")
Back to Journals » HIV/AIDS - Research and Palliative Care » Volume 8
Evaluating the role of atazanavir/cobicistat and darunavir/cobicistat fixed-dose combinations for the treatment of HIV-1 infection
Authors Crutchley RD, Guduru R, Cheng A
Received 24 October 2015
Accepted for publication 8 December 2015
Published 9 March 2016 Volume 2016:8 Pages 47—65
DOI https://doi.org/10.2147/HIV.S99063
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Bassel Sawaya
Rustin D Crutchley,1 Rakesh C Guduru,2 Amy M Cheng1
1Department of Pharmacy Practice and Translational Research, College of Pharmacy, University of Houston, 2CompanionDX, Houston, TX, USA
Abstract: Atazanavir/cobicistat (ATV/c) and darunavir/cobicistat (DRV/c) are newly approved once daily fixed-dose protease inhibitor combinations for the treatment of HIV-1 infection. Studies in healthy volunteers have established bioequivalence between cobicistat and ritonavir as pharmacoenhancers of both atazanavir (ATV) and darunavir (DRV). In addition, two randomized clinical trials (one Phase II and one Phase III noninferiority trial with a 144-week follow-up period) demonstrated that cobicistat had sustainable and comparable efficacy and safety to ritonavir as a pharmacoenhancer of ATV through 144 weeks of treatment in HIV-1-infected patients. Furthermore, one Phase III, open-label, single-arm, clinical trial reflected virologic and immunologic responses and safety outcomes consistent with prior published data for DRV/ritonavir 800/100 mg once daily, supporting the use of DRV/c 800/150 mg once daily for future treatment of treatment-naïve and -experienced HIV-1-infected patients with no DRV resistance-associated mutations. Low rates of virologic failure secondary to resistance to antiretroviral regimens were present in these clinical studies. Most notable adverse events in the ATV studies were hyperbilirubinemia and in the DRV study rash. Small increases in serum creatinine and minimally reduced estimated glomerular filtration rate Cockcroft–Gault calculation (eGFRCG) were observed in ATV/c and DRV/c clinical studies consistent with other studies evaluating elvitegravir/cobicistat/tenofovir/emtricitabine for the treatment of HIV-1 infection. These renal parameter changes occurred acutely in the first few weeks and plateaued off for the remaining study periods and are not necessarily clinically relevant. Cobicistat has numerous advantages compared to ritonavir such as fewer drug–drug interactions, being devoid of anti-HIV-1 activity, as well as it has better solubility affording coformulation with other antiretrovirals as simplified fixed-dose combinations. Overall, the recent approval of ATV/c and DRV/c offers HIV patients opportunities for improved adherence to lifelong treatment. Future studies are warranted to determine the efficacy and safety of ATV/c and DRV/c in treatment-experienced patients.
Keywords: atazanavir, darunavir, cobicistat, HIV protease inhibitors, treatment simplification
Introduction
Tremendous progress has been made over the past couple of decades in the treatment of HIV infection. The HIV population demographics are shifting more to an aging population in first-world countries; by 2015, more than 50% of all HIV-infected individuals in the US will be older than 50 years of age.1 This can be attributed to the availability of numerous different classes of antiretrovirals (ARVs) in addition to newer agents within these classes characterized by excellent efficacy and better tolerability. In particular, the current era of simplified highly active antiretroviral therapy (HAART) is a major contributor to the treatment success of HIV-infected individuals today.
Treatment of HIV in patients who are newly diagnosed with HIV (treatment-naïve) usually includes two nucleoside reverse transcriptase inhibitors (NRTIs) as backbone therapy in combination with either an integrase strand transfer inhibitor or nonnucleoside reverse transcriptase inhibitor (NNRTI) or a boosted protease inhibitor (PI). Recommendations on which ARV regimens are preferred for treatment-naïve HIV-infected individuals can be reviewed in the Department of Health and Human Services (DHHS) and British HIV Association guidelines.2,3 Fortunately, numerous combinations of these ARVs exist as once daily HAART regimens, facilitating improved adherence and better treatment outcomes. Choice of HAART regimens for HIV-infected patients depends on several factors. One factor that favors the choice of a PI-based regimen is when future potential adherence to HAART is questionable, since PIs possess a high genetic barrier (eg, usually have to accumulate several mutations for these to not work effectively as compared to some NNRTIs [efavirenz and rilpivirine] or integrase strand transfer inhibitors [raltegravir and elvitegravir] that can become ineffective with poor adherence and the development of a single mutation). For over a decade, lopinavir/ritonavir has been a PI commonly used for the treatment of HIV-infected patients worldwide. Coformulation of lopinavir with ritonavir has been reasonably favorable for treating the HIV pediatric population, since its use together has better palatability versus using a different PI such as atazanavir or darunavir boosted with ritonavir solution (possessing very poor palatability). However, disadvantages of lopinavir/ritonavir include increased pill burden, more adverse effects (eg, gastrointestinal), and twice daily dosing making it less desirable as a preferred treatment option for HIV-infected patients in general. For these reasons, lopinavir/ritonavir is recommended as an other treatment option according to DHHS guidelines.2
Because PIs undergo extensive metabolism in the liver and intestine predominately by CYP3A enzymes resulting in low systemic exposure and increased risk for the development of resistance, PIs are generally used together with subtherapeutic doses of a CYP3A inhibitor ritonavir (100, 200 mg/day) to increase their concentrations.4,5 Several limitations do exist with ritonavir. One of these includes multiple drug–drug interactions (DDIs); ritonavir is an inhibitor of CYP enzymes 3A and 2D6 (lesser degree), but also is an inducer of CYP enzymes 1A2, 2C9, 2C19, and glucuronyltransferases.5,6 Because of ritonavir’s poor solubility, it is difficult to coformulate with other ARVs, unless an advanced melt extrusion technology is used.7,8 Other limitations of ritonavir include its association with adverse events (AEs) (gastrointestinal and metabolic) and an increased risk of resistance if used in a non-PI-containing regimen.4–6
For these reasons, alternative pharmacoenhancers or boosters have been investigated. Cobicistat (Tybost®) was approved by the US Food and Drug Administration (FDA) in 2014. Cobicistat is now available in the US in once daily coformulated tablets such as Stribild® (elvitegravir 150 mg/cobicistat 150 mg/tenofovir [TDF]) 300 mg/emtricitabine ([FTC] 200 mg) and recent PI combinations Prezcobix® (darunavir 800 mg/cobicistat 150 mg) and Evotaz® (atazanavir 300 mg/cobicistat 150 mg). Cobicistat has numerous advantages compared to ritonavir (Table 1): it has better solubility affording increased opportunity for coformulation with other ARVs; fewer DDIs (inhibition of CYP3A and 2D6 [lesser degree], but is not an inducer of CYP enzymes); and it also does not have intrinsic anti-HIV-1 activity, thereby, potentially reducing the risk for resistance. Another benefit of cobicistat is the reduced pill burden because of its availability in fixed-dose combinations (FDC). For some HIV-infected patients, pill burden can be a major barrier to adherence to HAART. Development of these coformulated products has simplified convenience of daily administration, potentially improving adherence, and ensuring positive treatment outcomes for HIV-1-infected patients.
