")
Back to Journals » Clinical and Experimental Gastroenterology » Volume 12
Evaluating tofacitinib citrate in the treatment of moderate-to-severe active ulcerative colitis: design, development and positioning of therapy
Authors Tran V, Shammas RM, Sauk JS, Padua D
Received 13 December 2018
Accepted for publication 6 April 2019
Published 2 May 2019 Volume 2019:12 Pages 179—191
DOI https://doi.org/10.2147/CEG.S150908
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Anastasios Koulaouzidis
Vivy Tran,1 Rania M Shammas,2 Jenny S Sauk,1,3 David Padua1,3–4
1Department of Medicine, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA; 2Department of Rheumatology, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA; 3Tamar and Vatche Manoukian Division of Digestive Diseases, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA; 4Department of Medicine, VA Greater Los Angeles Healthcare System, Los Angeles, CA, USA
Abstract: The etiology of ulcerative colitis (UC) is complex and involves a host of genetic, epigenetic and environmental factors. Over the last thirty years, signaling pathways like the Janus kinase (JAK) signaling pathway have been implicated in its pathogenesis. Pharmacologic blockade of this pathway is available through several small molecule inhibitors, including tofacitinib. Tofacitinib is an orally administered pan-JAK inhibitor that was first approved by the Food and Drug Administration (FDA) for use in rheumatologic disorders such as rheumatoid arthritis and psoriatic arthritis. The FDA approved its use in moderate-to-severe active ulcerative colitis in 2018. The aim of this review will be to discuss the role of tofacitinib in ulcerative colitis. We will discuss the role of JAK-STAT signaling, clinical data available for tofacitinib, and the safety profile for this therapy. Tofacitinib’s place in the UC management algorithm is currently being debated. This effective oral therapy is poised to be a mainstay of UC therapeutics. This review will highlight the key clinical features and detail the UC experience to date.
Keywords: inflammatory bowel disease, Jak inhibitor, Xeljanz, ulcerative colitis, oral therapy
Introduction
Inflammatory bowel diseases (IBD) consists of both ulcerative colitis (UC) and Crohn’s disease (CD). These diseases are characterized by chronic gastrointestinal inflammation with alternating relapsing and remitting episodes of inflammation. In comparison to Crohn’s disease, ulcerative colitis is characterized by inflammation that is limited to the mucosal layer of the colon and typically extends proximally in a continuous fashion from the rectum. While the pathophysiology of IBD remains unclear, the origin is likely related to a combination of genetic susceptibility and environmental factors, which trigger inappropriate mucosal immune responses.1–3
The primary goals in therapy are to induce and maintain remission. Conventional therapies include corticosteroids, aminosalicylates, immunomodulatory agents (thiopurines, methotrexate), and biologics, including anti-tumor necrosis (anti-TNF) agents, anti-integrin agents and anti IL-12/IL-23 agents.4 While biologic agents have revolutionized the treatment of IBD showing favorable short- and long-term outcomes in decreasing clinical relapse and hospitalization rates, decreasing surgical intervention, and significantly improving quality of life,4–9 many patients fail to respond or have secondary loss of response characterized by re-emergence of symptoms or relapse of inflammation requiring other therapeutic interventions. Indeed, up to 30% of patients receiving biologic therapies are primary non-responders and 10–20% become secondary non-responders every year thereafter. Studies have shown specifically with anti-TNF agents that 12 months after starting therapy, 23–46% of patients are not sufficiently controlled and require dose intensification. An additional 5–13% of patients starting anti-TNF therapies will have their drug discontinued as a result of non-response.10 This may be related to inadequate drug concentrations, formation of anti-drug antibodies or having no mechanistic response to anti-TNF therapies.11 In addition, compliance rates to biologics have been shown to be as low as 66% in some IBD populations, which may be related to the method of administration as an injection or infusion.12 Many biologic therapies require access to infusion centers thereby adding another level of complexity to patient care. In the CHOOSE trial, convenience and time required for therapy influenced the patients’ selection of a specific anti-TNF drug, which can affect adherence to a certain treatment.13
Given the limitations of our current biologic therapies, novel treatments are needed to help our patient population. Oral treatment options that are safe and effective would offer great value to the management of IBD. Tofacitinib, an oral inhibitor of Janus Kinase 1–3, became available in 2018 for the treatment of adults with moderate-to-severe ulcerative colitis and has offered an option for patients who have failed other biologic therapies.14,15
JAK-STAT signaling pathway
JAK signaling pathway
The JAK-STAT signaling pathway is a chain of interactions between proteins in a cell that are involved in processes of immunity as well as cell division and cell death.16 The pathway communicates extracellular cytokine stimuli to the cell nucleus, which then results in activation of gene transcription. This pathway consists of Janus Kinases (JAKs), signal transducer and activator of transcription proteins (STATs) and receptors, which bind these chemical signals. There are four members of the JAK family (JAK1-3, and TYK2) along with seven members of the STAT family (STAT1-4, STAT5A/B, STAT6).
The binding of cytokines, such as interferons (IFN) and interleukins (IL), to cell-surface receptors cause the JAK-associated receptors to dimerize (Figure 1).17 The recruitment of JAK proteins results in trans- and/or auto-phosphorylation of associated JAKs, increasing the activity of these kinase domains and creating binding sites for STATs. STATs bind to the phosphotyrosines on the cytokine receptor tails. STATs are then phosphorylated by JAKs and then dissociated from the receptor, forming hetero- or homodimers with the opposite STAT and then translocated to the cell nucleus to induce or repress transcription of target genes. A large number of ligands can activate JAK-STAT signaling pathways. A wealth of research has shown that cytokines that interact with JAK-STAT, such as IL-6, IL-9, IL-10, IL-12/23, IL-22, granulocyte-macrophage colony-stimulating factor (GM-CSF) and interferon (IFN)-gamma, play key roles in the pathogenesis of both ulcerative colitis and Crohn’s disease.18
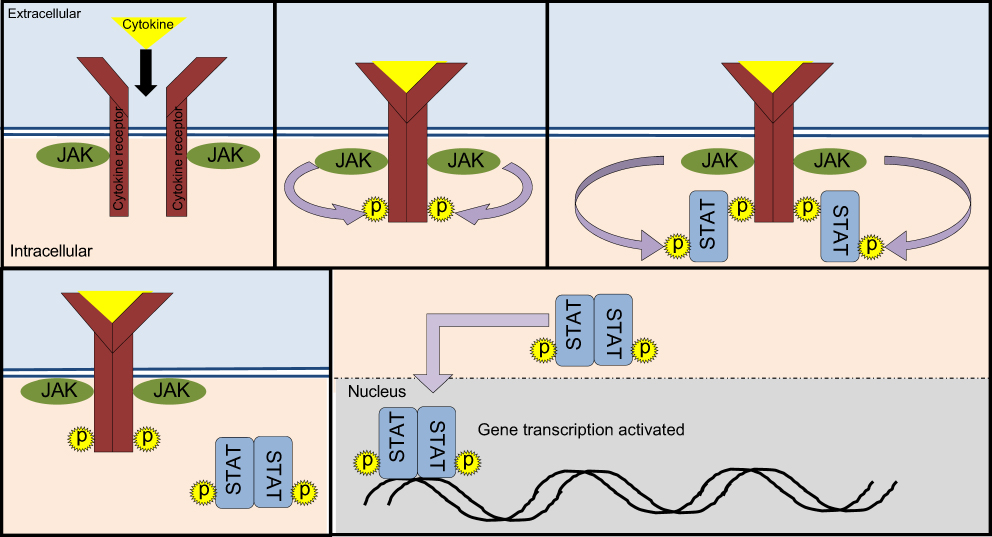 | Figure 1 JAK-STAT pathway. The JAK-STAT pathway involves of 1) cytokines which can include interleukins, interferon, granulocyte-macrophage colony stimulating factor 2) cytokine receptors 3) JAKs (JAK1-3, TYK2) and 4) STATs (STAT1-6). |
The importance of JAK-STAT signaling pathway is also demonstrated by the studies of primary immunodeficiencies. Severe combined immunodeficiency has been related to mutations in JAK3, and other conditions such as hyperimmunoglobulinemia E syndrome, mucocutaneous candidiasis, as well as viral susceptibility have been associated with several mutations of STAT proteins.19–22
JAK-STAT signaling in IBD
