")
Back to Journals » Journal of Pain Research » Volume 13
Evaluating the Role of CXCR3 in Pain Modulation: A Literature Review
Authors Aloyouny AY , Bepari A, Rahman I
Received 20 March 2020
Accepted for publication 17 July 2020
Published 6 August 2020 Volume 2020:13 Pages 1987—2001
DOI https://doi.org/10.2147/JPR.S254276
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Robert B. Raffa
Ashwag Yagoub Aloyouny,1 Asmatanzeem Bepari,2 Ishrat Rahman1
1College of Dentistry, Princess Nourah bint Abdulrahman University, Riyadh, Kingdom of Saudi Arabia; 2College of Medicine, Princess Nourah bint Abdulrahman University, Riyadh, Kingdom of Saudi Arabia
Correspondence: Ishrat Rahman
Department of Basic Dental Sciences, College of Dentistry, Princess Nourah bint Abdulrahman University, 5.4, Building Number 22, Airport Road, Al-Narjes, Riyadh 11671, Kingdom of Saudi Arabia
Tel +966 540451780
Email [email protected]
Abstract: CXCR3 is a well-known receptor involved in immune cell recruitment and inflammation. Pathological inflammation leads to pain stimulation and hence nociception. Therefore, we decided to review the recent research on CXCR3 to identify its precise role in the modulation of pain in a variety of clinical conditions targeting various regions of the body. Studies were selected from PubMed Medline, which relate CXCR3 to the progression of diseases with either bone cancer pain, neuropathic pain, cystitis pain, osteoarthritis and rheumatoid arthritis pain, dental pain, in particular, periodontitis and pulpitis. In all the diseases studied, a high prevalence of CXCR3 and/or its ligand were identified where CXCR3 is a key player in the pathophysiological process of many inflammatory conditions. CXCR3 and its ligands, particularly CXCL10, modulate nociception via actions in the dorsal root ganglia and dorsal horn of the spinal cord, in cases of bone cancer pain, neuropathic, and joint pain. However, with the other studied disease, no direct link to pain has been made, although it contributes to the pathological progression of the diseases and hence would be a causal factor for the pain. Furthermore, CXCR3 appears to play a role in desensitizing the opioid receptor in the descending modulatory pathway within the brain stem as well as modulating opioid-induced hyperalgesia in the dorsal horn of the spinal cord. Further research is required for understanding the exact mechanisms of CXCR3 in pain modulation centrally and peripherally. A greater understanding of the immunological activities and pharmacological consequence of CXCR3 and its ligands could help in the discovery of newer drugs for modulating pain arising from pathogenic or inflammatory sources. Given the significance of the CXCR3 for nociception, its utilization may prove to be beneficial as a target for analgesia.
Keywords: chemokines, algesia, analgesia, opioid receptor, inflammation
Introduction
Pain is an unpleasant emotional and sensory experience linked to the actual damage of tissue. Nociceptive pain is an acute physiological response to injury as a result of inflammation. The primary afferent sensory neurons have cell bodies in the dorsal root ganglion projecting through the periphery, with various nociceptors such as transient receptor potential (TRP) channel family of receptors, e.g. TRPV1, TRPM8, TRPA1. Many of these are polymodal such as TRPV1 being able to respond to heat and chemical stimuli.1 The signal synapses at the dorsal horn of the spinal cord, resulting in the release of neurotransmitter mostly glutamate binding to the NMDA receptor or substance P binding to NK1 receptor to activate the second-order neuron, in the ascending spinothalamic tract, which projects in the thalamus. From the thalamus, the third-order neuron projects signal into various areas of the higher centre, cerebral cortex, involving the frontal cortex, the sensory cortex and the limbic system, for processing of pain perception relating to emotions, behaviour and action.2,3
Morphine is a high-affinity efficacious agonist at the μ-opioid receptor (MOR) and is well known for its potent analgesic effects. It modulates the pain pathway by binding to receptors present in the dorsal horn and also in the PAG and RVM areas of the brain stem.4 One of the drawbacks of continuous morphine use is central sensitisation and opioid-induced hyperalgesia,5 which probably contributes to the development of opioid tolerance.6 In inflammation a cascade of inflammatory mediators contributes to altering primary afferent sensory neuronal responses, these changes bring about peripheral sensitization.7
Chemokines and their receptors are described as controlling agents of cell migration. They orchestrate the immune system controlling pathophysiological processes such as inflammation.8 Chemokines are small 17–16kDa proteins known as chemotactic cytokines.9 The activation of the seven-transmembrane domain mediates the biological activities of chemokines G-protein coupled cell-surface receptors.10 During hematopoiesis, innate, and adaptive immune responses, chemokines dominate all the leukocyte relocation. They play a crucial role in several critical conditions involving; HIV-infection, cancer, hematopoiesis, embryogenesis, atherosclerosis, and angiogenesis.9 The association of chemokines and its receptors is complex because chemokines can bind to several different receptors and multiple chemokines can activate a single chemokine receptor.10
Chemokines are classified by structure into two major sub-families; CC and the CXC families.11 The CC-chemokines usually appeal to T-lymphocytes, natural killer cells, eosinophil, monocytes, and basophils,12,13 whereas the CXC-chemokines mainly attract neutrophils and leukocytes.14,15 Activation of CXCR3 chemokine receptor by its natural ligands CXCL9, CXCL10, and CXCL11 is known to cause migration and activation of immune cells such as T lymphocytes, especially Th1 cells, natural killer cells, B cells, and dendritic cells.16 CXCR3 is also able to regulate the development and differentiation of effector and memory populations of T cells.17–19 The ligands for CXCR3; CXCL9, CXCL10 and CXL11 are IFN- γ inducible and IFN- γ is well known to be a pro-inflammatory cytokine, having a central role in pathological inflammation and autoimmune diseases.
CXCR3 is known to exist as three different spliced variants, CXCR3-A, CXCR3-B, and CXCR3-alt.20,21 Where CXCR3 is generally termed to be the CXCR3-A isoform. The CXCR3-B isoform is also a receptor for CXCL4. Both A and B variants are known to have distinct cell signalling events are thought to mediate opposing functional effects.20 CXCR3-alt appears to have limited biological activities.21 CXCR3 is expressed on a wide variety of tissues and other non-immune cells that include the brain, heart, skeletal muscle, colon, kidney, liver, small intestines, placenta, smooth muscle cells, and neurons.20 Therefore, it is highly conceivable that CXCR3 in different tissues have different effects arising from alterations in signal transduction pathways.22 Furthermore, chemokines appear to have promiscuous interactions between receptors, for instance, CCL21 not only binds to the CCR7 receptor but also has been shown to have functional effects through binding to the CXCR3 receptor. CCL21 caused activation of murine microglia, increasing Cl− currents and initiating chemotaxis through the CXCR3 receptor.23
The primary nociceptive afferents are responsible for initiating and mediating the sensation of pain in dorsal root ganglia and trigeminal ganglia. The expression of CXCR3 has been observed in the nervous tissues that include primary sensory neurons.24 The inflammatory and neuropathic pain conditions result in the up-regulation of CXCR3 signalling in sensory neurons, which is implicated in maintaining a chronic state of pain.24 The exact mechanisms underlying neuronal hyperexcitability remain largely unknown,25 but it is conceivable that nociceptive and pruritic impact may be directed by CXCR3 through the activation of immune cells, resulting in the release of inflammatory mediators targeting the sensory neurons.26
Blocking chemokine receptor activity by a drug is of therapeutic importance for inflammatory conditions,27 and it may also be valuable for producing analgesic effects. CT526284 and CT522274 are two small molecular weight compounds, which have previously shown to block CXCL11, CXCL10, and CXCL9 mediated G-protein activation in T-cell membranes.28 It would be interesting to identify if antagonists at CXCR3 could block neuronal responses. In the present study, we discuss the possible role of CXCR3 in the pain modulation of several diseases.
Methods
The study was aimed to discuss the possible role of CXCR3 in the pain modulation of several diseases. A brief literature review was conducted to identify studies that link CXCR3 to the pathogenesis of several diseases. For this purpose, Boolean operators were used, which help in connecting and defining the relationship between the search terms. The database PubMed Medline was used to search for original research articles in the English language. The time-period selected for this study was 2005–2020, to cover the last 15 years of research. Review articles were excluded. The information related to the article retrieval and its selection has been presented in Figure 1. The study was divided into following sub-themes; the role of CXCR3 in bone cancer pain, the role of CXCR3 in osteoarthritis and rheumatoid pain, the role of CXCR3 in neuropathic pain, the role of CXCR3 in cystitis/bladder pain syndrome, and role of CXCR3 in dental pain. The themes discuss the prevalence, and occurrence of the diseases as well as the essential role played by CXCR3 towards the progression of the disease and in modulating the pain associated with these diseases.