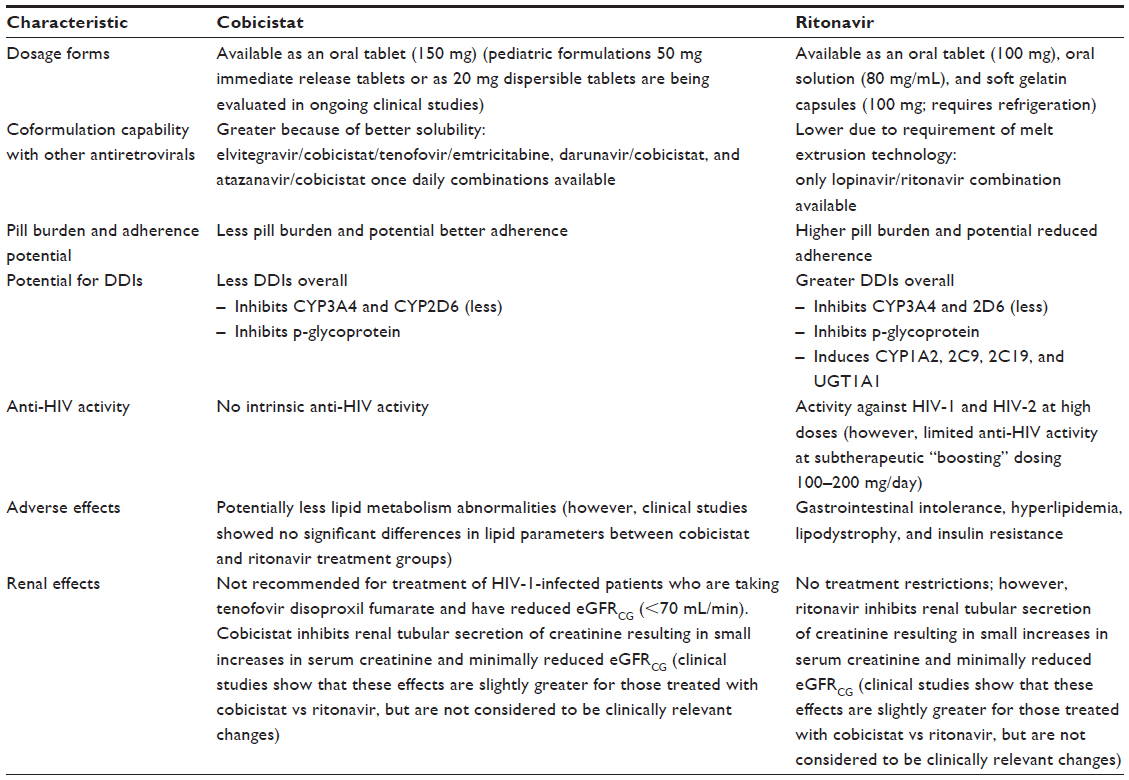 | Table 1 Differences between cobicistat and ritonavir as pharmacoenhancers |
This article reviews current literature on the pharmacodynamic and pharmacokinetic properties of cobicistat, including identification of clinically relevant DDIs with atazanavir/cobicistat (ATV/c) and darunavir/cobicistat (DRV/c), as well as summarizes bioequivalent studies in healthy volunteers and clinical trials evaluating efficacy and tolerability of ATV/c and DRV/c for the treatment of HIV-1-infected patients, and provides some important clinical considerations for HIV-1-infected patients using these combination therapies. A discussion of elvitegravir/cobicistat/TDF/FTC is beyond the scope of this review and the reader is encouraged to review data regarding this combination therapy discussed in other reviews.9–12
Clinical pharmacology
Pharmacodynamics
While both ritonavir and cobicistat are used to increase serum concentrations of other ARV agents, cobicistat differs in that it does not intrinsically have any ARV activity. Cobicistat does not inhibit recombinant HIV-1 protease and has no detectable antiviral activity in cell culture against HIV-1, hepatitis B virus (HBV), or hepatitis C virus (HCV).13 It is inactive against HIV-1 protease (50% maximal inhibitory concentration >30 μM) and has no inhibitory effect against HIV-1 replication (effective concentration [EC]50 >30 μM).14 Although cobicistat is structurally similar to ritonavir, its inactivity as an ARV may be attributed to a small chemical structural difference to ritonavir (Figure 1).15 Ritonavir has a key hydroxyl group moiety in its chemical structure unlike cobicistat, which allows ritonavir to exert its antiviral activity by attaching to the active site of HIV-1 protease.14 Cobicistat has greater aqueous solubility (0.1 mg/mL in water at 20°C) than ritonavir (considered practically insoluble in water) at both neutral (75 vs ~2.0 μg/mL at pH 7.4) and acidic conditions (>6,500 vs 3.1 μg/mL at pH 2.2).14 This unique physical characteristic of cobicistat is advantageous, since it allows for coformulation with other ARV agents.13,14,16
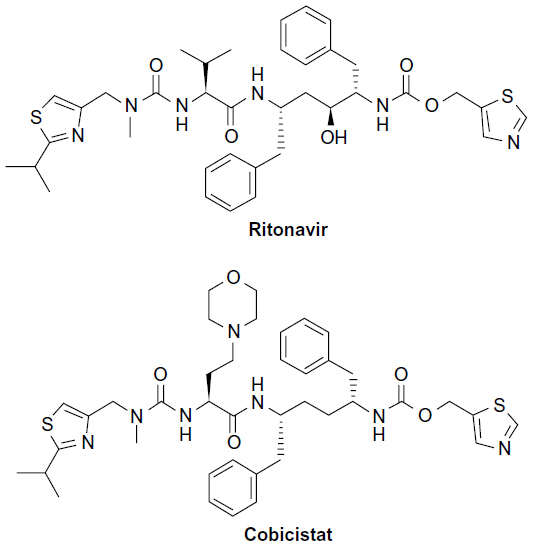 | Figure 1 Chemical structures of ritonavir and cobicistat. |
Both cobicistat and ritonavir are mechanism-based inhibitors of CYP3A and increase the systemic exposure of CYP3A substrates atazanavir (ATV) and darunavir (DRV).13 Cobicistat inactivates CYP3A in a time- and concentration-dependent manner similar to ritonavir at both low and high concentrations.14 The inhibitory potency of cobicistat is similar to that of ritonavir with respect to its activity on CYP3A.
Data suggest that cobicistat may have a lower potential for toxicity related to altered lipid metabolism as compared to ritonavir.14 Cobicistat affected normal adipocyte function less than ritonavir in vitro, since lipid accumulation in human adipocytes was characterized by EC50 >30 vs 16 μM, respectively. Furthermore, in an insulin-stimulated glucose uptake assay, ritonavir showed a more pronounced effect of mean inhibition at 10 μM in mouse adipocytes compared to cobicistat.14
Cobicistat is not interchangeable with ritonavir and is not recommended in combination with other PIs darunavir 600 mg bid, fosamprenavir, saquinavir, or tipranavir due to lack of exposure data. Furthermore, since coadministration of cobicistat with different ARV agents (other than ATV, DRV, and elvitegravir) has not been studied, its effect on the plasma concentrations of other ARVs is unpredictable, increasing the risk of therapeutic failure and development of resistance.13
Pharmacokinetics
Cobicistat is rapidly absorbed after oral administration with its maximum plasma concentration (Cmax) reached within 4 hours.13,17 Cobicistat is primarily metabolized by CYP3A and to a lesser degree by CYP2D6. Since it is also a CYP3A inhibitor, it follows nonlinear pharmacokinetics (PK) with increased dosing of cobicistat as evidenced by the mean area under the plasma concentration–time curve (AUC) over the dosing interval. For example, in one study in healthy volunteers, with increased dosing of cobicistat (50–300 mg/day), the AUC increased more than proportionally with higher doses (as high as 47-fold).18 It is recommended that cobicistat be taken with food since systemic exposure to cobicistat is greater in the fed versus fasted state. This recommendation is supported by the greater PK properties of cobicistat including Cmax, AUC, and Tmax in healthy volunteers who took a single dose of darunavir 800 with 150 mg of cobicistat in the fed versus fasted state.19 Cobicistat is highly protein bound to plasma (97%–98%), and its mean apparent volume of distribution at steady state was 152 and 100 L following doses of 100 and 200 mg/day in healthy volunteers, respectively.18 Cobicistat is eliminated primarily via feces (86%) and minimally via urine (8.2%).13 The apparent clearance of cobicistat was significantly decreased (up to 95%) with increased dosing in healthy volunteers who received doses of cobicistat ranging from 50 to 300 mg/day.18 The median plasma half-life of cobicistat is 3–4 hours.13
DDIs between PI combinations ATV/c and DRV/c
As mentioned earlier, cobicistat is metabolized by CYP3A and to a minor extent by CYP2D6 enzymes and does not undergo glucuronidation.13 Compared to ritonavir, cobicistat is a much weaker activator of the pregnane X receptor (activation increases CYP3A expression) and is less likely to cause DDIs through CYP3A induction.14,17 Ritonavir appears to induce CYP3A4, CYP1A2, CYP2B6, CYP2C9, CYP2C19, and CYP2C8, as well as Phase II enzyme uridine 5-diphospho-glucuronosyltransferase (UGT), increasing its potential for multiple DDIs.16 Of note, cobicistat is slightly more selective than ritonavir in its inhibition of CYP3A, and also exhibits reduced inhibitory activity toward CYP2D6, CYP2C8, and CYP2C9, further decreasing its potential for interactions with other drugs that are substrates of these specific CYP enzymes.14
In addition to cobicistat being a major CYP3A4 substrate, in vitro data suggest that it may also be a substrate for drug transporters p-glycoprotein and breast cancer resistance protein.20 Cobicistat may also inhibit p-glycoprotein, breast cancer resistance protein, and other drug transporters including organic anion-transporting polypeptide 1B1 and 1B3.13,20 In vitro data have also shown that cobicistat may inhibit proximal renal tubular cell drug transporters multidrug and toxin extrusion protein 1 (MATE-1) and organic cation transporter (OCT) novel type 1.21 It is anticipated that cobicistat will have similar DDIs to ritonavir with respect to their shared effects as CYP3A4 and p-glycoprotein inhibitors (refer to Table 2 which summarizes potentially clinically relevant DDIs between PI combinations ATV/c and DRV/c with selected coadministered agents). Many of these are theoretical DDIs; future studies are warranted to determine the clinical effects of cobicistat on the plasma concentrations of other drugs. The reader is encouraged to refer to up-to-date DDI information available on the HIV Drug Interactions website and DHHS guidelines.2,22
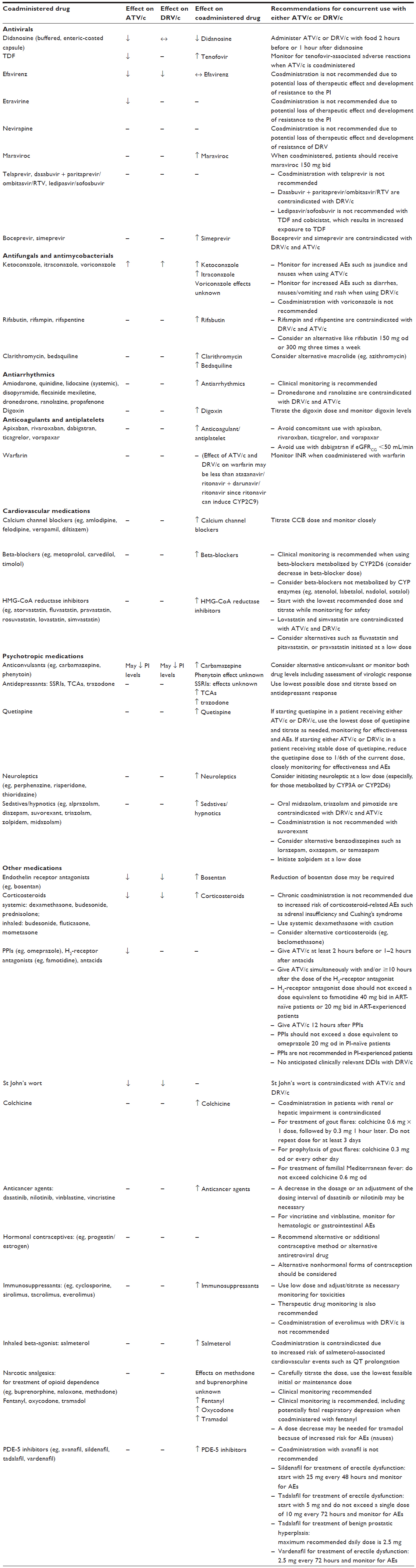 | Table 2 Summary of drug–drug interactions between protease inhibitor combinations atazanavir/cobicistat and darunavir/cobicistat with selected coadministered drugs2,54,55 |
Bioequivalence PK studies with ATV/c and DRV/c
When comparing ATV/c 300/150 mg as an FDC to ATV/ritonavir (ATV/r) 300/100 mg, one study showed bioequivalent ATV exposures based on geometric means ratio (GMR) for the ATV Cmax, Ctau, and AUCtau in the fed state (Table 3).23 Furthermore, another ATV study comparing ATV/c 300/150 mg as an FDC to their separate agents showed bioequivalent ATV exposures based on GMRs for the ATV Cmax, Ctau, and AUCtau in both fasted and fed states (Table 3).24 These two studies established bioequivalence between cobicistat and ritonavir as pharmacoenhancers of ATV, since the 90% confidence interval (CI) for the GMR for pertinent PK parameters was within the bioequivalence range of 80%–125%.
