")
Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 13
Evaluation of the Pharmacokinetics of Trazpiroben (TAK-906), a Peripherally Selective D2/D3 Dopamine Receptor Antagonist, in the Presence and Absence of Itraconazole, a Potent CYP 3A4 Inhibitor
Authors Chen C, Zhang W , Bari M, Almansa C, Baratta M, Rosario M
Received 13 March 2021
Accepted for publication 20 June 2021
Published 12 July 2021 Volume 2021:13 Pages 145—155
DOI https://doi.org/10.2147/CPAA.S310609
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Arthur E. Frankel
Chunlin Chen,1 Wenwen Zhang,1 Muhammad Bari,2 Cristina Almansa,1 Mike Baratta,1 Maria Rosario1
1Takeda Development Center Americas, Inc, Cambridge, MA, USA; 2Takeda International UK, Ltd, High Wycombe, UK
Correspondence: Chunlin Chen Email [email protected]
Purpose: Treatment options for gastroparesis, such as metoclopramide and domperidone, are limited because of safety concerns, which may be exacerbated in the presence of inhibitors of drug metabolism. This study evaluated the effect of itraconazole on the pharmacokinetics, safety, and tolerability of trazpiroben (previously TAK-906), a novel, peripherally selective D2/D3 dopamine receptor antagonist.
Methods: This was a phase 1, two-period, crossover trial in healthy participants (NCT03161405). On day 1, period 1 (days 1– 3), participants received a single oral dose of trazpiroben 25 mg. During period 2 (days 4– 9), participants received oral itraconazole 200 mg once daily (days 1– 5) and one oral dose of trazpiroben 25 mg post itraconazole on day 4. Trazpiroben pharmacokinetics were assessed. Safety assessments included triplicate electrocardiograms.
Results: Twelve healthy males (24– 45 years old) were studied. Co-administration of itraconazole increased trazpiroben area under the concentration–time curve from time 0 to infinity by 1.28-fold (90% confidence interval: 1.10, 1.49) and maximum plasma concentration (Cmax) by 1.98-fold (1.64, 2.39) versus trazpiroben alone. Placebo-corrected, change from baseline in corrected QT interval at the observed geometric mean Cmax for trazpiroben alone (9.53 ng/mL) and with itraconazole (18.00 ng/mL) was estimated at 1.31 ms (− 0.39, 3.01) and 1.54 ms (− 0.15, 3.24), respectively. There were no clinically relevant abnormalities in any safety parameters.
Conclusion: These results indicate that TAK‑906 is relatively insensitive to inhibition of cytochrome P450 3A4, and cardiovascular safety concerns associated with domperidone are unlikely to be elicited by trazpiroben under similar conditions.
Keywords: drug–drug interactions, D2/D3 dopamine receptor antagonist, gastroparesis, pharmacokinetics, QT effects
Introduction
Gastroparesis is a chronic symptomatic condition characterized by delayed gastric emptying without mechanical obstruction.1 Cardinal symptoms include nausea, vomiting, early satiety, abdominal pain, persistent anorexia, and postprandial fullness.2
Available therapies for the treatment of gastroparesis are limited, primarily owing to safety concerns. Cisapride was approved by the US Food and Drug Administration (FDA) in 1993 for gastro-esophageal reflux disease and was used off-label for the treatment of gastroparesis; however, it was withdrawn from the market in 2000 owing to the risk of cardiac side effects.3 The motilin agonist, erythromycin, has demonstrated efficacy in improving gastric emptying,4–6 but its use as a long-term treatment is limited owing to development of tolerance, concerns surrounding antibiotic resistance,6,7 and potential risk of adverse cardiovascular effects. Metoclopramide, a dopamine receptor antagonist, is approved by the FDA for short-term use (up to 12 weeks) in patients with gastroparesis and for up to 5 days by the European Medicines Agency (EMA), owing to its safety profile, particularly the increased risk of tardive dyskinesia.8–10
Peripherally selective D2/D3 dopamine receptor antagonists are promising treatment options for gastroparesis because beneficial effects would be expected by targeting D2 and D3 receptors in the stomach and in the vomiting center in the area postrema, outside the blood–brain barrier,11–13 in contrast with centrally penetrating agents such as metoclopramide.14
Domperidone, a peripherally selective D2/D3 dopamine receptor antagonist, is approved by the EMA for short-term treatment of nausea and vomiting in low doses.15,16 However, it has not been approved by the FDA because of the potential risk of QT interval prolongation leading to torsade de pointes and possible subsequent sudden cardiac death. This risk is increased by drug–drug interactions between domperidone and potent inhibitors of cytochrome P450 (CYP) 3A4 such as itraconazole, which can lead to reduced clearance of domperidone and increase the risk of QT interval prolongation.17 Although the molecular mechanism of QT interval prolongation has not yet been determined, it is believed that domperidone is one of several drugs that may elicit this effect by binding to potassium efflux channels.17
Trazpiroben (previously referred to as TAK-906 or ATC-1906M) is a peripherally active novel D2/D3 receptor antagonist in development for the treatment of chronic idiopathic and diabetic gastroparesis. Results from the first-in-human trial of trazpiroben (AT-01C study), exploring oral doses ranging from 5 mg to 300 mg in healthy US participants, showed exposure increased in a dose-proportional manner and limited accumulation after multiple dosing for 5 days. Continuous 12-lead triplicate electrocardiogram (ECG; Holter) monitoring from 1 hour pre-dose through 24 hours after dosing was performed in the single ascending dose phase, with the aim to exclude clinically relevant ECG effects of the drug.18 Trazpiroben was well tolerated and appeared to have a favorable safety profile.