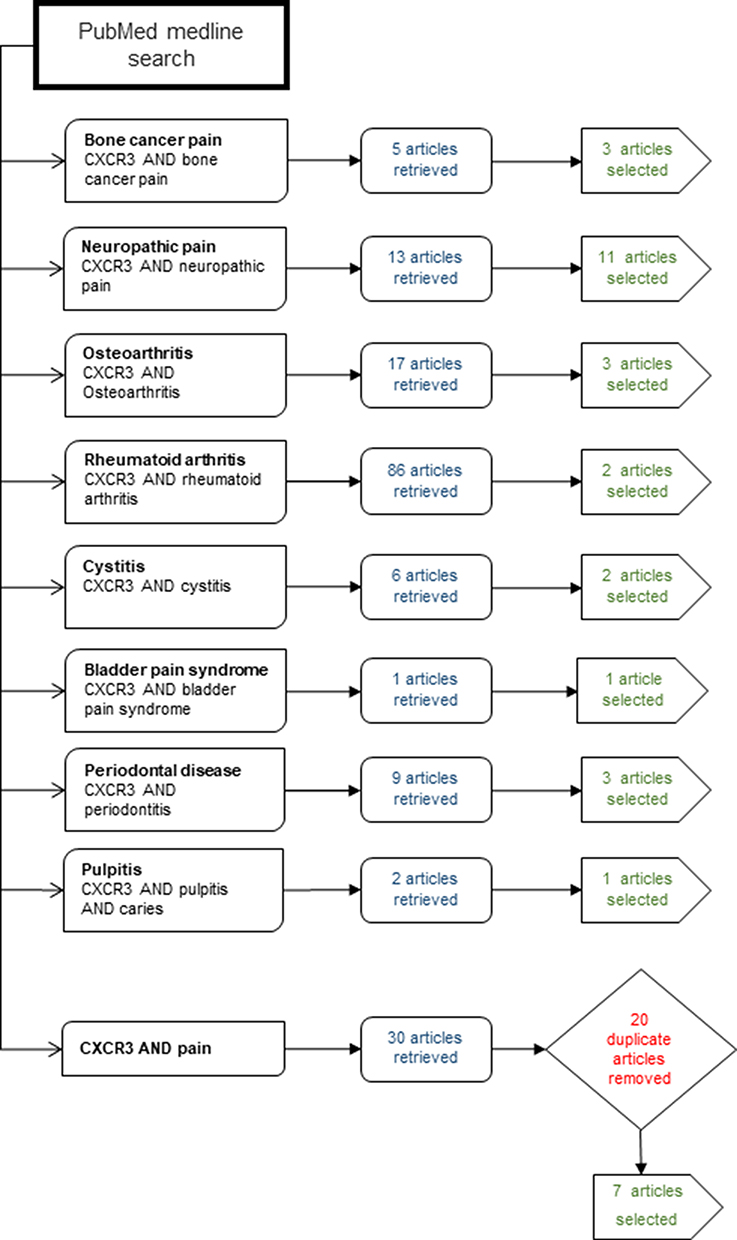 |
Figure 1 Flow chart identifying the themes of individual searches and the numbers of articles retrieved. |
Results and Discussion
Role of CXCR3 in Bone Cancer Pain
Cancer is defined as a group of diseases caused because of overgrowth and spread of abnormal cells in a part of the body. Lung, prostate, kidney, and breast cancer are the main culprits in which the cancer cells frequently metastasize and spread to the multiple bones of the body. As a result, approximately 90–95% of the patients suffer from life-altering or severe pain to metastatic cancer.29 Bone cancer pain is reported as the most common type of pain syndrome among patients who have cancer of lungs, prostate, or breast.13 However, despite its prevalence, very little research is available on cancer metastasized to bone.
Guan et al30 studied the role of CXCR3 in the induction and maintenance of bone cancer pain using a rat model of bone cancer pain. Walker-256 mammary gland carcinoma cells were inoculated into the rat’s tibia, to induce bone cancer pain. Mechanical paw withdrawal threshold (PWT) was used to assess the pain response/mechanical allodynia. Time-dependent and CXCR3 activation-dependent Akt and ERK1/2 phosphorylation occurred in the test rats. Immunohistochemistry and Western blot analysis revealed that CXCR3 was expressed in various types of neurons in the dorsal root ganglia as well in astrocytes and microglia in the spinal cord. In addition, CXCR3 was co-localised with pAkt or pERK1/2. CXCR3 inhibition using a CXCR3 antagonist, AMG487 could decrease mechanical allodynia and decrease pAkt and pERK1/2 levels. The additional use of a PI3K inhibitor, wortmannin, or MEK inhibitor, U0126, diminished the subsequent activation of Akt and ERK1/2 respectively also reducing mechanical allodynia in test mice but not in control. Together this indicated two branches of signalling both mediated via CXCR3 activation causing bone cancer pain; CXCR3-PI3K-pAKT and CXCR3-MEK-ERK1/2. Also, Wortmannin inhibited both pAkt and pERK1/2 protein levels, and conversely U0126 inhibited both pERK1/2 and pAKt. Thus, activation of the spinal chemokine receptor CXCR3 mediated bone cancer pain via crosstalk of downstream PI3K/Akt- Raf/MEK/ERK pathways. The results of Guan et al were also reviewed by Guo and Gao,31 further highlighting the importance of neuronal microglia communication for maintenance of bone cancer pain, and the need for it to be further explored.31 Hence, CXCR3 does appear to play an essential role in maintaining bone cancer pain. However, research unravelling the complexities of the physiological effects of CXCR3 in the spine is necessary in order for the development of target strategies against CXCR3 to be of therapeutic value.
An earlier study by Bu et al32 addressed whether CXCR3 had a role in bone cancer pain associated with metastatic breast cancer. Walker 256 rat mammary gland carcinoma cells were injected into the tibia of the rats. Mechanical allodynia was examined by von Frey filament stimulation by measuring the hind PWT. Results showed an up-regulation of CXCL10 and CXCR3 in rats injected with the carcinoma cells compared to control which also correlated with an increase in mechanical allodynia. Blockade of CXCR3 activation using anti-CXCL10 antibody, and AMG487, caused a reduction in mechanical allodynia and suppressed microglial activation, as assessed by immunohistochemical analysis of spinal sections detecting the activated microglial marker CD11b. In naïve rats, intrathecal administration of recombinant CXCL10 induced mechanical allodynia, and this was inhibited by AMG487. Results indicate that CXCL10-mediated activation of CXCR3 led to mechanical allodynia and the maintenance was dependent on microglial activation, since inhibiting microglial activation, using minocycline, attenuated the mechanical allodynia initially (day 7) but completely abolished it after 10–14 days. Furthermore, in naïve rats’ intrathecal injection of recombinant CXCL10 caused higher spinal CXCL10mRNA expression, which was partially attenuated by treatment with minocycline. Thus, the activation of spinal CXCL10/CXCR3 pathway was found to be crucial in mediating bone cancer pain in rats through activation of microglia. Hence, the CXCL10/CXCR3 pathway could be a novel clinical therapy for metastatic breast cancer-induced bone pain.32
Zhou et al33 evaluated chemokines and their receptors as potential therapeutic targets for bone cancer pain in a review highlighting that BCP prevalence is high due to the inefficiency of non-steroidal anti-inflammatory drug analgesics, such as ibuprofen, aspirin, naproxen, and fenoprofen.33 There is a need for different therapeutic methods and interventions to be developed for bone cancer pain and considering the role of the CXCR3 receptor in the development and maintenance of BCP it could be considered as a potential therapeutic target.
It is evident from both pieces of literature by Guan et al30 and Bu et al32 that spinal CXCR3 activation and/or its activation in the dorsal root ganglia is a critical event in the initiation and maintenance of bone cancer pain and is dependent on microglial activation. Additionally, it appears as though the mechanisms involved are cyclic in nature where the offending factor; bone cancer pain initiated in the tibia of the rats, leads to CXCR3 activation which activates downstream events as well as activating neighbouring microglial cells to further intensify the CXCR3 response and hence the pain. A schematic representation of the downstream signalling pathways and the mechanisms initiated by CXCR3 activation leading to bone cancer pain is illustrated in Figure 2.