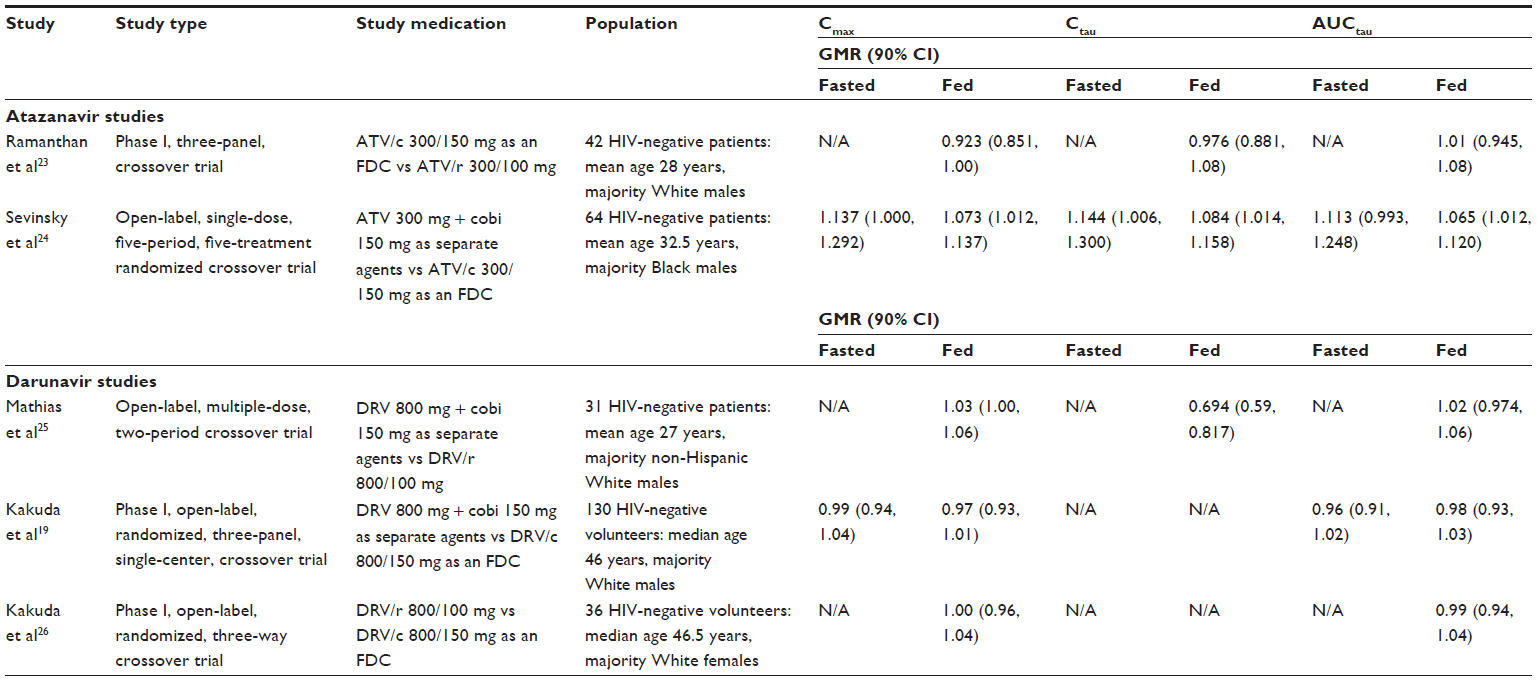 | Table 3 Summary of bioequivalence pharmacokinetics studies with ATV/c and DRV/c |
When comparing DRV 800 mg + cobicistat 150 mg as separate agents to DRV/ritonavir (DRV/r) 800/100 mg as separate agents, one study demonstrated bioequivalent DRV exposures based on the GMRs for the DRV Cmax and AUCtau (Table 3).25 Although bioequivalence for DRV exposures was not established for Ctau in this study, the mean concentration was greater than 18-fold above the protein-adjusted EC50 for wild-type virus (55 ng/mL) supporting sufficient ARV activity. Another study comparing DRV/c 800/150 mg as FDC to DRV 800 mg and cobicistat 150 mg as separate agents under either fasted or fed conditions showed bioequivalent DRV exposures based on GMRs for the DRV Cmax, Ctau, and AUCtau (Table 3).19 An additional study comparing DRV/r 800/100 mg as separate agents to DRV/c 800/150 mg as an FDC showed bioequivalent DRV exposures based on GMRs for the DRV Cmax, Ctau, and AUCtau in the fed state (Table 3).26 Overall, these three bioequivalence studies provide adequate support for using cobicistat as an alternative pharmacoenhancer to ritonavir when coadministered with DRV.
Clinical efficacy
Atazanavir with cobicistat
The clinical efficacy of cobicistat versus ritonavir as pharmacoenhancers for ATV both administered with a tenofovir disoproxil fumarate (TDF)/FTC NRTI backbone as initial treatment for HIV-1 infection has been evaluated in two randomized double-blind, multicenter clinical studies (one Phase II study and one Phase III study including a follow-up efficacy report from 144 weeks).27–29
In the Phase II study, eligible criteria for inclusion were those who were HIV-1-infected adults (≥18 years of age), screening plasma HIV-1 RNA ≥5,000 copies/mL, CD4 cell count >50 cells/μL, no prior use of ARVs (treatment-naïve), no primary PI genotypic resistance mutations, estimated glomerular filtration rate Cockcroft–Gault calculation (eGFRCG) ≥80 mL/min, aspartate amino transferase (AST) or alanine aminotransferase (ALT) ≤2.5 times upper limit of normal, total bilirubin ≤1.5 mg/dL, and a negative serum pregnancy test for women.27 Those excluded from this study were those with hepatitis coinfections B or C, new AIDS-defining condition within 30 days of screening, or vaccination within 90 days of starting treatment. Patients included in the study were randomized 2:1 to treatment with open-label ATV 300 mg and FTC/TDF 200/300 mg and either cobicistat 150 mg once daily (ATV/c group) or ritonavir 100 mg od (ATV/r group).27 The primary efficacy endpoint of this study was the proportion of patients with HIV-1 RNA <50 copies/mL at week 24. Secondary endpoints were the proportion of patients with HIV-1 RNA <50 copies/mL at week 48, and CD4 cell count changes from baseline to weeks 24 and 48.
Among 137 patients screened, 85 patients were randomized 2:1 to the ATV/c or ATV/r groups. Six patients randomized to the ATV/c group never received the study drug. Therefore, 79 randomized patients (ATV/c [n=50] and ATV/r [n=29]) made up the intent-to-treat analysis efficacy and safety populations. There were no significant differences in baseline demographics and disease characteristics between the two treatment groups. This population was considerably young (mean age 34–37), mostly male (86%–94%), and predominantly White (55%–62%) and Black (28%–36%). Mean HIV clinical parameters HIV-1 RNA (log10 copies/mL) and CD4 cell count (cells/mm3) for ATV/c and ATV/r groups at baseline were 4.56 and 4.69 and 365 and 343, respectively. Stratified by viral load, 76% patients had HIV-1 RNA <100,000 copies/mL and 24% HIV-1 RNA >100,000 copies/mL in the ATV/c group, whereas 62% and 38% fell within these viral load thresholds in the ATV/r group, respectively.
Results from the study showed that at week 24, 84% of ATV/c patients and 86% of ATV/r patients had HIV-1 RNA <50 copies/mL, and 82% and 86% at week 48, respectively. Mean CD4 cell count changes from baseline to week 24 increased by 203 and 199 cells/mm3 for ATV/c and ATV/r groups, respectively, and from baseline to week 48 increased by 230 and 206 cells/mm3. No patients experienced virologic failure in either treatment group.27
In the Phase III noninferiority study, inclusion criteria were mostly similar to the Phase II study except for the following eligibility standards required for the Phase III study: sensitivity to ATV and the NRTI backbone FTC/TDF (no resistance mutations), eGFRCG of ≥70 mL/min, AST/ALT levels of ≤5 times upper limit of normal, an absolute neutrophil count of ≥1,000 cells/mm3, a platelet count of ≥50,000 platelets/mm3, and a hemoglobin level of ≥8.5 g/dL.28 Worth noting, patients who had positivity for HBV surface antigen or HCV antibody were also allowed in this study. Patients were excluded from the study if they had any new AIDS-defining conditions or serious infections within 30 days of screening.28 Eligible patients were randomized at a 1:1 ratio to receive either cobicistat or ritonavir, each administered once daily with ATV and standard FTC/TDF combination. The primary efficacy endpoint of this study was the proportion of patients with HIV-1 RNA <50 copies/mL at week 48 (in accordance with the FDA-defined snapshot analysis defined as patients with HIV-1 RNA load of <50 copies/mL between days 309 and 378). Noninferiority of cobicistat to ritonavir treatment was assessed using the intention-to-treat (ITT) population and conventional 95% CI approach with a prespecified noninferiority 12% margin.28 Additional efficacy endpoints included several others: a per-protocol snapshot analysis, time to loss of virologic response algorithm, proportion of patients with virologic suppression (HIV-1 RNA load of <50 copies/mL) using methods such as missing-equals-excluded (M=E) and missing-equals-failure (M=F), and the changes in CD4 cell counts from baseline to week 48.28
During the screening period, 698 patients were randomly assigned to receive either cobicistat or ritonavir and matching placebo. Among these, 692 were included in the study and 344 were randomized to receive cobicistat and 348 to ritonavir. Baseline demographic characteristics were similar between the two treatment groups. Among the two groups, mean age was 37–38, primarily male 82.5%–83.4%, 10.6%–12.8% were Asian, 18.1%–18.9% were Black or had an African heritage, 26.4%–28.5% were Hispanic/Latino, and the majority were White 57.6%–61.8%. In terms of hepatitis coinfections, 2.6%–4.7% had HBsAg positivity and 4.6%–6.1% had HCV antibody positivity. Pertinent baseline HIV clinical parameters included median log10 copies/mL 4.78 and 4.84 in the ATV/c and ATV/r groups, respectively. Furthermore, 38.4% vs 41.1% had a HIV-1 RNA load >100,000 copies/mL, as well as mean (± standard deviation [SD]) CD4 cell count at baseline was 353±170.5 and 351±175.5 cells/mm3 in the ATV/c and ATV/r groups, respectively.