Polypharmacy is common in patients with gastroparesis, meaning there is an increased risk of drug–drug interactions because of overlap in metabolic pathways, such as the CYP superfamily.19 There remains a need for novel, efficacious treatments for chronic gastroparesis without the safety concerns associated with existing therapies, such as domperidone. Trazpiroben is mainly metabolized by CYP 3A4 and CYP 2C8, although non-CYP metabolism also occurs through cytosolic liver enzymes. This phase 1 study investigated the pharmacokinetics, safety, and tolerability of trazpiroben in the presence and absence of itraconazole, a potent CYP 3A4 inhibitor recommended by the FDA and EMA for use in drug–drug interaction studies.20,21
Methods
This was a phase 1, single-center, single-sequence, open-label, two-period, crossover trial in healthy individuals (NCT03161405). Participants were eligible for inclusion in the trial if they were: a man or women aged 18–55 years; had a body mass index (BMI) greater than 18 kg/m2 and no more than 30 kg/m2, and a body weight greater than 50 kg at screening; had no history of tobacco- or nicotine-containing product use in the 6 months before administration of the initial drug dose; and were judged to be in good health by the investigator based on clinical evaluations (including laboratory safety tests, medical history, physical examination, and 12-lead ECG and vital sign measurements performed during screening and prior to administration of the initial dose of study treatment). This trial was approved and conducted in compliance with the institutional review board regulations of the US Code of Federal Regulations, Good Clinical Practice regulations and guidelines, and all applicable local regulations, and in accordance with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. This trial was conducted in compliance with the informed consent regulations and all patients provided signed informed consent forms prior to enrollment in the trial. The clinical trial protocol, the investigator’s brochure, a sample informed consent form, and other trial-related documents were reviewed and approved by the Midlands Independent Review Board (Overland Park, KS).
Trial Design
On day 1 of period 1 (3 days’ duration), eligible participants received a single oral dose of trazpiroben 25 mg in the absence of itraconazole (Figure 1). During period 2 (6 days), participants received itraconazole 200 mg in oral solution once daily on days 1–5; they also received a single oral dose of trazpiroben 25 mg 1 hour after the itraconazole dose on day 4. There was a minimum washout period of 4 days between the trazpiroben dose on day 1 of period 1 and the beginning of period 2. Doses of trazpiroben 25 mg in both periods were administered after an overnight fast of at least 8 hours. All participants who completed the study attended a follow-up visit 10–14 days after their final dose of trazpiroben 25 mg.
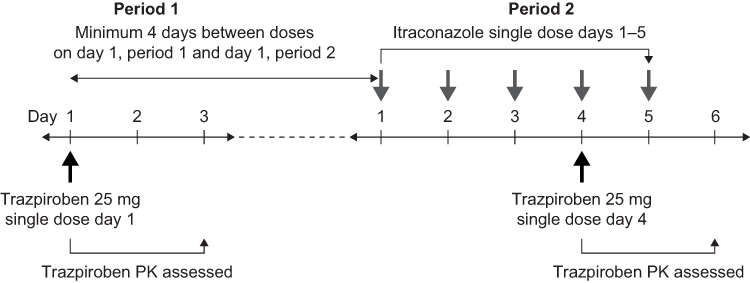 |
Figure 1 Study design. Abbreviation: PK, pharmacokinetics. |
Assessment of Plasma Pharmacokinetics of Trazpiroben and Metabolite M23
The plasma pharmacokinetics of both trazpiroben and M23, the primary human metabolite of trazpiroben, were analyzed in this study. Although pharmacologically inactive, evaluation of M23 was included to aid understanding of the impact of itraconazole on trazpiroben metabolism. Blood samples for assessment of the plasma pharmacokinetics of trazpiroben and M23 were collected at 0 (pre-dose), 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 16, 24, 36, and 48 hours after trazpiroben 25 mg administration in each period. The total blood volume drawn for the trial was approximately 180 mL.
The analysis of trazpiroben in plasma samples was performed by Covance Laboratories, Inc. (Madison, WI). The plasma concentrations of trazpiroben and M23 were generated using a validated analytical method employing liquid chromatography–tandem mass spectrometry with a linear assay range of 0.05–50.00 ng/mL. Accuracy and precision for trazpiroben were 1.3–5.0% and 2.2–6.7%, respectively, and accuracy and precision for M23 were −0.7–2.4% and 1.4–7.0%, respectively. Recovery for trazpiroben and M23 in plasma were in the ranges 99.1–102.2% and 101.3–106.0%, respectively. Trazpiroben and M23 were stable in plasma at ambient temperature for at least 24 hours and after five freeze–thaw cycles when both were stored at −20°C and −70°C. Trazpiroben and M23 were stable in whole blood at ambient temperature and in a wet ice bath for at least 2 hours. Accuracy and precision values for trazpiroben and M23 in lipemic and hemolyzed plasma met acceptance criteria. Selectivity was evaluated in six volumes of plasma for each analyte and met acceptance criteria. A matrix factor experiment for trazpiroben, M23, and respective internal standards was conducted in naive, hemolyzed, and lipemic plasma to assess suppression and enhancement in matrix. Processed sample stability and reinjection reproducibility were established at 143 and 157 hours, respectively. Both trazpiroben and M23 are stable in frozen plasma at −20°C and −70°C for at least 196 days. Trazpiroben and M23 in 50:50 (methanol/water, v/v) stock solutions were stable for at least 30 days when stored at 4°C.
The following plasma pharmacokinetic parameters of trazpiroben and M23 were calculated using non-compartmental analysis with Phoenix WinNonlin® version 6.3 (Certara, Princeton, NJ): maximum plasma concentration (Cmax), time to maximum plasma concentration (tmax), area under the concentration–time curve from time 0 to infinity (AUC0–∞), area under the concentration–time curve from time 0 to the last quantifiable concentration (AUC0–last), and terminal disposition phase half-life (t1/2z). AUC0–last and AUC0–∞ were calculated using a linear trapezoidal rule. Actual sampling times were used in all pharmacokinetic computations. No imputation of incomplete or missing data was performed for trazpiroben or M23, and plasma concentrations below a quantifiable level (<0.05 ng/mL) were treated as zero.
Safety Assessments
Safety assessments were performed throughout the study and included the recording of adverse events (AEs), hematology and chemistry laboratory measurements, and monitoring of vital signs (body temperature, respiratory rate, heart rate, and systolic and diastolic blood pressure). Physical examinations, including measurement of height, weight, and BMI, were conducted at protocol-specified timepoints. Triplicate ECG measurements were taken at 0 (pre-dose), 1, 2, 4, 8, and 48 hours after each dose of trazpiroben 25 mg and the mean of these measurements recorded at each time point.
Determination of Sample Size and Statistical Analyses
A sample size of 12 was determined based on an acceptance range of 50–200%, and the assumption that the intra-individual coefficient of variation for AUC0–last and Cmax of trazpiroben would not exceed 25%. This sample size provides 80% power to conclude that plasma concentration of trazpiroben does not increase more than twofold in the presence of itraconazole versus in its absence. The ratio of geometric means of the Cmax and AUCs (AUC0–last and AUC0–∞) with itraconazole relative to trazpiroben 25 mg alone (and associated 90% confidence intervals [CIs]) were determined by exponentiating the appropriate estimates for the difference between each regimen in the log-transformed parameters.
