")
Back to Journals » International Journal of Nanomedicine » Volume 15
Evaluation of Cytotoxic Effect of Cholesterol End-Capped Poly(N-Isopropylacrylamide)s on Selected Normal and Neoplastic Cells
Authors Misiak P , Niemirowicz-Laskowska K , Markiewicz KH , Misztalewska-Turkowicz I , Wielgat P, Kurowska I, Siemiaszko G, Destarac M, Car H, Wilczewska AZ
Received 21 May 2020
Accepted for publication 30 July 2020
Published 30 September 2020 Volume 2020:15 Pages 7263—7278
DOI https://doi.org/10.2147/IJN.S262582
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Pawel Misiak,1 Katarzyna Niemirowicz-Laskowska,2 Karolina H Markiewicz,1 Iwona Misztalewska-Turkowicz,1 Przemysław Wielgat,3 Izabela Kurowska,1,4 Gabriela Siemiaszko,1 Mathias Destarac,5 Halina Car,2 Agnieszka Z Wilczewska1
1Faculty of Chemistry, University of Bialystok, Bialystok, Poland; 2Department of Experimental Pharmacology, Medical University of Bialystok, Bialystok, Poland; 3Department of Clinical Pharmacology, Medical University of Bialystok, Bialystok, Poland; 4Doctoral School of Exact and Natural Sciences, University of Bialystok, Bialystok, Poland; 5IMRCP, CNRS UMR 5623, Université de Toulouse, Toulouse, France
Correspondence: Agnieszka Z Wilczewska; Karolina H Markiewicz Tel +48 85 7388037
Email [email protected]; [email protected]
Purpose: Efficient intracellular delivery of a therapeutic compound is an important feature of smart drug delivery systems (SDDS). Modification of a carrier structure with a cell-penetrating ligand, ie, cholesterol moiety, is a strategy to improve cellular uptake. Cholesterol end-capped poly(N-isopropylacrylamide)s offer a promising foundation for the design of efficient thermoresponsive drug delivery systems.
Methods: A series of cholesterol end-capped poly(N-isopropylacrylamide)s (PNIPAAm) with number-average molar masses ranging from 3200 to 11000 g·mol– 1 were synthesized by reversible addition-fragmentation chain transfer (RAFT) polymerization from original xanthate-functionalized cholesterol and self-assembled into micelles. The physicochemical characteristics and cytotoxicity of cholesterol end-capped poly(N-isopropylacrylamide)s have been thoroughly investigated.
Results: Phase transition temperature dependence on the molecular weight and hydrophilic/hydrophobic ratio in the polymers were observed in water. Biological test results showed that the obtained materials, both in disordered and micellar form, are non-hemolytic, highly compatible with fibroblasts, and toxic to glioblastoma cells. It was found that the polymer termini dictates the mode of action of the system.
Conclusion: The cholesteryl moiety acts as a cell-penetrating agent, which enables disruption of the plasma membrane and in effect leads to the restriction of the tumor growth. Cholesterol end-capped PNIPAAm showing in vitro anticancer efficacy can be developed not only as drug carriers but also as components of combined/synergistic therapy.
Keywords: cholesterol-end capped poly(N-isopropylacrylamide), cell-penetrating molecules, thermoresponsive polymer micelles, drug carriers
Introduction
Delivering a therapeutic compound to the target place, at the right time, and in the right amount is one of the major challenges of modern medicine. The idea of a targeting system that allows releasing drug molecules under specific conditions provides an excellent solution. Therefore, recently, a vast development in the field of smart drug delivery systems (SDDS) has been observed.1–3 Thermoresponsive polymers, which undergo a sharp change of their physical properties in response to the temperature alteration, are among the most often investigated smart drug carriers.4 It is due to the flexibility in the control of chemical composition and functions of macromolecules, and in the control of the stimulus. Additionally, the increase of the local or systemic temperature may enhance the sensitivity of cancer cells to chemotherapeutic drugs, modulate immunity, and quicken apoptosis of tumor cells.5,6 Therefore, thermosensitive drug delivery systems are particularly explored for application in cancer therapy. Poly(N-isopropylacrylamide) (PNIPAAm) is the most frequently used polymer for the preparation of thermo-sensitive SDDS due to its phase transition temperature (lower critical solution temperature, LCST), which is close to the physiological temperature of human body.7
Inefficient intracellular delivery is one of the main problems in drug delivery in general.8,9 One of the strategies to improve cellular uptake is the modification of a carrier structure with a cell-penetrating ligand.9 Cholesterol, an essential structural component of animal cell membranes, which has a great influence on the membrane fluidity, microdomain structure (lipid rafts), and permeability9,10 can be used for this purpose. Due to its hydroxyl group which can easily be derivatized, large-scale availability and relatively low cost, cholesterol has been used as a starting material for the synthesis of diverse steroid-based molecules.10,11
A variety of polymers comprising cholesterol moieties in the main chain or as side chains have been reported.12 Cholesterol end-capped polymers can be synthesized by polymerizations performed in the presence of cholesterol-based molecules that initiate polymerization of monomers. For instance, cholesterol-based atom transfer radical polymerization (ATRP) initiators were used to synthesize poly(N-isopropylacrylamide)13 and poly(poly(ethylene glycol) methacrylate),14 and a cholesterol-functional trithiocarbonate reversible addition-fragmentation chain transfer (RAFT) agent was employed to generate well-defined poly(poly(ethylene glycol) acrylate).15 Well-defined polymers bearing cholesterol side chains can be prepared by RAFT polymerization of cholesterol-containing monomers, eg, cholesteryl methacrylate.16 Cholesterol may be also introduced to the polymer backbone by post-modification. For instance, cholesterol-comprising PNIPAAm copolymers for thermosensitive drug delivery systems were obtained by post-polymerization reaction between the end-groups17 or side-chain groups18 of the copolymer and cholesterol derivatives.
In this regard, we present the synthesis of cholesterol end-capped poly(N-isopropylacrylamide)s as a foundation for future drug carriers. We propose an original approach to control PNIPAAm chain length and hydrophobic/hydrophilic balance by means of a cholesterol-functional dithiocarbonate RAFT agent. We show phase transition temperature dependence on the molecular weight and hydrophilic/hydrophobic ratio in water. We demonstrate that the obtained cholesterol end-capped PNIPAAm are non-hemolytic and non-toxic to fibroblasts, but toxic to glioblastoma cells. We aimed to understand the interactions between the proposed delivery systems and the plasma membranes. The essential role of the cholesteryl moiety as a ligand enabling glioblastoma cell membranes disruption is evidenced.
Materials and Methods
Materials
The initiator, 2,2′-azobis(2-methylpropionitrile) (AIBN, MERCK) was recrystallized from chloroform. Monomer, N-isopropylacrylamide (NIPAAm, 99%, ACROS) was recrystallized from toluene-hexane (60:40, v/v), prior to use. 2-Bromopropionylbromide (97%, ALDRICH), carbon disulfide (CS2, ≥99.9%, SIGMA-ALDRICH), potassium hydroxide (KOH, Pure, AVANTOR), triethylamine (Et3N, AVANTOR), n-propylamine (99%, Riedel-de Haen AG), tributylphosphine (TBP, >90% GC, FLUKA), Phosphate Buffer Solution (PBS, pH=7.4, GIBCO) and Dulbecco’s Modified Eagle Medium (DMEM, GIBCO) were used as received. All organic solvents were purchased from Avantor Performance Materials, Poland S.A., and were distilled prior to use.
Methods
Nuclear Magnetic Resonance (NMR)
The 1H and 13C NMR spectra were recorded on Bruker Avance II 400 or Avance DPX 200 spectrometers operating at 400 and 200 MHz, respectively, using CDCl3 or D2O (for potassium O-ethyl carbonodithioate) solutions.
Attenuated Total Reflectance Fourier Transform Infrared Spectroscopy (ATR-FTIR)
All ATR-FTIR spectra were recorded using Thermo Scientific Nicolet 6700 FTIR spectrophotometer equipped with ATR accessory. Spectra were ratioed against the spectra of background and collected in the wavenumber range 4000 to 500 cm−1 by co-adding 32 scans with a resolution of 4 cm−1.
Size Exclusion Chromatography (SEC)
Molecular weights and molecular weight distributions were determined by size exclusion chromatography (SEC) using THF as an eluent at a flow rate of 1.0 mL‧min−1 at 25 °C. Before analysis polymers were carefully dissolved in the eluent (final concentration was 5 mg mL−1) and filtrated through 0.45 μm PTFE filter. The samples were analyzed using two-column set Styragel HR3 and HR4 (Waters) coupled with a three detector system: refractometer thermostated at 35 °C (Optilab Rex, Wyatt technology), a UV detector (Prostar, Varian) set at 254 nm and a multi-angle laser light scattering detector (Mini Dawn, 3 angles, Wyatt technology). The dn/dc of the copolymers was assumed to be equal to the PNIPAAm homopolymer19 (0.087 mL g−1).
Fluorescence Measurements
Hitachi F-7000 Fluorescence Spectrophotometer was used to determine critical micelle concentration (CMC) by pyrene fluorescent probe method adapted from literature.20–22 Ten microliters of pyrene stock solution (0.15 mM) were poured into empty vials and acetone was evaporated by argon stream. Then, 3 mL of polymer aqueous solutions of various concentrations (10−4 to 2 mg/mL) were added into vials and shaken vigorously. The mixtures were stored overnight in the refrigerator for equilibrium. Emission spectra (range 360–460 nm, excitation wavelength 339 nm, the slit width 2.5 nm) were recorded. The ratios between intensities of emission peaks at 373 and 393 nm (I373/I393) were plotted as a function of the polymer concentration. The CMC values were determined as the curve breakpoint.