JAK-STAT signaling is known to be important in the regulation and modulation of the immune response. Genome-wide association studies have shown single nucleotide polymorphisms (SNPs) in cytokines (IL-12B), cytokine receptors (IL-23R), JAKs (JAK2, TYK2), and STAT proteins (STAT3) are associated with increased risk of IBD.3,23 The role of the JAK-STAT pathway in the mucosal immune system is central in IBD, a disease with imbalance of pro- and anti-inflammatory mediators that result in excess activation of the host immune response.24 JAK-STAT signaling plays a role in maintaining this balance through the development and differentiation of T helper cells (Th) and maintenance of the mucosal immune system.18
The JAK-STAT pathway is critical to the fate of Th cells, which in turn has a significant role in the initiation of immune responses. Imbalances of these T cells correspond with mucosal damage in IBD, with CD patients having increased Th1 cells with Th1/Th17 mucosal responses and UC patients with more mucosal Th2-mediated inflammation.25,26 The presence of Th17-related cytokines like IL-17A, IL-17F, IL-22 and IL-26 was also found to be involved in the pathogenesis of CD and UC.27–29 Th1 cell differentiation relies on TYK2, JAK2 and STAT4 for IL-12 signaling whereas Th2 cell differentiation relies on JAK1, JAK3 and STAT6 for IL-4 signal transduction.30 STAT5A and STAT5B are also involved in Th1 and Treg cells by promotion of T-bet and FoxP3 expression, respectively.16 Th17 cells differentiate in response to IL-6, IL-21, IL-23 activating STAT3.31 For instance, IL-6, which can activate the JAK1/STAT3 pathway, increases survival and proliferation of T cells on the lamina propria in IBD patients. Serum concentrations of IL-6 have been directly correlated to disease activity in IBD.32–34
Other key members of the JAK-STAT signaling pathway are found to have roles in maintaining intestinal homeostasis and decreasing disease activity. TYK2 in animal models were found to be significant in the pathogenesis of colitis through the regulation of the IL-12/IL-23 axis. In vivo experiments studying colitis with TYK2−/- mice had slower and lower disease activity compared to TYK2+/+ mice.35 IL-10, associated with JAK1 and TYK2 kinases which activate STAT3, has a critical role in maintaining gut homeostasis, which was evidenced in IL-10-deficient mice who developed severe spontaneous enterocolitis due to immune hyperactivation.36 In humans, genetic studies have identified SNPs in the IL-10 gene as a risk factor for IBD development.37 Animal models have also shown the role of STAT3 in maintenance of intestinal homeostasis. Mice with intraepithelial cell-specific (IEC) STAT3 deficiency were more susceptible to experimental colitis.38 And STAT3 knockout in IECs and macrophages had lethal enterocolitis after STAT3 deletion.39
These studies reveal multiple important components in the JAK-STAT pathway that are essential in the balance of immune mediators and intestinal homeostasis, playing a role in the pathogenesis of IBD. These components thus have provided a rationale for targeting this pathway in the development of new IBD therapies.
Tofacitinib on JAK inhibition
Tofacitinib is a reversible, competitive inhibitor that binds to the adenosine triphosphate (ATP) binding site in the catalytic cleft of the kinase domain of JAK.40 By binding to the ATP site, tofacitinib inhibits the phosphorylation and activation of JAK, thereby preventing phosphorylation and activation of STAT proteins and of corresponding gene transcription activation. Tofacitinib was developed by Pfizer as a JAK3 inhibitor to be used as an immunosuppressant in organ transplantation. However, in vitro kinase assays show tofacitinib inhibiting JAK1, JAK2, JAK3, and to a lesser extent, TYK2. In cellular settings where JAKs signal in pairs, tofacitinib preferentially inhibits signaling by cytokine receptors associated with JAK3 and JAK1 with 5-to-100 fold selectivity over JAK2.41,42 It efficiently blocks common γ-chain cytokines including IL-2, IL-4, IL-15 and IL-21. Since it also has activity against JAK1 and JAK2, tofacitinib also constrains signaling by IFN-γ, IL-6, and to a lesser extent IL-12 and IL-23. As a result of these activities, tofacitinib impairs differentiation of CD4+T helper cells, limits generation of pathogenic Th17 cells, blocks NK cell differentiation and limits production of TNF and other proinflammatory cytokines, thereby affecting both the innate and adaptive immune system.43
Tofacitinib pharmacokinetic profile
Tofacitinib is characterized by rapid absorption and elimination, with peak plasma concentration within 1 hr and terminal half-life of approximately 3 hrs. It is shown to be a well-absorbed drug with an oral bioavailability of about 93%.44 Clearance of tofacitinib is by 30% renal metabolism and 70% hepatic metabolism, mostly attributed to CYP3A4 activity.44 An extended release version of the drug is also available which was designed to provide a once-daily dosing option. This formulation of tofacitinib XR 11 mg daily has shown to have equivalence in bioavailability to 5 mg immediate release twice daily.45 The extended release formulation has a terminal half-life of 5.9 hrs and is currently approved for rheumatoid arthritis (RA) alone.
Tofacitinib and RA and other autoimmune diseases
Tofacitinib is most commonly used in the treatment of RA. In May 2012, the FDA approved the use of the first oral biologic drug, tofacitinib, for the use in adults with moderate to severe active RA. Current RA treatment guidelines from the American College of Rheumatology advocate for the use of tofacitinib after a patient who has failed or is intolerant to either methotrexate, other similar disease modifying medications or other biologic therapies.46 Several randomized, double-blind, multicenter clinical trials of 6–24 months duration showed that tofacitinib monotherapy (as first- or second-line treatment) and combination therapy with methotrexate (as second- and third-line treatment) was effective in reducing signs and symptoms of disease with improvement of health-related quality of life. These benefits were sustained during long-term therapy up to 96 months. Tofacitinib was well tolerated during 114 months of treatment with much of its adverse events generally similar to that of other biologic agents. Infections were the most common adverse event reported. In combination with methotrexate, neither tofacitinib nor adalimumab were superior in efficacy and both drugs had similar tolerability profiles.
By December 2017, the FDA approved tofacitinib for the treatment of active psoriatic arthritis. In two pivotal randomized, placebo-controlled, double-blind trials, tofacitinib was found to significantly reduce psoriatic arthritis disease activity within 3 months in patients who had failed conventional disease-modifying agents such as methotrexate, and reduce arthritis activity in those who had previously failed anti-TNF therapy when compared to placebo.47,48 In clinical practice, tofacitinib is generally used in patients with psoriatic arthritis who have failed or are intolerant to anti-TNF therapy or other biologic agents currently available.
Other autoimmune diseases may also potentially benefit from tofacitinib therapy. There are currently Phase II trials showing promise for tofacitinib use in the treatment of ankylosing spondylitis, as well as case series reports suggesting that tofacitinib may be useful in the treatment of autoimmune-mediated alopecia.49,50 However, more studies are needed prior to incorporating tofacitinib into the treatment paradigm for these other autoimmune conditions.
Clinical studies of tofacitinib in ulcerative colitis
In ulcerative colitis, tofacitinib efficacy has been evaluated in four multi-center, randomized, double-blind, placebo-controlled clinical trials. These studies investigated the use of tofacitinib in the treatment of adult patients with moderate-to-severe UC and found significant improvement in clinical and endoscopic outcomes when used for induction and maintenance of remission in UC.