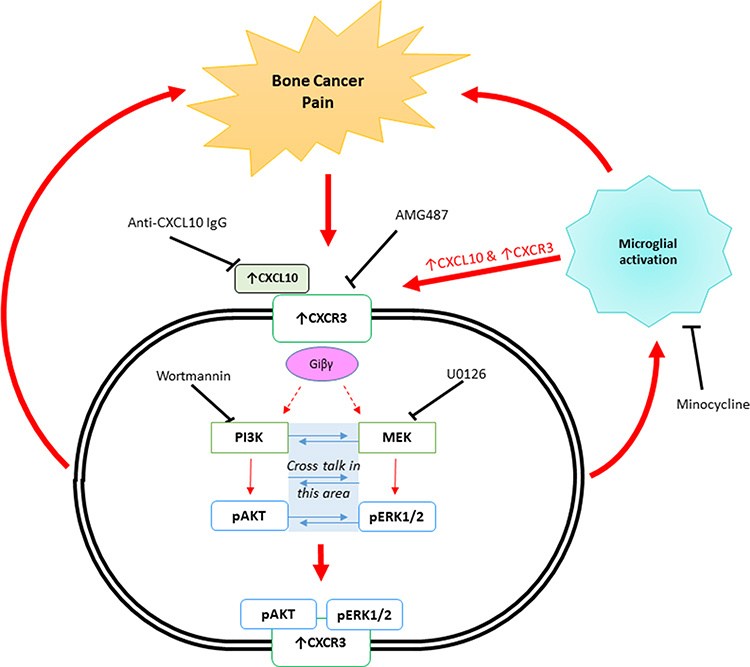 |
Figure 2 Schematic representation of the possible CXCR3-mediated signalling mechanisms involved in Bone Cancer Pain, occurring in the dorsal root ganglia and or the dorsal horn of the spinal cord. |
Role of CXCR3 in Neuropathic Pain
Any nerve injury or nerve disease promotes neuropathic pain. Nerve disease is a chronic condition that causes spontaneous pain, allodynia, hyperalgesia, and other side effects, such as; depression, anxiety and sleep deprivation. Neurological pain can also cause pruritis due to the compression of the nervous root system, and itching is a primary symptom of allergic contact dermatitis (ACD).27 It is important to note that acute itch is mediated via histaminergic neurons, whereas chronic itch is mediated via non-histaminergic neurons. White et al2 reviewed the role of the chemokines in the pathophysiology of chronic pain syndromes. Chemokines and their receptors can modulate pain hypersensitivity among animals. In particular, the dorsal root ganglia neurons were identified as having upregulated expression of CXCR3 and its associated chemokine ligands in states of pain hypersensitivity. The occurrence of persistent neuropathic pain appears to depend on the inflammatory cytokines and chemokines. Therefore, a detailed examination of the chemokine system in the human body is likely to present more favourable interventions. Furthermore, CXCR3 is well known to be involved in the pathogenesis of many neurological chronic disease states such as Multiple Sclerosis, Alzheimer’s Disease and other neurodegenerative and neuroinflammatory conditions.34 Thus, a greater understanding of the signalling mechanisms of CXCR3 in painful neuropathic conditions may greatly be beneficial for creating efficacious drug therapies.
A study conducted by Chen et al35 used the well-established rat chronic construction injury (CCI) model to examine the role of CXCR3 in the generation of neuropathic pain. Mechanical Allodynia was measured using the von-Frey filament stimulation and the Dixon’s up-down method assessing the PWT. Thermal sensitivity was also tested on the plantar surface of each hind paw by exposing a beam of radiant heat through a transparent glass surface. Western blot analysis and immunohistochemistry stains were used to detect and measure CXCR3 and other proteins. CCI of the sciatic nerve caused thermal hyperalgesia and mechanical allodynia as well as increased p-ERK levels. The feel of pain corresponded to a significant increase in CXCR3 and its ligand CXCL10 in the neurons of the spinal cord and dorsal root ganglia. Moreover, pain was attenuated through intrathecal injection of the CXCR3 inhibitor, AMG487, which also corresponded to a decrease in p-ERK levels. Overall, the study concluded that signalling of CXCR3 plays an important role in chronic constriction injury and that p-ERK appears to be a key downstream event for causing neuropathic pain.
Jiang et al36 used mice to investigate how CXCR3 worked in the spinal cord using spinal nerve ligation (SNL) induced neuropathic pain and how the mechanisms participated in pain regulation. As evidenced by the study, SNL caused demethylation of the CXCR3 gene promoter and increased binding of the CCAAT-enhancer-binding protein a (C/EBPa) to the promoter leading to increased CXCR3 expression in spinal neurons. Moreover, SNL also caused elevated levels of CXCL10 to be produced in the spinal nerves and astrocytes. The use of a CXCR3 antagonist and gene silencing methods (shRNA) showed that spinal inhibition of CXCR3 caused a reduction in neuropathic pain. In naïve mice, CXCL10 induced CXCR3-mediated allodynia, assessed using the von Frey and Dixon’s up-down method. The study showed that CXCL10 activates the CXCR3 receptor to enhance excitatory synaptic transmission in ascending spinal neurons; this is a process by which CXCR3 is involved in the maintenance of neuropathic pain.
A study conducted by Wang et al37 examined the association of spinal caspase-6 with remifentanil-induced hyperalgesia through CCL21 and its receptor CXCR3 in mice. Remifentanil is a potent and short-acting MOR agonist, its long-term therapeutic use is hindered due to its ability to cause post-operative hyperalgesia and hence a state of chronic pain. Remifentanil-induced hyperalgesia (RIH) was established through thermal paw withdrawal latency and mechanical PWT. RT-qPCR and Western blot were used to evaluate the expression of CXCR3 and caspase-6, which is an intracellular cysteine protease, highly associated with neuroinflammation and hence known to modulate microglia activation and chronic pain states.38 The results showed that remifentanil exposure caused thermal hyperalgesia and mechanical allodynia and also correlated with increased expression of CCL21, CXCR3 and spinal caspase-6 in the dorsal horn of the spinal cord. Interestingly, a reduction in the spinal expression of CCL21 and CXCR3 occurred as a result of intrathecal injection with a caspase-6 inhibitor; VEID-fmk, correlating with its effect at reducing RIH. In Naïve mice, Injection of exogenous caspase-6 led to mechanical allodynia and thermal hyperalgesia as well as increasing CXCR3 expression in the spinal cord, and both these responses were blocked with the co-administration of an anti-CCL21 antibody. Additionally, exogenous CCL21 injection promoted an acute hyperalgesic state in naïve mice which was then blocked with CXCR3 antagonist; NBI-74330 pretreatment. Furthermore, in RIH mice, intrathecal pretreatment with the anti-CCL21 antibody or NBI-74330, attenuated the RIH and pre-treatment with anti-CCL21 antibody also impaired upregulation of CXCR3 mrNA expression in the dorsal horn. These data strongly support that the interactions between CXCR3, CCL21 and caspase-6 are important in the neuroinflammatory pathogenesis of remifentanil-induced hyperalgesia.37
One particular study by Xu et al39 sheds light on the connection between spinal iron overload and CXCR3-mediated neuropathic pain in rats. CNS iron overload, mediated by iron-responsive element-negative divalent metal transporter 1 (IRE(-)DMT1), initiates neuroinflammatory damage and has been associated with several neurodegenerative diseases.40,41 Also, RIH was associated with higher levels of IRE(-)DMT1 and iron overload, leading to oxidative stress and neurotoxicity.42 As expected, CCI neuropathic pain induced CXCL10 and CXCR3 expression in the dorsal horn of the spinal cord. An increase in mechanical allodynia and thermal hyperalgesia correlated with increased expression of spinal IRE(-)DMT1 and spinal iron overload. Intrathecal injection of the CXCR3 antagonist, NBI-74330 attenuated the mechanical allodynia and hyperalgesia, therefore reducing neuropathic pain. Furthermore, chelation of the systemic iron using intraperitoneal deferoxamine caused a reduction in CXCL10 and CXCR3 expression as well as suppressing the mechanical allodynia and thermal hyperalgesia. Intrathecal administration of exogenous CXCL10 induced a state of hyperalgesia in naïve rats and interestingly caused an increase in iron concentration. In addition, co-administration of deferoxamine suppressed the hyperalgesic behavioural states in the naïve mice and reduced iron overload as well as caused a reduction in CXCR3 expression.39 Together the data strongly support the idea that iron overload caused by increased expression by IRE(-)DMT1 is associated with CXCL10-CXCR3 axis and both arms co-modulate hypernociception caused by CCI, or possibly even opioid-induced hyperalgesia.
Jing et al43 used CXCR3 knockout (KO) mice to ascertain the role of CXCR3 in neuropathic itch, in several different acute itch models and a chronic itch model, maintenance of formalin-induced acute pain, and Freund’s adjuvant-induced chronic inflammatory pain using behavioural studies to assess pain and itch. In the CXCR3 KO, mice normal itching was reported in the case of acute itch models but reduced chronic scratching in the case of allergic contact dermatitis (ACD) and the dry skin model. These results were reflected in the wild-type mouse models of experimental ACD and the dry skin induced itch model, where increased expression of CXCR3 and CXCL10 was observed in the dorsal horn of the spinal cord, compared to untreated. Intrathecal injection of CXCR3 antagonist, NBI-74330 reduced the chronic itch but not acute itch. Furthermore, CXCR3 KO mice experienced less acute and chronic inflammatory pain than wild type. Central sensitization to itch, alloknesis, was also decreased in the CXCR3 KO mice, compared to wild type. Thus, CXCR3 was required for the maintenance of neuropathic pain, and in the spinal cord, it helps in mediating alloknesis and chronic itching.
