")
Back to Journals » Drug Design, Development and Therapy » Volume 11
Evaluation of the antitumor activity of NOV202, a novel microtubule targeting and vascular disrupting agent
Authors Rickardson L , Kutvonen E , Orasniemi S, Högberg M, Kallio MJ , Rehnmark S
Received 25 January 2017
Accepted for publication 17 March 2017
Published 28 April 2017 Volume 2017:11 Pages 1335—1351
DOI https://doi.org/10.2147/DDDT.S133189
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Linda Rickardson,1 Emma Kutvonen,2 Satu Orasniemi,2 Marita Högberg,1 Marko J Kallio,2,3 Stefan Rehnmark1
1Noviga Research AB, Södertälje, Sweden; 2Department of Physiology, Institute of Biomedicine, University of Turku, 3Centre for Biotechnology, University of Turku, Turku, Finland
Purpose: Overall, ~65% of patients diagnosed with advanced ovarian cancer (OC) will relapse after primary surgery and adjuvant first-line platinum- and taxane-based chemotherapy. Significant improvements in the treatment of OC are expected from the development of novel compounds having combined cytotoxic and antiangiogenic properties that make them effective on refractory tumors.
Methods: Permeability of NOV202 was determined with Caco-2 monolayer assay. The compound’s pharmacokinetic profile and plasma:brain distribution were assessed in male C57Bl/6 mice. The compound’s impacts on tubulin, microtubules and cell cycle were investigated by using in vitro tubulin polymerization assay, cell-based immunofluorescence and live cell microscopy. The IC50 concentrations of NOV202 were assessed in a panel of eight cancer cell lines. Impact of the compound on vascular tube formation was determined using the StemKit and Chick chorioallantoic membrane assays. The in vivo efficacy of the compound was analyzed with an OC xenograft mouse model.
Results: NOV202 was found to suppress cancer cell proliferation at low nanomolar concentrations (IC50 2.3–12.0 nM) and showed equal efficacy between OC cell line A2780 (IC50 2.4 nM) and its multidrug-resistant subline A2780/Adr (IC50 2.3 nM). Mechanistically, NOV202 targeted tubulin polymerization in vitro in a dose-dependent manner and in cells induced an M phase arrest. In vivo, NOV202 caused a dose-dependent reduction of tumor mass in an A2780 xenograft model, which at the highest dose (40 mg/kg) was comparable to the effect of paclitaxel (24 mg/kg). Interestingly, NOV202 exhibited vascular disrupting properties that were similar to the effects of Combretastatin A4.
Conclusion: NOV202 is a novel tubulin and vascular targeting agent that shows strong anticancer efficacy in cells and OC xenograft models. The finding that the compound induced significantly more cell death in Pgp/MDR1 overexpressing OC cells compared to vincristine and paclitaxel warrants further development of the compound as a new therapy for OC patients with treatment refractory tumors and/or relapsing disease.
Keywords: ovarian cancer, NOV202, microtubule-targeting agent, vascular disruption, M phase arrest, multidrug resistance
Introduction
Ovarian cancer (OC) is the leading cause of death among all gynecologic cancers. About 1.3% of women will develop the disease during their life and currently the 5-year survival rate for all types of OC is ~45%.1 It should be noted that in patients with advanced disease (ie, stages III–IV), the 5-year survival rate is <30%. Despite many cancer drug innovations and clinical studies, the treatment of OC patients has not improved substantially over the last decade. Another factor that has overshadowed the therapeutic advancement is the fact that the origin and pathogenesis of OC remain poorly understood. The three main primary OC groups are epithelial, sex cord/stromal and germ cell tumors, of which the first group accounts for ~90% of all cases.2 However, according to the recent morphological, molecular biology and genetic data, OC is a heterogeneous disease composed of different subtypes of tumors exhibiting dissimilar clinicopathologic features and tumorigenic behavior. Due to this diversity, it is not surprising that even new targeted adjuvant therapies such as vascular endothelial growth factor (VEGF) and EGFR pathway inhibitors have shown limited improvement to the overall OC survival rate. This emphasizes the clinical need for potent cytotoxic agents in the treatment of OC, as well as novel combinations of cytotoxic agents with biologics (eg, checkpoint inhibitors and anti-VEGF therapeutics) to improve therapeutic outcome.
Taxanes, together with vinca alkaloids and epothilones, belong to the group of tubulin targeting agents that exert their antitumor activity by causing perturbation of microtubule dependent cellular reactions.3 Besides induction of cell cycle arrest and cell death, many of these drugs also have antivascular actions that likely contribute to their clinical efficacy.4 Critically, these agents alter the microtubule dynamics in all cells of the body, which causes significant dose-limiting toxicities on normal tissues.3 Regardless of their adverse effects, today throughout the world, the standard of care in OC encompasses cytoreductive surgery followed by the administration of paclitaxel/docetaxel-platinum-based chemotherapy.5 Moreover, taxanes are also used as part of combination chemotherapy for the treatment of breast and nonsmall cell lung cancers.6,7 Importantly, despite that about two-thirds of OC patients initially respond to the combined surgery-chemotherapy treatment of ~70% relapse at a median of 15 months from the original diagnosis.8 Moreover, the subsequent chemotherapy regimens typically fail due to the intrinsic and acquired chemoresistance. One contributing factor for tumor recurrence, metastasis and drug insensitivity is the fact that most of the women with OC are diagnosed at an advanced stage of the disease when the tumor aggressiveness is increased.9 Therefore, an urgent medical need exists for improved early detection of OC and development of more effective new therapies for the treatment of refractory tumors and relapsing disease.
One characteristic mechanism behind the acquired drug resistance is an accelerated drug efflux because of the enhanced expression of ATP binding cassette (ABC) transporter proteins, such as P-glycoprotein, Pgp/ABCB1/MDR1.10,11 The known substrates of ABC transporter proteins include many cytotoxic drugs, such as taxanes, vinca alkaloids, anthracyclines, actinomycin D and mitomycin C.12,13 To overcome this serious clinical problem a number of strategies can be considered. For example, the therapeutic effects of existing drugs may be enhanced by combining them with the next generation Pgp inhibitors under development.14 Alternatively, adjustment of the clinical concentration of the drug-of-choice to counteract the kinetic parameters of Pgp-mediated efflux can improve the therapy outcome.15 Finally, the development of novel anticancer compounds that escape the efflux pump is expected to lead to better treatment results. Here, we describe the characteristics and antitumor activity of a novel microtubule depolymerizing and vascular disrupting agent, NOV202, that exhibits significant cell proliferation suppressive effects in OC xenograft models and in cultured cancer cell lines, including a multidrug-resistant subline.
Materials and methods
Compounds
NOV202 (MW 413), synthesized at SP Process Development AB (Södertälje, Sweden), was used at a concentration range of 0.1 nM–10.0 μM depending on the assay. Paclitaxel (0.1 nM–3.0 μM; Selleckchem, Houston, TX, USA), vincristine (0.1 nM–3.0 μM; Selleckchem), vinblastine (5.0 nM–3.0 μM; Selleckchem), lexibulin (CYT997) (0.1 nM–3.0 μM, Selleckchem), Combretastatin A4 (0.8–50 nM; Selleckchem) and epothilone-B (5.0 nM–3.0 μM; Selleckchem) were used as reference compounds. All compounds were dissolved into dimethyl sulfoxide (DMSO, max 0.1% of the total volume).
Reagents and animals
All reagents are from Sigma-Aldrich Corp. (St Louis, MO, USA) unless otherwise noted. Rj:NMRI-Foxn1nu/Foxn1nu female mice (Janvier Laboratories, St Berthevin Cedex, France) were used in the maximal tolerated dose and xenograft assays, and C57Bl/6 male mice (Taconic Biosciences Inc., Ejby, Denmark) were used in the pharmacokinetic study. All animal assays and handling were approved by and conducted in compliance with the policies of the Institutional animal care committees of the Experimental Pharmacology & Oncology Berlin-Buch GmbH (Berlin, Germany) and In Vivo Design AB (Uppsala, Sweden) that fully meet the ethical and legal requirements as defined in the 2010/63/EU directive and national animal protection laws of German and Sweden. All mice were housed under strictly controlled and standardized barrier conditions (22°C±1°C temperature, 50%±10% relative humidity, 12 h light/dark rhythm) in individually ventilated Macrolon Type-II cages (Techniplast S.p.A., Buguggiate, Italy). The mice received autoclaved food (pellet chow) and acidified (pH 4.0) drinking water ad libitum.