Trazpiroben Concentration–QT Analysis
In the US AT-01C study, the first-in-human trial of trazpiroben, the core laboratory used TQT Plus (iCardiac Technologies), an advanced computer-assisted and statistical process, to extract ECGs from continuous 24-hour recordings collected in TQT studies. In the AT-01C study, high-precision QT analyses were performed, and the relationship between change from baseline QT interval corrected for heart rate by the Fridericia method (ΔQTcF)22 and trazpiroben plasma concentrations was investigated by a linear mixed-effects modelling approach, with ΔQTcF as the dependent variable, plasma concentration (Conc) of trazpiroben as the covariate, treatment (Treat; active or placebo), and timepoint (Time) as categorical factors, and a random intercept per participant:18
∆QTcF ~ Conc + Treat + Time
The linear mixed-effects model was based on placebo and placebo-controlled data from the AT-01C study (no placebo was evaluated in the current trial).18 The model was used to investigate the change from baseline in QTcF,18 and the placebo-corrected change from baseline QTcF (∆∆QTcF)22 of trazpiroben at Cmax in the presence and absence of itraconazole was predicted. M23 was not included in the model because it is a pharmacologically inactive metabolite of trazpiroben. Full details of the trazpiroben concentration-QT analysis have been described previously.18
Results
Participant Characteristics
In total, 46 individuals were screened at a single US center. Of these, 34 (30 male, 4 female) were not eligible for enrollment, primarily for not meeting the study inclusion criteria (16 individuals) or because the study cohort had already been filled (13 individuals). Participants were also considered ineligible for enrollment owing to loss to follow up during the screening period (1 participant), meeting study exclusion criteria (3 participants), or withdrawal from the screening period (1 participant). In total, 12 healthy men with a median age of 33 years (range: 24–45 years) and a median BMI of 26 kg/m2 (range: 22–30 kg/m2) were eligible for enrollment and were included in the study. Of these, 11 completed both study periods; one was withdrawn because of a positive recreational drug screen.
Plasma Pharmacokinetics of Trazpiroben
At the first assessment following administration of trazpiroben 25 mg alone and with itraconazole (0.5 hours), plasma concentrations were quantifiable (≥0.05 ng/mL) in all participants (Figure 2). At 24 hours post dose, trazpiroben concentrations were undetectable in nine of the 12 participants (75%) in the absence of itraconazole and in seven of the 11 participants (64%) in the presence of itraconazole. By 