Another study by Piotrowska et al44 and Piotrowska et al45 used a murine chronic constriction injury (CCI) model of neuropathic pain and mice behavioural studies as well as protein analysis using Western blots and immunohistochemistry to show that up-regulation of CXCR3 expression in the microglia was triggered by neuropathic conditions. An overexpression of CXCL10 in naive mice caused algesia and a decrease in the analgesic effect of morphine indicating CXCR3 is involved in neuropathic pain modulation. Further, the antibody blockade of CXCL10 enhanced the analgesic effects of morphine. Intrathecal administration of a CXCR3 antagonist; NBI-74330 reduced the occurrence of concomitant hypersensitivity and was marked by a reduction in the CXCR3 ligands; CXCL4, CXCL9 and CXCL10 at the DRG level and spinal cord and in addition it reduced microglia and astrocyte activation, determined using the markers Iba1 and GFAP, respectively. The findings highlight that CXCR3 antagonist is effective against neuropathic pain and could be therapeutically used possibly as an adjunct for reducing the use of morphine.
In the clinical practice, the long-term use of morphine antinociception is hindered as a result of its high tolerance level. There is an essential role of interaction between microglia and neuron in controlling the mechanism of morphine tolerance. Wang et al46 study was conducted to assess the signalling of CXCL10/CXCR3 in the PAG, mid-brain, that results in the development of morphine tolerance. Neuronal CXCR3 and microglial CXCL10 were expressed parallel to the activation of microglia and repeated morphine administration. A previous report by Bajova et al47 showed the exposure to chronic CXCL10 could increase CREB phosphorylation level in cultured hippocampal neurons. An increase in CAMII, CREB, p38 signalling could be required for mediating morphine tolerance.48 Thus, together the results suggest that the microglial induced neuronal CXCR3 activation could develop morphine tolerance possibly via CaMKII/CREB/p38 signalling.
Another study induced CCI of the sciatic nerve in rats to analyse the impact of CXCR3 activation in the spinal cord and the involvement of changes in blood-spinal cord carrier (BSCB) permeability (BSCB).49 The results showed that CCI caused an increase in CXCR3 expression in the dorsal horn of the spinal cord, further the expression was localised to neurons and not to microglia or astrocytes. CCI-induced CXCL10-mediated CXCR3 activation coupled with disruption of the BSCB which enabled the migration and infiltration of T cells into the spinal parenchyma. Intrathecal administration of an anti-CXCL10 antibody blocked the hyperalgesia as measured by hind PWT and also reduced the BSCB permeability. Together the findings suggest that CXCR3 activation regulates blood–spinal cord barrier (BSCB) permeability, which is further involved in pathogenesis of CCI-induced neuropathic pain and maintenance of hypersensitivity. This clearly suggests that neuroimmune interactions involved in CCI-induced neuropathic pain depend on the migration of T-cells.
Ye et al50 reported that overexpression of CXCL10/CXCR3 in the spinal cord of naive mice caused algesia and cancer-induced bone pain, which was also associated with a decrease in the analgesic effect of morphine. Both CXCR3 and MOR were co-localised at the lamina II of the spinal cord, and morphine treatment transiently increased CXCL-10 expression, a site known to be innervated by type C fibers,51 producing chronic dull pain. In a rat model of bone cancer pain, antibody blockade of CXCL10 inhibited the Gi protein-signalling cascade leading to enhanced analgesic effects of morphine. Results highlight that a single dose of morphine can lead to a transient increase in CXCL10 expression within the spinal cord. Thus, morphine antinociception can be enhanced by preventing CXCR3 activation by CXCL10. Conversely, CXCL10 overexpression can lead to acute algesia and it reduced the morphine analgesic effect. Overall findings indicate that, in the spinal cord, CXCL10 negatively regulates morphine analgesia suggesting that the CXCL10/CXCR3 activation can reduce the analgesic effects of morphine in relieving BCP.48 It is possible that the interactions between CXCR3 and MOR in the brain stem could be a critical step in causing morphine-induced hyperalgesia, central sensitisation and opioid tolerance. In these studies, CXCL10 appears to be a strong pain-producing agent, whereas the CXCR3 blockade using an antagonist could be an effective analgesic option and it would be interesting to measure its effects on morphine tolerance.
The literature described above form a compelling body of evidence that describes a complex mechanism involving CXCR3 and its ligands in particular CXCL10 as a proneuroinflammatory and pronociceptive chemokine receptor–chemokine pair which is key in the initiation, development and maintenance of neuropathic pain. The atypical ligand of CXCR3 (CCL21) as well as CXCL10 appear to modulate opioid-induced hyperalgesia and may also play a key role in morphine tolerance. Together the data can be used to describe a mechanism for neuropathic pain involving several key events that initiate from an inducing factor in what appears to be a vicious circle of neuroinflammation, leading to hyperalgesia and neuropathic pain. In many of the studies, CCI sciatic nerve injury was used to induce neuropathic pain. Subsequently, key events were highlighted all relating to CXCR3 activation such as disruption of the blood–spinal cord barrier (BSCB) facilitating neuroimmune interactions, iron overload in the CNS, interactions between CXCR3 and MOR affecting opioid-induced pain hypersensitivity, activation of intracellular inflammatory mediators such as caspase-6 and CXCR3 activation subsequently initiating downstream signalling pathways, involving ERK/AKT, further activating microglia and exacerbating the neuroinflammation and hence nociception. Indeed, there is some evidence in the literature to suggest that both CXCR3 activation and iron overload could alter the integrity of the BSCB, as has been described earlier.
A schematic representation of the possible signalling and mechanisms initiated by CXCR3 activation leading to neuropathic pain and opioid-induced hypersensitivity is illustrated in Figure 3.
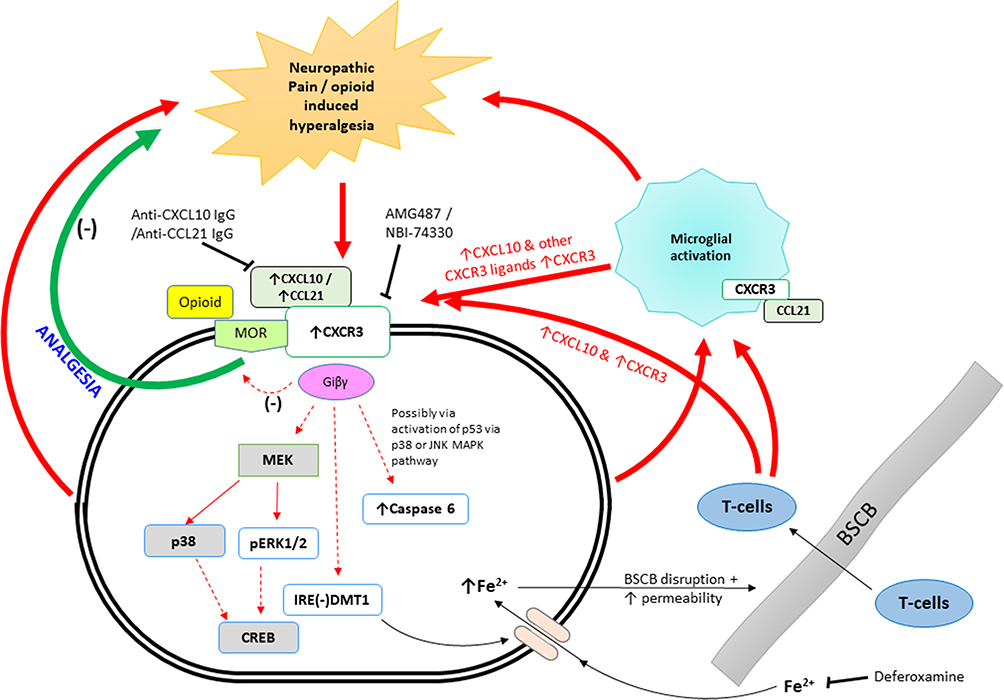 |
Figure 3 Schematic representation of the possible signalling events and mechanism involved in neuropathic pain occurring in the dorsal root ganglia and or the spinal cord and also opioid-induced hyperalgesia or morphine tolerance occurring in the spinal cord and in the PAG, mid brain. |
Role of CXCR3 in Osteoarthritis and Rheumatoid Pain
Osteoarthritis is a painful, chronic, and disabling condition leading to affect the entire joint. Despite the severity of this disease, its aetiology is still poorly understood.52 It is the most common disease among mammals and is considered to be a part of the ageing process. The disease is also considered to be the most common cause of chronic disability among elderly individuals and people of working age.19 There are several risk factors of osteoarthritis, such as having a previous history of joint trauma and obesity. Various normal healthy cells, such as; synovial cells, chondrocytes, and several other cells present in the joints produce and respond to chemokines.