Cell culture
The following cell lines and culture medium (indicated inside the brackets) were used in the study. The leukemia cell line CCRF-CEM (RPMI 1640), the ovarian carcinoma cell lines A2780 and A2780/Adr (RPMI 1640), the pancreatic adenocarcinoma cell line BXPC-3 (RPMI 1640), the lymphoma cell line U-937 (RPMI 1640), the myeloma cell line RPMI 8226 (RPMI 1640) and the colon carcinoma cell lines HT29 (McCoy’s medium) and HCT116 (McCoy’s medium) were purchased from European Collection of Cell Cultures (ECACC, Salisbury, UK). The bone metastatic breast adenocarcinoma cell line MDA-MB-231-SA (Dulbecco’s Modified Eagle’s Medium [DMEM]), the nontumorigenic breast luminal epithelial cell line MCF-10A (DMEM:HAM’s F12, 1:1), the human intestinal Caco-2 cell line (EMEM), and the ovarian adenocarcinoma cell lines OvCar-8 (RPMI 1640), OvCar-4 (RPMI 1640) and CaOv-3 (DMEM) were purchased from the American Type Culture Collection (Manassas, VA, USA). The media were supplemented with 10% fetal bovine serum (FBS), glutamine (Sigma-Aldrich Corp.) and penicillin/streptomycin (Sigma-Aldrich Corp.), except for the MDA-MB-231-SA where 5% horse serum and for Caco-2 20% FBS were used. The cells were maintained in a humidified atmosphere at 37°C, 5% CO2 and subcultured twice a week. For experiments, a density of 1,000–2,500 or 5,000–10,000 cells per well were seeded in 384-well or 96-well microplates, respectively, depending on the assay.
Pharmacokinetics and distribution
The pharmacokinetics and distribution of NOV202 were determined in male C57Bl/6 mice after intravenous (IV) (10 mg/kg, EtOH and 10% Tween 80 in 5% glucose in water) and oral (PO) (30 mg/kg, 70% PEG400, 0.25% Tween) administrations at 0, 0.5, 1, 2, 4, 6 and 24 h post-dosing. Blood samples were collected from three animals (tail vein or orbital sinus) per group in ethylenediaminetetraacetic acid-coated collection tubes (Sarstedt AB, Helsingborg, Sweden) and plasma was separated by centrifugation at 2,000× g for 5 min. Plasma samples were stored in −80°C until transportation on dry ice. In the pharmacokinetics study, the actual concentration was determined to 9.5 and 28.6 mg/kg for IV and PO, respectively.
Caco-2 permeability assay
For determination of NOV202 permeability, Caco-2 cell monolayers were grown to confluence on collagen-coated, microporous, polycarbonate membranes in 12-well transwell plates (Corning Inc., New York, USA). The permeability assay buffer was Hanks Balanced Salt Solution containing 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) and 15 mM glucose at pH 7.4. The buffer in the receiver chamber also contained 1% bovine serum albumin. The dosing solution concentration was 5 μM test compound in the assay buffer. Cell monolayers were dosed on the apical side (A-to-B) or basolateral side (B-to-A) and incubated at 37°C with 5% CO2 in a humidified incubator. Samples were taken from the donor and receiver chambers at 120 min. Each determination was performed in duplicate. The coapplied Lucifer yellow flux was also measured for each monolayer to out rule any cell monolayer damage during the flux period. All samples were assayed by liquid chromatography-mass spectrometry and tandem mass spectrometry using electrospray ionization.
The apparent permeability, Papp, and percent recovery were calculated as follows:
Papp = (dCr/dt) × Vr/(A × CA), |
where dCr/dt is the slope of the cumulative concentration in the receiver compartment versus time in μM/s; Vr is the volume of the receiver compartment in cm3; A is the area of the insert (1.13 cm2 for 12-well plate); CA is the average of the nominal dosing concentration and the measured 120 min donor concentration in μM.
Interpretations of the results were done based on the following thresholds: Permeability classification: (Papp A-to-B) <1.0×10−6 cm/s: low; (Papp A-to-B) ≥1.0×10−6 cm/s: high. Significant efflux: efflux ratio >3.0 and (Papp B-to-A) ≥1.0×10−6 cm/s.
In vitro tubulin polymerization assay
NOV202 (1.0–10 μM) was tested in a tubulin polymerization assay kit purchased from Cytoskeleton Inc. (Denver, CO, USA). Polymerization is followed by fluorescence enhancement due to the incorporation of a fluorescent reporter into microtubules as polymerization occurs. Vincristine and paclitaxel at 3.0 μM were used as positive controls for tubulin polymerization inhibition and stabilization, respectively. All compounds were dissolved in DMSO and further diluted with sterile water to obtain a maximum DMSO concentration of 0.1%, which was used as solvent control. NOV202 and control compounds were incubated with bovine tubulin protein in a cell-free environment and the fluorescence was then measured continuously using Fluostar Optima (BMG Labtech GmbH, Ortenberg, Germany) at 360/450 nm for 60 min.
Vascular tube formation assay
Concentration dependent vascular tube formation induced by VEGF and tube disruption (cord length, mm/mm2) were estimated using the StemKit (Essen BioScience Inc., Ann Arbor, MI, USA) and IncuCyte FLR (Essen BioScience) according to the manufacturer’s procedure.
In short, cocultures of GFP-labeled endothelial colony forming cells (ECFCs) and adipocyte derived stem cells (ADSCs) were created on 96-well assay plates. Compound studies were performed using a concentration range of 0.046–100 nM for NOV202 and 0.78–100 nM for the reference compound Combretastatin A4, a tubulin inhibitor and vascular disrupting agent currently in phase III clinical trials. The neoangiogenic (cord formation) assay was initiated on day 0 by coapplying the compounds with VEGF (20 ng/mL). For determination of disruption of the established cords, VEGF (20 ng/mL) was applied alone on day 0 to form cord networks and 4 days later the compounds were administrated in the continued presence of VEGF (20 ng/mL). Cord formation/disruption was monitored kinetically using IncuCyte live cell imaging system. Fluorescent and phase-contrast images (10×) were gathered every 6 h for up to 8 days and analyzed for cord length and number of branch points from 3 to 6 separate samples for each treatment condition.
The chicken embryo chorioallantoic membrane (CAM) assay
An ex ovo chicken embryo CAM assay was used to evaluate the compound’s impact on angiogenesis.16 In short, fertilized chicken eggs from a local commercial hatchery were incubated in an automated incubator at 37.5°C and 60% humidity. On incubation day 4, the eggs were broken gently to individual 10 cm petri dishes and incubated further. Two days later a small sterile filter paper (5 mm diameter) containing the experimental solution was placed on the CAM between two main blood vessels. The following drug concentrations, selected on the basis of a pilot test, were used; NOV202 (10, 50 and 200 nM) and Combretastatin A4 (10 and 50 nM). DMSO served as a negative control. The eggs were further incubated for 2 days and the blood vessel growth and integrity was recorded with a digital camera at 0, 24 and 48 h time points. The relative surface area (vasculature growth) of vessels touching the filter paper and their relative disruption index (amount of intact vessel segments longer than 10 mm) were measured from the photographs using the Metamorph software (Molecular Devices LLC, Sunnyvale, CA, USA).
Cytotoxicity assay
The fluorometric microculture cytotoxicity assay (FMCA) was used for the measurement of the cytotoxic and/or antiproliferative effect of compounds in vitro.17 For the experiments, 96- or 384-well microplates (Thermo Fisher Scientific, Waltham, MA, USA) were prepared with test compounds and stored at −20°C or −80°C until use. Cells were then seeded into the drug-prepared plates that were placed in an incubator for 72 h. On the last day of incubation, the plates were washed, and a buffer containing fluorescein diacetate was added and incubated with the cells for 45 min. The fluorescence per well was measured in a FLUOStar Optima (BMG Labtech GmbH) and a survival index % (SI) for each compound-treated well was calculated with the equation: Compound-treated cells minus blank divided by control cells minus blank. From the SI, graphs were plotted using GraphPadPrism 5.0 (GraphPad Software Inc., San Diego CA, USA) and IC50-values for each cell type and compound were determined from the curves.
Live cell microscopy
Cells treated with NOV202 (3.0 and 50 nM), or reference drug paclitaxel (5.0 and 50 nM), vinblastine (5.0 and 50 nM) or epothilone (5.0 and 50 nM), were filmed in three replicates with IncuCyte live cell imager (Essen BioScience) using 1 h interval for a total of 72 h after adding the compounds on cells. The IncuCyte images were used for generating time-lapse films and growth curves (the average cell confluence from six images per each time point). Time-lapse films were used for determining the duration of mitosis (10 cells per well, 30 cells per a treatment condition). Moreover, the fate of each mitotic cell that was used to calculate the length of cell division was recorded. Also the cell death index (CDI) was determined at 24 and 48 h time points.
