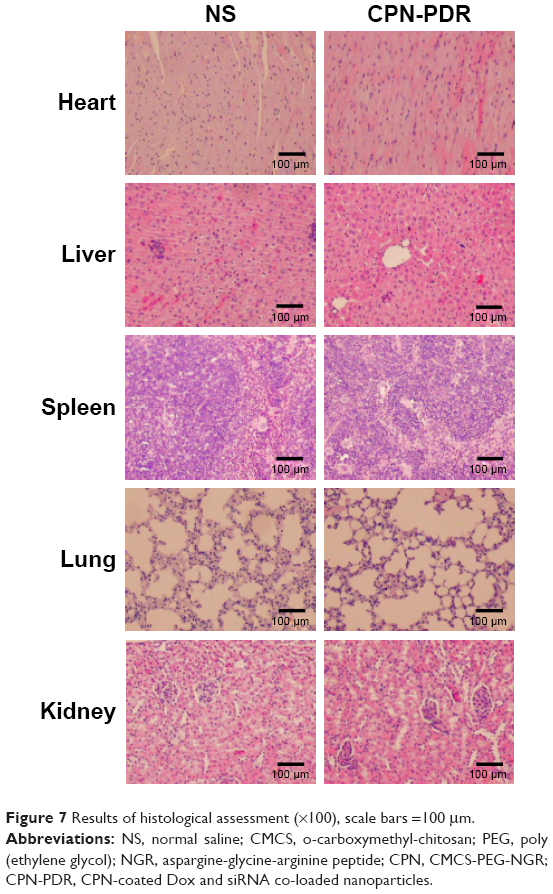 | Figure 7 Results of histological assessment (×100), scale bars =100 μm. |
Conclusion
In summary, the delivery system materials were demonstrated to be nontoxic and biocompatible. The nontoxic and negatively charged copolymer CPN coating could significantly decrease the cytotoxicity of the cationic core, bPDR. The obtained co-delivery system CPN-PDR was also confirmed with enhanced cytotoxicity against tumor cells. Gene transfection efficiency induced by CPN-PDR was fairly equal with that of the commercial product Lipofectamine 2000, implying the successful intracellular delivery and good transfection of siRNA. Moreover, the preliminary evaluation of safety showed CPN-PDR to have good biocompatibility, so it has great potential for further exploitation and clinical application. With the development of aptamer technology, the promising application prospects of this novel oligodeoxynucleotide-based co-loading platform will further increase.
Acknowledgments
This work was supported by the National Natural Science Foundation (number 81402867). The authors would like to express their thanks to the School of Pharmaceutical Science, Shandong University, for providing the required infrastructure to carry out this study.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. | ||
Gandhi NS, Tekade RK, Chougule MB. Nanocarrier mediated delivery of siRNA/miRNA in combination with chemotherapeutic agents for cancer therapy: current progress and advances. J Control Release. 2014;194:238–256. | ||
Li J, Wang Y, Zhu Y, Oupický D. Recent advances in delivery of drug–nucleic acid combinations for cancer treatment. J Control Release. 2013;172:589–600. | ||
Hu CM, Zhang L. Nanoparticle-based combination therapy toward overcoming drug resistance in cancer. Biochem Pharmacol. 2012;83:1104–1111. | ||
Nakamura K, Abu Lila AS, Matsunaga M, Doi Y, Ishida T, Kiwada H. A double-modulation strategy in cancer treatment with a chemotherapeutic agent and siRNA. Mol Ther. 2011;19:2040–2047. | ||
Davidson BL, McCray PB Jr. Current prospects for RNA interference-based therapies. Nat Rev Genet. 2011;12:329–340. | ||
Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001;15:188–200. | ||
Vilar E, Tabernero J. Molecular dissection of microsatellite instable colorectal cancer. Cancer Discov. 2013;3:502–511. | ||
Wu SY, Lopez-Berestein G, Calin GA, Sood AK. RNAi therapies: drugging the undruggable. Sci Transl Med. 2014;6:240ps7. | ||
Fatemian T, Othman I, Chowdhury EH. Strategies and validation for siRNA-based therapeutics for the reversal of multi-drug resistance in cancer. Drug Discov Today. 2014;19:71–78. | ||
Xiong XB, Lavasanifar A. Traceable multifunctional micellar nanocarriers for cancer-targeted co-delivery of MDR-1 siRNA and doxorubicin. ACS Nano. 2011;5:5202–5213. | ||
Salzano G, Riehle R, Navarro G, Perche F, De Rosa G, Torchilin VP. Polymeric micelles containing reversibly phospholipid-modified anti-survivin siRNA: a promising strategy to overcome drug resistance in cancer. Cancer Lett. 2014;343:224–231. | ||
Salzano G, Navarro G, Trivedi MS, De Rosa G, Torchilin VP. Multifunctional polymeric micelles co-loaded with anti-survivin siRNA and paclitaxel overcome drug resistance in an animal model of ovarian cancer. Mol Cancer Ther. 2015;14:1075–1084. | ||
Yu D, Wu F, Liu Y, Liu H, Xia Q. Bcl-2 gene silence enhances the sensitivity toward 5-Fluorouracil in gastric adenocarcinoma cells. Biomed Pharmacother. 2013;67:615–619. | ||
Tsouris V, Joo MK, Kim SH, Kwon IC, Won YY. Nano carriers that enable co-delivery of chemotherapy and RNAi agents for treatment of drug-resistant cancers. Biotechnol Adv. 2014;32:1037–1050. | ||