Dynamic Light Scattering (DLS) and ζ Potential
The colloidal stability of the obtained polymeric systems was studied using a Zetasizer Nano-ZS (Malvern Instruments, UK) equipped with a 4 mW helium/neon laser (l = 633 nm) and a thermoelectric temperature controller. All measurements were performed at 25 °C with a backscatter detection system at 173°. The chol-PNIPAAm-X(2–5) were dissolved in water, PBS, DMEM, and 0.9% NaCl to reach a final concentration of 1 mg‧mL−1. DLS measurements were performed after 24 hours, one week, and one month after micelle formation and zeta potential measurements were performed after one week. The particle sizes are expressed as the mean hydrodynamic diameter of 5 measurements. Polymer chol-PNIPAAm-X(1) was insoluble in aqueous solutions at room temperature.
Ultraviolet-Visible Spectroscopy (UV-Vis)
All UV-Vis spectra were recorded on a Jasco V-670 Spectrophotometer at a wavelength range of 190–800 nm. Spectra of O-ethyl-S-(1-cholesteroxycarbonyl)ethyldithiocarbonate (standard) and chol-PNIPAAm-X(1–5) were taken in ethanol solution at a concentration of 0.2 mg‧mL−1. A maximum absorption peak at 280 nm was used for analysis.
Turbidimetry
Cloud points were determined by thermo-regulated UV-Vis spectroscopy. Spectra of particle suspensions prepared in 4 different aqueous solutions (DMEM, PBS, deionized water, and 0.9% NaCl) at a concentration of 1 mg‧mL−1 were taken on Jasco V-670 Spectrophotometer in the absorbance mode at a wavelength of 400 nm and a heating rate of 0.5 °C/min.
Thermal Analysis
Thermogravimetric analyses (TGA) were performed on a Mettler Toledo Star TGA/DSC unit. Samples weighing 2–3 mg were placed in aluminum oxide crucibles and heated from 50 °C to 900 °C at a heating rate of 10 K‧min−1 under an argon flow rate of 40 mL‧min−1. Differential scanning calorimetry (DSC) measurements were performed on a Mettler Toledo Star DSC system. To measure glass transition temperature (Tg) the samples were first heated to 200 °C at 20 K‧min−1 heating rate and held at this temperature for 10 min to remove their thermal history, followed by quenching to −20 °C. A heating rate of 10 K‧min−1 was used for the second heating run. The Tg was taken as the midpoint of the heat capacity change in the second heating run. Differential scanning calorimetry was also used to determine the LCST of samples chol-PNIPAAm-X(2–5). An aqueous solution of a sample (10 mg‧mL−1, 15 μL) was sealed in an aluminum crucible and equilibrated at 25 °C for 10 minutes. Then, it was heated from 25 °C to 40 °C at a rate of 1K‧min−1 under an argon flow rate of 200 mL‧min−1.
Imaging Methods
Polymers for scanning electron microscopy (SEM) were placed on carbon tape glued to aluminum tables and coated with a 4 nm layer of gold by Leica ACE 200 coater to improve imaging. The SEM imaging was performed with a field emission gun (FEG) at 10 kV with a magnification of 1000 times. Samples were imaged on SEM TFP 2017/12 Inspect S50 FEI. For transmission electron microscopy (TEM) imaging, polymers were prepared by plunge-freeze-drying. At first, 0.1 mL‧min−1 aqueous solutions were prepared, then 3 µL of the sample was applied on a holey carbon copper grid (holey carbon-coated grids onto 200 mesh copper, SPI Supplies) and a surplus of the solution was removed with a tissue paper, the process was repeated 2 times for more convenient and better imaging. After that, grids were immersed into liquid nitrogen (LN2) and dried overnight using a vacuum pump. TEM images were taken using a Tecnai G2 X-Twin microscope. Images were captured at the accelerating voltage of 200kV using cryotrap (cooper trap equipped with LN2 Dewar) as a cooling tool for the microscope column. Optical microscopy imaging was performed on the KEYENCE VHX-6000 Digital Microscope equipped with high-resolution zoom lens VH-Z500R with a magnification of 500 times (Supplementary Fig 6).
Lyophilization
Samples were lyophilized on Christ Alpha 1–2 LDplus with double-chamber. Aqueous solutions of polymers were frozen with liquid nitrogen and then lyophilized for 24 hours under 0.013 mbar pressure.
Hemolysis Assay
The hemolytic activity was tested using human red blood cells (RBCs) suspended in phosphate-buffered saline (PBS) (hematocrit ~5%) with a concentration range of polymeric agents from 0.05 to 0.25 mg‧mL−1. In brief, the RBCs were incubated with tested agents for 1 h at 37 °C. In the next step, relative hemoglobin concentration in supernatants after centrifugation at 2500 g was monitored by measuring absorbance at 540 nm. One hundred percent hemolysis was taken from samples in which 1% Triton X-100 was added to disrupt all cell membranes.
Ethics Statement
The hemolytic activity of the tested agents was evaluated in blood samples from healthy adult volunteers under IRB approval: R-I-002/245/2019. This study was approved by the Institutional Review Board (IRB) of The Medical University of Bialystok. All subjects provided informed written consent and collected samples were anonymous.
Cell Culture
Mouse glioma GL261 cells (ACC802, DSMZ Germany) and human fibroblast cell line (ATCC, Manassas, USA) were cultured in Dulbecco’s Modified Eagle Medium/F12 (DMEM/F12) and Dulbecco’s Modified Eagle Medium (DMEM), respectively, supplemented with 10% heat-inactivated fetal bovine serum (all Gibco; Life Technologies, Germany) and 100 μg‧mL−1 penicillin/streptomycin (Merck, Germany). Cells were plated in 96 well plates at a seeding density 104/0.32cm2 and incubated at 37°C in a humified atmosphere containing 5% CO2. Both, glioma cells and fibroblasts with a passage number between 5 and 7 were used in the experiments. Naïve cells were cultured to reach confluency of 80% and then exposed to tested agents applied at the following concentrations: 0; 0.05; 0.1 and 0.25 mg‧mL−1.
Cytotoxicity Assessment
Cytotoxic effect was evaluated using the neutral red method, which is based on the ability of viable cells to take up the dye by active transport and incorporate into lysosomes, whereas non-viable cells will not take up the dye. In brief, mouse glioma GL261 and human cells were treated by tested polymers, were added in concentrations ranging from 0 to 0.25 mg‧mL−1 and left for further incubation for 24 hours at 37 °C. To quantify the viability of cells, the 0.33% solution of Neutral Red Solution was added and followed by further incubation for 2 hours. After the incubation period, the medium was carefully removed and the cells were rinsed with Neutral Red Assay Fixative. Next, the fixative solution was removed and the incorporated dye was solubilized in Neutral Red Assay Solubilization Solution. Absorbance was measured using a Biotek microplates reader at 540 nm wavelength.
In another set of experiments, the ability of tested polymers to interact with the plasma membrane was evaluated. For this purpose, the detection of lactate dehydrogenase (LDH) release from treated mouse glioma GL261 cells was performed following a commercially available protocol. In brief, after the addition of tested polymers and 24-hours incubation, the medium was transferred into 96 well plates, and then, 50 µL of Master Reaction mix was added. After 10–15 min of incubation, the absorbance was measured using a Biotek microplates reader at a wavelength of 450 nm.
All experiments were performed in triplicate and the results were shown as average ± SD.
Synthetic Procedures
Synthesis of Potassium O-Ethyl Carbonodithioate
Potassium hydroxide (21 g, 0.375 mol, 1 eq.) was dissolved in ethanol (76 mL) in a round-bottom flask. The flask was placed in an ice bath, then carbon disulfide (22.6 mL, 0.375 mol, 1 eq.) was added dropwise. A yellow precipitate was washed 3 times by diethyl ether and purified by crystallization from ethanol. Forty-one grams of yellow solid was obtained with a 68% yield. 1H NMR (400 MHz, D2O, δ, ppm): 4.53 (q, 2H), 1.41 (t, 3H). 13C NMR (100 MHz, D2O, δ, ppm): 235.5 (C=S), 73.0 (CH2), 16.3 (CH3).
Synthesis of 2-Bromo-O-(3-Cholesteryl)propanoate
Cholesterol (5 g, 12.93 mmol, 1 eq.) was dissolved in dried DCM (150 mL) and then Et3N (2.5 mL, 17.92 mmol, 1.4 eq.) was added, and reaction mixture was placed in an ice bath. Subsequently, 2-bromopropionyl bromide (1.88 mL, 17.92 mmol, 1.4 eq.) was added dropwise over 30 minutes. The mixture was stirred at 0 °C for 3 h. Next, it was poured into 100 mL of distilled water. To isolate the product, extraction was carried out with DCM. The product was purified by crystallization from ethanol. 5.8 g of white solid was obtained with 86% yield. 1H NMR (400 MHz, CDCl3, δ, ppm): 5.39 (s, 1H), 4.63 (m, 1H), 4.32 (q, 1H, J=7 Hz), 2.35 (d, 2H), 1.99–1.85 (m, 5H), 1.80 (d, 3H, J=7 Hz), 1.41–1.03 (m, 18H), 1.02 (s, 3H), 1.01–0.85 (m, 12H), 0.67 (s, 3H). 13C NMR (100MHz, CDCl3, δ, ppm): 169.6 (C=O), 139.3 (C=C), 123.0 (CH), 75.6 (CH), 56.7 (CH), 56.1 (CH), 50.0 (CH), 42.3 (C), 40.6 (CH), 39.7 (CH2), 39.5 (CH2), 37.7 (CH2), 36.9 (CH2), 36.6 (CH2), 36.2 (C), 35.8 (CH), 31.9 (CH2), 31.8 (CH3), 28.2 (CH2), 28.0 (CH), 27.5 (CH2), 27.3 (CH2), 24.3 (CH2), 23.8 (CH2), 22.8 (CH3), 22.5 (CH3), 21.0 (CH), 19.3 (CH3), 18.7 (CH3), 11.8 (CH3). FT-IR (ATR, ν) cm−1: 2935, 2866, 1734, 1466, 1443, 1377, 1333, 1268, 1222, 1159, 1054, 997, 944, 797, 734, 678.