Immunofluorescence labeling
Cells were grown on coverslips in 12-well plates until optimal cell confluence was obtained. Drugs were premixed with culture medium and added to cells for 24 or 48 h. The following drug concentrations were selected on the basis of a pilot test and were used in further assays; NOV202 (10–50 nM), paclitaxel (5.0–50 nM), vinblastine (5.0–50 nM) and epothilone-B (5.0–50 nM). The cells on coverslips were fixed for 15 min in 60 mM Pipes, pH 6.9, 25 mM HEPES, 10 mM ethylene glycol tetraacetic acid, 4 mM MgSO4 containing 2% paraformaldehyde, 0.2% glutaraldehyde and 0.5% Triton X-100. The coverslips were rinsed in 10 mM 3-(N-morpholino)propanesulfonic acid, pH 7.4, 150 mM NaCl, 0.05% Tween-20 (MBST), and blocked for overnight in 20% boiled normal goat serum in MBST. Cells were stained for 1 h at room temperature (RT) in 5% goat serum in MBST containing primary antibodies against pericentrin (ab4448, 1:500; Abcam, Cambridge, UK) and alpha-tubulin (ab7291, 1:300; Abcam) followed by Alexa Fluor 488 and 555 dyes against mouse and rabbit antigens (Thermo Fisher Scientific) diluted 1:400 in MBST with 5% goat serum. DNA was counter stained 10 min at RT with 4′,6-diamidino-2-phenylindole (50 ng/mL in H2O) before mounting in Vectashield (Vector laboratories, Burlingame, CA, USA).
Fluorescence microscopy, image capture and analysis
To visualize the microtubules, pericentrin and DNA, images were acquired using ScanR microscope (Olympus Corp., Tokyo, Japan) equipped with a Hamamatsu ORCA-ER CCD digital camera (Hamamatsu Photonics, Hamamatsu City, Japan). Image stacks (10 layers at 1.0 μm step size) were captured using a 20× objective. The mitotic index (MI) was quantitated in a population of a minimum of 500 cells per treatment condition. Higher resolution images were acquired using Zeiss Axiovert 200 M microscope (Carl Zeiss Corp., Oberkochen, Germany) equipped with a Hamamatsu ORCA-ER CCD camera (Hamamatsu Photonics) and Metamorph image analysis software (Molecular Devices). Image stacks (15–35 layers at 0.2–1.0 μm step size) were captured using a 63× oil immersion objective. Image stack was converted to maximum projections before the analysis of cell morphology.
Human OC cell xenograft study
A study of the maximal tolerated dose of the NOV202 in NMRI nu/nu female mice was performed before the xenograft study. The plasma levels of NOV202 were determined after PO administration at 1 and 4 h post-dosing. For xenografts, 8 weeks old NMRI nu/nu female nude mice were used as follows; 8 animals in the vehicle group, 6 animals in each dose group of NOV202 and 6 animals in the paclitaxel group. For tumor establishment A2780 cells (passage 5) were inoculated subcutaneously and the tumors were allowed to reach a size of ca. 0.08 mm3 (typically by the study day 8) prior to start of the treatments. NOV202 was administered PO at 3 different doses (10, 20 and 40 mg/kg) in a 5 days treatment and 2 days off treatment cycle. The treatment was continued until study day 18 for the control and dose groups 10 and 20 mg/kg, and terminated at day 20. The 40 mg/kg dose group was continued until study day 26, and terminated at day 29. The treatment was compared to mice treated IV with paclitaxel (24 mg/kg) once in a week until study day 22. The tumor volume was measured at regular intervals (day 11, 13, 15, 18, 20, 22, 25 and 28).
Statistical analysis
Statistical analyses were performed using paired two-tailed Student’s t-test. Statistical significance was defined as *P≤0.05, **P≤0.01 and ***P≤0.001. Values are presented as the average ± standard deviation (SD).
Results and discussion
Pharmacokinetics, permeability and distribution of NOV202
The pharmacokinetic profile of NOV202 was investigated after intravenous and oral administration in male C57Bl/6 mice. The nominal dose was 10 and 30 mg/kg for IV and PO administration, respectively, and the actual concentration was determined to be 9.5 and 28.6 mg/kg. The pharmacokinetics data are summarized in Table 1. The permeability of NOV202 was tested through Caco-2 monolayers in both the apical-to-basolateral and basolateral-to-apical directions. First, the compound passed the Lucifer yellow monolayer integrity test criteria (<0.8×10−6 cm/s). The compound’s permeability was ranked to be high and no significant efflux was observed (Table 2). The distribution of NOV202 in plasma and brain was determined in male C57Bl/6 mice after IV and PO administrations at 2, 6 and 24 h post-dosing (Table 3). At the 2 h time-point the concentration of NOV202 in brain markedly exceeded the plasma levels, indicating that the compound bypasses the blood–brain barrier and readily distributes into brain tissue. This suggests a possibility for the treatment of brain metastasis using NOV202. In conclusion, our data show that NOV202 has good pharmacokinetic properties and high cell permeability. To determine existence of possible sex differences in the pharmacokinetic profile of NOV202 we will extend the profiling to female mice in near future.
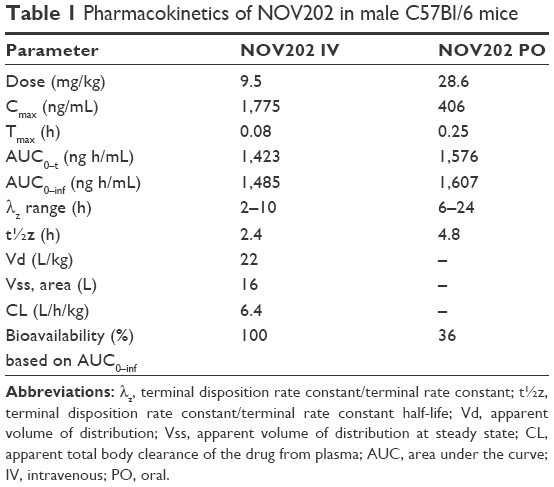 | Table 1 Pharmacokinetics of NOV202 in male C57BI/6 mice |
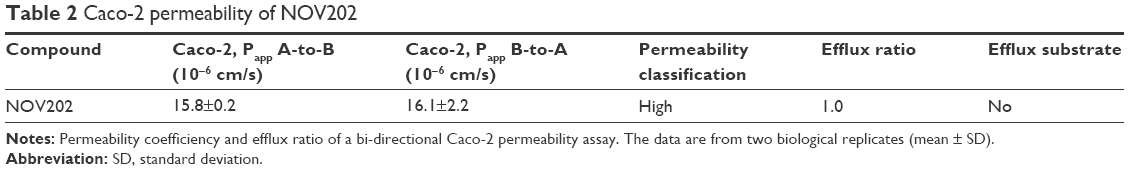 | Table 2 Caco-2 permeability of NOV202 |
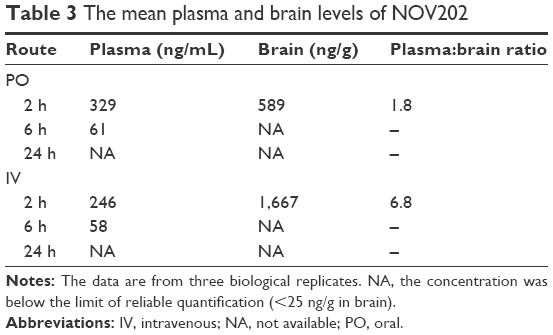 | Table 3 The mean plasma and brain levels of NOV202 |
Suppression of cell proliferation by NOV202
A 3-day nonclonogenic microplate-based cell viability FMCA setup was used for the measurement of the cytotoxic and/or antiproliferative effect of NOV202 and reference compounds paclitaxel, vincristine and CYT997 in vitro. Quantification of the cell survival shows that the compound possesses strong antiproliferative impact on all cell lines tested. In the panel of six cell lines that were analyzed first, the IC50 values for NOV202 were between 3.9 and 12 nM (Table 4). In order to study if NOV202 is a substrate for the Pgp/MDR1 efflux pump, the compound was evaluated in a pair of OC cell lines consisting of a drug sensitive parental A2780 and its multidrug-resistant subline A2780/Adr. Quantification of the FMCA data indicated that NOV202 was equally effective in suppressing cell proliferation in the drug resistant cells as the parental cells (Table 5); the ratio of IC50 values (A2780/Adr variant vs parental A2780) was 0.96, indicating similar potency between the drug resistant and sensitive cells. Notably, the reference drugs paclitaxel, vincristine and CYT997 were found to be notably less effective in inducing cell killing in the drug resistant variant cell line as compared to the parental cell line; the IC50 values were 810, 210 and 183 nM, and IC50 ratios 3,115, 955 and 3.35 for paclitaxel, vincristine and CYT997 in the A2780/Adr cells, respectively (Table 5). We conclude that NOV202 suppresses cell proliferation of various cancer cell lines of different tissue origin at low nanomolar concentrations. Moreover, the compound appears not to be a substrate of the Pgp/MDR1 efflux pump, which would be a significant clinical advance in comparison to paclitaxel in the treatment of OC patients with advanced or recurring disease.