Qian J, Xu M, Suo A, et al. Folate-decorated hydrophilic three-arm star-block terpolymer as a novel nanovehicle for targeted co-delivery of doxorubicin and Bcl-2 siRNA in breast cancer therapy. Acta Biomater. 2015;15:102–116. | ||
Dvorak HF. Tumor stroma, tumor blood vessels, and antiangiogenesis therapy. Cancer J. 2015;21:237–243. | ||
Gatson NN, Chiocca EA, Kaur B. Anti-angiogenic gene therapy in the treatment of malignant gliomas. Neurosci Lett. 2012;527:62–70. | ||
Chen S, Feng J, Ma L, Liu Z, Yuan W. RNA interference technology for anti-VEGF treatment. Expert Opin Drug Deliv. 2014;11:1471–1480. | ||
Zhao J, Zhang Z, Zhao N, Ma BA, Fan QY. VEGF silencing inhibits human osteosarcoma angiogenesis and promotes cell apoptosis via PI3K/AKT signaling pathway. Cell Biochem Biophys. 2015;73:519–525. | ||
Carmeliet P. VEGF as a key mediator of angiogenesis in cancer. Oncology. 2005;69:4–10. | ||
Ferrara N, Hillan KJ, Gerber HP, et al. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3:391–400. | ||
Chang JH, Garg NK, Lunde E, Han KY, Jain S, Azar DT. Corneal neovascularization: an anti-VEGF therapy review. Surv Ophthalmol. 2012;57:415–429. | ||
Chen Y, Bathula SR, Li J, Huang L. Multifunctional nanoparticles delivering small interfering RNA and doxorubicin overcome drug resistance in cancer. J Biol Chem. 2010;285:22639–22650. | ||
Liu T, Wang M, Wang T, Yao Y, Zhang N. Co-delivery of doxorubicin and siRNA by a simplified platform with oligodeoxynucleotides as a drug carrier. Colloids Surf B Biointerfaces. 2015;126:531–540. | ||
Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8:129–138. | ||
Panwar N, Yang C, Yin F, Yoon HS, Chuan TS, Yong KT. RNAi-based therapeutic nanostrategy: IL-8 gene silencing in pancreatic cancer cells using gold nanorods delivery vehicles. Nanotechnology. 2015;26:365101. | ||
Kumar K, Vulugundam G, Jaiswal PK, Shyamlal BRK, Chaudhary S. Efficacious cellular codelivery of doxorubicin and EGFP siRNA mediated by the composition of PLGA and PEI protected gold nanoparticles. Bioorg Med Chem Lett. 2017;27:4288–4293. | ||
Li Y, Liu R, Yang J, Ma G, Zhang Z, Zhang X. Dual sensitive and temporally controlled camptothecin prodrug liposomes codelivery of siRNA for high efficiency tumor therapy. Biomaterials. 2014;35:9731–9745. | ||
Butt AM, Amin MC, Katas H, Abdul Murad NA, Jamal R, Kesharwani P. Doxorubicin and siRNA codelivery via chitosan-coated pH-responsive mixed micellar polyplexes for enhanced cancer therapy in multidrug-resistant tumors. Mol Pharm. 2016;13:4179–4190. | ||
Han M, Lv Q, Tang X, et al. Overcoming drug resistance of MCF-7/ADR cells by altering intracellular distribution of doxorubicin via MVP knockdown with a novel siRNA polyamidoamine-hyaluronic acid complex. J Control Release. 2012;163:136–144. | ||
Zhang S, Zhao B, Jiang H, Wang B, Ma B. Cationic lipids and polymers mediated vectors for delivery of siRNA. J Control Release. 2007;123:1–10. | ||
Kanasty R, Dorkin JR, Vegas A, Anderson D. Delivery materials for siRNA therapeutics. Nat Mater. 2013;12:967–977. | ||
Xia Y, Xu T, Wang C, et al. Novel functionalized nanoparticles for tumor-targeting co-delivery of doxorubicin and siRNA to enhance cancer therapy. Int J Nanomedicine. 2017;13:143–159. | ||
Liu F, Feng L, Zhang L, Zhang X, Zhang N. Synthesis, characterization and antitumor evaluation of CMCS–DTX conjugates as novel delivery platform for docetaxel. Int J Pharm. 2013;451:41–49. | ||
Luo K, Li C, Li L, She W, Wang G, Gu Z. Arginine functionalized peptide dendrimers as potential gene delivery vehicles. Biomaterials. 2012;33:4917–4927. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.