Patients with osteoarthritis and rheumatoid arthritis display higher levels of chemokines, including ligands of CXCR3, CXCL10, and CXCL11, in the synovial fluid.52–54 The infiltration of CXCR3-positive T-cells led by a chemotactic gradient to sites of damage plays a crucial part in the pathophysiology of rheumatoid arthritis.9 Infiltration of activated immune cells leads to the production of intense inflammation and pain. One particular study performed in mice used a CXCR3 antagonist to block the CXCR3 receptor, resulting in reduced pathological signs of rheumatoid arthritis, which shows that blocking the infiltration of CXCR3-positive T-cells with an antagonist can lead to a reduction of inflammation55 and subsequently pain.
Lurati et al52 assessed the T cell distribution and Th CD4+ cells activation degree in the peripheral blood of patients with rheumatoid arthritis (RA), osteoarthritis (OA), and healthy donors. The study analysed 15 patients diagnosed with RA, 56 patients with hip or knee OA, and 20 healthy patients as matched controls. The T cell subpopulation was identified and evaluated using multi-channel flow cytometry with monoclonal antibodies against CD3, CD8, CD4, CCR6, CXCR3, CD38, and HLA DR. The findings showed that CD4+ CD38+ DR+ (i.e., CD4 T activated cells) and Th17 (CCR6+ CXCR3-) count was found higher for RA patients. However, they were lower for the osteoarthritis patients and the control group. However, osteoarthritis patients had higher levels of Th17 cells, and CD4 T activated cells than the control group. The findings of the study conclude that the OA pathogenesis disease might have immunological or inflammatory involvement similar to rheumatoid arthritis (RA). It showed that Th17 cell profiles differ quantitatively concerning the expression of activation markers between OA and RA patients.
Miller, Miller and Malfait53 provide a systematic review for the proalgesic effects of cytokines and chemokines which were identified in osteoarthritic joints, including TNF-α, chemokines, and fractalkine. Cytokines and chemokines may not only lead to joint destruction or synovitis in OA but may induce and directly activate innervating nociceptors. The authors emphasize an understanding of the production of these mediators at the DRG and spinal cord, during the initial changes that occur in a compromised joint may help in enhancing the pain pathway by sensitising the neuron.
Penatti et al54 evaluated the immune cells, CD4+T-cell subsets, and their cytokine profiles for rheumatoid arthritis (RA) and inflammatory osteoarthritis (OA) for defining particular immune signatures. They analysed the synovial membranes (SM), peripheral blood and synovial fluid (SF) in OA and RA patients. The frequency of the CD4+T-cell subset was studied using the flow cytometry, and ELISA was used for measuring the cytokine concentrations in serum and synovial fluid. The findings highlighted that in peripheral blood, the regulatory T-cell frequency of OA patients was altered, and having higher levels of Th17 and Th1/17 cell frequencies than in RA patients, with both subsets predominate with CCR6+ expression. In the synovial membranes of OA patients, CXCR3 expression predominated, as Th17 cells were few, and levels of Th1/Th17 cells were higher. Levels of T reg cells and Tfh cells were higher in RA patients than in OA patients, as is expected with the disease having an autoimmune inflammatory aetiology. IL-17 and B lymphocyte stimulator (Bly) levels were higher in RA patient’s SF and serum samples with an increasing correlation with the disease activity and the autoantibodies. Blys levels were also higher in RA patients who had a low disease score. They concluded that OA inflammatory patients had immune activation in the synovial compartment displaying different frequencies of the T-cell subset as well as cytokine profiles.
Jenh et al55 examined the in vitro pharmacological properties of the small molecule CXCR3 antagonist (SCH 546,738) by radioligand receptor binding as well as T cell chemotaxis assays. Mouse collagen-induced arthritis, rat and mouse experimental autoimmune encephalomyelitis, and rat cardiac transplantation models were used for determining the in vivo efficacy of SCH 546,738. The findings showed that the binding of SCH 546,738 with human CXCR3 occurred at a high affinity (0.4nM). Non-competitive displacement of radiolabelled CXCL10 and CXCL11 occurred using SCH 546,738, with an IC50 ranging from 0.8 to 2.2 nM. In activated T-cells, CXCR3-mediated chemotaxis was specifically inhibited by SCH 546,738 with an IC90 of approximately 10 nM. The study established that autoimmune disease development reduces with the administration of antagonist SCH 546,738 in the collagen-induced arthritis model by reducing the joint leukocyte infiltration and structural damage to cartilage and bone. The results indicated that therapy using a CXCR3 antagonist could help overcome the treatments of autoimmune diseases, by primarily preventing inflammatory cell recruitment and hence could attenuate the progressive nociception.
Benigni et al56 investigated the role of CXCR3 in the pathogenesis of osteoarthritis. Collagenase intraarticular injection was given to WT mice and CXCR3 KO mice. CXCR3 KO mice were injected and observed throughout 8 weeks at day 21, +1 and +3. Synovial thickening, cartilage damage, and osteophyte formation were histologically assessed in order to ascertain the progression of osteoarthritis. Synovial fluid was collected following induction of the disease to identify the inflammatory cells and quantify cytokines using flow cytometry and ELISA assays. Results show that osteoarthritis developed only in WT mice and not in CXCR3 KO mice. Neutrophils and NK cells were among the first to accumulate within the synovium during osteoarthritis. The study highlighted that the CXCL10/CXCR3 axis plays a crucial role in the development of osteoarthritis with increased levels of CXCL10 in the synovial fluid in WT mice but not in the CXCR3 KO mice. Hence, neutrophils, NK cells and CXCR3/CXL10 axis appear to be involved in the induction and progression of osteoarthritis. Therefore, CXCL10-mediated CXCR3 activation appears to be essential for the pathogenesis of osteoarthritis.
Two distinct descending pain modulatory pathways originating from the RVM that contribute to pain hypersensitivity in chronic pain states have been described. First, the 5-HT (serotonin)-positive cell bodies, which release 5-HT into the dorsal horn.57 The second are cell bodies in the RVM, which express the MOR.58 Carr et al59 studied the descending modulatory pain pathway in a mouse joint pain model using microarray analysis. Abasement of the neuronal MOR using dermorphin-saporin, confirmed by immunohistochemistry, caused a reduction in the PWT, thus revealing that the MOR itself was necessary for maintaining hypersensitivity to joint pain. The abasement of MOR in the neuronal cell bodies of the RVM, the originating point of the descending nociceptive pathway terminating in the dorsal horn of the spinal cord, led to the downregulation of genes; Cxcr3 and its ligands Cxcl9 and Cxcl10 in the dorsal horn, 7 days post joint inflammation. Therefore, the signalling interactions between MOR and CXCR3 in the descending modulatory pathway appear to be crucial in maintaining pain hypersensitivity. Thus, CXCR3 and its ligands CXCL9 and CXCL10 may play a role in the modulation of nociception in cases of joint inflammation.
The data above support CXCR3 to be a potential drug target for the prevention of immune cell recruitment, which decreases inflammation, leading to a decrease in the ascending nociceptive pathway stimulation but also may modulate descending nociceptive pathway signalling possibly through the actions of MOR.
Role of CXCR3 in Cystitis/Bladder Pain Syndrome
Bladder Pain Syndrome (BPS) results in severe urinary symptoms and pelvic pain. It is considered to be one of the causes of pain, affecting millions of people globally. Moreover, the lack of reliable treatments results in impairing the quality of life to a great extent. It is one of the debilitating chronic diseases that are classified by lower urinary tract symptoms and suprapubic pain.60 Also, the etiology of the disease is still largely unknown or poorly understood. Furthermore, the treatments are primarily chosen based on the experience and preference of the practitioner.61
A study conducted by Kim62 focused on the histological findings characterized by bladder discomfort, urinary frequency, urgency, and pelvic pain. Inflammatory Urinary markers were investigated based on the function and molecular structure. The identified inflammatory markers were; histamine, mast cells, IL-6, methyl histamine, CXCR3, chemokine, PAP, HIP, AIBG, ORM1, tyramine, TNFSF14, and 2-oxoglutarate. Another study performed by Akiyama et al63 used immunofluorescence analysis to identify the expression and localization of CXCR3 and other immune cells in the Hunner type interstitial cystitis (HIC) bladder compared to control bladder samples (non-diseased). Results showed there was increased infiltration of CXCR3-positive plasma cells into the lamina propia of the Hunner type interstitial cystitis compared to that of the non-HIC bladder.
The studies above identify CXCR3 to play a significant role in the chronic inflammation associated with cystitis; therefore, it may be considered as a causal factor for the pain.