 | Table 4 Antiproliferative activity (IC50, nM) of NOV202 and reference compounds in various cancer cell lines |
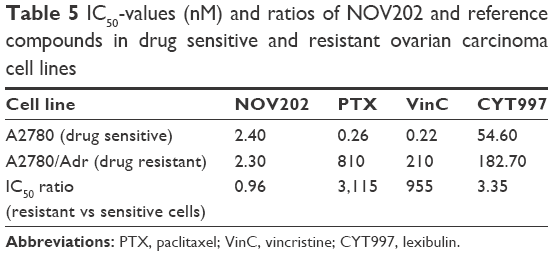 | Table 5 IC50-values (nM) and ratios of NOV202 and reference compounds in drug sensitive and resistant ovarian carcinoma cell lines |
NOV202 induces M phase arrest and cell death in dividing cancer cells
The M phase effects of NOV202 were investigated in a several human cancer cell lines of different origin. Paclitaxel, vinblastine and epothilone-B were used as reference compounds. First, HCT116, HCT8, and MCF10A cells cultured in the presence of NOV202 (50 nM) or a reference compound (50 nM) were time-lapse filmed for 3 days immediately after supplementation of the culture medium with a drug. Cell population growth curvatures were determined using the IncuCyte software based on the cell confluency. The analysis showed strong growth suppression in all three cell lines treated with NOV202, which was comparable to the efficacy of paclitaxel, vinblastine and epothilone-B (Figure 1A). From the same time-lapse videos we also measured the length of M phase arrest and recorded the fate of 30 mitotic cells for each cell line treated with the compounds at 50 nM concentration. As expected, treatment of cells with NOV202 caused a transient mitotic delay that lasted for several hours; the average duration of mitosis in HCT116, HCT8 and MCF10A cell lines was 26.2±7.1 h, 11.8±3.1 h and 16.4±5.2 h, respectively (Figure 1B), which all were significantly longer than in the vehicle-treated controls (P<0.001). Similarly, treatment of HCT116, HCT8 and MCF10A cells with paclitaxel, vinblastine or epothilone-B induced a long M phase arrest that was comparable to the NOV202 treatment (Figure 1B). The HCT116 cells that have an intact mitotic checkpoint18 and normal p53 response19 exhibited onefold longer M phase arrest upon the compound treatments when compared to the HCT8 cells (Figure 1B). Also, the fate of the mitotically arrested cells was found to be different between HCT116 and HCT8 cells; half or more of the HCT116 cells died from the M phase arrest although the majority of the HCT8 cells underwent mitotic slippage (Figure 1C). The fate of nontransformed MCF10A cells resembled more that of the HCT116 cells than HCT8 cells (Figure 1C).
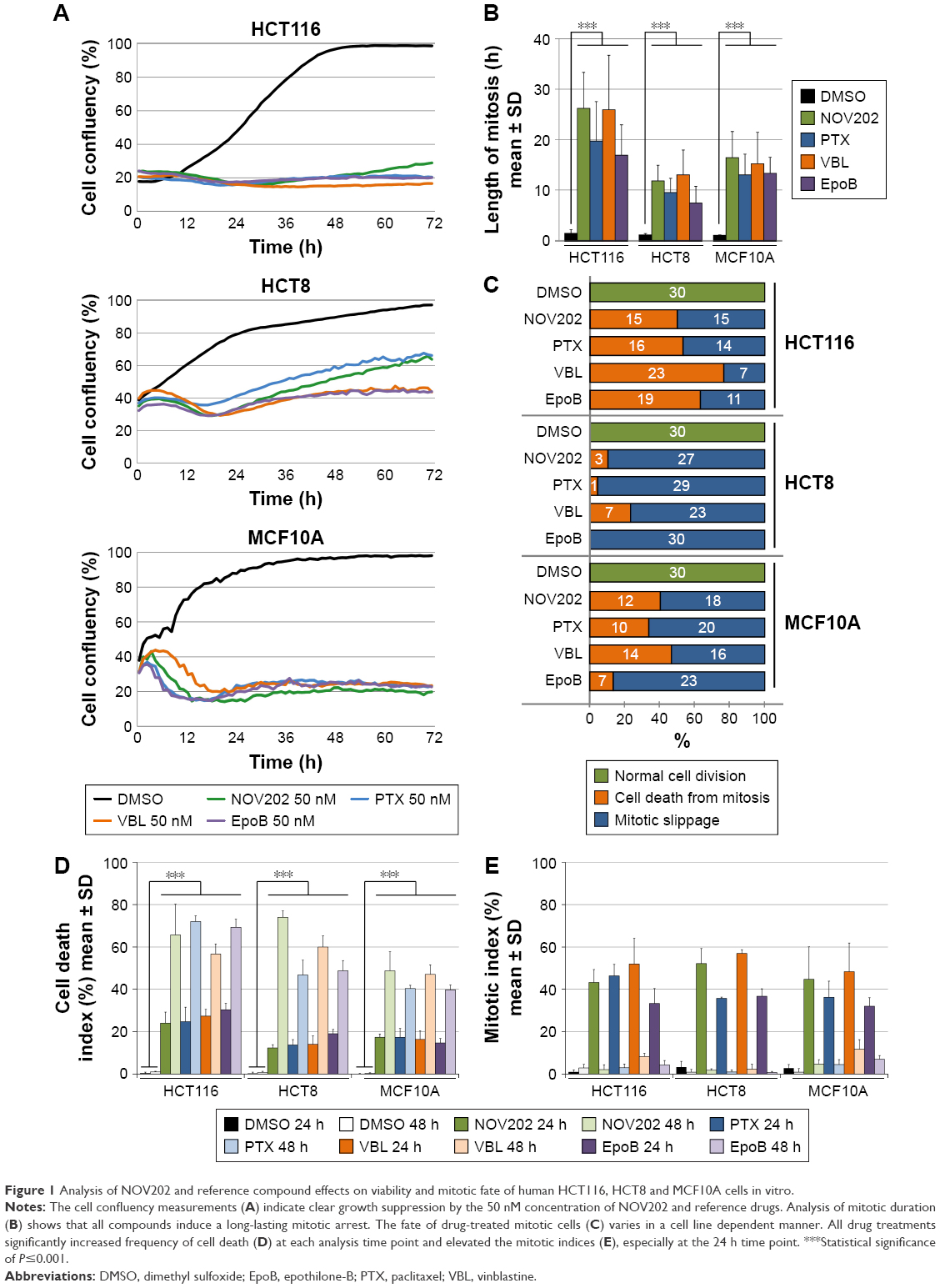 | Figure 1 Analysis of NOV202 and reference compound effects on viability and mitotic fate of human HCT116, HCT8 and MCF10A cells in vitro. |
Next, we determined the CDI from the IncuCyte films for each treatment at 24 and 48 h time points using morphological criteria of apoptosis such as cell rounding and condensation, plasma membrane blebbing and cytosolic vacuolization.20 Quantification of the results indicated a significant increase in cell death by the compounds (50 nM) at both time points in each cell line in comparison to controls (P≤0.001, Figure 1D). Especially, at the 48 h time point the frequency of cell death was massive; the HCT116 cells were the most sensitive and exhibited an average CDI between 57% and 72% pending on the treatment while the MCF10A cells were the least sensitive of the three cell lines showing an average CDI between 40% and 49% pending on the compound (Figure 1D). Finally, the MI was determined from cell samples that were fixed and DNA stained at 24 and 48 h time points posttreatment and imaged using HC-fluorescent microscope enabling accurate identification of mitotic cells based on chromatin condensation. As expected, at the 24 h time point the average MI was significantly elevated in each compound treated cell line (50 nM) in comparison to the respective DMSO control (P≤0.001, Figure 1E) while at the 48 h time point the average MI was only between 1% and 12% due to the high cytotoxicity of the compounds. The data show that NOV202 treatment phenocopies typical cellular effects of microtubule targeting drugs in vitro. Indeed, 50 nM NOV202 had equal efficacy as paclitaxel in each three cell lines. The compound induces a long-lasting M phase arrest that in the mitotic checkpoint efficient cells is followed by death of the arrested cells and in the checkpoint deficient cells by death of interphase cells after mitotic slippage. At this time, no additional cell cycle perturbing impacts were observed, which suggests that the compound has very little, if any, off-target effects in cultured cancer cells.