Results of the study showed that 293/344 patients (85.2%) versus 304/348 patients (87.4%) achieved HIV-1 RNA load of <50 copies/mL at week 48 according to the FDA snapshot ITT analysis (difference, −2.2% [95% CI, −7.4%–3.0%]) in the ATV/c and ATV/r groups, respectively. These results satisfied the criteria for noninferiority of cobicistat versus ritonavir both in combination with ATV and FTC/TDF. This finding is further supported by similar results in the two treatment groups regarding virologic response using other efficacy parameters. In addition, in a subgroup analysis, virologic response was similar between the two groups for patients with a HIV-1 RNA viral load of >100,000 copies/mL (~86%). Mean changes in CD4 cell counts from baseline to 48 weeks were similar between the two groups (+213 cells/mm3 in the ATV/c group and +219 cells/mm3 in the ATV/r group). Rates of virologic failure were comparable between the two treatment groups (5.6% [n=20] in the ATV/c group and 4% [n=14] in the ATV/r group). In the overall population (n=692), 3.5% of patients (12 in ATV/c and 12 in ATV/r group) met the criteria for resistance testing. Among 10 patients in the ATV/c group with available data, two developed resistance mutations to FTC (M184V) and no resistance to TDF and PIs. None in the ATV/r group developed resistance mutations to any of the ARV agents. Lastly, virologic responses (HIV-1 RNA viral load <50 copies/mL at week 48) in the ATV/c group were comparable to those in the ATV/r group across all various patient subgroups including covariates age, sex, race, adherence to HAART, baseline CD4 cell count, and baseline HIV-1 RNA viral load.
Follow-up efficacy data regarding this Phase III study through week 144 was recently published.29 Primary efficacy outcome of this study was similar to Phase III study (proportion of patients with virologic suppression at week 144 using US FDA-defined snapshot analysis).29 At week 144, virologic responses in the ATV/c group were comparable to the ATV/r group (72.1% vs 74.1%, respectively). Mean changes in CD4 cell counts from baseline to 144 weeks were similar between the two groups (+310 cells/mm3 in the ATV/c group and +332 cells/mm3 in the ATV/r group). Development of resistance to the two treatment regimens through week 144 was infrequent; three patients in the ATV/c group and one in the ATV/r group developed resistance to FTC, with no detection of resistance to either PI or TDF in any of the study subjects.29
Overall, these two randomized clinical trials demonstrate that cobicistat had sustained and comparable efficacy to ritonavir as a pharmacoenhancer of ATV through 144 weeks of treatment.27–29
Darunavir with cobicistat
The clinical efficacy of cobicistat as a pharmacoenhancer for DRV administered with a background regimen of two, fully active, investigator selected NRTIs as treatment for both treatment-naïve and -experienced HIV-1-infected patients (with no DRV mutations) has been evaluated in one Phase IIIb open-label, single-arm, US multicenter study, including a subgroup analysis over 48 weeks.30,31 Most of the impetus for moving forward in evaluating DRV/c in this clinical study stemmed from bioequivalent studies including fasting and fed conditions discussed earlier.19,26
In the Phase III study, patients were eligible for inclusion if they met the following criteria: adult HIV-1-infected, treatment-naïve or treatment-experienced (on stable ARV regimen for at least 12 weeks prior to screening), no DRV resistance-associated mutations (RAMs) present at baseline (these were defined as having any one of the following mutations such as V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V, or L89V in the HIV-1 protease gene), plasma HIV-1 RNA ≥1,000 copies/mL at screening, eGFRCG ≥80 mL/min, and genotypic sensitivity to two NRTIs chosen by the investigator. Exclusion criteria were previous or current use of DRV, a newly diagnosed AIDS-defining condition, proven/suspected acute hepatitis or treatment for hepatitis C, and females who were pregnant or breastfeeding.30 This Phase III study was conducted at 56 sites throughout the US. Since this was an open-label, single arm clinical trial, all patients received darunavir 800 mg od (2×400 mg tablets) plus cobicistat (150 mg tablet od), together with two NRTIs, all administered with food. The primary endpoint of this study was any treatment emergent grade 3 (severe) or grade 4 (life threatening) AEs that occurred through week 24.30 Secondary outcome endpoints were any treatment-emergent AEs through weeks 24 and 48, including those who discontinued the study drug because of an AE, and ARV efficacy at weeks 24 and 48 (defined as proportion of patients with HIV-1 RNA <50 copies/mL during week 24 or week 48 window according to FDA Snapshot analysis).30
Among 397 patients screened, 313 were enrolled and treated (included in the ITT population). One patient was never treated and out of the 83 screening failures most were due to screening eGFRCG <80 mL/min or HIV-1 RNA <1,000 copies/mL. Among the 313 ITT patients, 295 were treatment-naïve and 18 were treatment-experienced. No patients had any DRV RAMs. Overall, 86% (268/313) completed the study through week 48, and 14% (45/313) discontinued, mainly because of AEs (n=15) and loss to follow-up (n=13).30 Since no comparative treatment groups are included in this study, the demographics describing this population (N=313) as a whole are as follows: 89% were male, 60% White, 35% African Heritage, and median age 35 (range 18–72). Among HIV clinical parameters, median baseline HIV-1 RNA was 4.8 log10 copies/mL (with 42% having a HIV-1 RNA >100,000 copies/mL) and median CD4 cell count was 361 cells/mm3. Overall, 2% of patients were coinfected with HBV and 3% with HCV. Other important baseline treatment considerations included median eGFRCG 114 mL/min and most of the population were treated with a background FTC/TDF NRTI regimen (96% [n=301]).
The results of the study showed that median adherence to DRV/c in the overall population through week 48 was 100% as measured by pill count. These excellent adherence rates support the relatively good virologic responses achieved by the ITT population. Overall, the virologic response rate was 82% (258/313; 95% CI 78%–87%) at week 24 and 81% (253/313; 95% CI 76%–85%) through week 48. In the treatment-naïve patients, virologic response rate at week 48 was 83% (244/295; 95% CI 78%–87%). Rates of virologic failure (11% vs 8%) and missing virologic data (9% vs 9%) at week 48 were similar in the total population versus treatment-naïve population, respectively. Virologic response rates at week 48 were also comparative in patients who had baseline HIV-1 RNA ≤100,000 copies/mL (81%) and >100,000 copies/mL (80%) suggesting durable efficacy of DRV/c regimen regardless of baseline viral load. These rates were also similar among treatment-naïve patients (84% vs 81%, respectively). In terms of immunologic response, the overall population (M=E) experienced an increase in median change in CD4 cell count from baseline to week 24 of 131 cells/mm3 and from baseline to week 48 of 167 cells/mm3. In treatment-naïve patients, the median change in CD4 cell count from baseline to week 48 increased by 169 cells/mm3.
In this study, 15 patients had samples eligible for resistance analysis.30 In the postbaseline genotypic analysis through week 48 in the 15 patients, one treatment-experienced patient developed a DRV RAM at position I84 as a mixture with wild-type (I84I/V), which was not associated with DRV phenotypic resistance. Two of the 15 patients (one treatment-experienced and one treatment-naïve) developed the M184V RAM associated with phenotypic resistance to both lamivudine and FTC; both of these patients were receiving FTC as part of their NRTI background regimen. In the other 12 patients evaluated for resistance, no new primary RAMs were detected in any of their respective ARV regimens. Overall, these low rates of resistance confirm the efficacy of DRV boosted with cobicistat through its characteristically high genetic barrier.
This Phase III study, including a follow-up subgroup analysis of treatment-naïve HIV-1-infected patients, reflected virologic and immunologic responses consistent with prior published data for DRV/r 800/100 mg od, supporting the use of DRV/c 800/150 mg od in combination with two NRTIs for future treatment of treatment-naïve and -experienced HIV-1-infected patients with no DRV RAMs.30,31
Safety and tolerability
Atazanavir with cobicistat
In the Phase II study, multiple study time assessments occurred at screening, baseline, weeks 2, 4, 8, 12, 16, and then every 8 weeks through week 48 to capture relevant safety data.27 Treatment-related AEs were similar among both ATV/c and ATV/r groups, except nausea was experienced by more patients in the ATV/c group (Table 4). Mean (SD) baseline total bilirubin (mg/dL) was 0.6 (0.37) and 0.5 (0.20) for ATV/c and ATV/r groups, respectively. During the 48-week study period, mean indirect bilirubin (mg/dL) was higher among patients in the ATV/c group versus the ATV/r group, but was only statistically significant at week 2 (3.2 vs 2.1, respectively; P=0.05). At weeks 24 and 48, there were no significant differences between the two treatment groups with respect to mean changes in fasting lipids from baseline. Mean (SD) baseline eGFRCG (mL/min) was 117 and 122 for ATV/c and ATV/r groups, respectively. Mean percentage change from baseline in eGFRCG was numerically decreased more in the ATV/c group versus the ATV/r group throughout the 48 weeks, but was only significant at week 2 (108 mL/min [−8%] vs 117 mL/min [−3%]; P=0.02), remaining comparable at 48 weeks (104 mL/min [−12%] vs 111 mL/min [−11%]; P=0.8). Among the 4% (3/79) of patients who discontinued treatment prematurely due to AEs related to study drugs, 4% (2/50) in the ATV/c group (one experienced moderate vomiting on day 3, and the other patient had a severe generalized maculopapular rash on day 11) and 3% (1/29) in the ATV/r group experienced a mild ocular icterus on day 21.