Synthesis of O-Ethyl-S-(1-Cholesteroxycarbonyl)ethyldithiocarbonate
2-Bromo-O-(3-cholesteryl)propanoate (5 g, 9.6 mmol, 1 eq.) was dissolved in ethyl acetate (100 mL). Then, potassium O-ethyl carbonodithioate (2.5 g, 15.6 mmol, 1.6 eq.) solution in acetonitrile (10 mL) was added dropwise. The mixture was stirred in an ice bath for 24 h. The product was purified by crystallization from ethanol. 4.38 g of yellowish solid was obtained with 81% yield. 1H NMR (400 MHz, CDCl3, δ, ppm): 5.38 (s, 1H), 4.63 (m, 3H, J=7.1 Hz), 4.34 (q, 1H, J=7.3 Hz), 2.33 (m, 2H), 2.02–1.85 (m, 5H), 1.56 (d, 3H, J=7.3 Hz), 1.42 (t, 3H, J=7.1 Hz), 1.41–1.03 (m, 18H), 1.02 (s, 3H), 1.01–0.85 (m, 12H), 0.67 (s, 3H). 13C NMR (100MHz, CDCl3, δ, ppm): 212.2 (C=S), 170.7 (C=O), 139.4 (C=C), 122.9 (CH), 75.4 (CH), 70.1 (CH2), 56.6 (CH), 56.1 (CH), 50.0 (CH), 47.4 (CH), 42.3 (C), 39.7 (CH2), 39.5 (CH2), 37.9 (CH2), 36.9 (CH2), 36.6 (CH2), 36.2 (C), 35.8 (C), 31.9 (CH2), 31.8 (CH3), 28.2 (CH2), 28.0 (CH), 27.6 (2xCH2), 24.3 (CH2), 23.8 (CH2), 22.8 (CH3), 22.5 (CH3), 21.0 (CH), 19.3 (CH3), 18.7 (CH3), 13.7 (CH3), 11.8 (CH3). FT-IR (ATR, ν) cm−1: 2941, 2890, 2865, 1733, 1465, 1365, 1287,1208, 1151, 1111 (C=S), 1050, 994, 972, 895, 799, 736.
General Procedure for RAFT Polymerization with O-Ethyl-S-(1-Cholesteroxycarbonyl)ethyldithiocarbonate
Appropriate amounts (Table SI1) of O-ethyl-S-(1-cholesteroxycarbonyl)ethyldithiocarbonate), NIPAAm and THF were placed in a flask and the mixture was degassed by bubbling with argon for 15 min. After heating to 70 °C, AIBN was added and the solution was stirred for 24 h. Then, THF was evaporated on a rotary evaporator, yellowish (chol-PNIPAAm-X(1)) or white (chol-PNIPAAm-X(2–5)) products were dissolved in DCM, precipitated in cold hexane, filtered and dried in a furnace (60 °C). chol-PNIPAAm-X(2): 1H NMR (400 MHz, CDCl3, δ, ppm): 4.62 (m, 3H, cholX), 4.00 (CH(CH3)2, NIPAAm), 1.14 (CH(CH3)2, NIPAAm), 1.32–2.30 (polymer backbone), 1.01 (s, 3H, chol-X), 0.92–0.85 (m, 12H, chol-X), 0.67 (s, 3H, chol-X). FT-IR (ATR, ν) cm−1: 3429 (N-H), 3282 (N-H), 3062, 2966, 2926, 2872, 1637 (C=O amide), 1534 (N-H), 1454, 1383, 1365, 1172, 1129, 1041, 924, 879, 833.
General Procedure for Gentle Aminolysis of the Xanthate ω-End Group at the End of the Polymeric Chain23–25
Chol-PNIPAAm-X(2–5) (0.01 mmol) were dissolved in THF (1 mL) and the mixture was degassed by bubbling with argon for 15 min. Then, n-propylamine (7.4 µL, 0.09 mmol) and tributylphosphine (4.9 µL, 0.02 mmol) were added. The mixture was stirred overnight. Products (chol-PNIPAAm-SH(2–5)) were precipitated in cold hexane, centrifugated, and dried in a furnace (60 °C).
Free Radical Polymerization of NIPAAm
The polymer without a cholesterol-based CTA agent was prepared for biological properties comparison. NIPAAm (200 mg, 1.77 mmol) and THF (2 mL) were placed in a flask and the mixture was degassed by bubbling with argon for 15 min. After heating to 70 °C, AIBN (1 mg, 0.006 mmol) was added and the solution was stirred for 24 h, reaching 100% conversion. Next, THF was evaporated on a rotary evaporator, white PNIPAAm product was dissolved in DCM and precipitated in cold hexane, filtered, and dried in a furnace (60 °C). PNIPAAm: 1H NMR (400 MHz, CDCl3, δ, ppm): 4.00 (CH(CH3)2), 2.40–1.40 (polymer backbone), 1.13 (CH(CH3)2).
Micelles Formation (Method 2)
Ten milligrams of the polymer was dissolved in 1 mL of THF, and the obtained solution was added dropwise to 10 mL of deionized water and shaken for 24 h. Then, the solution (11 mL) was lyophilized. To facilitate the description of the results, the polymers obtained by this method were marked with prefix M.
Results and Discussion
Synthesis and Characterization of Chol-PNIPAAm-X Polymers
PNIPAAm containing a cholesterol moiety at the end of chains (chol-PNIPAAm-X) were synthesized by RAFT polymerization from xanthate-functionalized cholesterol. O-ethyl-S-(1-cholesteroxycarbonyl)ethyldithiocarbonate (RAFT agent, chol-X) was synthesized in two steps (Figure 1). 2-Bromo-O-(3-cholesteryl)propanoate (chol-Br) was obtained by the reaction of the cholesterol hydroxyl group with 2-bromopropionyl bromide. Nucleophilic substitution of bromine with potassium O-ethyl carbonodithioate led to the formation of xanthate-functionalized cholesterol (chol-X). The product was characterized by 1H and 13C NMR, and FT IR spectroscopy. To our knowledge, such a cholesterol-based RAFT agent has not been described in the literature.
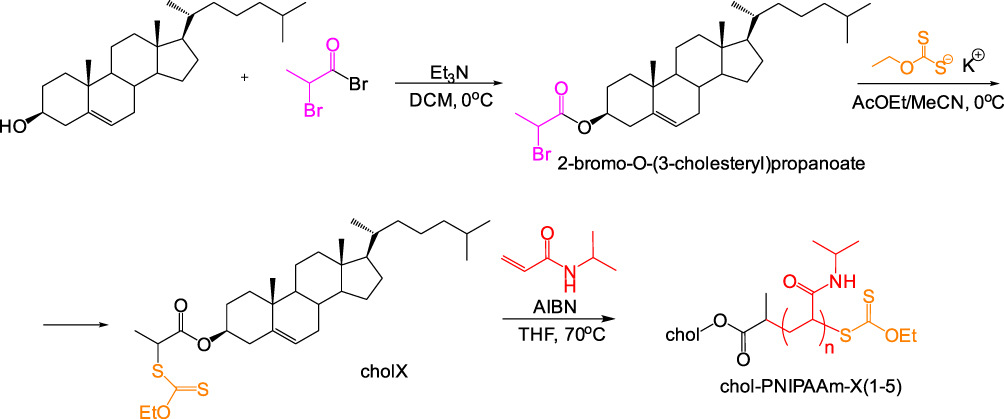 |
Figure 1 Synthesis of cholesterol-based RAFT agent and chol-PNIPAAm-X homopolymers. |
The detailed conditions for NIPAAm polymerizations from chol-X are presented in Table 1 and Supplementary Table SI1. To minimize the contribution of dead chains coming from AIBN initiation, the relative concentrations of AIBN to chol-X remained low (ratio between 0.005 for (1) and 0.15 for (5)). In all cases, the polymerizations reached more than 96% conversion after 24 h of reaction at 70 °C. The products were purified by precipitation from hexane. The polymers were characterized by 1H NMR, FT IR (Supplementary Figures SI1–6), and size exclusion chromatography (SEC) (Table 1, Supplementary Figure SI7). For the lowest theoretical number-average molecular weights (Mn,th) (entries 1 and 2), molecular weight distributions were narrow, and good agreement was observed between Mn,th and the apparent number-average molecular weight (Mn) obtained from SEC (Table 1). However, in the cases of chol-PNIPAAm-X(3–5), Mn values level off and differ from the theoretical ones. It is most likely due to chain transfer to solvent, which dictates an upper limit in accessible molecular weight, and also tends to broaden molecular weight distribution.26,27
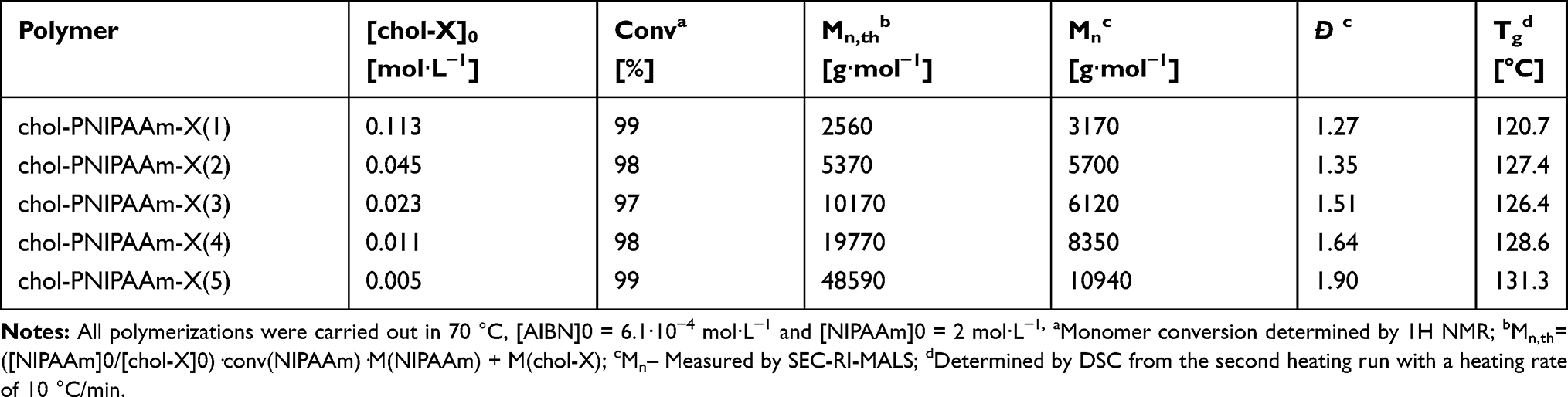 |
Table 1 Synthetic Details and Results of Chol-X-Mediated Polymerizations of NIPAAm |
A comparative analysis of the number of dithiocarbonate groups at the end of the polymer chains was carried out by UV spectroscopy. UV spectra obtained for samples at the same concentration confirmed the occurrence of dithiocarbonate groups at the end of the polymer chains (Supplementary Figure SI8). The absorption band at 280 nm related to the π–π* transition of the OC(S)S moiety28,29 was present in the UV spectra of all samples. As expected, the intensity of this band was reduced with increased chain length.