36 hours post dose, trazpiroben concentrations were undetectable in all participants. The median tmax of trazpiroben 25 mg was 1.00 hour in both the presence and absence of itraconazole. The mean t1/2z of trazpiroben 25 mg alone was 2.32 hours compared with 5.00 hours after co-administration of trazpiroben 25 mg with itraconazole (Table 1). Compared with administration of trazpiroben 25 mg alone, co-administration with itraconazole increased total systemic exposure to trazpiroben (AUC0–∞) by 1.28-fold (90% CI: 1.10, 1.49) and Cmax by 1.98-fold (90% CI: 1.64, 2.39; Table 2; Figure 3). The percentage of AUC extrapolated was small (<5%), with minimum and maximum values of 0.43, 3.42 for trazpiroben alone and 0.36, 2.32 for trazpiroben co-administered with itraconazole.
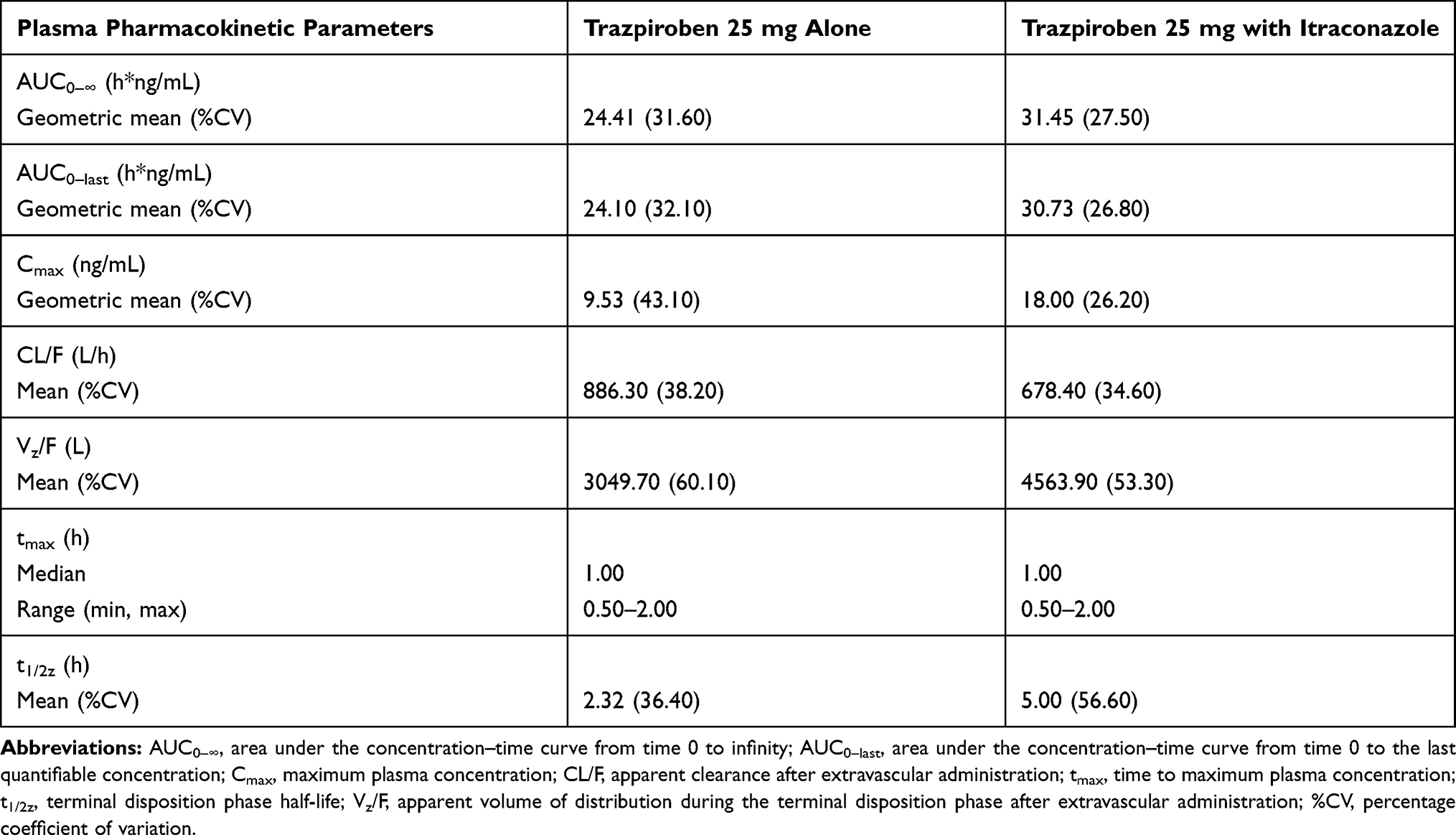 |
Table 1 Summary of the Plasma Pharmacokinetic Parameters of Trazpiroben 25 mg in the Presence and Absence of Itraconazole |
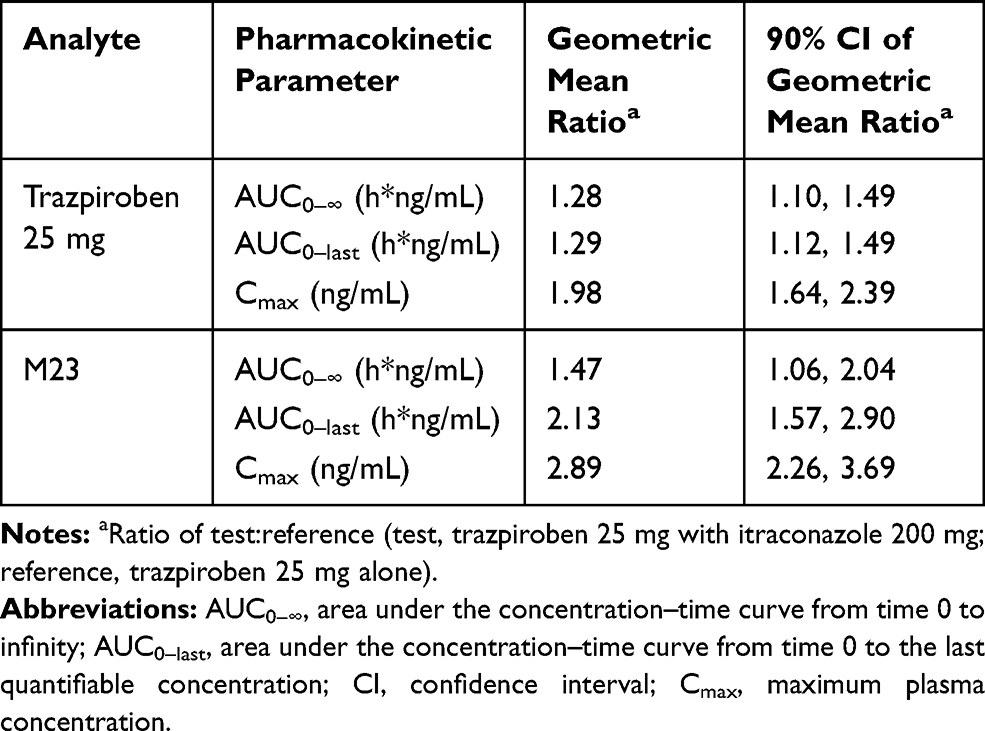 |
Table 2 Summary of the Plasma Pharmacokinetic Parameters of Trazpiroben and M23 After Administration of a Single Oral Dose of Trazpiroben 25 mg in the Presence and Absence of Itraconazole |
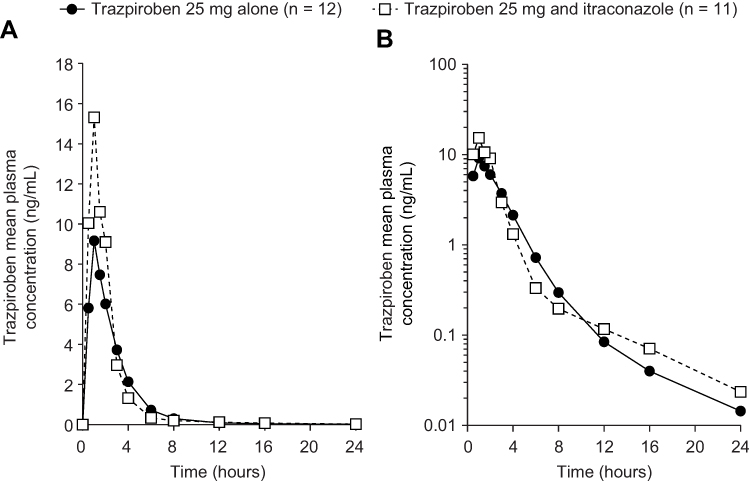 |
Figure 2 Mean plasma concentration–time curves of trazpiroben after administration of a single oral dose of trazpiroben 25 mg in the presence and absence of itraconazole on (A) a linear scale and (B) a semi-log scale. |
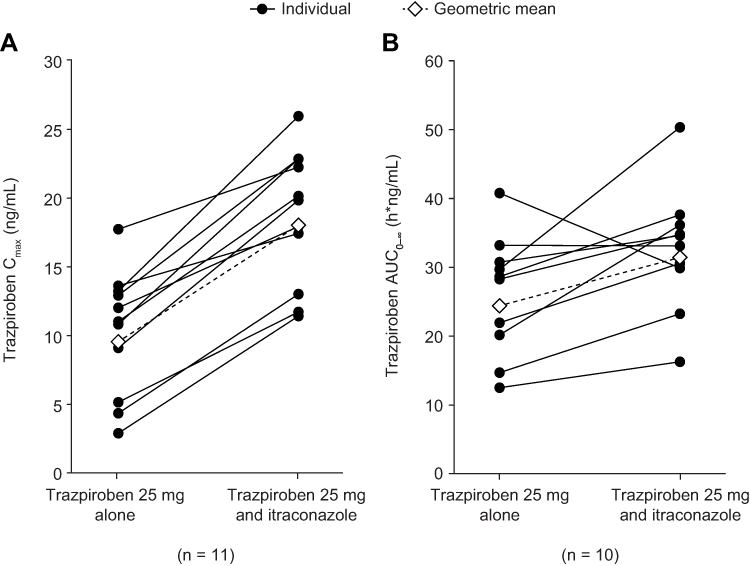 |
Figure 3 (A) Cmax and (B) AUC0–∞ of trazpiroben after administration of a single oral dose of trazpiroben 25 mg in the presence and absence of itraconazole. Abbreviations: AUC0–∞, area under the concentration–time curve from time 0 to infinity; Cmax, maximum plasma concentration. |
Plasma Pharmacokinetics of Metabolite M23
At 0.5 hours after the administration of trazpiroben 25 mg alone, plasma concentrations of M23 were quantifiable (≥0.05 ng/mL) in eight of the 12 participants (67%) and nine of the 11 participants (82%) receiving trazpiroben 25 mg in the presence of itraconazole (Figure 4). At 3 hours post dose, M23 concentrations were similar between individuals receiving trazpiroben 25 mg alone and trazpiroben 25 mg in the presence of itraconazole. Eight hours after dosing, plasma concentrations of M23 were detectable in four of the 12 participants (33%) who received trazpiroben 25 mg in the absence of itraconazole and four of the 11 participants (36%) who received trazpiroben 25 mg in the presence of itraconazole. By 12 hours after administration of trazpiroben 25 mg, concentrations of M23 were undetectable in all participants in both the presence and absence of itraconazole.