Effects of NOV202 in different OC cell lines
To analyze the effects of NOV202 in OC models in vitro we selected three OC cell lines that according to our earlier data exhibit different response to microtubule targeting agents (unpublished data, M Kallio, personal communication, August 2015). Ovarian adenocarcinoma OvCar-8, OvCar-4 and CaOv-3 cells cultured in the presence of 12.5 nM of NOV202 or 12.5 nM of paclitaxel were time-lapse filmed for 3 days immediately after supplementation of the culture medium with a drug (Figure S1). Control cells were treated with the vehicle only. The MI and CDI were determined from the time-lapse films at 12, 24, 48 and 72 h time points. The ovarian adenocarcinoma OvCar-8 and OvCar-4 cells responded moderately to the NOV202 treatment (12.5 nM) and exhibited a peak 27.0%±4.0% and 24.0%±9.5% MI at 12 h time point, respectively (Figure 2A). This was significantly more in comparison to the vehicle-treated control cells exhibiting MI of 6.7%±1.2% and 5.3%±2.5% at 12 h time point, respectively (P<0.001). In the later time points, the MI of NOV202 treated OvCar-8 and OvCar-4 cell populations dropped dramatically that was likely due to the short transient M phase arrest and increasing cell death. In contrast, the ovarian adenocarcinoma CaOv-3 cells exhibited much lower MI as only 12.7%±2.5% and 7.6%±2.9% of the cells were at M phase arrest after 12 h treatment with 12.5 nM NOV202 or DMSO, respectively (Figure 2A). Overall the results were similar after treatment of the three cell lines with 12.5 nM paclitaxel (Figure 2A); OvCar-8 and OvCar-4 cells exhibited a moderate mitotic arrest at 12 h time point while the CaOv-3 cells did not markedly accumulate to M phase (Figure 2A).
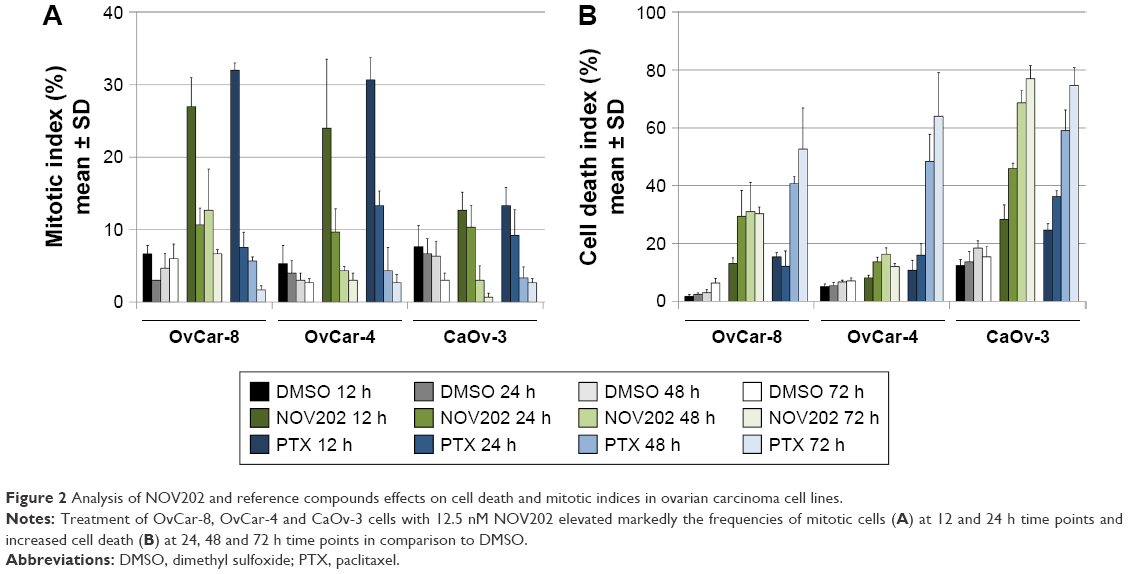 | Figure 2 Analysis of NOV202 and reference compounds effects on cell death and mitotic indices in ovarian carcinoma cell lines. |
Also, the fate of the mitotic cells varied markedly between the three cell lines and drug treatments. The average frequency of cell death started to rise significantly (P<0.001) in OvCar-8 (29.0%±9.0%) and CaOv-3 cells (46.0%±1.7%) 24 h after the treatment with 12.5 nM of NOV202 in comparison to the controls (2.3%±0.6% and 13.7%±2.5%, respectively, Figure 2B). However, the OvCar-4 cells survived much better than the NOV202 treatment as only 13.7%±1.5% of the cells were dead at the 24 h time point in comparison to the CDI of 5.3%±1.1% in the controls (Figure 2B). At the 48 and 72 h time points the average CDI remained high in the NOV202-treated OvCar-8 (31.0%±10.5% and 30.0%±2.3%, respectively) and CaOv-3 cells (68.7%±4.2% and 77.0%±4.6%, respectively) while in the OvCar-4 cells CDI remained low (16.3%±2.1% and 12.0%±1.0%, respectively, Figure 2B). Moreover, the OvCar-4 cells were more sensitive to 12.5 nM of paclitaxel at 48 and 72 h time points when compared to same concentration of NOV202 (Figure 2B). It should, however, be noted that OvCar-4 cells treated with a higher 50 nM concentration of NOV202 underwent a massive cell death; at 24, 48 and 72 h time points the average CDI was 28.7%±6.8%, 48.3%±10.7% and 59.3%±13.2%, respectively. Treatment of OvCar-8 and CaOv-3 cells with 12.5 nM of paclitaxel resulted in CDI profiles that closely resembled the outcome of exposure to 12.5 nM of NOV202 (Figure 2B). Finally, the three cell lines were also subjected to a combination treatment with NOV202 and paclitaxel but no synergistic effects were observed when compared to single drug dosing (data not shown). We conclude that NOV202 and paclitaxel possess similar capacity to induce M phase arrest in different OC cell lines but the outcome of the cell cycle delay varies in a cell line dependent manner. The data reflect underlying high heterogeneity of OC cancer lines and tumors, which causes challenges in the treatment of patients. In future, the clinical decision making on OC chemotherapy could benefit from the assessment of ovarian tumor cells’ mitotic checkpoint activity prior to drug selection and start of the treatments.
NOV202 inhibits tubulin polymerization in vitro in a dose-dependent manner
NOV202 was designed to interfere with the colchicine binding site of α-tubulin and thereby suppresses the microtubule dynamics by inhibiting tubulin polymerization. The observations made in cultured human cells support the notion that the compound perturbs normal microtubule dynamics and cause a cell cycle arrest at M phase. To confirm the mechanism of action of NOV202 in vitro the compound was tested in cell-free tubulin polymerization assay. Paclitaxel and vincristine were used as controls with known high tubulin affinity. NOV202 was found to suppress tubulin heterodimer polymerization to microtubules at 3.0 μM concentration similar to 3.0 μM of vincristine (Figure 3). At the 10 μM concentration NOV202 was sufficient to block the polymerization almost completely while at 1.0 μM concentration the compound slowed down the rate of the polymerization, but could not suppress it. As expected, paclitaxel was an effective stabilizer of tubulin polymerization at the 3.0 μM concentration tested. The effects of NOV202 on interphase and mitotic microtubules were confirmed to be very robust in cultured HCT116, HCT8 and MCF10A cells; the microtubule networks were markedly disrupted or completely wiped out after exposure to low 15–50 nM concentrations of NOV202 (data not shown). We conclude that NOV202 efficiently perturbs normal tubulin polymerization in vitro and in cells.
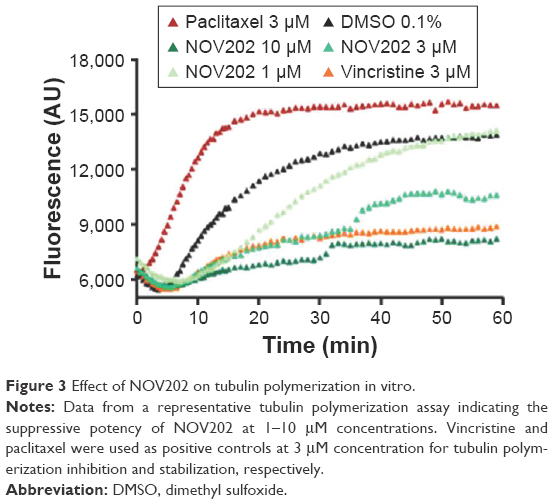 | Figure 3 Effect of NOV202 on tubulin polymerization in vitro. |
NOV202 perturbs vascular cord formation and disrupts established cord structures equally to Combretastatin A4
A number of experimental tubulin inhibitors as, for example, Combretastatin A4 (fosbretabulin), CYT997 and ENMD-1198 have been reported to exhibit antiangiogenetic and/or vascular disrupting effects.21–23 To determine the potential impact of NOV202 on tumor vasculature in vitro we used cocultures of human endothelial colony forming cells and adipose-derived stem cells. The impacts of NOV202, Combretastatin A4 and suramin, a known inhibitor of VEGF, on vascular cord formation and preformed cord structures were measured on VEFG stimulated cocultures (Figure 4, for controls see Figure S2). In the presence of basal media, a peak mean cord length of 4.1±0.8 mm/mm2 and a peak mean branch point of 17.5±3.8 mm/mm2 were observed (Figure S2). Addition of VEGF (20 ng/mL +0.1% DMSO) increased the peak mean cord length to 8.7±0.5 mm/mm2 and peak mean branch point to 47.6±4.4 mm/mm2 (Figures S2 and 4A). NOV202 was found to completely inhibit tube formation at the lowest 4.6 nM concentration tested (Figure 4A). This effect was comparable to the Combretastatin A4 treatment (Figure 4A). For the established cords, the presence of assay media alone yielded a peak mean cord length of 5.2±0.1 mm/mm2 and a peak mean branch point of 21.9±1.2 mm/mm2 (Figure S2) VEGF (20 ng/mL +0.1% DMSO) evoked a rapid time-dependent formation of vascular cords, as evidenced by an increase in the peak mean cord length and peak mean branch points (9.6±0.4 mm/mm2 and 58.4±4.4 mm/mm2, respectively, Figure S2). Cord formation stabilized around the 120 h time point (data not shown). NOV202 (0.046–100 nM) and Combretastatin A4 (0.78–100 nM) both caused a concentration dependent disruption of cords with maximal cord disruption (>96%) observed at the highest concentration tested (Figure 4C). A four-parameter logistic fit of the area under the curve data between 96 and 186 h time point gave pIC50 values of 8.3 and 8.7 for NOV202 and Combretastatin A4, respectively (Figure 4D). Importantly, despite the significant impact on cord formation and cord structures the viability of the endothelial colony forming cells and adipocyte derived stem cells was not impaired by the NOV202, Combretastatin A4 and control treatments (Figure 4B, Figure S3).