The TGA curves of all samples show a small weight loss (around 2%) in the temperature range up to 100 °C, most likely due to the removal of water (Supplementary Figure SI9). The samples show one significant weight loss (~95%) in the temperature range characteristic of PNIPAAm decomposition (380–420 °C). The degradation temperature increases with the weight fraction of PNIPAAm. The maximum of the degradation rate shifts to higher T values with increasing weight fraction of PNIPAAm (Figure SI 9). DSC measurements were performed to evaluate Tg values of the polymers (Table 1, Supplementary Figure SI10). As expected, Tg increases with an increasing average molecular weight of the PNIPAAm chain.30
CMC Determination
Hydrophilic chains ended with cholesterol moiety are amphiphilic polymers capable of forming polymeric micelles by self-assembly in aqueous solutions above critical micelle concentration (CMC). Polymer micelles offer advantages as drug carriers, primarily because of their small sizes which can markedly improve the in vivo performance of encapsulated drugs.31 CMC is one of the key parameters for the preparation of polymer micelles as DDSs. CMCs of the chol-PNIPAAm-X(2–5), investigated by standard fluorescence spectroscopy method using pyrene as a probe (Supplementary Figure SI11), were equal to 4.6 × 10−3, 6.2 × 10−3, 10−2, and 3.3 × 10−2 mg·mL−1, respectively. The CMC increases as the hydrophilic/hydrophobic ratio (or hydrophilic block length) increases, which is consistent with the previously reported data.14,17
DLS and ζ Potential Measurements
For further study, the chol-PNIPAAm-X(2–5) polymers were prepared in two ways: by direct dissolution in water (method 1), or by precipitation from organic solvent followed by freeze-drying and dissolution in water (method 2). The chol-PNIPAAm-X(1) sample was insoluble in water due to a too low hydrophilic/lipophilic balance, hence its further analysis was not carried out. DLS measurements of samples prepared by methods 1 and 2 were performed 24 h and one week after dissolution in water (at a concentration of 1 mg/mL). The results indicated a large heterogeneity and instability of all the systems prepared by method 1, making determination of their sizes impossible. The polymers prepared by method 2 (M_chol-PNIPAAm-X(2–5)) act differently in aqueous solution (Supplementary Figure SI12). DLS measurements taken 24h after dissolution in water confirmed the formation of micelles of diameters ranging from 15 to 26 nm, which changed insignificantly after one week (Figure 2A). Additionally, ζ potential was measured to determine the stability of the systems in water after one week (Figure 2B). For the chol-PNIPAAm-X(2–5) prepared by method 1, the observed zeta potential values were close to zero. For the micelles obtained by method 2, the values ranged from −14 to −8 mV. The preparation method of the polymers significantly influenced the colloidal stability of the systems. However, no clear dependence of zeta potential on PNIPAAm chain length was observed.
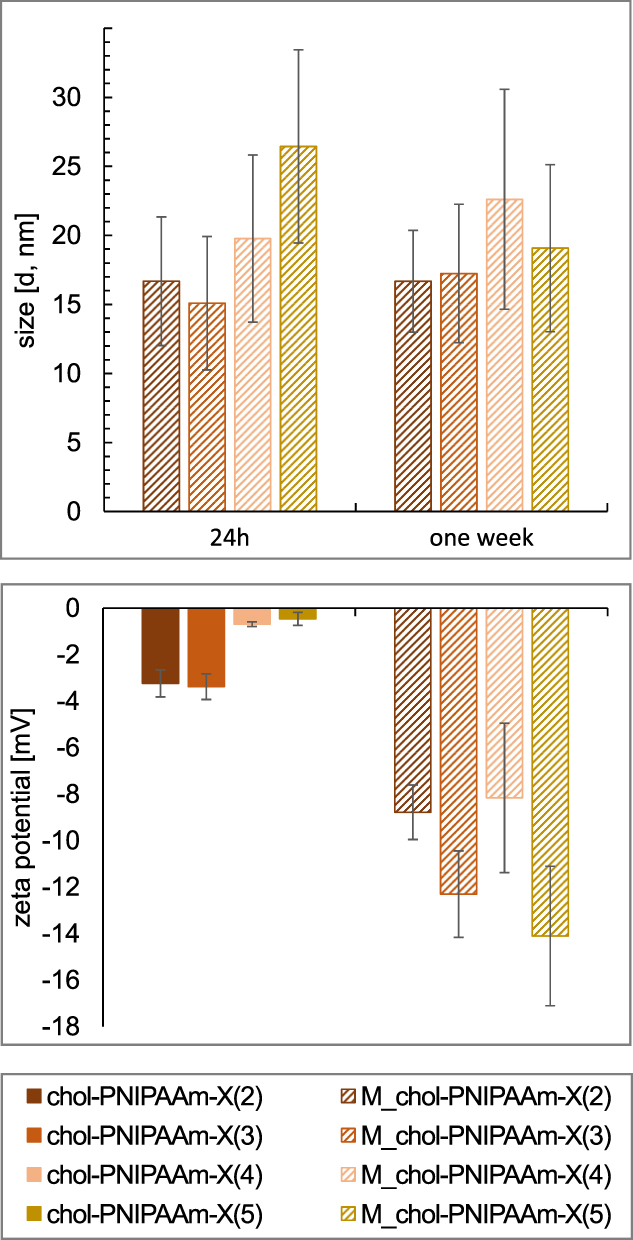 |
Figure 2 DLS data (size distribution by number) (top), and zeta potential measurement results (bottom). |
TEM and SEM Analysis
To investigate the morphology of the obtained products TEM measurements were conducted. In Figure 3, the TEM images were taken for the chol-PNIPAAm-X(2) and M_chol-PNIPAAm-X(2) samples are presented. In images A, B, and C it is impossible to determine specific shapes, the whole resembles a crumpled piece of cloth. In contrast, in pictures D, E, F one can see spherical shapes with dimensions of 20–30 nm. Also, SEM images (Supplementary Figure SI13) show differences in the topography of the studied homopolymers. In the cases of the polymers prepared by method 1, the surface is flat, and the edges are sharp, while for the systems obtained by the second method, the surface is lumpy.