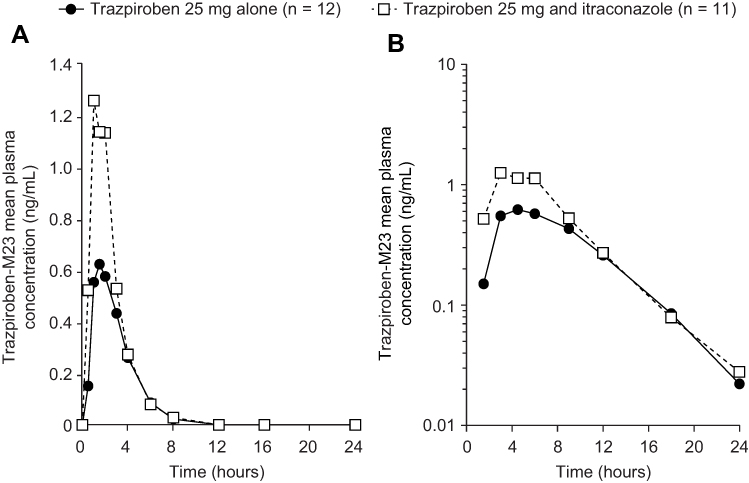 |
Figure 4 Mean plasma concentration–time curves of M23 (a pharmacologically inactive metabolite of trazpiroben) after administration of a single oral dose of trazpiroben 25 mg in the presence and absence of itraconazole on (A) a linear scale and (B) a semi-log scale. |
Compared with administration of trazpiroben 25 mg alone, co-administration with itraconazole increased total systemic exposure (AUC0–∞) of M23 by 1.47-fold (90% CI: 1.06, 2.04) and Cmax by 2.89-fold (90% CI: 2.26, 3.69; Table 2). The mean t1/2z of M23 was 1.52 hours with trazpiroben 25 mg alone and 1.30 hours following co-administration with itraconazole.
Metabolite (M23) to parent (trazpiroben) ratios based on AUC0–∞ were consistent in both the absence and presence of itraconazole, with a mean of 10.6% and 11.5%, respectively.
Safety Assessments of Trazpiroben 25 Mg
Baseline observed QTcF was similar during both study periods. The mean change from baseline in the observed QTcF after trazpiroben 25 mg alone was negative at all post-dose time points and the mean change from baseline in the observed QTcF after co-administration of trazpiroben 25 mg with itraconazole was less than 2 ms (Table 3). Using the linear mixed-effects model, the predicted ∆∆QTcF at the observed geometric mean Cmax for trazpiroben 25 mg alone (9.53 ng/mL) and trazpiroben 25 mg with itraconazole (18.00 ng/mL) were estimated to be 1.31 ms (90% CI: −0.39, 3.01) and 1.54 ms (90% CI: −0.15, 3.24), respectively (Figure 5). ∆∆QTcF at the highest observed individual Cmax of trazpiroben 25 mg with itraconazole (25.90 ng/mL) was estimated to be 1.77 ms (90% CI: 0.06, 3.47; Figure 5).
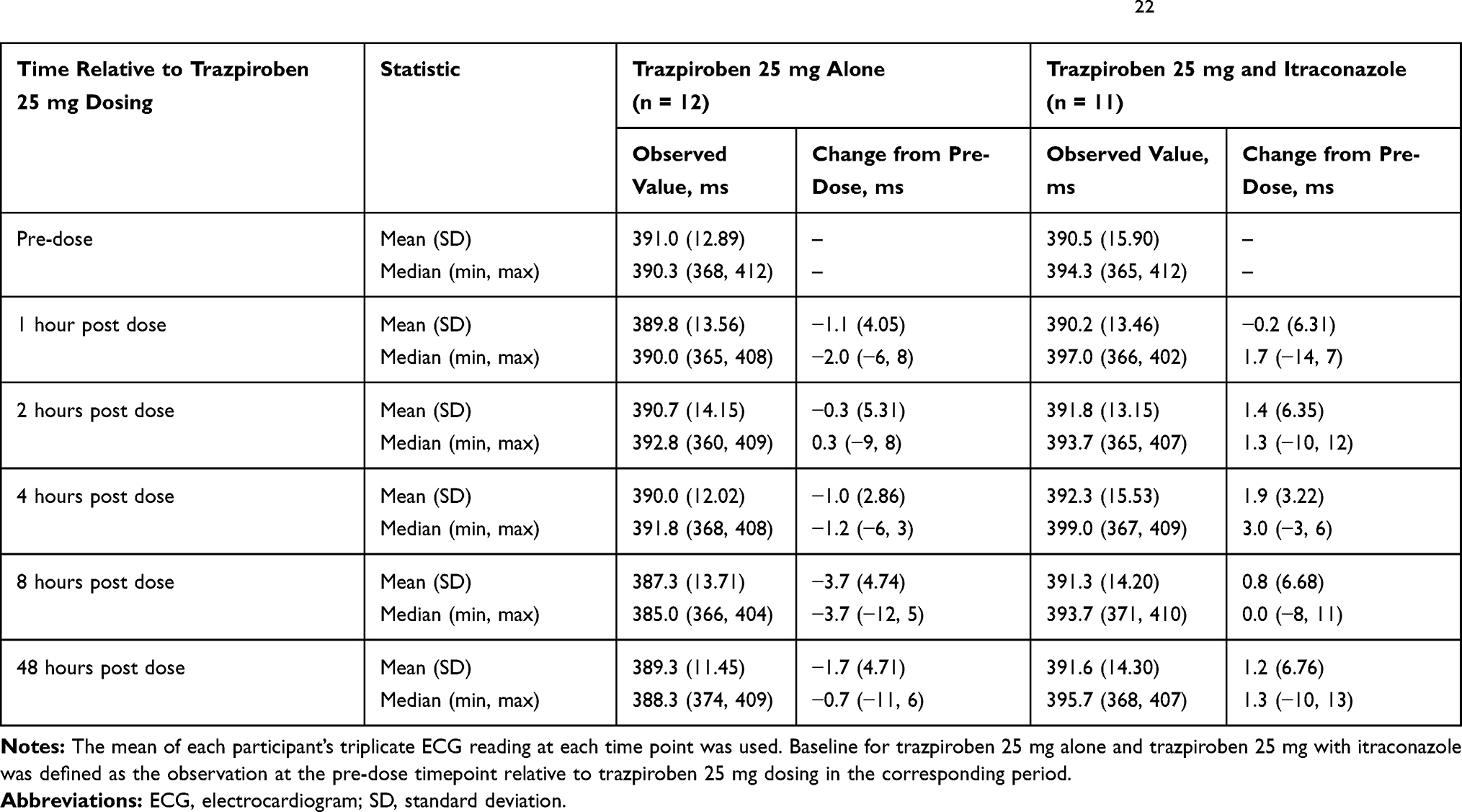 |
Table 3 Summary of Mean 12-Lead Triplicate ECG QT Intervals Corrected Using Fridericia’s Formula22 |
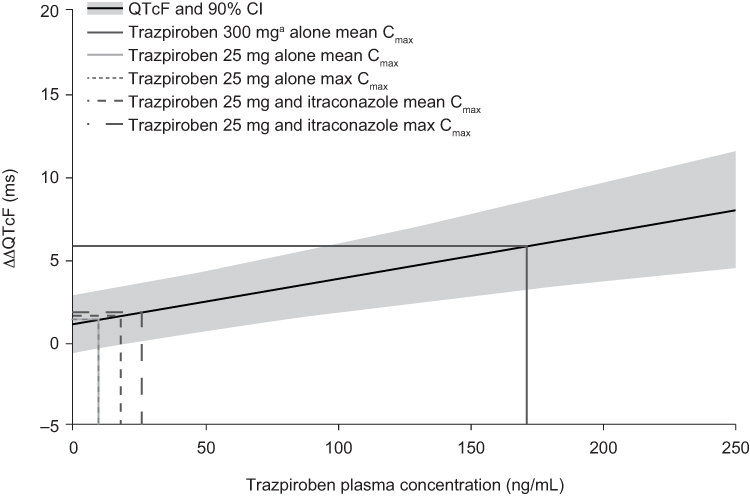 |
Figure 5 Linear mixed-effects model of the predicted ΔΔQTcF at different trazpiroben plasma concentrations after administration of a single oral dose of trazpiroben 25 mg in the presence and absence of itraconazole. Abbreviations: ΔΔQTcF, placebo-corrected change from baseline in QTcF; CI, confidence interval; Cmax, maximum plasma concentration; QTcF, QT interval corrected for heart rate by the Fridericia method. Notes: The solid black line with grey shading denotes the model-predicted mean ∆∆QTcF with 90% CI. a300 mg was the maximum oral dose in the single ascending dose study. |
There were no AEs reported in period 1 of the study. Two AEs in period 2 were reported (swelling of the face prior to trazpiroben 25 mg dosing in one individual and constipation after dosing in another); both were mild in intensity and considered unrelated to trazpiroben 25 mg by the investigator. During the study, there were no deaths and no AEs led to the discontinuation of trazpiroben 25 mg. There were no clinically relevant abnormalities observed in vital signs, ECGs (including QTcF interval; Table 3), physical examinations, hematology and chemistry parameters, urinalysis, or other safety parameters.
Discussion