Use as induction therapy
Clinical response and remission
Tofacitinib has been evaluated as an induction agent for UC patients in three clinical trials (Table 1). The Phase II clinical trial included 194 patients with moderate-to-severe UC, defined as a total Mayo score of 6 to 12 with endoscopic sub-score of 2 or 3.14 Patients were then randomly assigned to receive oral tofacitinib at a dose of 0.5 mg, 3 mg, 10 mg, 15 mg or placebo, which was administered twice daily for 8 weeks. Primary endpoint was clinical response at 8 weeks, defined as an absolute decrease from baseline in the Mayo score of 3 or more, relative decrease from baseline of 30% or more, and with an accompanying decrease in the rectal bleeding sub-score of 1 point or more with an absolute rectal bleeding sub-score of 0 or 1. Clinical response was achieved by 42% in the placebo group and 32%, 48%, 61% and 78% of patients receiving tofacitinib at a dose of 0.5 mg (p=0.39), 3 mg (p=0.55), 10 mg (p=0.10), 15 mg (p<0.001), respectively. Clinical remission (defined as Mayo score ≤2 with no subscore >1) at 8 weeks occurred in 10% in the placebo group and 13%, 33%, 48%, 41% of patients receiving tofacitinib at a dose of 0.5 mg (p=0.76), 3 mg (p=0.01), 10 mg (p<0.001), 15 mg (p<0.001), respectively.
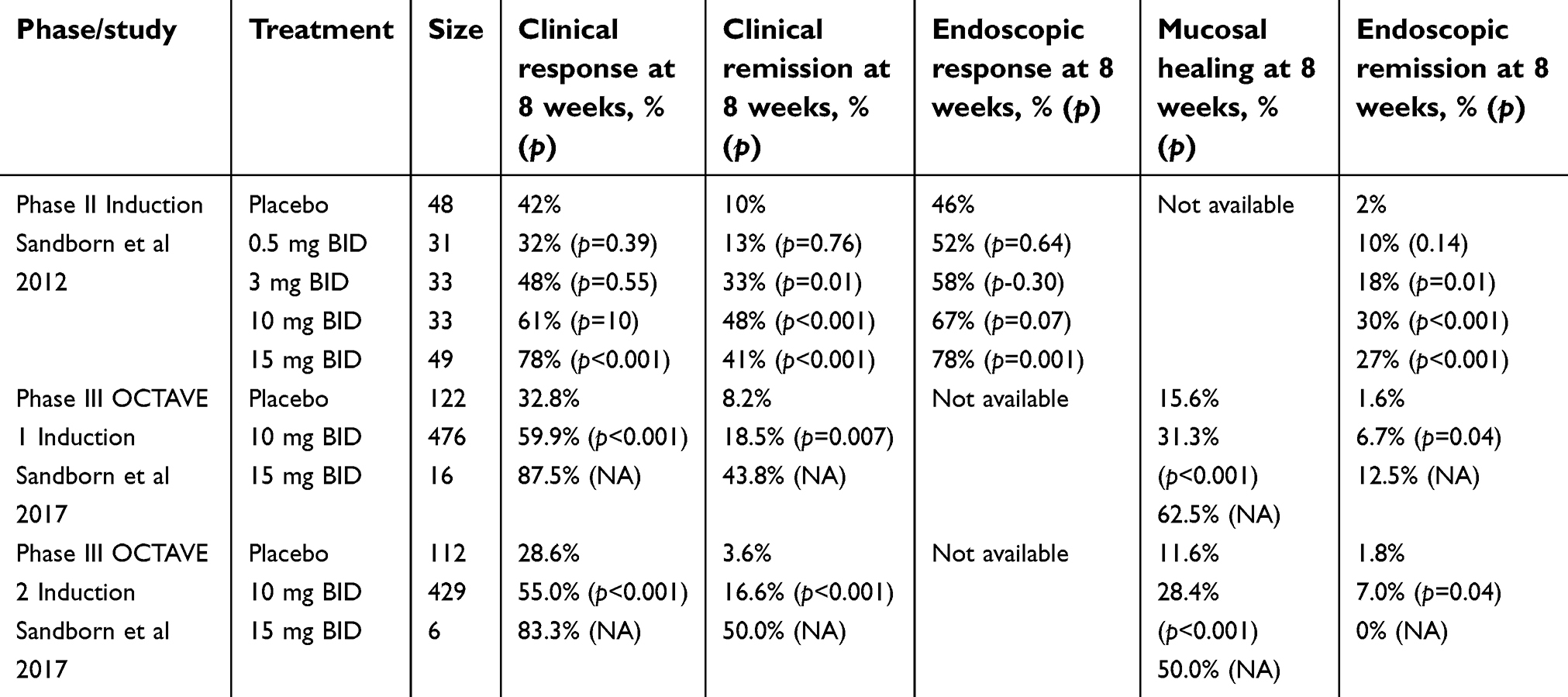 | Table 1 Tofacitinib as induction therapy in ulcerative colitis |
OCTAVE 1 and OCTAVE 2 were identical Phase III clinical trials that evaluated the efficacy of tofacitinib as induction therapy in 1139 adult patients with moderate-to-severe UC.15 Patients were randomly assigned to receive oral tofacitinib at a dose of 10 mg, 15 mg or placebo twice daily. However, Pfizer decided to discontinue further exploration of the dose of 15 mg twice daily due to feedback from RA regulatory authorities. Randomization was halted after 42 patients were assigned to this group. The primary endpoint was clinical remission at 8 weeks defined as a total Mayo score of ≤2 with no sub-score >1 and a rectal bleeding sub-score of 0. Clinical remission occurred in 18.5% of patients in the tofacitinib group versus 8.2% in the placebo group (p=0.007) in OCTAVE 1 trial and in 16.6% versus 3.6% (p<0.001) in OCTAVE 2. The rates of clinical response at 8 weeks were also higher with improved health-related quality of life in those with tofacitinib than placebo. The onset of action was rapid, with improvement of the partial Mayo score in as early as 2 weeks. These trials differ from the Phase II trial by the additional inclusion criteria that patients have rectal bleeding with sub-score 1 to 3 and its subsequent resolution as part of clinical remission. Interestingly, in these induction trials, there was no difference in tofacitinib efficacy in anti-TNF naïve patients compared to those previously exposed to anti-TNF agents. While more clinical remission was reported in anti-TNF naïve patients compared to those exposed to anti-TNF agents, these numbers were not statistically significant (25.2% and 22.1% versus 12.6% and 12%) making tofacitinib potentially an attractive treatment option in anti-TNF refractory patients.
Endoscopic response
Along with clinical response, detection of mucosal healing with endoscopic evaluation is also an important treatment target. In the Phase II trial, endoscopic response was defined as a decrease from baseline in the endoscopy sub-score by ≥1 and endoscopic remission defined as endoscopy sub-score of 0.14 An endoscopic response at 8 weeks was identified in 46% of patients receiving placebo compared to 52% receiving 0.5 mg (p=0.64), 58% receiving 3 mg (p=0.30), 67% receiving 10 mg (p=0.07) and 78% receiving 15 mg (p=0.001). Endoscopic remission occurred in 2% receiving placebo compared to 10% receiving 0.5 mg (p=0.01), 18% receiving 3 mg (p=0.01), 30% receiving 10 mg (p<0.001) and 27% receiving 15 mg (p<0.001). In Phase III trials, mucosal healing was defined as endoscopic sub-score of ≤1 at 8 weeks.15 In comparison to Phase II trials, these endoscopic Mayo scores were evaluated by central reading. Mucosal healing occurred in 31.3% of the patients of the tofacitinib group versus 15.6% in the placebo group (p<0.001) in OCTAVE 1 trial and in 28.4% versus 11.6% (p<0.001) in OCTAVE 2.