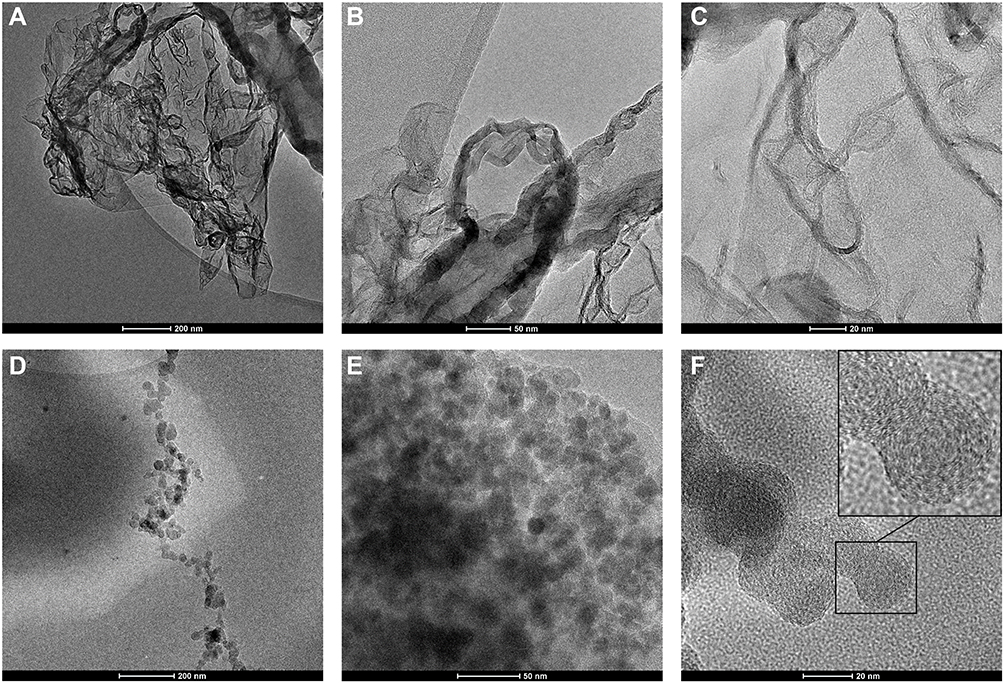 |
Figure 3 TEM images of chol-PNIPAAm-X(2) (A–C) and M_chol-PNIPAAm-X(2) (D–F) with different magnifications. |
The Influence of Molecular Weight and End Groups on Cloud Point Temperatures
In general, it is established that the LCST of PNIPAAm in water is independent of molecular weight (MW) for polymers of MW > 50 kDa.32–34 For lower molecular weight samples, changes in phase transition temperature values measured in water were observed.32,35,36 The cloud point temperature (TCP) of well-defined low molecular weight PNIPAAm showed an inversed molecular weight dependence: the shorter the polymer, the higher the cloud point.37,38 Changes in TCP were also correlated with hydrophobicity/hydrophilicity of the polymer end groups, whose contribution increases as the molecular weight decreases. For polymers of the same molecular weights, TCP values increased with the hydrophilicity of the end group.39
To determine phase transition temperatures of the obtained polymers in water, turbidimetry and calorimetry were used. The data collected by these two methods are consistent, only in the case of the longest polymer chain (chol-PNIPAAm-X(5)), the difference is above 2°C (Table 2, Figure 4, Supplementary Figure SI14). The preparation method of the aqueous polymer dispersions did not affect TCP values. The phase transition temperature increases with molecular weight and hydrophilic/hydrophobic ratio in the polymer. This is in line with literature data as the molecular weights of the studied polymers are in the range where the LCST depends on MW and nature of end groups.
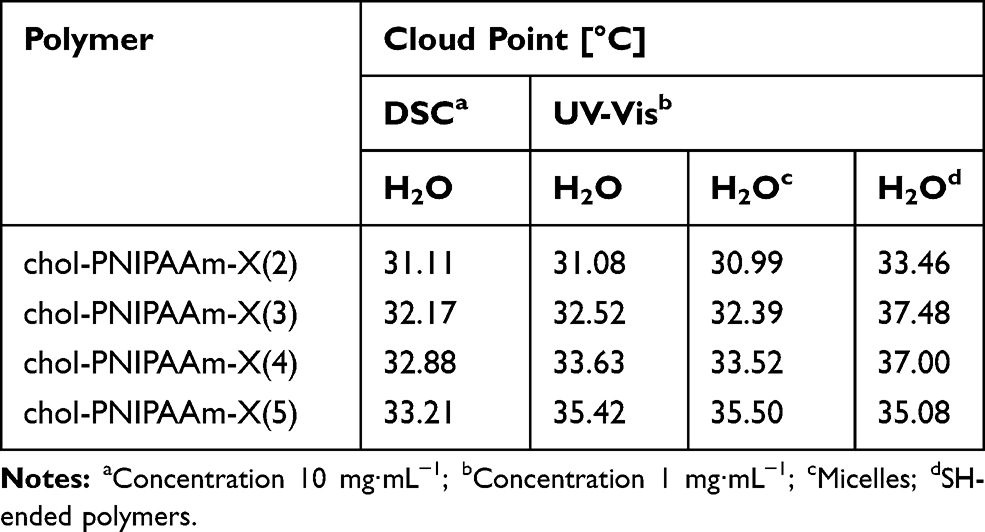 |
Table 2 UV-Vis and DSC Data for Chol-PNIPAAm-X(2–5) |
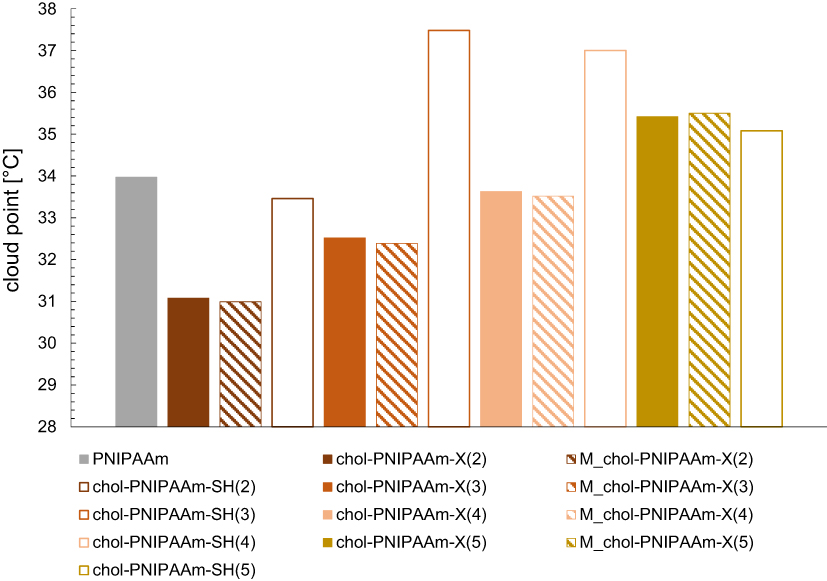 |
Figure 4 Cloud points dependence on molecular weight and polymer end groups. |
The observed end-group effect is due to both end groups contribution: cholesteryl and dithiocarbonate. To investigate the possibility of tuning phase transition temperatures, the reduction of dithiocarbonate groups was carried out leading to thiol-ended polymers chol-PNIPAAm-SH(2–5) (Supplementary Figure SI15). As expected, TCP values of SH-ended systems (2, 3, and 4) measured in water were noticeably higher compared to their dithiocarbonate-containing counterparts (Table 2, Figure 4).23,40 However, the influence of the end-group becomes less apparent when the MW increases, as for the longest polymer chain (entry 5) similar TCP values were observed regardless of polymer termini.
Biological Studies
Hemocompatibility Studies of PNIPAAm Derivatives
Hemocompatibility is one of the key parameters limiting the clinical applicability of blood-contacting materials.41 As proposed by Totea et al, depending on the hemolysis percentage, materials can be classified into three categories: materials resulting in over 5% hemolysis are hemolytic, between 5% and 2% are slightly hemolytic, and below 2% are non-hemolytic.42 Hemolytic activity measurements revealed that independently of the concentration used, all tested polymers show hemolytic activity around 1% (Figure 5A). This makes them very attractive materials for the formation of drug carriers applied via intravenous injection.
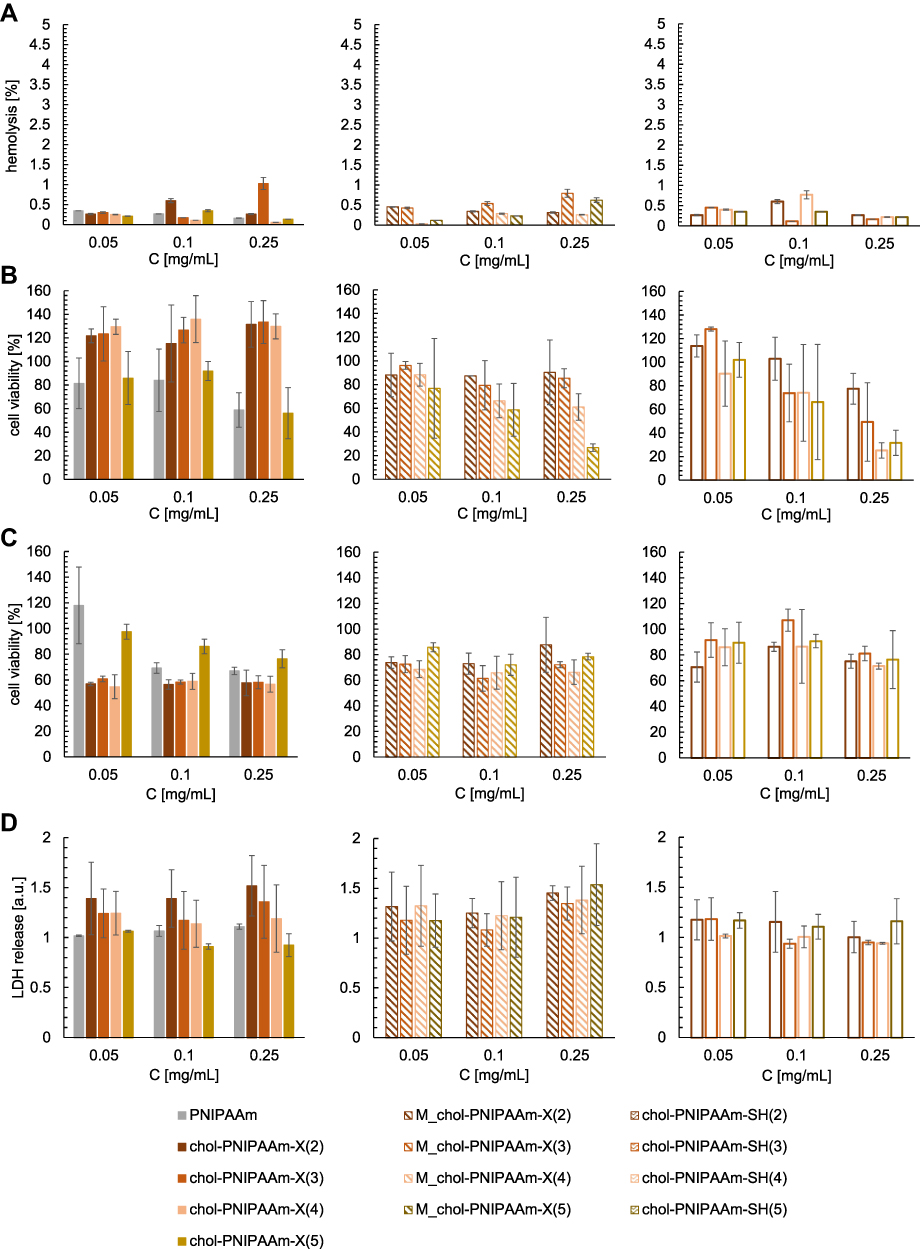 |
Figure 5 (A) Hemolytic activity measurements results. Fibroblasts (B) and glioblastoma cells (C) viability determined by neutral red uptake assay. (D) LDH release assay results. |
Cytotoxicity Studies of Cholesterol End-Capped PNIPAAm
To investigate the potential of the synthesized polymers with cholesteryl moiety as drug carriers, fibroblasts (Figure 5B) and glioblastoma cells (Figure 5C) were treated with different doses of the studied materials. The influence of the molecular weight, sample preparation method (1 or 2), and polymer end groups on their compatibility with the representative of host cells and cytotoxic efficacy against cancer cells were studied. The results provide information about the relationship between structure/properties and biological activity (structure-activity relationship – SAR) of tested agents.