Role of CXCR3 in Dental Pain
Periodontal diseases are the most common cause of oral inflammation among the human population. It is a chronic inflammatory disease-causing pain especially, on chewing.29 T lymphocytes are part of a complex network of heightened immune responses resulting in the pathogenesis of periodontal disease.64,65 Indeed, CXCR3 and its ligand CXCL10 are highly expressed in diseased periodontal tissue64 and are now well known to be one of the mediators of this pathogenic process. Much literature is available associating CXCR3 and its role in T-cell migration to the pathology of periodontal disease.65
Hiyari et al66 performed a study to identify genes that are significantly associated with periodontal disease using a model of LPS induced periodontal disease in mice. The FAST-LMM program was used to perform genome-wide association studies (GWAS). Results showed that the CXCL10 ligand is one of the pro-inflammatory cytokines that highly and exclusively expressed the C57BL/6J strain of mice (strain with high bone loss). Mice with the CXCR3 gene deletion displayed a 50% reduction in bone loss and reduced osteoclast activity. The CXCR3 antagonist AMG-487 caused a 45% reduction in bone loss in wild-type mice, which were injected with LPS.
A study performed in 2009 by Hosokawa et al67 used ELISA assays to measure the production of CXCL10 by human gingival fibroblasts that had been subjected to stimulation by an array for different cytokines and their combinations. The cytokines used were pro-inflammatory cytokines (IL-1 and TNF-α), T-helper 1 cytokine (IFN-γ), Th2 cytokines (IL-4 and IL-3), T-helper 17 cytokines and Regulatory T-cells cytokines. IL-1, TNF, and IFN-γ caused expression of CXCL10 in the gingival fibroblasts, and Th2 cytokines inhibited the production of IL-1, TNF and IFN- γ induced CXCL10 production.
The cytokine, chemokine, and chemokine receptor expression profiles were determined through the course of periodontitis using a mouse model of Actinobacillus actinomycetemcomitans-induced periodontal disease. Pro-inflammatory cytokines (TNF-α, IFN- γ, and IL-12) were observed soon after the injection of periodontal disease-causing bacteria and their expression remained through the course of the disease. Various Th1 chemokines and their receptors, including CXCL10 and CXCR3, also followed a similar pattern of expression. Th2 chemokine expression started at day 30 after injection of the microbes. Intense bone loss and inflammation were predominantly expressed through the Th1 phase.68
Irreversible pulpitis can cause severe continuous or paroxysmal pain as well as thermal hypersensitivity.27,69 A study by Adachi et al69 found a significantly higher expression of CXCL10 mRNA in inflamed dental pulp compared to the healthy dental pulp; this expression was localized to macrophages, endothelial cells, and fibroblasts. CXCR3 was mostly exclusively expressed on T-cells. Cultured dental pulp fibroblasts also secreted CXCL10 in response to stimulation by live caries-related bacteria but not to heat-killed bacteria.69 These results suggest that CXCR3-positive-activated T-cells and its ligand CXCL10 may have a significant role in the pathogenesis of pulpitis. Hence, it is highly conceivable that endodontic pain may be modulated by the pharmacological interactions between CXCR3 and CXL10. Dental caries also initiates the expression of several cytokines and pro-inflammatory mediators, including CXCL11 and CXCL10 in the odontogenic layer of the tooth, as well as the pulp itself.70 Thus, contributing to the pathophysiology of pulpal inflammation.
CXCR3 plays an important role in modulating pain related to dental pathologies such as; periodontitis and pulpitis due to its capability of causing immune cell migration and thus contributing to the progression of the disease.
Conclusion
The current study was conducted to evaluate if CXCR3 plays any role in the diseases associated with severe pain. A cohort of literature was examined to collect information regarding the role of CXCR3 in modulating the pain in several parts of the body. All the data suggest that CXCR3 is a critical player in the progression of inflammatory diseases, and the inflammatory mediators can cause activation of nociceptors in the primary afferent nerves.28 An emerging body of literature indicates that CXCR3 activation in neuronal cells and microglia of the spinal cord can modulate ascending nociceptive pathways resulting in hypersensitization and algesia. Therefore, it may be postulated that CXCR3 can modulate pain in two ways; first, by preventing the recruitment of immune cells, thus reducing the inflammatory response, the disease and hence the pain. Second, affecting nociception in the CNS tissue via its actions in the spinal cord, particularly, the dorsal root ganglion and the dorsal horn of the spinal cord as well as altering μ-opioid-induced pain modulation in the ascending and descending modulatory pathway at points in the brain stem, such as the RVM and PAG. Figure 4 shows a systematic representation of all the supporting studies, which identify CXCR3 to have a role in paid modulation at specific site in the pain pathway.
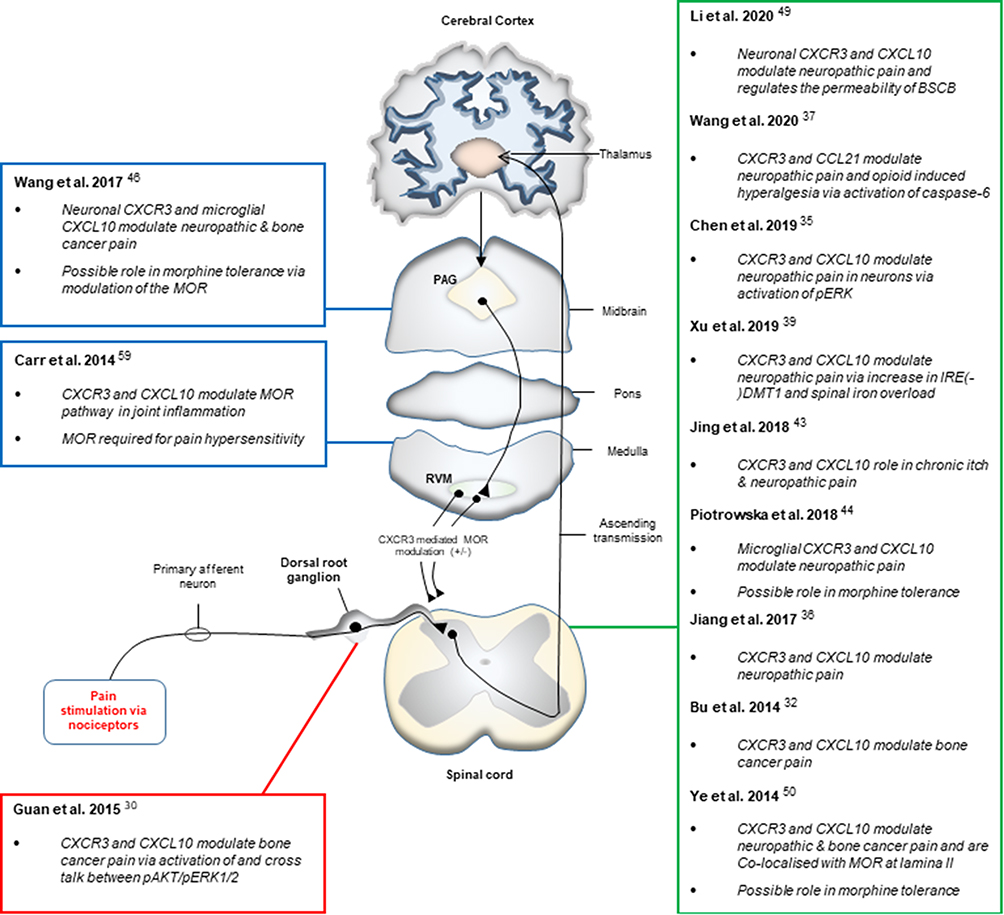 |
Figure 4 Schematic representation of the studies identifying CXCR3 to be involved in pain modulation at specific sites in the pain pathway. |
As reported, CXCR3 was found to play a crucial role in cases related to bone cancer pain, neuropathic pain, osteoarthritis/rheumatoid arthritis pain. The literature evaluated with regards to pain associated with cystitis/bladder pain syndrome, pulpitis and periodontitis identify CXCR3 to be part of the pathogenesis of the disease and hence be a causal factor for the pain. To this end, further research delving into the mechanisms of CXCR3 induced pain centrally, and peripherally is warranted to be able to develop novel potential therapies. Hence, the knowledge of CXCR3 can prove to be beneficial for the researchers and clinicians in developing interventions that can modulate pain. However, future studies detailing the CXCR3-mediated signalling pathways are required to effectively design therapeutic pain targets.
Abbreviations
ENG, etonogestrel; FDA, Food and Drug Administration; TRPV1, transient receptor potential vallinoid 1; BCP, bone cancer pain; ACD, allergic contact dermatitis; SNL, spinal nerve ligation; shRNA, gene silencing methods; MOR, μ-opioid receptor; KO, knockout; RA, rheumatoid arthritis; OA, osteoarthritis; BPS, bladder pain syndrome; HIC, Hunner type interstitial cystitis; GWAS, genome-wide association studies; BSCB, blood–spinal cord barrier; CCI, chronic constriction injury; RIH, remifentanil-induced Hyperalgesia.