Use as long-term therapy
OCTAVE Sustain evaluated long-term efficacy of tofacitinib treatment in patients with moderate-to-severe UC (Table 2).15 In this trial, 593 patients who had clinical response to induction therapy were randomly assigned to receive tofacitinib maintenance therapy with 5 mg (n=198), 10 mg (n=197) or placebo (n=198) twice daily for 52 weeks. Primary end point was clinical remission at 52 weeks. The majority of patients in the OCTAVE Sustain trial received tofacitinib during the induction trial (88%) and 30% were in remission at maintenance-trial entry. Clinical remission occurred in 34.3% of the 5 mg and 40.6% in the 10 mg compared to the 11.1% in the placebo group (p<0.001 for both comparisons with placebo). Mucosal healing defined as endoscopic Mayo score <2 at 52 weeks occurred significantly more in patients in the 5 mg group (37.4%) and 10 mg group (45.7%) than in the placebo group (13.1%, p<0.001 for both comparisons). Overall, tofacitinib had long-term efficacy, increasing clinical and endoscopic remission among patients that had both initially responded to therapy.
 | Table 2 Tofacitinib as maintenance therapy in ulcerative colitis |
OCTAVE Sustain also evaluated for those who have achieved remission during induction whether tofacitinib could be used to maintain remission (Table 2).15 A secondary end point was remission that was sustained defined as occurring at both weeks 24 and 52 and without the administration of glucocorticoids for ≥4 weeks before assessment (“glucocorticoid-free”). For those already in remission at maintenance-trial entry, sustained and glucocorticoid-free remission occurred in 35.4% in the 5 mg and 47.3% in the 10 mg compared to the 5.1% in the placebo group (p<0.001 for both comparisons). Sustained mucosal healing among these patients occurred in 33.3% in the 5 mg and 49.4% in the 10 mg compared to the 8.9% in the placebo group (p<0.001 in both comparisons). Thus, for those with remission, long-term use of tofacitinib is shown to maintain clinical and endoscopic remission.
Tofacitinib and CD
Given tofacitinib’s effects on inflammatory signaling, testing in Crohn’s disease was also evaluated via three multicenter, randomized, double-blind, placebo-controlled Phase II clinical trials.51,52 These trials looked at moderate-to-severe Crohn’s disease (defined as CDAI ≥220 to ≤450 with intestinal ulceration documented by colonoscopy) and patients were randomized to receive placebo or doses between 1 mg to 15 mg twice daily. These studies did not find significant difference in clinical remission in patients treated with tofacitinib compared to placebo (Table 3). The proportion of patients achieving clinical response was only significant for tofacitinib 5 mg twice daily compared to placebo in one trial.51 In the earlier studies, the proportion of patients in the placebo group achieving clinical remission or clinical response was substantially higher than that reported in other trials assessing CD with other agents. This high placebo response may have prevented a thorough evaluation of dose–response relationship of tofacitinib. High placebo rates in these trials could be due to inadvertent selection bias towards enrollment of patients with more benign history. Although the subsequent clinical trial required demonstration of visible ulceration,51 there was no requirement for centrally read endoscopy and no protocol-defined minimal requirement for the extent or severity of ulceration.
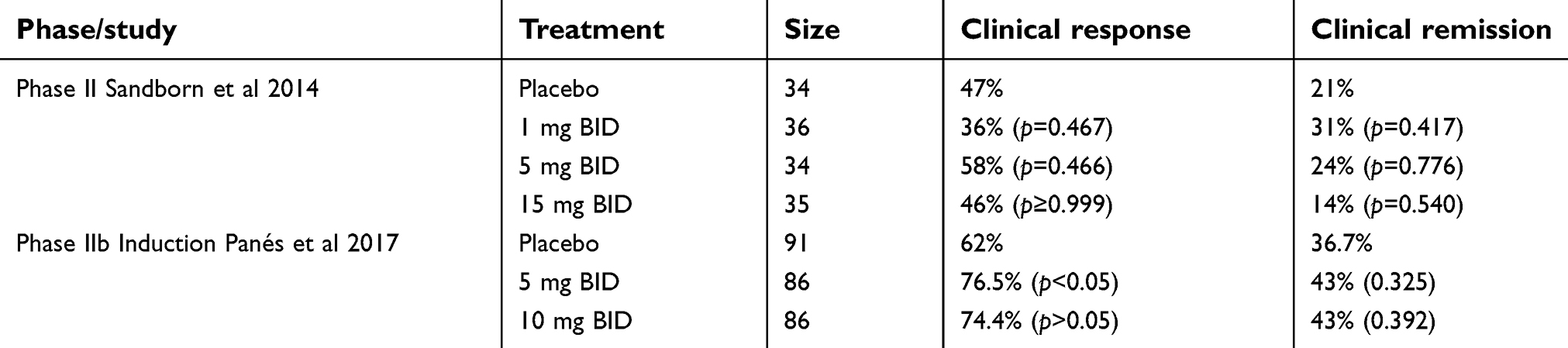 | Table 3 Tofacitinib as induction therapy in Crohn’s disease |
In secondary endpoints in these trials, there was a statistically significant change of C-reactive protein (CRP) and fecal calprotectin from baseline in those treated with 10 mg and 15 mg twice daily.51,52 The reduction in these biomarkers suggests some efficacy of tofacitinib in treating inflammation in Crohn’s disease. Further studies should explore the role of JAK-STAT inhibition in Crohn’s disease management. Filgotinib, a JAK1 selective inhibitor, has been shown in the FITZROY trial to induce clinical remission in patients with active Crohn’s disease with a reasonable safety profile.53 Another oral JAK1 inhibitor, upadacitinib (CELEST trial), also remains a promising therapeutic option for both induction and maintenance of remission for active Crohn’s disease.54
Safety profile
Much of the evidence for safety of tofacitinib comes from long-term studies conducted in RA. The recent use of tofacitinib in IBD clinical trials has precluded extensive long-term follow-up in this context. However, the RA trials and follow-up clinical data provide insight into the rates of potential events like malignancy, infections and cardiovascular disease. Given this information, the safety profile of tofacitinib in IBD and RA will be discussed.
IBD experience
Overall, tofacitinib was well tolerated in IBD studies and had a consistent safety profile in the seven clinical trials conducted in patients with IBD (Table 4). Between all seven IBD trials, the incidence of adverse events in those treated with tofacitinib between 0.5 mg to 15 mg were between 33% and 83%. Across these trials, nasopharyngitis (2–18.3%) and headaches (3.1–9.3%) were commonly reported adverse events. In Crohn’s disease trials, abdominal pain and nausea were also between 1–11.1% and 2.9–11.8%, respectively, but these results may be related to an insufficient clinical response to tofacitinib. Rare cases of intestinal perforation were included in serious adverse events, occurring in one CD patient receiving tofacitinib as maintenance therapy and in two UC patients in the induction trial (placebo group, 10 mg tofacitinib group).51 Discontinuation rates due to adverse events range 0–11.7%. In the UC trials, these numbers were similar between the placebo and treatment group but in OCTAVE Sustain the proportion was larger in the placebo group. There is an unmet need for long-term studies to further understand the long-term safety profile of tofacitinib.