Chol-PNIPAAm-X(2–4) directly dissolved in a medium (method 1) do not reduce fibroblasts viability at all tested concentrations. Moreover, a moderate increase in the fibroblasts viability (from 10% to 30% compared to the control culture) suggests possibility of their application in cell regeneration systems. In the case of chol-PNIPAAm-X(5) applied at the concentration of 0.05 and 0.25 mg‧mL−1, the fibroblasts viability at the level of 80% and 60%, respectively, was observed. A similar level of cytotoxicity on fibroblasts was detected after treatment with PNIPAAm obtained by free radical polymerization (not comprising cholesterol moiety).
Significant variability in the fibroblasts viability was detected after treatment with different doses of the carriers formed by method 2. At the lowest applied concentration (0.05 mg‧mL−1) limited inhibition of cell growth (viability at the level ~ 85–95%) was observed for all tested carriers. After treatment with M_chol-PNIPAAm-X(2 and 3), the viability of fibroblasts was maintained at the same level (90–80%) regardless of the applied dose. In the case of M_chol-PNIPAAm-X(4) and M_chol-PNIPAAm-X(5), the dose increased to 0.1 mg‧mL−1 caused reduction in the cell viability to 70% and 60%, respectively. For M_chol-PNIPAAm-X(5) the dose escalation to 0.25 mg‧mL−1 led to a significant decrease in the fibroblasts viability (up to 30%). In the case of thiol-ended polymers, the dose-dependent effect on the viability of fibroblasts was observed. At the lowest applied concentration (0.05 mg‧mL−1), the cell viability was classified around 100%. Double dose caused only a slight decrease in the fibroblasts viability. However, the addition of polymers at the highest concentration resulted in reduction in the cell viability to 80% in the case of chol-PNIPAAm-SH(2), 50% for chol-PNIPAAm-SH(3) and 30% for chol-PNIPAAm-SH(4–5).
The following experiments were performed on glioblastoma cells (Figure 5C). Gliomas belongs to the most frequent primary intracranial tumors that arise from the three types of glial cells of the brain. Their strong cellular and molecular heterogeneity, highly separative function of the blood-brain barrier (BBB) and impaired immune surveillance underlie the aggressive diffuse infiltration, high biological activity and failure of standard targeted pharmacological strategies.43–45 The studies show that chol-PNIPAAm-X(2–4) can deplete the tumor cells viability up to 60% regardless of the applied dose. Intriguingly, chol-PNIPAAm-X(5) did not affect the cell viability significantly, and at the highest applied concentration (0.25 mg‧mL−1) the inhibition of growth at the level of 20% was observed. However, it should be pointed out, that such a concentration of chol-PNIPAAm-X(5) was toxic to normal cells, while in the case of polymers of lower molecular weights chol-PNIPAAm-X(2–3) lack of cytotoxicity to fibroblasts has been observed. In the case of PNIPAAm, at the lowest applied dose, 100% of viable cells were detected. An increase of the concentration caused reduction in the cell viability up to 70%, with a similar effect against fibroblasts. A slightly lower decrease in the cell viability (~65% vs control) was observed after treatment with the M_chol-PNIPAAm-X(2–5) micelles. However, in the case of M_chol-PNIPAAm-X(5), at the lowest applied concentration, 20% depletion of viable cells number was detected in comparison to the chol-PNIPAAm-X(5) sample prepared by method 1. Thiol-terminated polymers caused only a slight decrease in glioblastoma cells viability.
Influence of Cholesteryl Moiety and Molecular Weight of Polymer on the Cell Membrane Stability
The presence of the steroid moiety in the structure of the tested macromolecules suggests the membrane-based mechanism of action. To investigate whether the polymers disrupt cell membranes, LDH release assay was performed. LDH is a soluble cytoplasmic enzyme that is present in almost all cells and is released into extracellular space through the impaired plasma membrane. The method enables the evaluation of necrotic-based cell death by determining the amount of this enzyme in the culture medium. It is important since cytoplasmic leakage from necrotic cells causes the release of intracellular contents into the extracellular milieu and evokes inflammatory responses.46 All carriers containing the cholesteryl moiety caused leakage of LDH from the treated cells (Figure 5D). These results confirm their ability to insert into the plasma membrane. Interestingly, in the case of the carriers obtained by method 1, the chain length effect and dose effect were observed. The highest content of LDH in the medium was detected after treatment with the polymer of the lowest molecular weight. Moreover, in the cases of chol-PNIPAAm-X(2–4), increasing concentration caused enhancement of LDH release. The reversed effect was observed for chol-PNIPAAm-X(5). For thiol-terminated polymers, slightly lower LDH release was detected, which correlates with the lower toxicity to the tested cells. Meaningly, no LDH release was observed from cells treated with PNIPAAm. This supports the hypothesis that the presence of cholesteryl moiety determines the ability of carriers to damage cellular membranes. In the cases of M_chol-PNIPAAm-X(2–5), the release of LDH was generally independent of the size of the tested carriers. All of them showed an increasing ability to impair the plasma membrane via the progressive release of LDH content to the medium of treated cells. This suggests that the micellar form of carriers enhances the ability to interact with the plasma membrane (Figure 6).
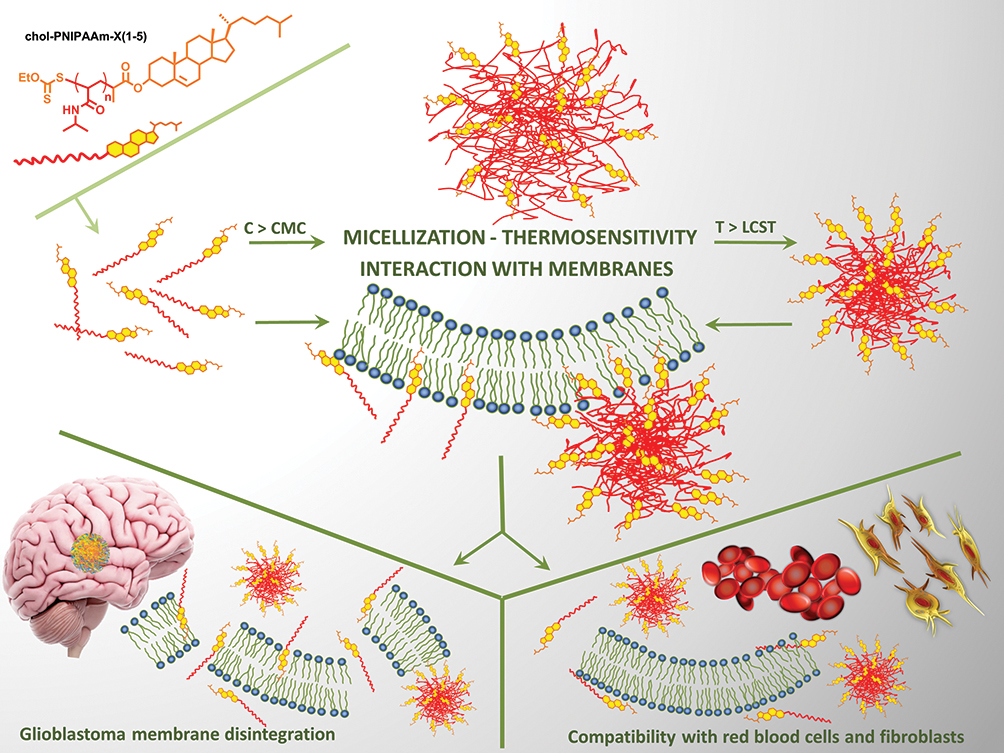 |
Figure 6 Graphical presentation of the idea of the research. |
Suggested Mode of Action: Cholesteryl Moiety Enables Two Ways of Interaction Between Carriers and Tested Cells
The plasma membrane is a crucial cell component that regulates the transport of active substances from the external environment into the cytoplasm. It is established that in cancer cell alterations in lipid metabolism, and consequently, composition and asymmetry of plasma membranes, occur. This leads to the decrease in membrane permeability to anticancer agents by one order of magnitude in comparison to the membranes of normal cells. The basic and preclinical studies on a new anticancer treatment are focused on the possibilities of cell membranes modulation aimed at their destruction and/or activation of immune cells in the tumor microenvironment. The application of lipid-based drug carriers, that may affect the composition of membrane, its properties, and associated functions, is a strategy to sensitize cancer cells to anticancer agents.47,48 Cholesterol is an important lipid molecule in animal cells as an essential constituent of the cell membrane and as a precursor for bile acids and steroid hormones.49 The concentration of cholesterol within the plasma membrane, the cholesterol-to-phospholipid (C:PL) molar ratio, correlates with membrane microviscosity, and is influenced by cholesterol content in the surrounding environment.50 Cholesterol as well as sphingomyelins are critically important in the formation of small, ordered lipid domains (“lipid rafts”) inside the cell membrane which are held together by hydrogen bonds, charge pairing, van der Waals and hydrophobic forces.51 Therefore, the first hypothesis is that the cholesterol moiety introduced to the carrier structure acts as a cell-penetrating agent. Wang et al observed that modification of fluorescent quantum dots (QDs) by cholesterol molecules provides plasma membrane enrichment and enhanced uptake into cancer cells via lipid raft-dependent endocytosis.52 The second hypothesis assumes that cholesterol-decorated carriers might interact with steroid receptors on the cell surface. Recent studies have shown that estrogen receptor ER‐α36 is highly expressed in glioblastoma specimens. It was found out that ER‐α36 knockdown increased the sensitivity of glioblastoma U87 cells to Tamoxifen (TAM).53 The estrogen receptors (ERα and ERβ) are also expressed on the surface of fibroblast cells. Both ERα and ERβ take part in collagen biosynthesis in the skin and play an important role in maintaining proper tissue homeostasis.54 In effect, two ways of interaction of synthesized cholesterol-based carriers with the plasma membranes of the tested cells might be considered.