Acknowledgments
The author is very thankful to all the associated personnel in any reference that contributed in/for this research. The research was funded by the Deanship of Scientific Research at Princess Nourah bint Abdulrahman University through the Fast-track Research Funding Program.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Julius D. TRP channels and pain. Annu Rev Cell Dev Biol. 2013;29(1):355–384. doi:10.1146/annurev-cellbio-101011-155833
2. White FA, Jung H, Miller RJ. Chemokines and the pathophysiology of neuropathic pain. Proc Natl Acad Sci U S A. 2007;104(51):20151–20158. doi:10.1073/pnas.0709250104
3. Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139(2):267–284. doi:10.1016/j.cell.2009.09.028
4. Kalyuzhny AE, Arvidsson U, Wu W, Wessendorf MW. μ-Opioid and δ-opioid receptors are expressed in brainstem antinociceptive circuits: studies using immunocytochemistry and retrograde tract-tracing. J Neurosci. 1996;16(20):6490–6503. doi:10.1523/JNEUROSCI.16-20-06490.1996
5. Colvin LA, Bull F, Hales TG. Perioperative opioid analgesia—when is enough too much? A review of opioid-induced tolerance and hyperalgesia. Lancet. 2019;393(10180):1558–1568. doi:10.1016/S0140-6736(19)30430-1
6. Mao J, Price DD, Mayer DJ. Mechanisms of hyperalgesian and morphine tolerance: a current view of their possible interactions. Pain. 1995;62(3):259–274. doi:10.1016/0304-3959(95)00073-2
7. Verma V, Sheikh Z, Ahmed AS. Nociception and role of immune system in pain. Acta Neurol Belg. 2015;115(3):213–220. doi:10.1007/s13760-014-0411-y
8. Qin L, Kufareva I, Holden LG, et al. Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science. 2015;347(6226):1117–1122. doi:10.1126/science.1261064
9. Raucci R, Colonna G, Giovane A, Castello G, Costantini S. N-terminal region of human chemokine receptor CXCR3: structural analysis of CXCR3 (1–48) by experimental and computational studies. Biochim Biophys Acta Bioenerg. 2014;1844(10):1868–1880. doi:10.1016/j.bbapap.2014.08.004
10. Burg JS, Ingram JR, Venkatakrishnan A, et al. Structural basis for chemokine recognition and activation of a viral G protein–coupled receptor. Science. 2015;347(6226):1113–1117. doi:10.1126/science.aaa5026
11. Nguyen AF. Purification and Study of CC Chemokine-Based Strategies to Combat Chronic Inflammation and HIV. UC Merced; 2017.
12. Bondeson J, Blom AB, Wainwright S, Hughes C, Caterson B, Van Den Berg WB. The role of synovial macrophages and macrophage‐produced mediators in driving inflammatory and destructive responses in osteoarthritis. Arthritis Rheum. 2010;62(3):647–657. doi:10.1002/art.27290
13. Breser ML, Motrich RD, Sanchez LR, Mackern-Oberti JP, Rivero VE. Expression of CXCR3 on specific T cells is essential for homing to the prostate gland in an experimental model of chronic prostatitis/chronic pelvic pain syndrome. J Immunol. 2013;190(7):3121–3133. doi:10.4049/jimmunol.1202482
14. Loetscher M, Gerber B, Loetscher P, et al. Chemokine receptor specific for IP10 and mig: structure, function, and expression in activated T-lymphocytes. J Exp Med. 1996;184(3):963–969. doi:10.1084/jem.184.3.963
15. Qin S, Rottman JB, Myers P, et al. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J Clin Invest. 1998;101(4):746–754. doi:10.1172/jci1422
16. Lacotte S, Brun S, Muller S, Dumortier H. CXCR3, Inflammation, and autoimmune diseases. Contemp Challenge Autoimmun. 2009;1173:310. doi:10.1111/j.1749-6632.2009.04813.x
17. Liu Q-Z, Ma W-T, Yang J-B, et al. The CXC chemokine receptor 3 inhibits autoimmune cholangitis via CD8+ T cells but promotes colitis via CD4+ T cells. Front Immunol. 2018;9:1090. doi:10.3389/fimmu.2018.01090
18. Chen W, Ukah TK, Miller MM, Zaghouani H, Wan X. PI3K-AKT-mTOR pathway mediates trafficking of diabetogenic T cells by modulating CXCR3 via T-bet. Am Assoc Immunol. 2016.
19. Oghumu S, Stock JC, Varikuti S, et al. Transgenic expression of CXCR3 on T cells enhances susceptibility to cutaneous Leishmania major infection by inhibiting monocyte maturation and promoting a Th2 response. Infect Immun. 2015;83(1):67–76. doi:10.1128/IAI.02540-14
20. Lasagni L, Francalanci M, Annunziato F, et al. An alternatively spliced variant of CXCR3 mediates the inhibition of endothelial cell growth induced by IP-10, Mig, and I-TAC, and acts as functional receptor for platelet factor 4. J Exp Med. 2003;197(11):1537–1549. doi:10.1084/jem.20021897
21. Ehlert JE, Addison CA, Burdick MD, Kunkel SL, Strieter RM. Identification and partial characterization of a variant of human CXCR3 generated by posttranscriptional exon skipping. J Immunol. 2004;173(10):6234–6240. doi:10.4049/jimmunol.173.10.6234
22. Smith HS. Current Therapy in Pain: 1st ed. Philadelphia: Saunders. Elsevier Health Sciences. 2009.
23. Rappert A, Biber K, Nolte C, et al. Secondary lymphoid tissue chemokine (CCL21) activates CXCR3 to trigger a Cl− current and chemotaxis in murine microglia. J Immunol. 2002;168(7):3221–3226. doi:10.4049/jimmunol.168.7.3221
24. Bhangoo S, Ren D, Miller RJ, et al. Delayed functional expression of neuronal chemokine receptors following focal nerve demyelination in the rat: a mechanism for the development of chronic sensitization of peripheral nociceptors. Mol Pain. 2007;3:
25. Qu L, Fu K, Yang J, Shimada SG, LaMotte RH. CXCR3 chemokine receptor signaling mediates itch in experimental allergic contact dermatitis. Pain. 2015;156(9):1737. doi:10.1097/j.pain.0000000000000208
26. Van den Beuken-van Everdingen M, De Rijke J, Kessels A, Schouten H, Van Kleef M, Patijn J. Prevalence of pain in patients with cancer: a systematic review of the past 40 years. Ann Oncol. 2007;18(9):1437–1449. doi:10.1093/annonc/mdm056
27. Terricabras E, Benjamim C, Godessart N. Drug discovery and chemokine receptor antagonists: eppur si muove! Autoimmun Rev. 2004;3(7–8):550–556. doi:10.1016/j.autrev.2004.07.037
28. Rahman I. Functional Analysis of the G-Protein Coupled Chemokine Receptor CXCR3-A and Its Ligands. University of Reading; 2007.