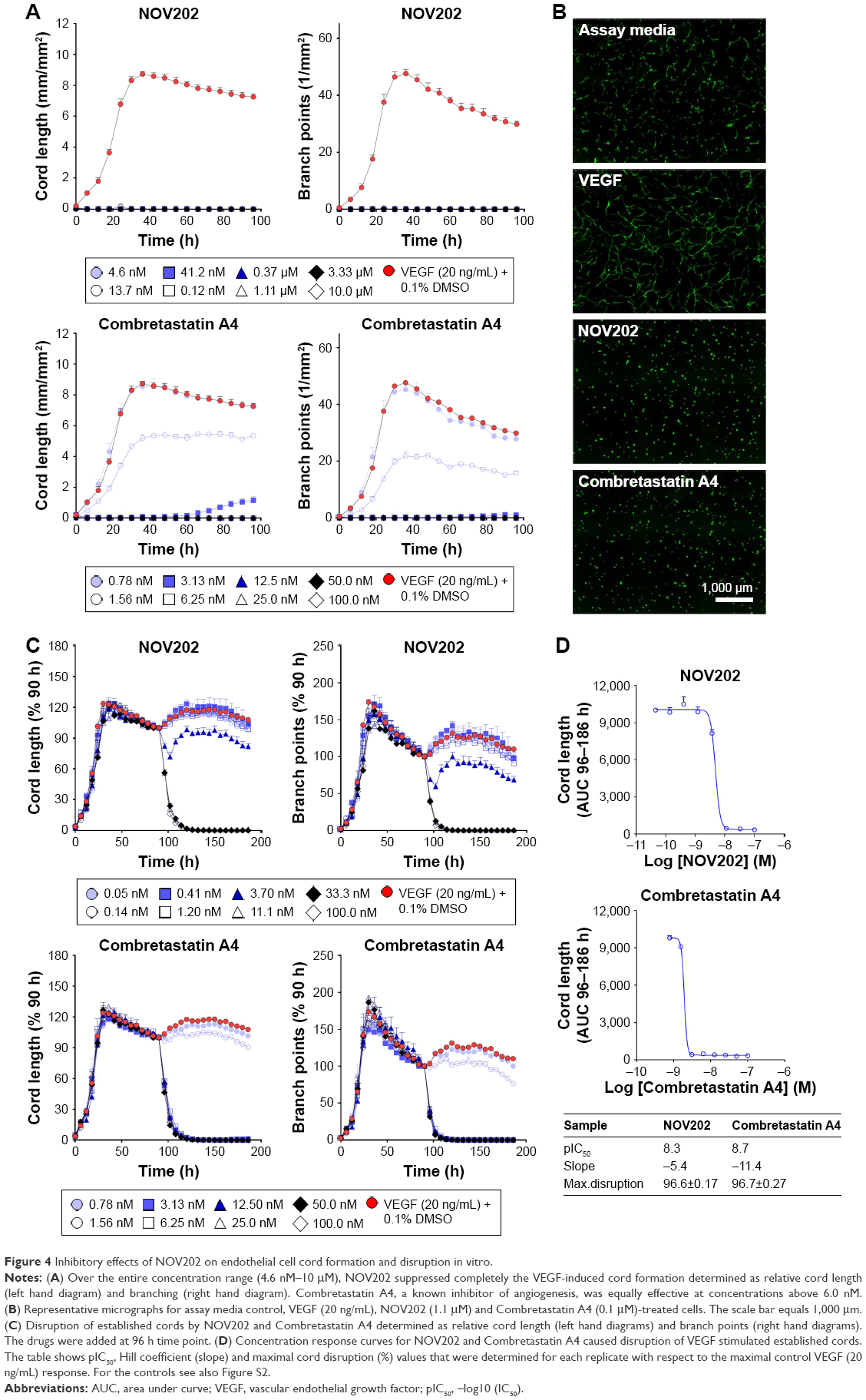 | Figure 4 Inhibitory effects of NOV202 on endothelial cell cord formation and disruption in vitro. |
NOV202 shows antiangiogenesis effects in the chicken embryo CAM assay
An ex ovo chick CAM assay was employed to confirm the observed antivascular effect of NOV202 in vitro. Analysis of the developing blood vessel in the chick embryos indicated that growth of the blood vessels, determined as change in the relative surface area of blood vessels, and the vascular integrity, determined as discontinuity of the vessels, were both significantly suppressed by 50 nM NOV202 and 50 nM Combretastatin A4 at 24 and 48 h time points posttreatment in comparison to controls (Figure 5A–C). At the 10 nM concentration both drugs were ineffective when compared to DMSO vehicle.
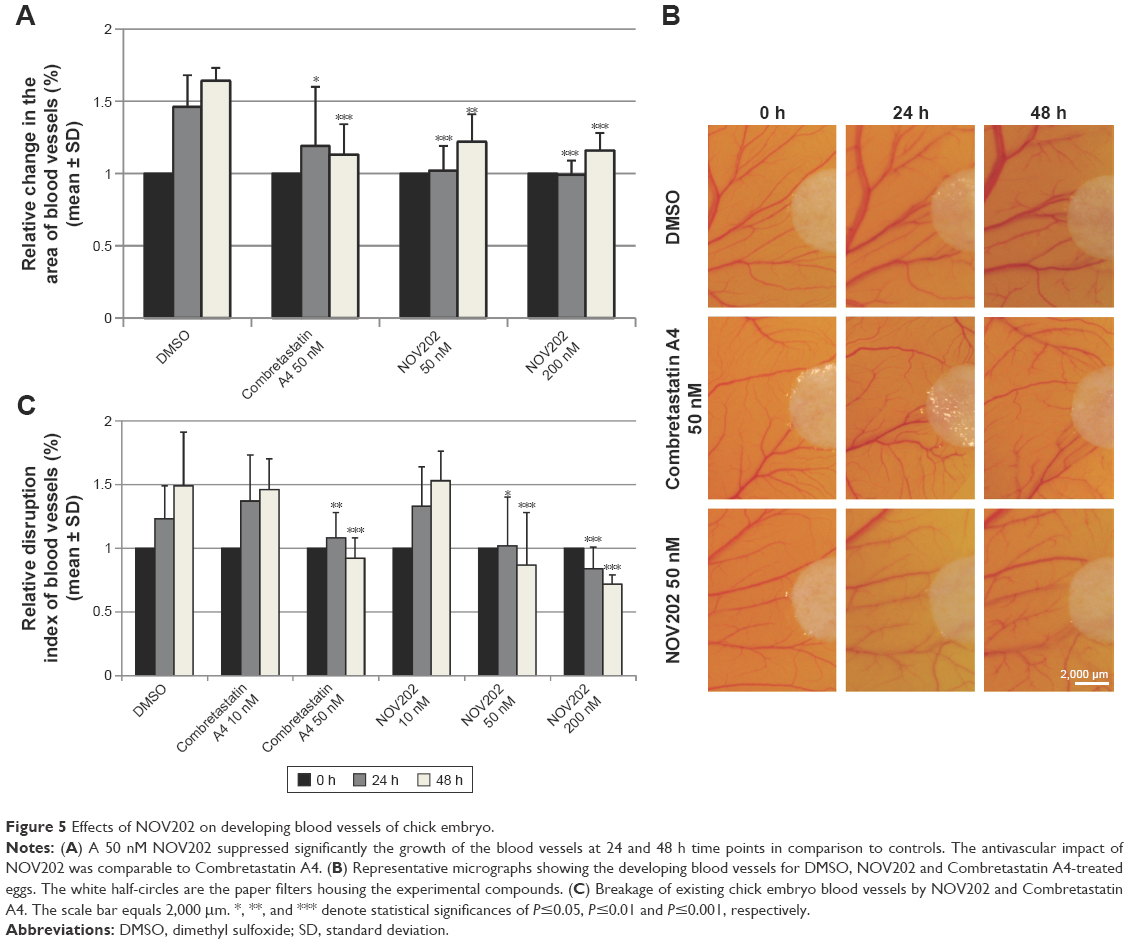 | Figure 5 Effects of NOV202 on developing blood vessels of chick embryo. |
NOV202 shows high in vivo efficacy in OC xenograft model
Encouraged by the in vitro results we next determined the in vivo efficacy of NOV202 using an ovarian carcinoma A2780 xenograft model. First, in the study of maximum tolerated dose the plasma concentration at the dose 40 mg/kg was determined to be 309 nM at 1 h and 76 nM at 4 h post-dosing, at the dose 20 mg/kg; 54 and 29 nM, and at the dose 10 mg/kg; 21 and 17 nM, respectively. As shown in Figure 6A, NOV202 caused a dose-dependent reduction in tumor volume, and at the highest dose (40 mg/kg) a clear antitumor effect that was comparable to the impact generated with paclitaxel at 24 mg/kg. At day 20 the antitumor effect was 83% compared to control group determined as reduced relative tumor volume. NOV202 was well tolerated during the treatment as determined by the body weight measurements (Figure 6B). We conclude that NOV202 exerts a clear inhibition of A2780 tumor growth at the highest dose, comparable to the inhibitory effect of paclitaxel. Notably, NOV202 was administered PO and paclitaxel IV, which shows the outstanding effects of this candidate drug. The IC50 value of NOV202 in the A2780 OC cell line was 2, 4 nM and therefore the plasma concentration in the efficacy study, especially at the 40 mg/kg dose, should be high enough for a potent reduction of tumor volumes in vivo, as shown in Figure 6A. The effect of NOV202 in multidrug resistant cells and the possible reduced toxicity is a key advantage of NOV202 and warrants further development of this anticancer compound toward the clinic.