Conclusion
The cholesterol end-capped poly(N-isopropylacrylamide)s of different molecular weights were synthesized from an original cholesterol-functional RAFT agent. The obtained polymers were further analyzed in two forms: (1) directly dissolved in the aqueous medium, and (2) precipitated from an organic solvent, freeze-dried, and dissolved in an aqueous medium. DLS and zeta potential studies results showed that the direct dissolution of polymers in aqueous media leads to unstable systems comprising free polymer chains and their aggregates. In contrast, the second method caused the formation of micelles of diameters around 20 nm. It was demonstrated that the phase transition temperature increases with molecular weight and hydrophilic/hydrophobic ratio in the polymer. Hemolytic activity tests showed that none of the tested systems disrupt red blood cell membranes at concentrations up to 0.25 mg‧mL−1 (hemolysis rates were lower than 1%). Fibroblasts and glioblastoma cells viability measurements showed that there is a relationship between the form, size, and concentration of the carriers and their cytotoxic activity. It was found that polymers of lower molecular weights (2 and 3) can reduce glioblastoma cells viability up to 60%, but are not toxic to fibroblasts. The LDH release assay results confirmed the essential role of cholesterol moiety in the proposed systems and the membrane-based mechanism of action: it enables penetration and disruption of glioblastoma plasma membranes, which leads to the restriction of the tumor growth. Due to the interesting biological properties, including in vitro anticancer efficacy, further development of cholesterol-ended PNIPAAm not only as drug carriers but also as components of synergistic therapy should be considered. The effects of treatment of fibroblasts and glioblastoma cells with polymers in disordered and micellar forms suggest that after micelles destabilization polymers might be compatible with healthy cells and maintain cytotoxic effect against cancer cells. This conclusion is particularly important from a pharmacological point of view as the disintegration of micelles (due to their dilution in the blood) is a major limitation of micelles as drug carriers. Hence the presented polymers offer a promising foundation for the design of efficient drug delivery systems showing high compatibility with the representative healthy cells and toxicity to neoplastically transformed ones.
Abbreviations
AIBN, 2.2′-azobis(2-methylpropionitrile); ATR-FTIR, attenuated total reflectance Fourier transform infrared spectroscopy; ATRP, atom transfer radical polymerization; BBB, blood-brain barrier; chol-Br, 2-bromo-O-(3-cholesteryl)propanoate; chol-X, O-ethyl-S-(1-cholesteroxycarbonyl)ethyldithiocarbonate; CMC, critical micelle concentration; CTA, chain transfer agent; DCM, dichloromethane; DLS, dynamic light scattering; DMEM, Dulbecco’s modified eagle medium; DSC, differential scanning calorimetry; IRB, institutional review board; LCST, lower critical solution temperature; LDH, lactate dehydrogenase; MW, molecular weight; NMR, nuclear magnetic resonance; PBS, phosphate buffer solution; PNIPAAm, poly(N-isopropylacrylamide); PTFE, polytetrafluoroethylene; RAFT, reversible addition-fragmentation chain transfer; RBCs, red blood cells; SDDS, smart drug delivery systems; SEC, size exclusion chromatography; SEM, scanning electron microscopy; TCP, cloud point temperature; TEM, transmission electron microscopy; Tg, glass transition temperature; TGA, thermogravimetric analyses; THF, tetrahydrofuran; UV-Vis, ultraviolet-visible spectroscopy.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
This work was financially supported by the National Science Centre, Poland, grant no. NCN/2016/21/B/ST5/01365 (AZW). Analyses were performed in the Centre of Synthesis and Analysis BioNanoTechno of the University of Bialystok. The equipment in the Centre was funded by the EU as a part of the Operational Program Development of Eastern Poland 2007–2013, projects: POPW.01.03.00-20-034/09-00 and POPW.01.03.00-20-004/11. This manuscript is dedicated to Professor Jacek W. Morzycki from the University of Bialystok on the occasion of his 70th birthday.
Disclosure
All authors report no conflicts of interest in this work.
References
1. Kalaydina R-V, Bajwa K, Qorri B, DeCarlo A, Szewczuk MR. Recent advances in “smart” delivery systems for extended drug release in cancer therapy. Int J Nanomedicine. 2018;13:4727–4745. doi:10.2147/IJN.S168053
2. Liu D, Yang F, Xiong F, Gu N. The smart drug delivery system and its clinical potential. Theranostics. 2016;6(9):1306–1323. doi:10.7150/thno.14858
3. Wilczewska AZ, Niemirowicz K, Markiewicz KH, Car H. Nanoparticles as drug delivery systems. Pharmacol Rep. 2012;64(5):1020–1037. doi:10.1016/s1734-1140(12)70901-5
4. Zhou Q, Zhang L, Yang T, Wu H. Stimuli-responsive polymeric micelles for drug delivery and cancer therapy. Int J Nanomedicine. 2018;13:2921–2942. doi:10.2147/IJN.S158696
5. Yamada Y, Itoh Y, Aoki S, et al. Preliminary results of M-VAC chemotherapy combined with mild hyperthermia, a new therapeutic strategy for advanced or metastatic transitional cell carcinoma of the urothelium. Cancer Chemother Pharmacol. 2009;64(6):1079–1083. doi:10.1007/s00280-009-0964-2
6. Dieing A, Ahlers O, Hildebrandt B, et al. The effect of induced hyperthermia on the immune system. Prog Brain Res. 2007;162:137–152. doi:10.1016/S0079-6123(06)62008-6
7. Chee CK, Rimmer S, Soutar I, Swanson L. Manipulating the thermoresponsive behavior of poly(N-isopropylacrylamide). In: McCormick CL, editor. Stimuli-Responsive Water Soluble and Amphiphilic Polymers. Vol. 780. ACS Symposium Series, American Chemical Society; 2000:223–237. doi:10.1021/bk-2001-0780.ch013
8. Zhang R, Qin X, Kong F, Chen P, Pan G. Improving cellular uptake of therapeutic entities through interaction with components of cell membrane. Drug Deliv. 2019;26(1):328–342. doi:10.1080/10717544.2019.1582730
9. Stewart MP, Langer R, Jensen KF. Intracellular delivery by membrane disruption: mechanisms, strategies, and concepts. Chem Rev. 2018;118(16):7409–7531. doi:10.1021/acs.chemrev.7b00678
10. Albuquerque H, Santos C, Silva A. Cholesterol-based compounds: recent advances in synthesis and applications. Molecules. 2018;24(1):116. doi:10.3390/molecules24010116
11. Morzycki JW. Recent advances in cholesterol chemistry. Steroids. 2014;83:62–79. doi:10.1016/j.steroids.2014.02.001
12. Zhou Y, Briand V, Sharma N, Ahn S, Kasi R. Polymers comprising cholesterol: synthesis, self-assembly, and applications. Materials. 2009;2(2):636–660. doi:10.3390/ma2020636
13. Tan S, Han C, Wang H, et al. Preparation and characterization of thermo-sensitive mixed micelles and in vitro drug release. Acta Polym Sin. 2011;11(11):1237–1243. doi:10.3724/SP.J.1105.2011.10301
14. Gao M, Yang Y, Bergfel A, Huang L, Zheng L, Bowden TM. Self-assembly of cholesterol end-capped polymer micelles for controlled drug delivery. J Nanobiotechnology. 2020;18(1):13. doi:10.1186/s12951-020-0575-y
15. Liu J, Setijadi E, Liu Y, Whittaker MR, Boyer C, Davis TP. PEGylated gold nanoparticles functionalized with β-cyclodextrin inclusion complexes: towards metal nanoparticle - polymer - carbohydrate cluster biohybrid materials. Aust J Chem. 2010;63(8):1245. doi:10.1071/CH10091
16. Zhou Y, Kasi RM. Synthesis and characterization of polycholesteryl methacrylate-polyhydroxyethyl methyacrylate block copolymers. J Polym Sci Part Polym Chem. 2008;46(20):6801–6809. doi:10.1002/pola.22988
17. Liu XM, Yang YY, Leong KW. Thermally responsive polymeric micellar nanoparticles self-assembled from cholesteryl end-capped random poly(N-isopropylacrylamide-co-N,N-dimethylacrylamide): synthesis, temperature-sensitivity, and morphologies. J Colloid Interface Sci. 2003;266(2):295–303. doi:10.1016/S0021-9797(03)00691-X