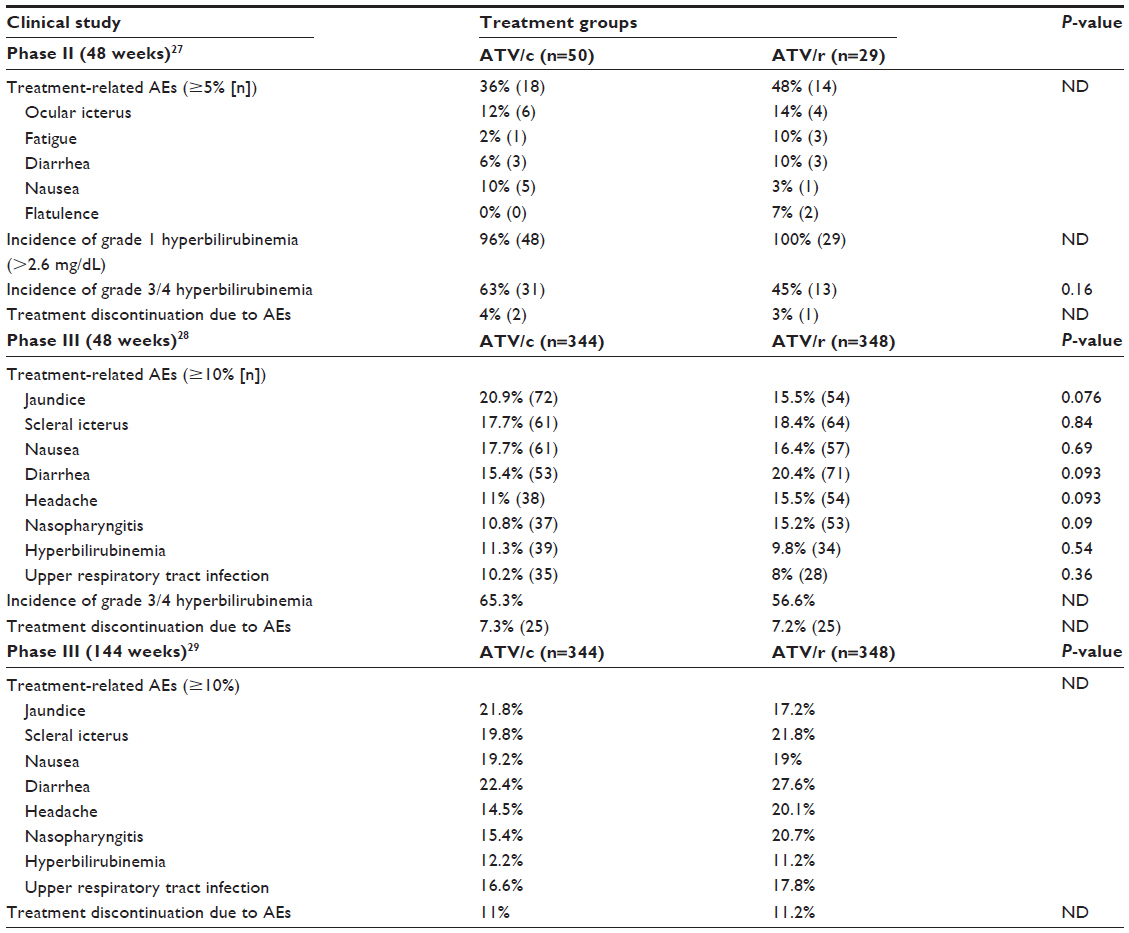 | Table 4 Summary of studies comparing tolerability of ATV/c to ATV/r |
In the Phase III study, additional safety data were collected through standard patient study visits occurring at weeks 2, 4, 8, 12, 16, 24, 32, 40, and 48.28 Treatment-related AEs were similar among both ATV/c and ATV/r groups and were considered mild or moderate (Table 4). Jaundice, scleral icterus, and hyperbilirubinemia were some of the most common AEs occurring similarly between the ATV/c and ATV/r groups (40.7% and 36.2%, respectively); these were also the primary AEs leading to low rates of discontinuation of treatment in both groups (3.5% and 3.2%). There were no significant differences in baseline eGFRCG between treatment groups. Renal AEs resulting in treatment discontinuation were reported for six patients (1.7%) in the ATV/c group and five patients (1.4%) in the ATV/r group. In the ATV/c group, one patient had treatment discontinued because of an increase in serum creatinine of 0.4 mg/dL, which led to an eGFRCG of less than 50 mL/min. Baseline serum creatinine for this patient was 0.86 mg/dL (eGFRCG 70 mL/min) returning to 0.95 mg/dL after discontinuation of treatment. The remaining five ATV/c patients had proximal tubulopathy manifestations (hypophosphatemia, proteinuria, or normoglycemic glycosuria). Among these five patients, four had available follow-up data showing improvement in renal laboratory parameters such as serum creatinine, serum phosphate, urine protein, and urine glucose levels after discontinuation of ATV/c.28 The one patient without follow-up data in the ATV/c group who developed proximal tubulopathy had Enterobacter sepsis and acute renal failure.28 In the ATV/r group, 3/5 patients had increases in serum creatinine without proximal tubulopathy and the other two patients had proximal tubulopathy with improvement in serum creatinine and complete reversal of proximal tubulopathy markers upon discontinuation of ATV/r. A small significant increase in median serum creatinine occurred between baseline and week 48 for the ATV/c group versus ATV/r group (+0.13 and +0.09 mg/dL, respectively; P<0.001) with a corresponding significant decrease in median eGFRCG during this study period (−12.9 and −9.1 mL/min, respectively; P<0.001). These changes developed initially in the first few weeks after starting treatment and plateaued off for the remaining study period through week 48.
Since TDF is associated with bone abnormalities, fracture incidence was evaluated in this study with two patients (0.6%) in ATV/c group and four patients (1.1%) in ATV/r group experiencing this AE. Most of these fractures were trauma-related except one patient in the ATV/r group who had a nontraumatic spinal compression fracture (considered an old and not acute fracture). Incidence of grade 3/4 elevations in liver enzymes AST and ALT were similar between ATV/c and ATV/r groups (2.9%/3.2% vs 2%/2%, respectively). One patient from each group had grade 3/4 elevations in AST or ALT, as well as hyperbilirubinemia (direct bilirubin level >1.5 mg/dL) possibly suggesting that hyperbilirubinemia was not associated with atazanavir. These clinical sequelae may have resulted from hepatitis coinfection, since the patient in the ATV/c group had acute HCV and the subject in the ATV/r group had suspected acute HBV. There were no significant differences between the two treatment groups with respect to mean changes in fasting lipids from baseline to week 48. Gastrointestinal adverse-related events were comparable between both treatment groups rarely leading to discontinuation with only one patient in the ATV/c group discontinuing treatment because of vomiting, one in the ATV/r group discontinuing treatment because of nausea, and none discontinuing because of diarrhea.
Follow-up safety data regarding this Phase III study showed that the tolerability profile of ATV/c compared to ATV/r remained similar long-term through 144 weeks.29 Most commonly reported AEs for both treatment groups are shown in Table 4.
The most common AEs in the ATV/c and ATV/r groups were due to elevated bilirubin (43.6% and 41.4%, respectively); these were also the primary AEs leading to discontinuation in both treatment groups (4.9% vs 4.0%). Rates of nausea and diarrhea were similar between the two groups. A small increase in median serum creatinine occurred between baseline and week 144 for the ATV/c group versus ATV/r group (+0.13 and +0.07 mg/dL, respectively) with a corresponding decrease in median eGFRCG during this study period (−15.1 and −7.5 mL/min, respectively). These changes in renal parameters developed initially in the first 4 weeks after starting treatment with minimal progression between weeks 48 and 144. A similar number of patients (n=7; 2%) in each group developed proximal renal tubulopathy. In the ATV/c group, 6/7 had reversibility of renal laboratory abnormalities (proteinuria, glycosuria, or hypophosphatemia) and improvements in serum creatinine after discontinuation of treatment (with one patient not discontinuing treatment). Among the six patients, three patients had renal laboratory parameters that normalized/returned to baseline, whereas in the three other patients with persistent renal laboratory abnormalities after discontinuation there was not a sufficient follow-up period to assess full reversibility in two patients and the third patient was switched to a ATV/r with TDF/FTC regimen.29 There were no significant differences in fasting lipid profiles between ATV/c and ATV/r groups from baseline to week 144.
In both Phase II and III studies, ATV/c appeared to be well tolerated displaying a tolerability profile similar to ATV/r suggesting that cobicistat has comparable safety to ritonavir as a pharmacoenhancer of ATV through 144 weeks of treatment.27–29
Darunavir with cobicistat
In the Phase IIIb single-arm, open-label study, the safety of DRV/c was evaluated at multiple time points including baseline and at weeks 2, 4, 8, 12, 16, 24, 36, and 48.30 Common AEs through week 48 that were considered study drug-related included diarrhea, nausea, headache, and flatulence (Table 5); most of these were mild in severity (grades 1 and 2). The incidence of any treatment-emergent grade 3 or 4 AEs regardless of causality was low through weeks 24 and 48. Among the overall population at 24 weeks, 16 patients had grade 3 AEs (5%) and two patients had grade 4 AEs (0.6%). In the treatment-naïve group through week 48, the most common grade 3/4 AEs regardless of causality were hypersensitivity (two patients had grade 3 and one patient had grade 4), maculopapular rash (two patients had grade 3), and peripheral neuropathy (two patients had grade 3).30 Study drug-related grade 3 AEs leading to study drug discontinuation occurred in five patients (2%); one patient had immune reconstitution syndrome, two patients had hypersensitivity, one patient had maculopapular rash, and one patient had allergic dermatitis, maculopapular rash, and vesicular rash. Serious AEs leading to study drug discontinuation but not study-drug-related included one patient who had grade 4 thrombocytopenic purpura and one patient who had grade 4 hypersensitivity (assumed to be related to concomitant lisinopril use). There were three serious AEs potentially related to study drug that were experienced by treatment-naïve patients (one patient reported immune reconstitution syndrome, one patient a rash, and the other patient a maculopapular rash). Most common AEs related to study drug and leading to discontinuation through week 48 were rash and maculopapular rash, nausea, and hypersensitivity.30 For patients who experienced these AEs, they resolved upon discontinuation of study drug. Among the overall population, only one patient (in the treatment-naïve group) discontinued because of a renal tubular disorder; this was considered mild in severity and resolved when the patient was switched to a different HAART regimen comprising DRV/r, abacavir, and lamivudine. Among the treatment-emergent grade 3–4 laboratory abnormalities, an increased creatine kinase was reported most commonly: 6% (18/310) in overall population at week 24, 7% (22/310) at week 48, and 8% (22/292) in treatment-naïve patients at week 48.