This study investigated the pharmacokinetics, safety, and tolerability of trazpiroben 25 mg (expected therapeutic dose) in the presence and absence of itraconazole in healthy participants. Following co-administration with itraconazole, mean trazpiroben Cmax was increased by approximately twofold and AUC0–∞ by approximately 1.3-fold. Guidance from the FDA and EMA defines moderately sensitive substrates as demonstrating ≥2- to <5-fold increases in AUC on co-administration with strong inhibitors of a given metabolic pathway in clinical drug–drug interaction studies, and so these findings indicate that trazpiroben is relatively insensitive to the inhibition of CYP 3A4-mediated metabolism.23–25 There is evidence that trazpiroben is also a substrate for P-glycoprotein (P-GP; data on file), which, given that itraconazole inhibits both CYP 3A4 and P-GP,20,21,26 means it is plausible that the increase of trazpiroben Cmax by twofold on co-administration with itraconazole reflects a complex interaction between CYP 3A4 and P-GP inhibition in the intestine. Median tmax was 1.00 hour in both the presence and absence of itraconazole, and the mean t1/2z was 5.00 hours in the presence of itraconazole and 2.32 hours in the absence of itraconazole. The mean t1/2z in the absence of itraconazole was consistent with the short half-life seen in the US AT-01C study. There were two mild AEs, both considered unrelated to treatment with trazpiroben 25 mg.
There remains an unmet medical need for effective treatments with favorable safety profiles for the long-term treatment of gastroparesis,17,27 as current therapies have significant safety concerns limiting the duration of their use. A retrospective case–control study of domperidone of 83,212 individuals with gastroparesis over a 15-year period found that the adjusted odds ratio for sudden cardiac death or serious ventricular arrhythmia in patients receiving domperidone was 1.59 (95% CI: 1.28, 1.98) versus those not receiving the drug.28 In addition, a case–control study performed using a primary care practice observational database covering over 500,000 patients with gastroparesis found an odds ratio of 3.8 (95% CI: 1.5, 9.7) for sudden cardiac death during current domperidone exposure versus no previous exposure.29 The safety concerns with domperidone may be exacerbated by inhibition of CYP 3A4. In a study of the pharmacokinetic interaction between domperidone and ketoconazole (another potent CYP 3A4 inhibitor), the area under the concentration–time curve from time 0 to 24 hours at steady state and the Cmax at steady state were increased by 3.57-fold (90% CI: 3.31, 3.86) and 2.93-fold (90% CI: 2.65, 3.25), respectively, in the presence versus the absence of ketoconazole. The corresponding mean placebo-adjusted QTcF with domperidone in the presence and absence of ketoconazole was 4.20 ms (95% CI: 0.77, 7.63) and 15.90 ms (95% CI: 12.47, 19.33), respectively; increases higher than those seen with trazpiroben 25 mg in the presence and absence of itraconazole.30 Although this is a crude comparison due to the use of a different CYP 3A4 inhibitor, this indicates that cardiovascular concerns seen with domperidone are unlikely to be elicited by trazpiroben under similar conditions.