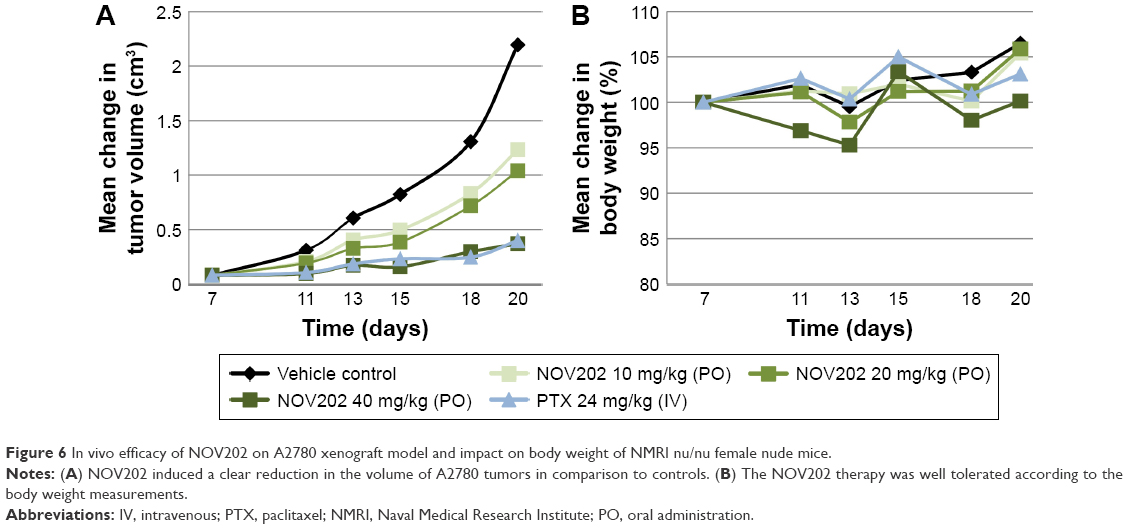 | Figure 6 In vivo efficacy of NOV202 on A2780 xenograft model and impact on body weight of NMRI nu/nu female nude mice. |
Conclusion
Tubulin targeting agents are widely used as antineoplastic drugs. Their optimal efficacy is, however, often compromised by the dose-limiting adverse effects and intrinsic/acquired drug resistance. By the development of NOV202 we aimed at overcoming one or more of these issues. The compound displayed a significant antitumor activity in vitro against a panel of human cancer cells of different origin. Importantly, the multidrug-resistant A2780/Adr cells were highly sensitive to the growth suppression by low concentrations of NOV202, which is a significant advance over the current first-line chemotherapeutic paclitaxel. In vivo, NOV202 induced marked tumor growth retardation in an A2780 xenograft model with good tolerability. The efficacy of the compound in other cancer models such as breast cancer will be explored in further studies. Interestingly, the compound exhibited a strong antiangiogenic and vascular disrupting action in vitro and in CAM assay ex vivo, which likely contributes to its antineoplastic efficacy Agents with vascular disrupting properties represent a new class of promising anticancer agents, which exhibit synergistic and/or additive effects in combination with many conventional cytotoxics. Novel combinations need to be explored in the clinic with a strong focus on finding optimal mixture of antivascular agents and other small molecule anticancer drugs, as well as, combinations with anticancer biologics. One of the biggest advantages of these agents, including NOV202, over commonly used chemotherapeutics is their ability to bind reversibly to the colchicine binding pocket, which reduces their toxicity. Our data suggest that NOV202 presents a promising approach for the treatment of refractory OC.
Disclosure
SR and MH are employees and shareholders in Noviga Research AB. The authors report no other conflicts of interest in this work.
References
Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975–2011, National Cancer Institute. Bethesda, MD. Available from: http://seer.cancer.gov/csr/1975_2011/ based on 2013 SEER data submission. Accessed June 10, 2016. | ||
Schorge JO, Modesitt SC, Coleman RL, et al. SGO White Paper on ovarian cancer: etiology, screening and surveillance. Gynecol Oncol. 2010;119(1):7–17. | ||
Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4(4):253–265. | ||
Schwartz EL. Antivascular actions of microtubule-binding drugs. Clin Cancer Res. 2009;15(8):2594–2601. | ||
McGuire WP, Rowinsky EK, Rosenhein NB, et al. Taxol: a unique antineoplastic agent with significant activity in advanced ovarian epithelial neoplasms. Ann Intern Med. 1989;111(4):273–279. | ||
Sledge GW Jr, Robert N, Sparano JA, et al. Paclitaxel (Taxol)/doxorubicin combinations in advanced breast cancer: the Eastern Cooperative Oncology Group experience. Semin Oncol. 1994;21(5 Suppl 8):15–18. | ||