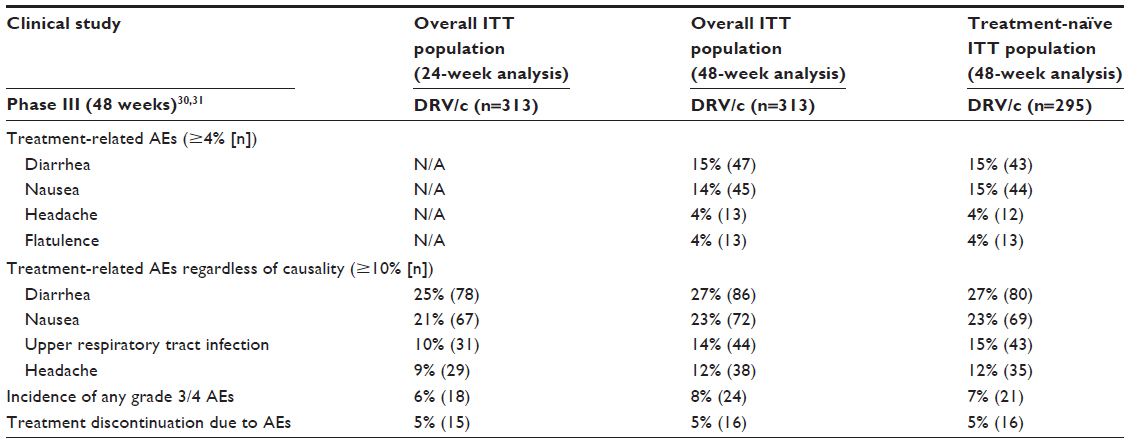 | Table 5 Summary of studies demonstrating tolerability of DRV/c |
Since most patients included in this study (99%) were taking TDF as one of the NRTIs in their backbone therapy, both renal AEs and incidence of bone fractures were evaluated. Through week 48, eight bone fractures (secondary to traumatic mechanisms) were reported. Baseline eGFRCG mL/min (median [range]) was 114 (67–321) and 115 (67–321) for overall population and treatment-naïve patients, respectively. An increase in median serum creatinine level (+0.10 mg/dL) from baseline to week 2 was reported among both the overall and treatment-naïve groups, remaining stable through week 48 (+0.09 and +0.08 mg/dL, respectively).
DRV/c was well tolerated in this study having a tolerability profile similar to published data for DRV/r 800/100 mg od, thus, supporting the use of DRV with cobicistat as an alternative pharmacoenhancer to ritonavir in both treatment-naïve and treatment-experienced HIV patients with no darunavir RAMs.30,31
Discussion/clinical implications
Clinical studies have shown that cobicistat has comparable and sustained efficacy and safety versus ritonavir as a pharmacoenhancer for ATV.27–29 In addition, one recent clinical study including a follow-up subgroup analysis of treatment-naïve HIV-1-infected patients showed efficacy and safety of cobicistat as a pharmacoenhancer for DRV.30,31 Rates of virologic suppression in these studies are comparable to other clinical studies comparing ATV/r to alternative HAART including different PIs such as fosamprenavir/ritonavir and lopinavir/ritonavir and the NNRTI efavirenz, as well as in clinical studies comparing DRV/r to alternative HAART including the PI lopinavir/ritonavir in both treatment-naïve and treatment-experienced patients with no DRV RAMs.32–36 Furthermore, studies in healthy volunteers have established bioequivalence between cobicistat and ritonavir as pharmacoenhancers of both ATV and DRV.19,23–26 Together, all of these studies support the use of cobicistat in combination with either ATV or DRV as novel, simplified treatment options for HIV-1 infection.
One of the advantages of starting treatment in HIV-1-infected patients with either ATV/c or DRV/c is their characteristically high genetic barrier compared to other ARV classes such as NNRTIs and integrase strand transfer inhibitors. This is supported further by comparable efficacy of ATV/c and DRV/c (>80% had a HIV RNA <50 copies/mL) after 48 weeks of treatment with low rates of virologic failure secondary to the development of resistance mutations.28,31 Only two patients in each of these clinical studies developed resistance to FTC (M184V RAM) and none developed resistance to either TDF or PI (ATV or DRV). It is unclear whether ATV/c is effective in treatment-experienced patients, since clinical studies only included those who were treatment-naïve. One limitation of the DRV/c Phase III open-label study is that it only included a single arm DRV/c treatment group with no comparator group such as DRV/r, but it did include both treatment-naïve and treatment-experienced patients demonstrating efficacy in both groups.30,31 Furthermore, when choosing between ATV/c and DRV/c as an initial or alternative treatment option for HIV-1 patients, DRV/c does have a higher genetic barrier to resistance than ATV/c and might be a more effective choice in patients suspected of having reduced adherence to future treatment and no darunavir RAMs at baseline.
In the ATV clinical trial (144 weeks), the most common AEs in the ATV/c and ATV/r groups were due to elevated bilirubin (43.6% and 41.4%, respectively); these were also the primary AEs leading to discontinuation in both treatment groups (4.9% vs 4.0%, respectively).29 Results from a large comparative clinical trial (AIDS Clinical Trials Group A5257 study) comparing ATV/r + TDF/FTC to DRV/r or raltegravir + TDF/FTC showed similar virologic suppression rates, but a greater rate of toxicities and treatment-related discontinuation in the ATV/r arm secondary to hyperbilirubinemia.37 For this reason, the DHHS guidelines recently updated their recommendations moving ATV/r + TDF/FTC combination from a “recommended” to an “alternative” treatment option for treatment-naïve HIV-1-infected patients.2 Elevation of indirect bilirubin is a common side effect of ATV that is caused by competitive inhibition of UGT1A1 enzyme leading to reduced glucuronidation of total bilirubin and increased serum concentrations. In addition, UGT1A1 genetics can play a role in influencing predisposition to patients experiencing varying degrees of hyperbilirubinemia.38 Although mean indirect bilirubin levels throughout the Phase II study period were slightly higher in patients receiving ATV/c compared with those treated with ATV/r, these differences did not translate into clinically significant differences (eg, manifestations of elevated indirect bilirubin levels such as ocular icterus and jaundice were comparable between two groups).27 Clinically, if a subject is treated with ATV/c and has an undetectable viral load, it is not necessary to change therapy unless hyperbilirubinemia presents a cosmetic problem to the patient. A potential solution in reversing hyperbilirubinemia in this case could be a simple switch from treatment with ATV/c to DRV/c.