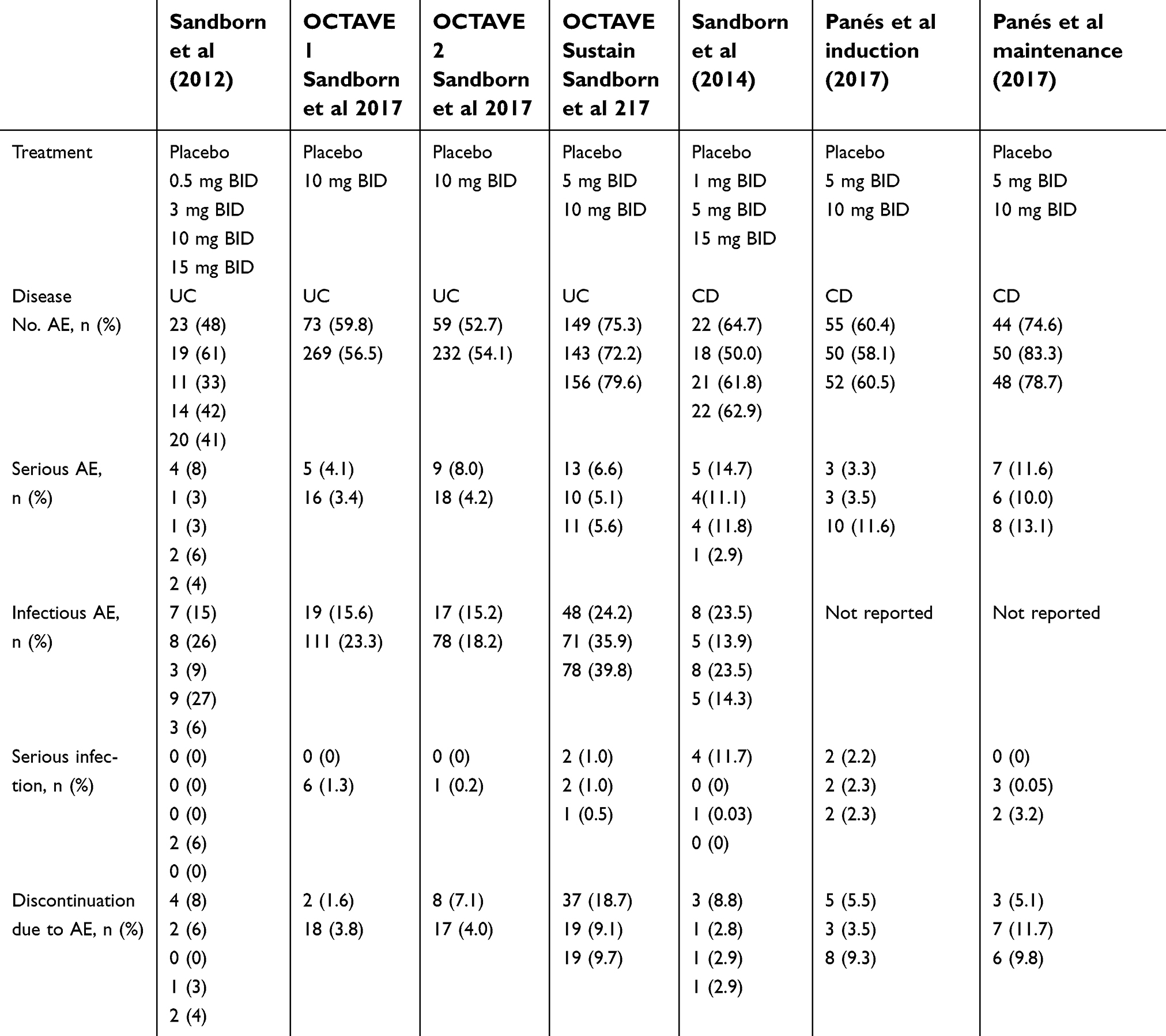 | Table 4 Tofacitinib safety profile in inflammatory bowel disease |
Infectious adverse events
The overall rate of infectious adverse events ranged 6–39.8% between all trials. Of these infections, nasopharyngitis and urinary tract infections were the most common infections. In OCTAVE 1 and 2, the percentage of patients with infections of all severity was higher in the 10 mg treatment group (23.3% and 18.2%) than in the placebo group (15.6% and 15.2%). In OCTAVE Sustain, the percentage of infections is also higher in both treatment groups of 5 mg and 10 mg (35.9% and 39.8%) than the placebo group (24.2%). The majority of these infections were mild or moderate in severity. Serious infections were minor in all trials, ranging 0–11.7%. Serious infections include cases of post-operative abscess, anal abscess, pneumonia, sepsis, Clostridium difficile colitis and cellulitis.
In OCTAVE Sustain, herpetic zoster infections also occurred in a total of 14 patients, 3 (1.5%) in the 5 mg group, 10 (5.1%) in the 10 mg group and 1 (0.5%) in the placebo group. In the Crohn’s maintenance study, there were two cases of non-serious herpes zoster in the 10 mg treatment group reported. No cases of herpes zoster were serious adverse events or resulted in discontinuation. This is important as the risk of herpes zoster is higher in those with immune-mediated disease like IBD, and this risk is increased with use of immunosuppressive therapy including the use of tofactanib.55,56
Overall, the incidence of opportunistic infections was also very low. In IBD cases, there were no cases of tuberculosis reported in these trials. For cytomegalovirus infections, two cases were reported, one in a patient with UC treated with 10 mg tofacitinib as induction and one in a patient with CD treated with placebo. Longer term trial data are available from the RA literature which showed incidence rates per 100 patient-years for tuberculosis of 0.2 and non-tuberculosis opportunistic infections of 0.3 and 0.2.57
Malignancy
Throughout all seven clinical trials, non-melanoma skin cancer (NMSC) was the most frequently diagnosed malignancy, occurring in six UC patients in the OCTAVE trials. All of these patients had prior exposure to thiopurines and most had a history of prior diagnosis of NMSC. Two cases of breast cancer were diagnosed, one UC patient receiving placebo and the second a CD patient receiving induction tofacitinib 10 mg. In longer-term RA trials, the incidence rate per 100 patient-years for NMSC and other malignancies excluding NMSC was 0.6 and 0.9, respectively.57
Hyperlipidemia and cardiovascular events
A dose-dependent increase in high-density lipoprotein (HDL), low-density lipoprotein (LDL) and total cholesterol (TC) has been observed throughout all studies. In IBD trials, these lipid levels increased with tofacitinib therapy and plateaued at 4 weeks. After drug discontinuation, these levels trended back towards normal. These changes in the lipid profile could potentially increase the risk of cardiovascular events, which has been a concern in the study of tofacitinib in other autoimmune diseases. In long-term plaque psoriasis, major adverse cardiovascular events (MACE) had an incidence rate of 0.26 per 100 person-years.58 In RA, pooled data from Phase III trials and long-term extension trials report incidence rates for MACE as 0.37 and 0.58 per 100 person-years.59 These rates were found to be comparable to placebo and did not increase in time.59 In IBD trials, only the UC OCTAVE trials reported cardiovascular events. These events include palpitations, tachycardia, bradycardia, acute coronary syndrome, myocardial infarction and congestive heart failure.
Despite these alterations to the lipid profile, tofacitinib has been shown in trials to decrease atherosclerosis.60 Its protective effect against atherosclerosis is not fully understood. Some studies suggest that upregulation of ATP binding cassette sub-family A member 1 (ABCA1), a key gene in cholesterol efflux, along with decreased formation of foam cells and inflammation may be responsible.61 Tofacitinib reduced the lipid content within synovial macrophages, increasing plasma concentration but without changes of CV-related lipid parameters that are suggestive of increased CV risk.62,63 There were no significant changes in lipid ratios more related to cardiovascular risk (LDL/HDL and TC/HDL ratios). Although LDL may be increased, no changes were observed in small dense LDL particles, which are thought to be more atherogenic than large particles and thus more accurate at predicting CV events.64
A recent statement issued by Pfizer in early 2019 announced modifications to the FDA post-marketing study, A3921133, in patients with RA transitioning patients from 10 mg twice a day to 5 mg twice a day due to concerns for safety signals from the Rheumatology Data Safety Monitoring Board of the 10 mg twice a day treatment arm. In this post-marketing study, evaluating safety of tofacitinib at two doses versus a tumor necrosis factor inhibitor(TNFi) control group in patients older than 50 years of age with at least one CV risk factor all on stable baseline doses of methotrexate, statistically important differences in occurrences of pulmonary embolism were noted in patients on tofacitinib at the 10mg twice a day dose compared to the 5mg twice daily dose and TNFi treatment arm. Further data will be required to elucidate this safety signal in IBD patients.