18. Liu XM, Pramoda KP, Yang YY, Chow SY, He C. Cholesteryl-grafted functional amphiphilic poly(N-isopropylacrylamide-co-N-hydroxylmethylacrylamide): synthesis, temperature-sensitivity, self-assembly and encapsulation of a hydrophobic agent. Biomaterials. 2004;25(13):2619–2628. doi:10.1016/j.biomaterials.2003.09.028
19. Despax L, Fitremann J, Destarac M, Harrisson S. Low concentration thermoresponsive hydrogels from readily accessible triblock copolymers. Polym Chem. 2016;7(20):3375–3377. doi:10.1039/C6PY00499G
20. Wang YM, Zheng SX, Chang HI, Tsai HY, Liang M. Microwave-assisted synthesis of thermo- and pH-responsive antitumor drug carrier through reversible addition-fragmentation chain transfer polymerization. Express Polym Lett. 2017;11(4):293–307. doi:10.3144/expresspolymlett.2017.29
21. Wang H, Li Z, Lu S, et al. Nano micelles of cellulose-graft-poly (l-lactic acid) anchored with epithelial cell adhesion antibody for enhanced drug loading and anti-tumor effect. Mater Today Commun. 2020;22:100764. doi:10.1016/j.mtcomm.2019.100764
22. Cammas S, Suzuki K, Sone C, Sakurai Y, Kataoka K, Okano T. Thermo-responsive polymer nanoparticles with a core-shell micelle structure as site-specific drug carriers. J Control Release. 1997;48(2–3):157–164. doi:10.1016/S0168-3659(97)00040-0
23. Glaria A, Beija M, Bordes R, Destarac M, Marty J-D. Understanding the role of ω-end groups and molecular weight in the interaction of PNIPAM with gold surfaces. Chem Mater. 2013;25(9):1868–1876. doi:10.1021/cm400480p
24. Patton DL, Mullings M, Fulghum T, Advincula RC. A facile synthesis route to thiol-functionalized α,ω-telechelic polymers via reversible addition fragmentation chain transfer polymerization. Macromolecules. 2005;38(20):8597–8602. doi:10.1021/ma051035s
25. Lima V, Jiang X, Brokken-Zijp J, Schoenmakers PJ, Klumperman B, Van Der Linde R. Synthesis and characterization of telechelic polymethacrylates via RAFT polymerization. J Polym Sci Part Polym Chem. 2005;43(5):959–973. doi:10.1002/pola.20558
26. Min Y, Jun L, Hongfei H. Radiation preparation of the water-soluble, temperature sensitive polymers in organic solvents. Radiat Phys Chem. 1995;46(4–6):855–858. doi:10.1016/0969-806X(95)00277-5
27. Sugihara Y, O’Connor P, Zetterlund PB, Aldabbagh F. Chain transfer to solvent in the radical polymerization of N-isopropylacrylamide. J Polym Sci Part Polym Chem. 2011;49(8):1856–1864. doi:10.1002/pola.24612
28. Juncal LC, Tobón YA, Piro OE, Della Védova CO, Romano RM. Structural, spectroscopic and theoretical studies on dixanthogens: (ROC(S)S)2, with R = n-propyl and isopropyl. New J Chem. 2014;38(8):3708–3716. doi:10.1039/C4NJ00708E
29. Mikhlin Y, Vorobyev S, Saikova S, et al. Preparation and characterization of colloidal copper xanthate nanoparticles. New J Chem. 2016;40(4):3059–3065. doi:10.1039/C6NJ00098C
30. Biswas CS, Patel VK, Vishwakarma NK, et al. Effects of tacticity and molecular weight of poly(N-isopropylacrylamide) on its glass transition temperature. Macromolecules. 2011;44(14):5822–5824. doi:10.1021/ma200735k
31. Lu Y, Zhang E, Yang J, Cao Z. Strategies to improve micelle stability for drug delivery. Nano Res. 2018;11(10):4985–4998. doi:10.1007/s12274-018-2152-3
32. Furyk S, Zhang Y, Ortiz-Acosta D, Cremer PS, Bergbreiter DE. Effects of end group polarity and molecular weight on the lower critical solution temperature of poly(N-isopropylacrylamide). J Polym Sci Part Polym Chem. 2006;44(4):1492–1501. doi:10.1002/pola.21256
33. Schild HG. Poly(N-isopropylacrylamide): experiment, theory and application. Prog Polym Sci. 1992;17(2):163–249. doi:10.1016/0079-6700(92)90023-R
34. Okada Y, Tanaka F. Cooperative hydration, chain collapse, and flat LCST behavior in aqueous poly(N-isopropylacrylamide) solutions. Macromolecules. 2005;38(10):4465–4471. doi:10.1021/ma0502497
35. Chung JE, Yokoyama M, Aoyagi T, Sakurai Y, Okano T. Effect of molecular architecture of hydrophobically modified poly(N-isopropylacrylamide) on the formation of thermoresponsive core-shell micellar drug carriers. J Control Release. 1998;53(1–3):119–130. doi:10.1016/S0168-3659(97)00244-7
36. Qiu X, Koga T, Tanaka F, Winnik FM. New insights into the effects of molecular weight and end group on the temperature-induced phase transition of poly(N-isopropylacrylamide) in water. Sci China Chem. 2013;56(1):56–64. doi:10.1007/s11426-012-4781-9
37. Pamies R, Zhu K, Kjøniksen A-L, Nyström B. Thermal response of low molecular weight poly-(N-isopropylacrylamide) polymers in aqueous solution. Polym Bull. 2009;62(4):487–502. doi:10.1007/s00289-008-0029-4
38. Xia Y, Burke NAD, Stöver HDH. End group effect on the thermal response of narrow-disperse poly(N-isopropylacrylamide) prepared by atom transfer radical polymerization. Macromolecules. 2006;39(6):2275–2283. doi:10.1021/ma0519617
39. Narumi A, Fuchise K, Kakuchi R, et al. A versatile method for adjusting thermoresponsivity: synthesis and ‘Click’ reaction of an azido end-functionalized poly(N-isopropylacrylamide). Macromol Rapid Commun. 2008;29(12–13):1126–1133. doi:10.1002/marc.200800055
40. Li X, ShamsiJazeyi H, Pesek SL, Agrawal A, Hammouda B, Verduzco R. Thermoresponsive PNIPAAM bottlebrush polymers with tailored side-chain length and end-group structure. Soft Matter. 2014;10(12):2008. doi:10.1039/c3sm52614c
41. Weber M, Steinle H, Golombek S, et al. Blood-contacting biomaterials. Front Bioeng Biotechnol. 2018;6:99. doi:10.3389/fbioe.2018.00099
42. Totea G, Ionita D, Demetrescu I, Mitache M. In vitro hemocompatibility and corrosion behavior of new Zr-binary alloys in whole human blood. Open Chem. 2014;12:
43. Bosma I, Reijneveld JC, Douw L, et al. Health-related quality of life of long-term high-grade glioma survivors. Neuro-Oncol. 2009;11(1):51–58. doi:10.1215/15228517-2008-049
44. Batash R, Asna N, Schaffer P, Francis N, Schaffer M. Glioblastoma multiforme, diagnosis and treatment; recent literature review. Curr Med Chem. 2017;24(27). doi:10.2174/0929867324666170516123206
45. Qazi MA, Vora P, Venugopal C, et al. Intratumoral heterogeneity: pathways to treatment resistance and relapse in human glioblastoma. Ann Oncol. 2017;28(7):1448–1456. doi:10.1093/annonc/mdx169
46. Chan FK-M, Moriwaki K, De Rosa MJ. Detection of necrosis by release of lactate dehydrogenase activity. In: Snow AL, Lenardo MJ, editors. Immune Homeostasis. Humana Press; 2013:65–70. doi:10.1007/978-1-62703-290-2_7
47. Rivel T, Ramseyer C, Yesylevskyy S. The asymmetry of plasma membranes and their cholesterol content influence the uptake of cisplatin. Sci Rep. 2019;9(1):5627. doi:10.1038/s41598-019-41903-w
48. Zalba S, Ten Hagen TLM. Cell membrane modulation as adjuvant in cancer therapy. Cancer Treat Rev. 2017;52:48–57. doi:10.1016/j.ctrv.2016.10.008
49. Yokoyama S. Release of cellular cholesterol: molecular mechanism for cholesterol homeostasis in cells and in the body. Biochim Biophys Acta. 2000;1529(1–3):231–244. doi:10.1016/S1388-1981(00)00152-9
50. Mason RP. Differential effect of cholesterol on membrane interaction of charged versus uncharged 1,4-dihydropyridine calcium channel antagonists: a biophysical analysis. Cardiovasc Drug Ther. 1995;9(S1):45–54. doi:10.1007/BF00878572
51. Nicolson GL. Cell membrane fluid–mosaic structure and cancer metastasis. Cancer Res. 2015;75(7):1169–1176. doi:10.1158/0008-5472.CAN-14-3216
52. Wang HY, Hua X-W, Jia HR, et al. Enhanced cell membrane enrichment and subsequent cellular internalization of quantum dots via cell surface engineering: illuminating plasma membranes with quantum dots. J Mater Chem B. 2016;4(5):834–843. doi:10.1039/C5TB02183A
53. Qu C, Ma J, Zhang Y, et al. Estrogen receptor variant ER-Α36 promotes tamoxifen agonist activity in glioblastoma cells. Cancer Sci. 2019;110(1):221–234. doi:10.1111/cas.13868
54. Markiewicz M, Znoyko S, Stawski L, Ghatnekar A, Gilkeson G, Trojanowska M. A role for estrogen receptor-α and estrogen receptor-β in collagen biosynthesis in mouse skin. J Invest Dermatol. 2013;133(1):120–127. doi:10.1038/jid.2012.264
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.