29. Chow E, Ding K, Parulekar WR, et al. Predictive model for survival in patients having repeat radiation treatment for painful bone metastases. Radiother Oncol. 2016;118(3):547–551. doi:10.1016/j.radonc.2015.10.018
30. Guan X-H, Fu Q-C, Shi D, et al. Activation of spinal chemokine receptor CXCR3 mediates bone cancer pain through an Akt-ERK crosstalk pathway in rats. Exp Neurol. 2015;263:39–49. doi:10.1016/j.expneurol.2014.09.019
31. Guo G, Gao F. CXCR3: latest evidence for the involvement of chemokine signaling in bone cancer pain. Exp Neurol. 2015;265:176–179. doi:10.1016/j.expneurol.2015.02.003
32. Bu H, Shu B, Gao F, et al. Spinal IFN-γ-induced protein-10 (CXCL10) mediates metastatic breast cancer-induced bone pain by activation of microglia in rat models. Breast Cancer Res Treat. 2014;143(2):255–263. doi:10.1007/s10549-013-2807-4
33. Zhou Y-Q, Gao H-Y, Guan X-H, et al. Chemokines and their receptors: potential therapeutic targets for bone cancer pain. Curr Pharm Des. 2015;21(34):5029–5033. doi:10.2174/1381612821666150831141931
34. Zhou YQ, Liu DQ, Chen SP, et al. The role of CXCR3 in neurological diseases. Curr Neuropharmacol. 2019;17(2):142–150. doi:10.2174/1570159X15666171109161140
35. Chen Y, Yin D, Fan B, et al. Chemokine CXCL10/CXCR3 signaling contributes to neuropathic pain in spinal cord and dorsal root ganglia after chronic constriction injury in rats. Neurosci Lett. 2019;694:20–28. doi:10.1016/j.neulet.2018.11.021
36. Jiang B-C, He L-N, Wu X-B, et al. Promoted interaction of C/EBPα with demethylated Cxcr3 gene promoter contributes to neuropathic pain in mice. J Neurosci. 2017;37(3):685–700. doi:10.1523/JNEUROSCI.2262-16.2016
37. Wang C, Li Q, Jia Z, et al. Spinal caspase-6 contributes to remifentanil-induced hyperalgesia via regulating CCL21/CXCR3 pathway in rats. Neurosci Lett. 2020;721:134802. doi:10.1016/j.neulet.2020.134802
38. Burguillos MA, Deierborg T, Kavanagh E, et al. Caspase signalling controls microglia activation and neurotoxicity. Nature. 2011;472(7343):319–324. doi:10.1038/nature09788
39. Xu W, Liu W, Yu W. The involvement of iron responsive element (-) divalent metal transporter 1-mediated the spinal iron overload via CXCL10/CXCR3 pathway in neuropathic pain in rats. Neurosci Lett. 2019;694:154–160. doi:10.1016/j.neulet.2018.12.001
40. Altamura S, Muckenthaler MU. Iron toxicity in diseases of aging: Alzheimer’s disease, Parkinson’s disease and atherosclerosis. J Alzheimers Dis. 2009;16(4):879–895. doi:10.3233/JAD-2009-1010
41. Ingrassia R, Garavaglia B, Memo M. DMT1 expression and iron levels at the crossroads between aging and neurodegeneration. Front Neurosci. 2019;13(575). doi:10.3389/fnins.2019.00575
42. Shu RC, Zhang LL, Wang CY, et al. Spinal peroxynitrite contributes to remifentanil-induced postoperative hyperalgesia via enhancement of divalent metal transporter 1 without iron-responsive element–mediated iron accumulation in rats. Anesthesiology. 2015;122(4):908–920. doi:10.1097/ALN.0000000000000562
43. Jing P-B, Cao D-L, Li -S-S, et al. Chemokine receptor CXCR3 in the spinal cord contributes to chronic itch in mice. Neurosci Bull. 2018;34(1):54–63. doi:10.1007/s12264-017-0128-z
44. Piotrowska A, Rojewska E, Pawlik K, et al. Pharmacological blockade of CXCR3 by (±)-NBI-74330 reduces neuropathic pain and enhances opioid effectiveness-evidence from in vivo and in vitro studies. Biochim Biophys Acta Biomembr. 2018;1864(10):3418–3437. doi:10.1016/j.bbadis.2018.07.032
45. Piotrowska A, Rojewska E, Pawlik K, et al. Dataset of (±)-NBI-74330 (CXCR3 antagonist) influence on chemokines under neuropathic pain. Data Brief. 2018;21:1145–1150. doi:10.1016/j.dib.2018.10.091
46. Wang W, Peng Y, Yang H, et al. Potential role of CXCL10/CXCR3 signaling in the development of morphine tolerance in periaqueductal gray. Neuropeptides. 2017;65:120–127. doi:10.1016/j.npep.2017.07.004
47. Bajova H, Nelson TE, Gruol DL. Chronic CXCL10 alters the level of activated ERK1/2 and transcriptional factors CREB and NF-κB in hippocampal neuronal cell culture. J Neuroimmunol. 2008;195(1–2):36–46. doi:10.1016/j.jneuroim.2008.01.003
48. Wang A, Bibb JA. Is CREB the angry bird that releases memory in alzheimer’s? Neuropsychopharmacology. 2011;36(11):2153–2154. doi:10.1038/npp.2011.126
49. Li HL, Huang Y, Zhou YL, et al. CXC motif chemokine 10 contributes to the development of neuropathic pain by increasing the permeability of the blood–spinal cord barrier. Front Immunol. 2020;11:477. doi:10.3389/fimmu.2020.00477
50. Ye D, Bu H, Guo G, et al. Activation of CXCL10/CXCR3 signaling attenuates morphine analgesia: involvement of Gi protein. J Mol Neurosci. 2014;53(4):571–579. doi:10.1007/s12031-013-0223-1
51. Todd AJ. Neuronal circuitry for pain processing in the dorsal horn. Nat Rev Neurosci. 2010;11(12):823–836. doi:10.1038/nrn2947
52. Lurati A, Laria A, Gatti A, Brando B, Scarpellini M. Different T cells’ distribution and activation degree of Th17 CD4+ cells in peripheral blood in patients with osteoarthritis, rheumatoid arthritis, and healthy donors: preliminary results of the MAGENTA CLICAO study. Open Access Rheumatol. 2015;7:63. doi:10.2147/OARRR.S81905
53. Miller RE, Miller RJ, Malfait A-M. Osteoarthritis joint pain: the cytokine connection. Cytokine. 2014;70(2):185–193. doi:10.1016/j.cyto.2014.06.019
54. Penatti A, Facciotti F, De Matteis R, et al. Differences in serum and synovial CD4+ T cells and cytokine profiles to stratify patients with inflammatory osteoarthritis and rheumatoid arthritis. Arthritis Res Ther. 2017;19(1):103. doi:10.1186/s13075-017-1305-1
55. Jenh C-H, Cox MA, Cui L, et al. A selective and potent CXCR3 antagonist SCH 546738 attenuates the development of autoimmune diseases and delays graft rejection. BMC Immunol. 2012;13(1):1–14. doi:10.1186/1471-2172-13-2
56. Benigni G, Dimitrova P, Antonangeli F, et al. CXCR3/CXCL10 axis regulates neutrophil–NK cell cross-talk determining the severity of experimental osteoarthritis. J Immunol. 2017;198(5):2115–2124. doi:10.4049/jimmunol.1601359
57. Ossipov MH, Dussor GO, Porreca F. Central modulation of pain. J Clin Invest. 2010;120(11):3779–3787. doi:10.1172/JCI43766
58. Heinricher M, Morgan M, Tortorici V, Fields H. Disinhibition of off-cells and antinociception produced by an opioid action within the rostral ventromedial medulla. Neuroscience. 1994;63(1):279–288. doi:10.1016/0306-4522(94)90022-1
59. Carr FB, Géranten SM, Hunt SP. Descending controls modulate inflammatory joint pain and regulate CXC chemokine and iNOS expression in the dorsal horn. Mol Pain. 2014;10:
60. Ogawa T, Ishizuka O, Ueda T, Tyagi P, Chancellor MB, Yoshimura N. Current and emerging drugs for interstitial cystitis/bladder pain syndrome (IC/BPS). Expert Opin Emerg Drugs. 2015;20(4):555–570. doi:10.1517/14728214.2015.1105216
61. Silva T, Garlet GP, Fukada SY, JSd S, Cunha F. Chemokines in oral inflammatory diseases: apical periodontitis and periodontal disease. J Dent Res. 2007;86(4):306–319. doi:10.1177/154405910708600403
62. Kim H-J. Update on the pathology and diagnosis of interstitial cystitis/bladder pain syndrome: a review. Int Neurourol J. 2016;20(1):13. doi:10.5213/inj.1632522.261
63. Akiyama Y, Morikawa T, Maeda D, et al. Increased CXCR3 expression of infiltrating plasma cells in Hunner type interstitial cystitis. Sci Rep. 2016;6(1):1–7. doi:10.1038/srep28652
64. Cekici A, Kantarci A, Hasturk H, Van Dyke TE. Inflammatory and immune pathways in the pathogenesis of periodontal disease. Periodontol 2000. 2014;64(1):57–80. doi:10.1111/prd.12002
65. Meyle J, Chapple I. Molecular aspects of the pathogenesis of periodontitis. Periodontol 2000. 2015;69(1):7–17. doi:10.1111/prd.12104
66. Hiyari S, Green E, Pan C, et al. Genomewide association study identifies Cxcl family members as partial mediators of LPS‐induced periodontitis. J Bone Miner Res. 2018;33(8):1450–1463. doi:10.1002/jbmr.3440
67. Hosokawa Y, Hosokawa I, Ozaki K, Nakanishi T, Nakae H, Matsuo T. Catechins inhibit CCL20 production in IL-17A-stimulated human gingival fibroblasts. Cell Physiol Biochem. 2009;24(5–6):391–396. doi:10.1159/000257431
68. Garlet GP, Avila-Campos MJ, Milanezi CM, Ferreira BR, Silva JS. Actinobacillus actinomycetemcomitans-induced periodontal disease in mice: patterns of cytokine, chemokine, and chemokine receptor expression and leukocyte migration. Microbes Infect. 2005;7(4):738–747. doi:10.1016/j.micinf.2005.01.012
69. Adachi T, Nakanishi T, Yumoto H, et al. Caries-related bacteria and cytokines induce CXCL10 in dental pulp. J Dent Res. 2007;86(12):1217–1222. doi:10.1177/154405910708601215
70. Horst OV, Horst JA, Samudrala R, Dale BA. Caries induced cytokine network in the odontoblast layer of human teeth. BMC Immunol. 2011;12(1):9. doi:10.1186/1471-2172-12-9
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.