Clinical laboratory changes
RA trials have reported increases in creatinine with subsequent decreases in measured glomerular filtration rates (mGFR) and decreases in neutrophil and lymphocyte counts.65,66 Despite these findings, across all seven IBD trials there were no significant changes in liver, renal and hematologic tests. In OCTAVE trials, two patients who had received tofacitinib in induction had absolute lymphocyte counts less than 500 cubic millimeter, but had low absolute lymphocyte counts at baseline (640 and 650 per cubic millimeter). Three other UC patients had absolute neutrophil counts (ANC) less than 1500 cubic millimeter, and none had less than 1000 cubic millimeter. In OCTAVE trials, creatinine kinase levels were higher in treatment groups than placebo groups but had no cases of myopathy or rhabdomyolysis.
Positioning tofacitinib in the treatment paradigm
Tofacitinib offers an exciting new opportunity for IBD, and in particular UC patients. By targeting a new pathway implicated in IBD pathogenesis, the hope is to expand the armamentarium of therapeutic agents and extend our ability to maintain remission in our patients. However, one of the challenges as we expand the therapeutic options is to organize the treatment algorithms and understand which agents would be most effective for our individual patients. The most befuddling aspect of IBD care for providers is the seemingly random pattern to which some patients respond to therapies while others fail. Future research into personalized medicine and precision health will be critical in helping physicians tailor the appropriate therapies to the correct patients. Until that time, care providers will have to take into account a variety of factors when deciding which therapies to start and how to transition between medications when a patient either fails to respond or loses response over time.
As a rapid-acting small molecule with low immunogenicity and as an effective therapy in refractory UC patients, tofacitinib is a promising therapeutic option. The clinical trial data has shown that tofacitinib is effective in achieving clinical remission, mucosal healing and improving patient quality parameters. Additionally, post-hoc analysis of the OCTAVE 1 and 2 trials highlighted the rapid onset of action with response as early as Day 3 in some of the patients.67 Early response times are attractive not only for patient satisfaction but can also enable patients to more quickly taper off steroids. Additionally, tofacitinib is a small molecule with a relative lack of immunogenicity as opposed to a large protein-based structure typical of the biologic therapies. Given this relative lack of immunogenicity, patients with a prior response to tofacitinib respond to retreatment following a period of treatment interruption without significant adverse events.68 Furthermore, subgroup analyses demonstrate a consistent treatment effect of tofacitinib regardless of prior anti-TNF failure, baseline corticosteroid use and baseline CRP levels.67 Considering these properties, tofacitinib may be an appropriate early therapeutic option for UC patients and may be a preferable second-line agent for previous anti-TNF non-responders.69
Another consideration a physician faces is the insurance coverage for each of the various IBD medications. The annual total costs of therapeutic management of UC per patient are estimated to range from $14,686 to $19,949 in the United States.70 Much of these high treatment costs are attributed to biologic therapies. Biologics such as the anti-TNF agents and anti-integrin agents have complex production processes that lend themselves to high production costs. Tofacitinib production is relatively less elaborate and could eventually allow for lower drug cost and higher cost-effectiveness ratio.71,72 Current research is evaluating the cost-effectiveness of tofacitinib therapy. Indeed, recent studies are beginning to explore and report tofacitinib’s advantage in these cost-effectiveness analyses.72
The positioning of tofacitinib into our treatment algorithms will involve comparative analyses with current therapies. Comparative evidence of tofacitinib and other biologic therapies for UC can only come from indirect treatment comparisons. Currently, there are limited data on direct head-to-head trials making comparisons between different agents difficult.73 Meta-analyses have demonstrated a comparable efficacy with other treatments including approved biologics adalimumab, golimumab, infliximab and vedolizumab in patients with no previous exposure to anti-TNF agents.74 Based on network meta-analyses comparing tofacitinib with other biologics, tofacitinib appears to be effective among patients previously exposed to anti-TNF agents, making it an attractive option for this subpopulation of patients.69 Ultimately, the use of tofacitinib in future therapeutic algorithms may depend on its overall safety profile. The pipeline of JAK-inhibitors includes more selective JAK-inhibitors, such as filgotinib and upadacitinib. Future studies are required to determine if these more selective agents will be more effective and have safer therapeutic profiles. As more data are gathered, we will have a clearer view as to the positioning of tofacitinib and other more selective JAK inhibitors in our treatment algorithms. For now, use of tofacitinib will require case-by-case evaluation, taking into account a variety of factors individualized to each patient.
Conclusion
Tofacitinib is a promising therapy for the induction and maintenance of remission in UC. It blocks the JAK-STAT pathway, interfering with the activity of many cytokines in the inflammatory cascade implicated in IBD. It also has many advantages being an orally administered small molecule with rapid absorption, short serum-half life, with no concern for immunogenicity and rapid onset of action. As evaluated in these randomized placebo-controlled trials, tofacitinib is an effective induction and maintenance agent capable of mucosal healing in UC.
Further research with head-to-head trials between tofacitinib and approved biologic therapies for UC could help determine where tofacitinib fits into the therapeutic algorithm of patients with UC. Continual research into more specific JAK inhibition can also expand the potential for more treatment options in the future. This pharmacologic class provides the option to treat UC with an oral medication, which can increase compliance, change management and improve the quality of life of these patients.
Disclosure
Dr Jenny S Sauk is a consultant for Corrona llc and received speaking fees from Abbvie, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annual Review of Immunology. 2010;28:573–621. doi:10.1146/annurev-immunol-030409-101225
2. Sartor RB. Mechanisms of disease: pathogenesis of Crohn‘s disease and ulcerative colitis. Nat Clin Practice Gastroenterol Hepatol. 2006;3(7):390–407. doi:10.1038/ncpgasthep0528
3. Anderson CA, Boucher G, Lees CW, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nature Genetics. 2011;43(3):246–252. doi:10.1038/ng.764
4.