Trazpiroben is a very weak inhibitor of the human ether-a-go-go-related gene (hERG) channel (50% inhibitory concentration [IC50] = 15.6 μM), unlike domperidone, thus reducing the potential for fatal arrhythmias.15 In the current study, the mean change from baseline in the observed QTcF after trazpiroben 25 mg alone was negative at all post-dose time points, and mean changes from baseline after co-administration with itraconazole were small. There was minimal change in the predicted ∆∆QTcF of trazpiroben 25 mg in the presence and absence of itraconazole, based on the linear mixed-effects model from the US AT-01C study.
As with most drug–drug interaction studies, this trial was performed in healthy individuals, and in the current study only male participants were eligible for enrollment following screening. The absence of female participants from the current trial may have a number of implications which must be considered. Women have been found to experience adverse drug reactions (ADRs) almost twice as frequently as men, with sex-related differences in pharmacokinetics strongly predicting sex-specific ADRs in females, meaning inclusion of women in clinical trials is vital for understanding how the disposition and AE profile of a medication may vary with gender.31 In addition, gastroparesis has been found to be more prevalent in women than in men, meaning that evaluation of trazpiroben pharmacokinetics and safety profile in both male and female participants is of great clinical relevance for this disease.32 Therefore, further testing of trazpiroben in wider patient populations is required to supplement the evidence presented here, and to provide further data supporting the efficacy and safety profile of trazpiroben for long-term use in patients with gastroparesis.
Conclusion
Compared with trazpiroben 25 mg alone, co-administration of itraconazole with trazpiroben 25 mg increased the Cmax of trazpiroben by only 1.98-fold (90% CI 1.64, 2.39) and AUC0–∞ by 1.28-fold (90% CI 1.10, 1.49), indicating that trazpiroben is relatively insensitive to the inhibition of CYP 3A4 as defined by guidance from both the FDA and EMA. These findings indicate that the cardiovascular safety concerns associated with domperidone, particularly in the presence of other drugs metabolized by CYP 3A4, are unlikely to be elicited by trazpiroben under similar conditions.
Abbreviations
ADR, adverse drug reaction; AE, adverse event; AUC0–∞, area under the concentration–time curve from time 0 to infinity; AUC0–last, area under the concentration–time curve from time 0 to the last quantifiable concentration; BMI, body mass index; CI, confidence interval; Cmax, maximum plasma concentration; CYP, cytochrome P450; ECG, electrocardiogram; EMA, European Medicines Agency; FDA, Food and Drug Administration; P-GP, P-glycoprotein; QTcF, QT interval corrected for heart rate by the Fridericia method; t1/2z, terminal disposition phase half-life; tmax, time to maximum plasma concentration.
Data Sharing Statement
Takeda makes patient-level, de-identified data sets and associated documents available after applicable marketing approvals and commercial availability have been received, an opportunity for the primary publication of the research has been allowed, and other criteria have been met as set forth in Takeda’s Data Sharing Policy (see https://www.takedaclinicaltrials.com for details). To obtain access, researchers must submit a legitimate academic research proposal for adjudication by an independent review panel, who will review the scientific merit of the research and the requestor’s qualifications and conflict of interest that can result in potential bias. Once approved, qualified researchers who sign a data sharing agreement are provided access to these data in a secure research environment. The datasets, including the redacted study protocol, redacted statistical analysis plan, and individual participant data supporting the results reported in this manuscript, will be available three months from initial request, to researchers who provide a methodologically sound proposal. The data will be provided after its de-identification, in compliance with applicable privacy laws, data protection and requirements for consent and anonymization.
Ethics Approval
All procedures performed in this trial were approved and conducted in compliance with the institutional review board regulations of the US Code of Federal Regulations, Good Clinical Practice regulations and guidelines, and all applicable local regulations, and in accordance with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Consent to Participate
This trial was conducted in compliance with the informed consent regulations and all patients provided signed informed consent forms prior to enrolment in the trial standards.
Acknowledgments
Medical writing assistance was provided by Fraser Harris and Alex Kisbey of Oxford PharmaGenesis Ltd, Oxford, UK, and was supported by Takeda Pharmaceutical Company, Ltd.
Author Contributions
C Chen, W Zhang, M Bari, C Almansa, M Baratta, and M Rosario designed the trial, analyzed the data, wrote, and reviewed the manuscript. All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
This study was sponsored by Takeda Pharmaceutical Company, Ltd.
Disclosure
C Chen was an employee of Takeda Development Center Americas, Inc. at the time the study was performed and received stock or stock options; he is currently an employee of Bayer Pharmaceuticals, Whippany, NJ, USA. W Zhang and M Baratta are employees of Takeda Development Center Americas, Inc. and receive stock or stock options. M Bari was an employee of Takeda International UK, Ltd. at the time the study was performed and received stock or stock options; he is currently Medical Director at New Medicines Ltd., Manchester, UK. C Almansa was an employee of Takeda Pharmaceuticals and received stock or stock options at the time of the study, and is currently an employee of Ironwood Pharmaceuticals, Boston, MA, USA. M Rosario was an employee of Takeda Pharmaceuticals and received stock or stock options at the time of the study, and is currently an employee of Syros Pharmaceuticals Inc., Cambridge, MA, USA. The authors report no other conflicts of interest in this work.
References
1. Masaoka T, Tack J. Gastroparesis: current concepts and management. Gut Liver. 2009;3(3):166–173. doi:10.5009/gnl.2009.3.3.166
2. Parkman HP, Hasler WL, Fisher RS; American Gastroenterological Association. American Gastroenterological Association technical review on the diagnosis and treatment of gastroparesis. Gastroenterology. 2004;127(5):1592–1622.