Cobicistat has been associated with small increases in serum creatinine and corresponding decreases in eGFRCG because of its effects on tubular secretion rather than glomerular filtration of serum creatinine.39 Iohexol is considered an acceptable standard for assessing actual glomerular filtration rate of the kidneys; it is not secreted or reabsorbed.40 A PK/pharmacodynamic study in healthy HIV-uninfected adults showed that cobicistat did not affect iohexol-measured (actual) glomerular filtration rate.39 Clinical studies evaluating ATV/c versus ATV/r showed that mean eGFRCG decreased from baseline in both treatment groups, with more of an acute effect after 2 weeks in the ATV/c group versus ATV/r group with similar differences after 144 weeks.27–29 The magnitude of these changes in eGFRCG is not considered to be clinically significant, since they are relatively small and did not progress through 144 weeks. Similar findings of the effects of cobicistat on serum creatinine and eGFRCG were observed in the DRV/c clinical studies.30,31 These renal findings are also consistent with results from studies evaluating elvitegravir/cobicistat/TDF/FTC for treatment of HIV-1 infection, further supporting the effects of cobicistat on tubular secretion of serum creatinine.41–43
One proposed mechanism that might explain why small increases in serum creatinine and reduced eGFRCG were also present in patients treated with ATV/r in clinical studies might be due to ritonavir’s inhibition of a renal transporter MATE-1 responsible for tubular secretion of creatinine.20,27,29,44 In vitro data have demonstrated that cobicistat is a weak inhibitor of human renal transporters OCT2 and MATE2-K and is a more potent inhibitor of OCTN1 and MATE1 (Figure 2).39 Overall, it seems that the inhibition of MATE 1 (whereby creatinine is a substrate) by both cobicistat and ritonavir may provide a plausible biological explanation for the small increases in serum creatinine observed in clinical studies. It is important to note that TFV is a substrate for different renal transporters organic anion transporter 1/3 and MRP4 in the proximal tubule and is not expected to have DDIs with cobicistat (Figure 2).21
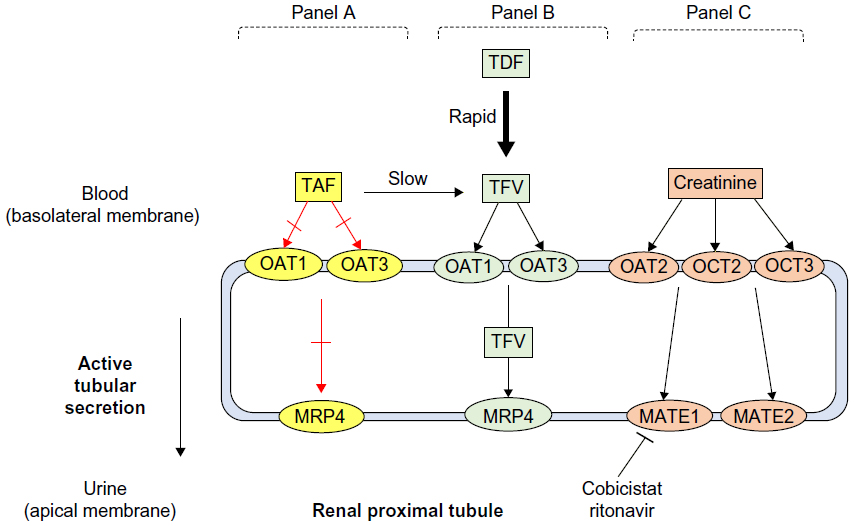 | Figure 2 Effect of TAF, TDF, cobicistat, and ritonavir on transporters in the renal proximal tubule. |
Rates of treatment discontinuation due to renal AEs in the Phase III ATV/c study (1.7% vs 1.4% in the ATV/r group) are consistent with previous studies.28,33 Additional clinical studies using TDF-containing boosted PI regimens showed similar rates of treatment discontinuation secondary to renal AEs (0%–3%).45–51 In the Phase III ATV/c study, five recipients in the ATV/c group and two in the ATV/r group discontinued treatment because of proximal tubulopathy; these findings are consistent with the TDF safety profile associated with proximal tubulopathy (also referred to as Fanconi syndrome).28 Furthermore, all patients who developed proximal tubulopathy had a reversal in their presentation (proteinuria, glycosuria, or hypophosphatemia) with improvements in serum creatinine when discontinuing treatment.28 Similar findings were observed in this study extended out to 144 weeks in which a similar number of patients (n=7; 2%) in each group (ATV/c vs ATV/r) developed proximal renal tubulopathy, and almost all patients had reversibility of renal laboratory abnormalities and improvements in serum creatinine after discontinuation of treatment.29
Because clinical studies using ATV/c and DRV/c restricted eligibility to including only those patients who had an eGFRCG >70 mL/min in most of these studies, it is unclear whether cobicistat can be used as a pharmacoenhancer for both ATV and DRV below these renal thresholds. However, one Phase III, noncomparative, open-label clinical trial showed that patients who had an eGFRCG between 50 and 89 mL/min and were switched from a stable ARV regimen containing DRV/r and ATV/r to DRV/c and ATV/c, respectively, maintained virologic suppression and well tolerated in terms of renal AEs (no cases of proximal renal tubulopathy and small changes in eGFRCG occurred as early as week 2 and nonprogressive through week 48).52 Similar renal findings were observed in HIV-1-infected treatment-naïve patients with mild-to-moderate renal impairment (eGFRCG 50–89 mL/min) who were treated with elvitegravir/cobicistat/TFV/FTC through 48 weeks.53
The upcoming approval of tenofovir alafenamide in the US (TAF, formerly GS-7340) might expand the clinical use of recent approvals ATV/c and DRV/c. Currently, ATV/c and DRV/c in combination with TDF is not recommended for patients who have an eGFRCG <70 mL/min.54,55 However, TAF is a next generation oral prodrug of TFV developed with improved safety and efficacy compared to TDF. TDF is metabolized to TFV, which is then phosphorylated twice intracellularly to the active moiety TFV diphosphate.56 TAF is more stable in plasma and is metabolized inside target cells via hydrolysis by intracellular cathepsin A, which results in higher intracellular levels of the active metabolite TFV diphosphate and lower plasma levels of TFV.56,57 TAF does not interact with renal transporters organic anion transporter 1 or 3 and is unlikely to accumulate in renal proximal tubules, potentially supporting its improved renal safety profile (Figure 2).56 Clinical studies using TAF have shown smaller changes in creatinine clearance and less tubular proteinuria as compared to those receiving TDF with the same background regimen.57–59 Worth noting, the clinical use of DRV/c might be increased in the future with the current development of darunavir/cobicistat/FTC/TAF as a once daily combination single tablet regimen.58
Gastrointestinal tolerability including nausea, vomiting, and diarrhea were considered to be of mild severity and comparable between both ATV/c and ATV/r groups in the Phase III study.28 Incidence of diarrhea appeared to be higher in the ATV/r group versus ATV/c group after 48 weeks (20.4% and 15.4% respectively; P=0.093), as well as in the expanded 144-week study (27.6% and 22.4%).28,29 However, no patients discontinued treatment because of diarrhea. Treatment-related AEs such as diarrhea were slightly higher (27%) in patients treated with DRV/c after 48 weeks compared to those in the ATV/c clinical studies.30,31 Most common AEs related to treatment with DRV/c and leading to discontinuation through week 48 were rash and maculopapular rash, nausea, and hypersensitivity.30 For patients who experienced these AEs, they resolved upon discontinuation of the study drug. Given that DRV possesses a sulfonamide moiety, patients who start treatment with DRV/c should be counseled on the potential for developing a rash.
One strength of the ATV/c and DRV/c Phase III clinical studies is the heterogeneity of the overall populations, including diverse ethnicities and patients who had either HBV or HCV coinfection (unlike most other clinical studies evaluating HAART which exclude patients with these coinfections).28,31 Another important demographic in these clinical studies was that the mean age was considerably young (mid-30s). Given that the HIV-1 population is aging, especially, in first world countries, additional clinical studies are warranted to determine the efficacy and safety of the use of cobicistat as a pharmacoenhancer of ATV and DRV in elderly populations.
Adherence to HAART is critical for achieving positive treatment outcomes. Numerous barriers to adherence to HAART exist including AEs and complexity of regimens (eg, pill burden and frequent administration of dosing). One major advantage of cobicistat is its ability to be coformulated with other ARVs allowing for reduced pill burden through the development of once daily FDC regimens. Numerous studies have shown the benefits of using once daily HAART regimens including improved rates of adherence.60,61 Simplicity and convenience are important factors for ensuring that HIV patients have sustained and adequate adherence to lifelong HAART. Other advantages of cobicistat as an alternative pharmacoenhancer to ritonavir include fewer DDIs and lack of anti-HIV-1 activity.
Another major barrier to adherence to HAART is palatability of ARV liquid formulations, especially ritonavir and lopinavir/ritonavir solutions (although ritonavir seems worse) for use in treatment of HIV-infected children. One study compared the PK and bioequivalence of different pediatric dosage formulations of cobicistat to the reference adult tablet formulation.62 In this study, the pediatric formulations of cobicistat administered either as 50 mg immediate release tablets or as 20 mg dispersible tablets were each bioequivalent to the cobicistat adult tablet formulation. All pediatric formulations were also considered to be palatable, thus, supporting future evaluation of these formulations for the treatment of HIV-1-infected children. If successful, ATV/c and DRV/c might also become more commonly prescribed ARV treatments compared to lopinavir/ritonavir, especially in younger HIV-infected children who have problems with adhering to ARVs due to poor palatability of ARV liquid formulations.
Conclusion
In conclusion, clinical trials have shown that cobicistat has a comparable efficacy and safety profile to ritonavir as a pharmacoenhancer of both ATV and DRV. Since ATV/c and DRV/c are available as once daily fixed-dose combinations, they offer numerous advantages in addition to simplified, reduced pill burden, including fewer DDIs versus boosting of ATV and DRV with ritonavir. This is especially important in an aging HIV-1 population with increasingly prevalent comorbidities and potential for polypharmacy. Additional studies are warranted to determine the efficacy and safety of ATV/c and DRV/c in treatment-experienced patients, although DRV/c use seems to be an acceptable option in treatment-experienced patients who have no darunavir RAMs.
Disclosure
The authors report no conflicts of interest in this work.