5. Hanauer SB, Feagan BG, Lichtenstein GR, et al. Maintenance infliximab for Crohn‘s disease: the ACCENT I randomised trial. Lancet (London, England). 2002;359(9317):1541–1549. doi:10.1016/S0140-6736(02)08512-4
6. Rutgeerts P, Sandborn WJ, Feagan BG, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. New Engl J Med. 2005;353(23):2462–2476. doi:10.1056/NEJMoa050516
7. Sandborn WJ, Feagan BG, Marano C, et al. Subcutaneous golimumab induces clinical response and remission in patients with moderate-to-severe ulcerative colitis. Gastroenterology. 2014;146(1):
8. Feagan BG, Rutgeerts P, Sands BE, et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. New Engl J Med. 2013;369(8):699–710. doi:10.1056/NEJMoa1215734
9. Holdam AS, Bager P, Dahlerup JF. Biological therapy increases the health-related quality of life in patients with inflammatory bowel disease in a clinical setting. Scand J Gastroenterology. 2016;51(6):706–711. doi:10.3109/00365521.2015.1136352
10. Ben-Horin S, Chowers Y. Review article: loss of response to anti-TNF treatments in Crohn‘s disease. Aliment Pharmacol Ther. 2011;33(9):987–995. doi:10.1111/j.1365-2036.2011.04612.x
11. Roda G, Jharap B, Neeraj N, Colombel JF. Loss of response to anti-TNFs: definition, epidemiology, and management. Clin Transl Gastroenterol. 2016;7:e135. doi:10.1038/ctg.2015.63
12. Wentworth BJ, Buerlein RCD, Tuskey AG, Overby MA, Smolkin ME, Behm BW. Nonadherence to biologic therapies in inflammatory bowel disease. Inflammatory Bowel Dis. 2018. doi:10.1093/ibd/izy102
13. Vavricka SR, Bentele N, Scharl M, et al. Systematic assessment of factors influencing preferences of Crohn‘s disease patients in selecting an anti-tumor necrosis factor agent (CHOOSE TNF TRIAL). Inflammatory Bowel Dis. 2012;18(8):1523–1530. doi:10.1002/ibd.21888
14. Sandborn WJ, Ghosh S, Panes J, et al. Tofacitinib, an oral Janus kinase inhibitor, in active ulcerative colitis. N Engl J Med. 2012;367(7):616–624. doi:10.1056/NEJMoa1112168
15. Sandborn WJ, Su C, Sands BE, et al. Tofacitinib as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2017;376(18):1723–1736. doi:10.1056/NEJMoa1606910
16. O‘Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity. 2012;36(4):542–550. doi:10.1016/j.immuni.2012.03.014
17. Yamaoka K, Saharinen P, Pesu M, Holt VE, Silvennoinen O, O‘Shea JJ. The Janus kinases (Jaks). Genome Biology. 2004;5(12):253. doi:10.1186/gb-2004-5-12-253
18. Coskun M, Salem M, Pedersen J, Nielsen OH. Involvement of JAK/STAT signaling in the pathogenesis of inflammatory bowel disease. Pharmacol Res. 2013;76:1–8. doi:10.1016/j.phrs.2013.06.007
19. Cacalano NA, Migone TS, Bazan F, et al. Autosomal SCID caused by a point mutation in the N-terminus of Jak3: mapping of the Jak3-receptor interaction domain. The EMBO J. 1999;18(6):1549–1558. doi:10.1093/emboj/18.6.1549
20. Hambleton S, Goodbourn S, Young DF, et al. STAT2 deficiency and susceptibility to viral illness in humans. Pro Nat Acad Sci USA. 2013;110(8):3053–3058. doi:10.1073/pnas.1220098110
21. Leonard WJ, O‘Shea JJ. Jaks and STATs: biological implications. Ann Rev Immunol. 1998;16:293–322. doi:10.1146/annurev.immunol.16.1.293
22. Mogensen TH. STAT3 and the Hyper-IgE syndrome: clinical presentation, genetic origin, pathogenesis, novel findings and remaining uncertainties. Jak-stat. 2013;2(2):e23435. doi:10.4161/jkst.23435
23. Cho JH, Gregersen PK. Genomics and the multifactorial nature of human autoimmune disease. N Engl J Med. 2011;365(17):1612–1623. doi:10.1056/NEJMra1100030
24. Goethel A, Croitoru K, Philpott DJ. The interplay between microbes and the immune response in inflammatory bowel disease. J Physiol. 2018;596(17):3869–3882. doi:10.1113/JP275396
25. Fuss IJ, Neurath M, Boirivant M, et al. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn‘s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol (Baltimore, Md: 1950). 1996;157(3):1261–1270.
26. Brand S. Crohn‘s disease: th1, Th17 or both? The change of a paradigm: new immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn‘s disease. Gut. 2009;58(8):1152–1167. doi:10.1136/gut.2008.163667
27. Brand S, Beigel F, Olszak T, et al. IL-22 is increased in active Crohn‘s disease and promotes proinflammatory gene expression and intestinal epithelial cell migration. Am J Physiol Gastrointestinal Liver Physiol. 2006;290(4):G827–G838. doi:10.1152/ajpgi.00513.2005
28. Fujino S, Andoh A, Bamba S, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52(1):65–70.
29. Dambacher J, Beigel F, Zitzmann K, et al. The role of the novel Th17 cytokine IL-26 in intestinal inflammation. Gut. 2009;58(9):1207–1217. doi:10.1136/gut.2007.130112
30. O‘Shea JJ, Murray PJ. Cytokine signaling modules in inflammatory responses. Immunity. 2008;28(4):477–487. doi:10.1016/j.immuni.2008.03.002
31. Durant L, Watford WT, Ramos HL, et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity. 2010;32(5):605–615. doi:10.1016/j.immuni.2010.05.003
32. Hyams JS, Fitzgerald JE, Treem WR, Wyzga N, Kreutzer DL. Relationship of functional and antigenic interleukin 6 to disease activity in inflammatory bowel disease. Gastroenterology. 1993;104(5):1285–1292.
33. Gross V, Andus T, Caesar I, Roth M, Scholmerich J. Evidence for continuous stimulation of interleukin-6 production in Crohn‘s disease. Gastroenterology. 1992;102(2):514–519.
34. Parisinos CA, Serghiou S, Katsoulis M, et al. Variation in interleukin 6 receptor gene associates with risk of Crohn‘s disease and ulcerative colitis. Gastroenterology. 2018;155(2):303–306.e302. doi:10.1053/j.gastro.2018.05.022
35. Ishizaki M, Akimoto T, Muromoto R, et al. Involvement of tyrosine kinase-2 in both the IL-12/Th1 and IL-23/Th17 axes in vivo. J Immunol (Baltimore, Md: 1950). 2011;187(1):181–189. doi:10.4049/jimmunol.1003244
36. Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75(2):263–274.
37. Lees CW, Barrett JC, Parkes M, Satsangi J. New IBD genetics: common pathways with other diseases. Gut. 2011;60(12):1739–1753. doi:10.1136/gut.2009.199679
38. Bollrath J, Phesse TJ, von Burstin VA, et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell. 2009;15(2):91–102. doi:10.1016/j.ccr.2009.01.002
39. Alonzi T, Newton IP, Bryce PJ, et al. Induced somatic inactivation of STAT3 in mice triggers the development of a fulminant form of enterocolitis. Cytokine. 2004;26(2):45–56. doi:10.1016/j.cyto.2003.12.002
40. Furumoto Y, Gadina M. The arrival of JAK inhibitors: advancing the treatment of immune and hematologic disorders. BioDrugs. 2013;27(5):431–438. doi:10.1007/s40259-013-0040-7
41. Hodge JA, Kawabata TT, Krishnaswami S, et al. The mechanism of action of tofacitinib – an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis. Clin Exp Rheumatol. 2016;34(2):318–328.