Bonomi P, Kim KB, Kugler J. Comparison of survival for stage IIIB versus stage IV non-small cell lung cancer (NSCLC) patients with etoposide-cisplatin versus taxol-cisplatin: an Eastern Cooperative Oncology Group (ECOG) trial. Proc Am Soc Clin Oncol. 1997;16:454a. | ||
Vasey PA, Jayson GC, Gordon A, et al; Scottish Gynaecological Cancer Trials Group. Phase III randomized trial of docetaxel-carboplatin versus paclitaxel-carboplatin as first-line chemotherapy for ovarian carcinoma. J Nat Cancer Inst. 2004;96(22):1682–1691. | ||
Davidson B, Tropé CG. Ovarian cancer: diagnostic, biological and prognostic aspects. Womens Health (Lond). 2014;10(5):519–533. | ||
Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2(1):48–58. | ||
Thomas H, Coley HM. Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting p-glycoprotein. Cancer Contl. 2003;10(2):159–165. | ||
Mickisch GH, Merlino GT, Galski H, Gottesman MM, Pastan I. Transgenic mice that express the human multidrug-resistance gene in bone marrow enable a rapid identification of agents that reverse drug resistance. Proc Natl Acad Sci U S A. 1991;88(2):547–551. | ||
Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Ann Rev Pharmacol Toxicol. 1999;39:361–398. | ||
Palmeira A, Sousa E, Vasconcelos MH, Pinto MM. Three decades of P-gp inhibitors: skimming through several generations and scaffolds. Curr Med Chem. 2012;19(13):1946–2025. | ||
Jang SH, Wientjes MG, Au JL. Kinetics of P-glycoprotein-mediated efflux of paclitaxel. J Pharmacol Exp Ther. 2001;298(3):1236–1242. | ||
Dohle DS, Pasa SD, Gustmann S, et al. Chick ex ovo culture and ex ovo CAM assay; how it really works. J Vis Exp. 2009;33:e1620. | ||
Lindhagen E, Nygren P, Larsson R. The fluorometric microculture cytotoxicity assay. Nat Protoc. 2008;3(8):1364–1369. | ||
Tighe A, Johnson VL, Albertella M, Taylor SS. Aneuploid colon cancer cells have a robust spindle checkpoint. EMBO Rep. 2001;2(7):609–614. | ||
Waldman T, Kinzler KW, Vogelstein B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 1995;55(22):5187–5190. | ||
Loo DT, Rillema JR. Measurement of cell death. Methods Cell Biol. 1998;57:251–264. | ||
Galbraith SM, Chaplin DJ, Lee F, et al. Effects of combretastatin A4 phosphate on endothelial cell morphology in vitro and relationship to tumour vascular targeting activity in vivo. Anticancer Res. 2001;21(1A):93–102. | ||
Burns CJ, Fantino E, Phillips ID, et al. CYT997: a novel orally active tubulin polymerization inhibitor with potent cytotoxic and vascular disrupting activity in vitro and in vivo. Mol Cancer Ther. 2009;8(11):3036–3045. | ||
Pasquier E, Sinnappan S, Munoz MA, Kavallaris M. ENMD-1198, a new analogue of 2-methoxyestradiol, displays both antiangiogenic and vascular-disrupting properties. Mol Cancer Ther. 2010;9(5):1408–1418. |
Supplementary materials
 | Figure S1 Cell population growth suppression by NOV202 and PTX in OC cell lines in vitro. |
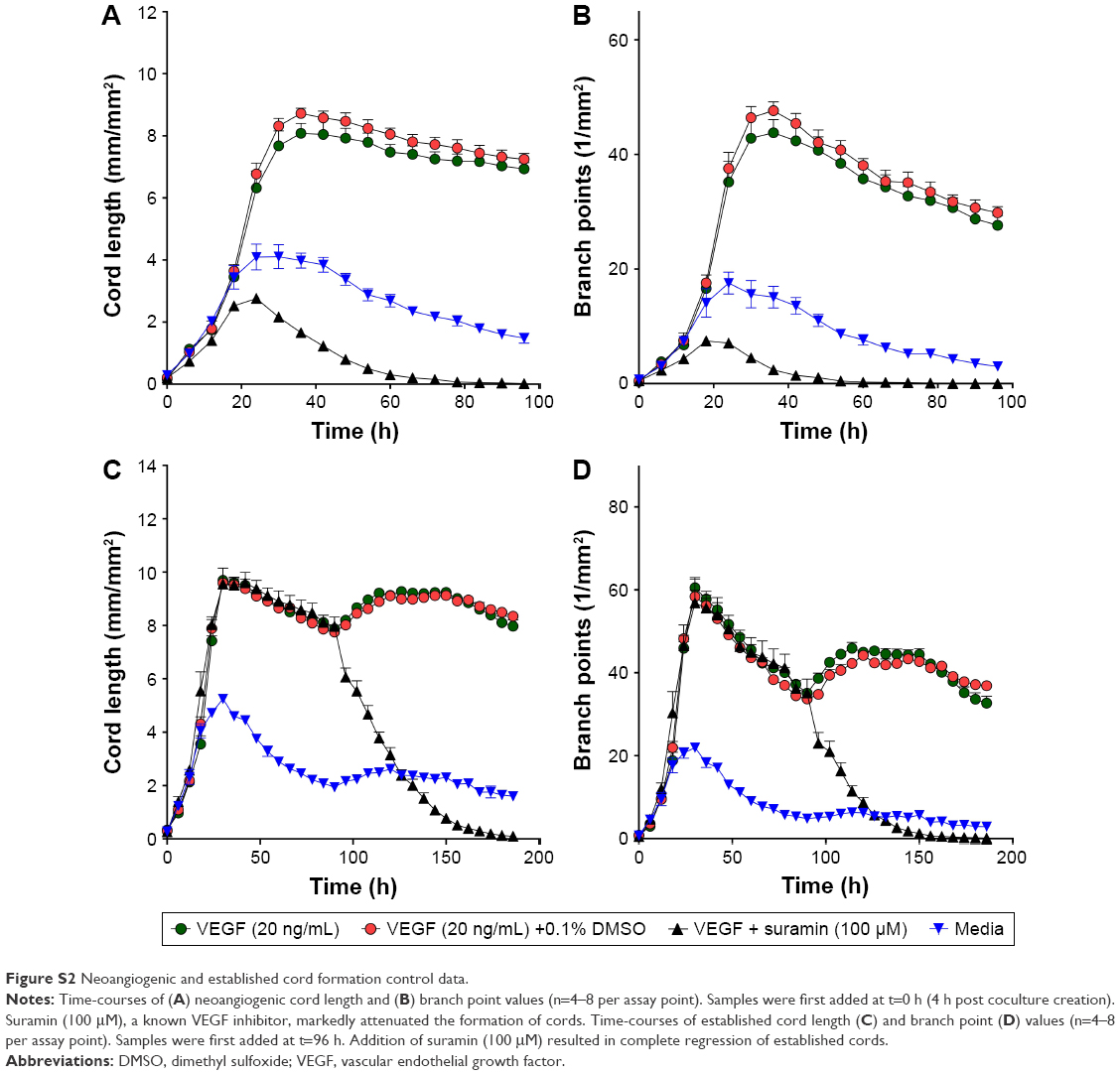 | Figure S2 Neoangiogenic and established cord formation control data. |
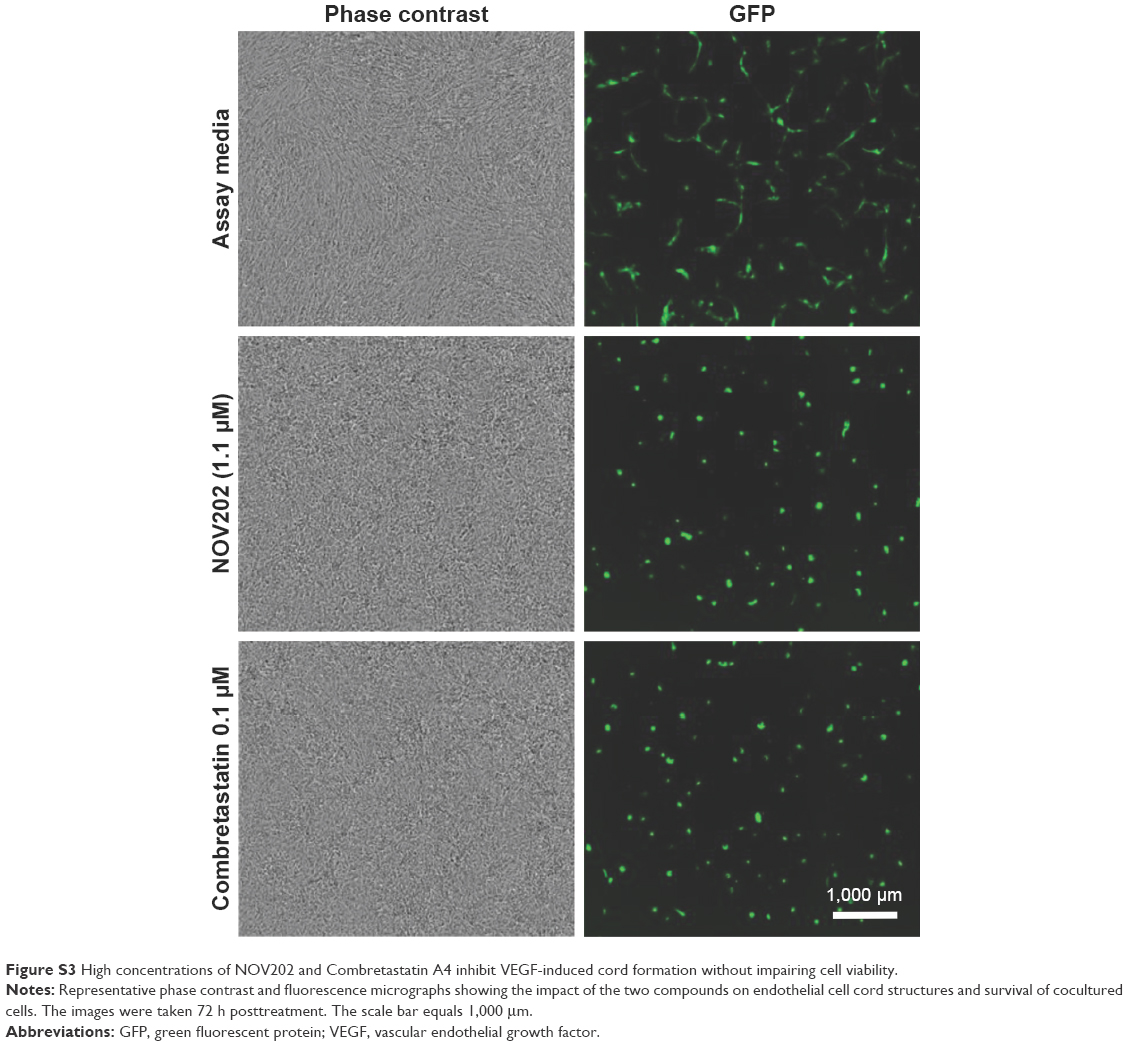 | Figure S3 High concentrations of NOV202 and Combretastatin A4 inhibit VEGF-induced cord formation without impairing cell viability. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.