3. Abrahamsson H. Treatment options for patients with severe gastroparesis. Gut. 2007;56(6):877–883. doi:10.1136/gut.2005.078121
4. Erbas T, Varoglu E, Erbas B, Tastekin G, Akalin S. Comparison of metoclopramide and erythromycin in the treatment of diabetic gastroparesis. Diabetes Care. 1993;16(11):1511–1514. doi:10.2337/diacare.16.11.1511
5. Janssens J, Peeters TL, Vantrappen G, et al. Improvement of gastric emptying in diabetic gastroparesis by erythromycin. Preliminary studies. N Engl J Med. 1990;322(15):1028–1031. doi:10.1056/NEJM199004123221502
6. Richards RD, Davenport K, McCallum RW. The treatment of idiopathic and diabetic gastroparesis with acute intravenous and chronic oral erythromycin. Am J Gastroenterol. 1993;88(2):203–207.
7. Hawkyard CV, Koerner RJ. The use of erythromycin as a gastrointestinal prokinetic agent in adult critical care: benefits versus risks. J Antimicrob Chemother. 2007;59(3):347–358. doi:10.1093/jac/dkl537
8. Rao AS, Camilleri M. Review article: metoclopramide and tardive dyskinesia. Aliment Pharmacol Ther. 2010;31(1):11–19. doi:10.1111/j.1365-2036.2009.04189.x
9. Rumore MM. Cardiovascular adverse effects of metoclopramide: review of literature. Int J Case Rep Images. 2012;3(5):1–10. doi:10.5348/ijcri-2012-05-116-RA-1
10. European Medicines Agency (EMA). European Medicines Agency recommends changes to the use of metoclopramide; 2013. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Referrals_document/Metoclopramide_31/WC500146610.pdf.
11. Darmani NA, Zhao W, Ahmad B. The role of D2 and D3 dopamine receptors in the mediation of emesis in Cryptotis parva (the least shrew). J Neural Transm. 1999;106(11–12):1045–1061. doi:10.1007/s007020050222
12. Kashyap P, Micci MA, Pasricha S, Pasricha PJ. The D2/D3 agonist PD128907 (R-(+)-trans-3,4a,10b-tetrahydro-4-propyl-2H,5H-[1]benzopyrano[4,3-b]-1,4-oxazin- 9-ol) inhibits stimulated pyloric relaxation and spontaneous gastric emptying. Dig Dis Sci. 2009;54(1):57–62. doi:10.1007/s10620-008-0335-6
13. Lee A, Kuo B. Metoclopramide in the treatment of diabetic gastroparesis. Expert Rev Endocrinol Metab. 2010;5(5):653–662. doi:10.1586/eem.10.41
14. Barone JA. Domperidone: a peripherally acting dopamine2-receptor antagonist. Ann Pharmacother. 1999;33(4):429–440. doi:10.1345/aph.18003
15. Michaud V, Turgeon J. Domperidone and sudden cardiac death: how much longer should we wait? J Cardiovasc Pharmacol. 2013;61(3):215–217. doi:10.1097/FJC.0b013e31827e2573
16. European Medicines Agency (EMA). Domperidone, Summary of product characteristics; 2021. Available from: https://www.medicines.org.uk/emc/product/556/smpc#POSOLOGY.
17. Rossi M, Giorgi G. Domperidone and long QT syndrome. Curr Drug Saf. 2010;5(3):257–262. doi:10.2174/157488610791698334
18. Whiting RL, Darpo B, Chen C, et al. Safety, pharmacokinetics, and pharmacodynamics of trazpiroben (TAK-906), a novel selective D2 /D3 receptor antagonist: a phase 1 randomized, placebo-controlled single- and multiple-dose escalation study in healthy participants. Clin Pharmacol Drug Dev. 2021. doi:10.1002/cpdd.906
19. Guengerich FP. Role of cytochrome P450 enzymes in drug-drug interactions. Adv Pharmacol. 1997;43:7–35.
20. The United States Food and Drug Administration (FDA). SPORANOX® (itraconazole). Prescribing information; 2010. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/022484S000lbl.pdf.
21. European Medicines Agency (EMA). SPORANOX® (itraconazole). Summary of product characteristics; 2020. Available from: https://www.medicines.org.uk/emc/product/1513/smpc.
22. Vandenberk B, Vandael E, Robyns T, et al. Which QT correction formulae to use for QT monitoring? J Am Heart Assoc. 2016;5(6):e003264. doi:10.1161/JAHA.116.003264
23. The United States Food and Drug Administration (FDA). Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers; 2020. Available from: https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers.
24. European Medicines Agency (EMA). Guideline on the investigation of drug interactions; 2012. Available from https://www.emaeuropaeu/en/documents/scientific-guideline/guideline-investigation-drug-interactions-revision-1_enpdf.
25. U.S. Food and Drug Administration. Clinical Drug Interaction Studies – cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions: guidance for Industry; 2020. Available from https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions.
26. Wang EJ, Lew K, Casciano CN, Clement RP, Johnson WW. Interaction of common azole antifungals with P glycoprotein. Antimicrob Agents Chemother. 2002;46(1):160–165. doi:10.1128/AAC.46.1.160-165.2002
27. Collins KK, Sondheimer JM. Domperidone-induced QT prolongation: add another drug to the list. J Pediatr. 2008;153(5):596–598. doi:10.1016/j.jpeds.2008.06.009
28. Johannes CB, Varas‐Lorenzo C, McQuay LJ, Midkiff KD, Fife D. Risk of serious ventricular arrhythmia and sudden cardiac death in a cohort of users of domperidone: a nested case‐control study. Pharmacoepidemiol Drug Saf. 2010;19(9):881–888. doi:10.1002/pds.2016
29. Straus SMJM, Sturkenboom MCJM, Bleumink G, et al. Non-cardiac QTc-prolonging drugs and the risk of sudden cardiac death. Eur Heart J. 2005;26(19):2007–2012. doi:10.1093/eurheartj/ehi312
30. Boyce MJ, Baisley KJ, Warrington SJ. Pharmacokinetic interaction between domperidone and ketoconazole leads to QT prolongation in healthy volunteers: a randomized, placebo-controlled, double-blind, crossover study. Br J Clin Pharmacol. 2012;73(3):411–421. doi:10.1111/j.1365-2125.2011.04093.x
31. Zucker I, Prendergast BJ. Sex differences in pharmacokinetics predict adverse drug reactions in women. Biol Sex Differ. 2020;11:1–14. doi:10.1186/s13293-020-00308-5
32. Camilleri M, Chedid V, Ford AC, et al. Gastroparesis. Nature Rev Dis Primers. 2018;4(1):1–19. doi:10.1038/s41572-018-0038-z
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.