42. Meyer DM, Jesson MI, Li X, et al. Anti-inflammatory activity and neutrophil reductions mediated by the JAK1/JAK3 inhibitor, CP-690,550, in rat adjuvant-induced arthritis. J Inflammation (London, England). 2010;7:41. doi:10.1186/1476-9255-7-59
43. Ghoreschi K, Jesson MI, Li X, et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J Immunol (Baltimore, Md: 1950). 2011;186(7):4234–4243. doi:10.4049/jimmunol.1003668
44. Dowty ME, Lin J, Ryder TF, et al. The pharmacokinetics, metabolism, and clearance mechanisms of tofacitinib, a janus kinase inhibitor, in humans. Drug Metab . 2014;42(4):759–773. doi:10.1124/dmd.113.054940
45. Lamba M, Wang R, Fletcher T, Alvey C, Kushner J, Stock TC. Extended-release once-daily formulation of tofacitinib: evaluation of pharmacokinetics compared with immediate-release tofacitinib and impact of food. J Clin Pharmacol. 2016;56(11):1362–1371. doi:10.1002/jcph.734
46. Singh JA, Saag KG, Bridges SL
47. Mease P, Hall S, FitzGerald O, et al. Tofacitinib or adalimumab versus placebo for psoriatic arthritis. N Engl J Med. 2017;377(16):1537–1550. doi:10.1056/NEJMoa1615975
48. Gladman D, Rigby W, Azevedo VF, et al. Tofacitinib for psoriatic arthritis in patients with an inadequate response to TNF inhibitors. N Engl J Med. 2017;377(16):1525–1536. doi:10.1056/NEJMoa1615977
49. van der Heijde D, Deodhar A, Wei JC, et al. Tofacitinib in patients with ankylosing spondylitis: a Phase II, 16-week, randomised, placebo-controlled, dose-ranging study. Ann Rheumatic Dis. 2017;76(8):1340–1347. doi:10.1136/annrheumdis-2016-210322
50. Shivanna CB, Shenoy C, Priya RA. Tofacitinib (Selective Janus Kinase Inhibitor 1 and 3): a promising therapy for the treatment of alopecia areata: a case report of six patients. Int J Trichol. 2018;10(3):103–107. doi:10.4103/ijt.ijt_21_18
51. Panes J, Sandborn WJ, Schreiber S, et al. Tofacitinib for induction and maintenance therapy of Crohn‘s disease: results of two Phase IIb randomised placebo-controlled trials. Gut. 2017;66(6):1049–1059. doi:10.1136/gutjnl-2016-312735
52. Sandborn WJ, Ghosh S, Panes J, Vranic I, Wang W, Niezychowski W. A Phase 2 study of tofacitinib, an oral Janus kinase inhibitor, in patients with Crohn‘s disease. Clin Gastroenterol Hepatol. 2014;12(9):1485–1493.e1482. doi:10.1016/j.cgh.2014.01.029
53. Vermeire S, Schreiber S, Petryka R, et al. Clinical remission in patients with moderate-to-severe Crohn‘s disease treated with filgotinib (the FITZROY study): results from a Phase 2, double-blind, randomised, placebo-controlled trial. Lancet (London, England). 2017;389(10066):266–275. doi:10.1016/S0140-6736(16)32537-5
54. Sandborn WJ, Feagan BG, Panes J, et al. Safety and efficacy of ABT-494 (Upadacitinib), an oral Jak1 inhibitor, as induction therapy in patients with Crohn‘s disease: results from celest. Gastroenterol. 2017;152(5):S1308–S1309. doi:10.1016/S0016-5085(17)34357-3
55. Marra F, Lo E, Kalashnikov V, Richardson K. Risk of herpes zoster in individuals on biologics, disease-modifying antirheumatic drugs, and/or corticosteroids for autoimmune diseases: a systematic review and meta-analysis. Open Forum Infectious Dis. 2016;3(4):ofw205. doi:10.1093/ofid/ofw205
56. Colombel J-F. Herpes zoster in patients receiving JAK inhibitors for ulcerative colitis: mechanism, epidemiology, management, and prevention. Inflammatory Bowel Dis. 2018;24(10):2173–2182. doi:10.1093/ibd/izy150
57. Cohen SB, Tanaka Y, Mariette X, et al. Long-term safety of tofacitinib for the treatment of rheumatoid arthritis up to 8.5 years: integrated analysis of data from the global clinical trials. Ann Rheumatic Dis. 2017;76(7):1253–1262. doi:10.1136/annrheumdis-2016-210457
58. Valenzuela F, Korman NJ, Bissonnette R, et al. Tofacitinib in patients with moderate-to-severe chronic plaque psoriasis: long-term safety and efficacy in an open-label extension study. Brit J Dermatol. 2018. doi:10.1111/bjd.16798
59. Charles-Schoeman C, Wicker P, Gonzalez-Gay MA, et al. Cardiovascular safety findings in patients with rheumatoid arthritis treated with tofacitinib, an oral Janus kinase inhibitor. Sem Arthritis Rheumatism. 2016;46(3):261–271. doi:10.1016/j.semarthrit.2016.05.014
60. Kume K, Amano K, Yamada S, et al. Tofacitinib improves atherosclerosis despite up-regulating serum cholesterol in patients with active rheumatoid arthritis: a cohort study. Rheumatol Int. 2017;37(12):2079–2085. doi:10.1007/s00296-017-3844-9
61. Wang Z, Wang S, Yun T, Wang C, Wang H. Tofacitinib ameliorates atherosclerosis and reduces foam cell formation in apoE deficient mice. Biochem Biophys Res Comm. 2017;490(2):194–201. doi:10.1016/j.bbrc.2017.06.020
62. Wolk R, Armstrong EJ, Hansen PR, et al. Effect of tofacitinib on lipid levels and lipid-related parameters in patients with moderate to severe psoriasis. J Clin Lipidol. 2017;11(5):1243–1256. doi:10.1016/j.jacl.2017.06.012
63. Perez-Baos S, Barrasa JI, Gratal P, et al. Tofacitinib restores the inhibition of reverse cholesterol transport induced by inflammation: understanding the lipid paradox associated with rheumatoid arthritis. Bri J Pharmacol. 2017;174(18):3018–3031. doi:10.1111/bph.13932
64. Rajman I, Eacho PI, Chowienczyk PJ, Ritter JM. LDL particle size: an important drug target? Bri J Pharmacol. 1999;48(2):125–133.
65. Kremer JM, Kivitz AJ, Simon-Campos JA, et al. Evaluation of the effect of tofacitinib on measured glomerular filtration rate in patients with active rheumatoid arthritis: results from a randomised controlled trial. Arthritis Res Ther. 2015;17:95. doi:10.1186/s13075-015-0612-7
66. Schulze-Koops H, Strand V, Nduaka C, et al. Analysis of haematological changes in tofacitinib-treated patients with rheumatoid arthritis across Phase 3 and long-term extension studies. Rheumatol (Oxford, England). 2017;56(1):46–57. doi:10.1093/rheumatology/kew329
67. Hanauer S, Panaccione R, Danese S, et al. Tofacitinib induction therapy reduces symptoms within 3 days for patients with ulcerative colitis. Clin Gastroenterol Hepatol. 2018;17(1):139–147.
68. Panes J, Bressler B, Colombel JF, et al. 905 – efficacy and safety of tofacitinib retreatment for ulcerative colitis after treatment interruption: results from the octave clinical trials. Gastroenterol. 2018;154(6):S–178. doi:10.1016/S0016-5085(18)31007-2
69. Singh S, Fumery M, Sandborn WJ, Murad MH. Systematic review with network meta-analysis: first- and second-line pharmacotherapy for moderate-severe ulcerative colitis. Aliment Pharmacol Ther. 2018;47(2):162–175. doi:10.1111/apt.14422
70. Cohen RD, Yu AP, Wu EQ, Xie J, Mulani PM, Chao J. Systematic review: the costs of ulcerative colitis in Western countries. Aliment Pharmacol Ther. 2010;31(7):693–707. doi:10.1111/j.1365-2036.2010.04234.x
71. Olivera P, Danese S, Peyrin-Biroulet L. Next generation of small molecules in inflammatory bowel disease. Gut. 2017;66(2):199–209. doi:10.1136/gutjnl-2016-312912
72. Wu B, Wang Z, Zhang Q. Cost-effectiveness of different strategies for the treatment of moderate-to-severe ulcerative colitis. Inflammatory Bowel Diseases. 2018;24(11):2291–2302.
73. Danese S, D‘Amico F, Bonovas S, Peyrin-Biroulet L. Positioning tofacitinib in the treatment algorithm of moderate to severe ulcerative colitis. Inflammatory Bowel Dis. 2018;24(10):2106–2112. doi:10.1093/ibd/izy076
74. Bonovas S, Lytras T, Nikolopoulos G, Peyrin-Biroulet L, Danese S. Systematic review with network meta-analysis: comparative assessment of tofacitinib and biological therapies for moderate-to-severe ulcerative colitis. Aliment Pharmacol Ther. 2018;47(4):454–465. doi:10.1111/apt.14449
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.