References
Gleason LJ, Luque AE, Shah K. Polypharmacy in the HIV-infected older adult population. Clin Interv Aging. 2013;8:749–763. | |
Department of Health and Human Services. Guidelines for the use of antiretroviral agents in HIV-1 infected adults and adolescents; April 2015. Available from: https://aidsinfo.nih.gov/guidelines/html/1/adult-and-adolescent-treatment-guidelines/0. Accessed September 16, 2015. | |
Williams I, Churchill D, Anderson J, et al. British HIV Association guidelines for the treatment of HIV-1-positive adults with antiretroviral therapy 2015. HIV Med; 2015. Available from: http://www.bhiva.org/documents/Guidelines/Treatment/2015/2015-treatment-guidelines.pdf. Accessed October 21, 2015. | |
Arya V, Robertson SM, Struble KA, Murray JS. Scientific considerations for pharmacoenhancers in antiretroviral therapy. J Clin Pharmacol. 2012;52(8):1128–1133. | |
Josephson F. Drug-drug interactions in the treatment of HIV infection: focus on pharmacokinetic enhancement through CYP3A inhibition. J Intern Med. 2010;268(6):530–539. | |
Shah BM, Schafer JJ, Priano J, Squires KE. Cobicistat: a new boost for the treatment of human immunodeficiency virus infection. Pharmacotherapy. 2013;33(10):1107–1116. | |
Klein CE, Chiu YL, et al. The tablet formulation of lopinavir/ritonavir provides similar bioavailability to the soft-gelatin capsule formulation with less pharmacokinetic variability and diminished food effect. J Acquir Immune Defic Syndr. 2007;44(4):401–410. | |
Breitenbach J. Melt extrusion: from process to drug delivery technology. Eur J Pharm Biopharm. 2002;54(2):107–117. | |
Manzardo C, Gatell JM. Stribild ® (elvitegravir/cobicistat/emtricitabine/tenofovir disoproxil fumarate): a new paradigm for HIV-1 treatment. AIDS Rev. 2014;16(1):35–42. | |
Olin JL, Spooner LM, Klibanov OM. Elvitegravir/cobicistat/emtricitabine/tenofovir disoproxil fumarate single tablet for HIV-1 infection treatment. Ann Pharmacother. 2012;46(12):1671–1677. | |
Perry CM. Elvitegravir/cobicistat/emtricitabine/tenofovir disoproxil fumarate single-tablet regimen (Stribild(R)): a review of its use in the management of HIV-1 infection in adults. Drugs. 2014;74(1):75–97. | |
Raffe S, Fisher M. The pharmacokinetics, pharmacodynamics and clinical efficacy of elvitegravir + cobicistat + emtricitabine + tenofovir combination therapy for the treatment of HIV. Expert Opin Drug Metab Toxicol. 2015;11(3):427–435. | |
Tybost [package insert]. Foster City, CA: Gilead Sciences; 2014. | |
Xu L, Liu H, Murray BP, et al. Cobicistat (GS-9350): A potent and selective inhibitor of human CYP3A as a novel pharmacoenhancer. ACS Med Chem Lett. 2010;1:209–213. | |
Johnson EF, Connick JP, Reed JR, et al. Correlating structure and function of drug-metabolizing enzymes: progress and ongoing challenges. Drug Metab Dispos. 2014;42(1):9–22. | |
Norvir [package insert]. North Chicago, IL: AbbVie Inc.; 2015. | |
Deeks ED. Cobicistat: a review of its use as a pharmacokinetic enhancer of atazanavir and darunavir in patients with HIV-1 infection. Drugs. 2014;74(2):195–206. | |
Mathias AA, German P, Murray BP, et al. Pharmacokinetics and pharmacodynamics of GS-9350: a novel pharmacokinetic enhancer without anti-HIV activity. Clin Pharmacol Ther. 2010;87(3):322–329. | |
Kakuda TN, Van De Casteele T, Petrovic R, et al. Bioequivalence of a darunavir/cobicistat fixed-dose combination tablet versus single agents and food effect in healthy volunteers. Antivir Ther. 2014;19(6):597–606. | |
Lepist EI, Phan TK, Roy A, et al. Cobicistat boosts the intestinal absorption of transport substrates, including HIV protease inhibitors and GS-7340, in vitro. Antimicrob Agents Chemother. 2012;56(10):5409–5413. | |
Stray KM, Bam RA, Birkus G, et al. Evaluation of the effect of cobicistat on the in vitro renal transport and cytotoxicity potential of tenofovir. Antimicrob Agents Chemother. 2013;57(10):4982–4989. | |
HIV Drug Interactions. The University of Liverpool and eMedFusion. [Last reviewed October 7, 2015]. Available from: http://www.HIV-druginteractions.org. Accessed October 22, 2015. | |
Ramanathan S, Warren D, Wei L, Kearney BP. Pharmacokinetic boosting of atazanavir with the pharmcoenhancer FS-9350 versus ritonavir. In: 49th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC); September 12–15, 2009; San Francisco, CA. | |
Sevinsky H, Tao X, Wang R, et al. A randomized trial in healthy subjects to assess the bioequivalence of an atazanavir/cobicistat fixed-dose combination tablet versus administration as separate agents. Antivir Ther. 2015; 20(5):493–500. | |
Mathias A LH, Warren D, Sekar V, Kearney BP. Relative bioavailability and pharmacokinetics of darunavir when boosted with the pharmacoenhancer GS-9350 versus ritonavir. In: 11th International Workshop on Clinical Pharmacology of HIV Therapy; April 7–9, 2010; Sorrento, Italy. | |
Kakuda TN, Opsomer M, Timmers M, et al. Pharmacokinetics of darunavir in fixed-dose combination with cobicistat compared with coadministration of darunavir and ritonavir as single agents in healthy volunteers. J Clin Pharmacol. 2014;54(8):949–957. | |
Elion R, Cohen C, Gathe J, et al. Phase 2 study of cobicistat versus ritonavir each with once-daily atazanavir and fixed-dose emtricitabine/tenofovir df in the initial treatment of HIV infection. AIDS. 2011;25(15):1881–1886. | |
Gallant JE, Koenig E, Andrade-Villanueva J, et al. Cobicistat versus ritonavir as a pharmacoenhancer of atazanavir plus emtricitabine/tenofovir disoproxil fumarate in treatment-naive HIV type 1-infected patients: week 48 results. J Infect Dis. 2013;208(1):32–39. | |
Gallant JE, Koenig E, Andrade-Villanueva JF, et al. Brief report: cobicistat compared with ritonavir as a pharmacoenhancer for atazanavir in combination with emtricitabine/tenofovir disoproxil fumarate: week 144 results. J Acquir Immune Defic Syndr. 2015;69(3):338–340. | |
Tashima K, Crofoot G, Tomaka FL, et al. Cobicistat-boosted darunavir in HIV-1-infected adults: week 48 results of a Phase IIIb, open-label single-arm trial. AIDS Res Ther. 2014;11:39. | |
Tashima K, Crofoot G, Tomaka FL, et al. Phase IIIb, open-label single-arm trial of darunavir/cobicistat (DRV/COBI): Week 48 subgroup analysis of HIV-1-infected treatment-naive adults. J Int AIDS Soc. 2014; 17(4 Suppl 3):19772. | |
Albrecht H. Abacavir/3TC vs tenofovir/FTC: interim results from ACTG 5202. AIDS Clin Care. 2008;20(4):28. | |
Daar ES, Tierney C, Fischl MA, et al. Atazanavir plus ritonavir or efavirenz as part of a 3-drug regimen for initial treatment of HIV-1. Ann Intern Med. 2011;154(7):445–456. | |
Molina JM, Andrade-Villanueva J, Echevarria J, et al. Once-daily atazanavir/ritonavir versus twice-daily lopinavir/ritonavir, each in combination with tenofovir and emtricitabine, for management of antiretroviral-naive HIV-1-infected patients: 48 week efficacy and safety results of the CASTLE study. Lancet. 2008;372(9639):646–655. | |
Cahn P, Fourie J, Grinsztejn B, et al. Week 48 analysis of once-daily vs twice-daily darunavir/ritonavir in treatment-experienced HIV-1-infected patients. AIDS. 2011;25(7):929–939. | |
Ortiz R, Dejesus E, Khanlou H, et al. Efficacy and safety of once-daily darunavir/ritonavir versus lopinavir/ritonavir in treatment-naive HIV-1-infected patients at week 48. AIDS. 2008;22(12):1389–1397. | |
Lennox JL, Landovitz RJ, Ribaudo HJ, et al. Efficacy and tolerability of 3 nonnucleoside reverse transcriptase inhibitor-sparing antiretroviral regimens for treatment-naive volunteers infected with HIV-1: a randomized, controlled equivalence trial. Ann Intern Med. 2014;161(7):461–471. | |
Johnson DH, Venuto C, Ritchie MD, et al. Genomewide association study of atazanavir pharmacokinetics and hyperbilirubinemia in AIDS Clinical Trials Group protocol A5202. Pharmacogenet Genomics. 2014;24(4):195–203. | |
German P, Liu HC, Szwarcberg J, et al. Effect of cobicistat on glomerular filtration rate in subjects with normal and impaired renal function. J Acquir Immune Defic Syndr. 2012;61(1):32–40. | |
Krutzen E, Back SE, Nilsson-Ehle I, Nilsson-Ehle P. Plasma clearance of a new contrast agent, iohexol: a method for the assessment of glomerular filtration rate. J Lab Clin Med. 1984;104(6):955–961. | |
DeJesus E, Rockstroh JK, Henry K, et al. Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir disoproxil fumarate versus ritonavir-boosted atazanavir plus co-formulated emtricitabine and tenofovir disoproxil fumarate for initial treatment of HIV-1 infection: a randomised, double-blind, phase 3, non-inferiority trial. Lancet. 2012;379(9835):2429–2438. | |
Rockstroh JK, DeJesus E, Henry K, et al. A randomized, double-blind comparison of coformulated elvitegravir/cobicistat/emtricitabine/tenofovir DF vs ritonavir-boosted atazanavir plus coformulated emtricitabine and tenofovir DF for initial treatment of HIV-1 infection: analysis of week 96 results. J Acquir Immune Defic Syndr. 2013;62(5):483–486. | |
Wohl DA, Cohen C, Gallant JE, et al. A randomized, double-blind comparison of single-tablet regimen elvitegravir/cobicistat/emtricitabine/tenofovir DF versus single-tablet regimen efavirenz/emtricitabine/tenofovir DF for initial treatment of HIV-1 infection: analysis of week 144 results. J Acquir Immune Defic Syndr. 2014;65(3):e118–e120. | |
Lepist EI, Zhang X, Hao J, et al. Contribution of the organic anion transporter OAT2 to the renal active tubular secretion of creatinine and mechanism for serum creatinine elevations caused by cobicistat. Kidney Int. 2014;86(2):350–357. | |
Elion R, Cohen C, Ward D, et al. Evaluation of efficacy, safety, pharmacokinetics, and adherence in HIV-1-infected, antiretroviral-naive patients treated with ritonavir-boosted atazanavir plus fixed-dose tenofovir DF/emtricitabine given once daily. HIV Clin Trials. 2008;9(4):213–224. | |
Johnson MA, Gathe JC Jr, Podzamczer D, et al. A once-daily lopinavir/ritonavir-based regimen provides noninferior antiviral activity compared with a twice-daily regimen. J Acquir Immune Defic Syndr. 2006;43(2):153–160. | |
Mills AM, Nelson M, Jayaweera D, et al. Once-daily darunavir/ritonavir vs lopinavir/ritonavir in treatment-naive, HIV-1-infected patients: 96-week analysis. AIDS. 2009;23(13):1679–1688. | |
Smith KY, Patel P, Fine D, et al. Randomized, double-blind, placebo-matched, multicenter trial of abacavir/lamivudine or tenofovir/emtricitabine with lopinavir/ritonavir for initial HIV treatment. AIDS. 2009;23(12):1547–1556. | |
Smith KY, Weinberg WG, Dejesus E, et al. Fosamprenavir or atazanavir once daily boosted with ritonavir 100 mg, plus tenofovir/emtricitabine, for the initial treatment of HIV infection: 48-week results of ALERT. AIDS Res Ther. 2008;5:5. | |
Soriano V, Arasteh K, Migrone H, et al. Nevirapine versus atazanavir/ritonavir, each combined with tenofovir disoproxil fumarate/emtricitabine, in antiretroviral-naive HIV-1 patients: the ARTEN Trial. Antivir Ther. 2011;16(3):339–348. | |
Walmsley S, Avihingsanon A, Slim J, et al. Gemini: a noninferiority study of saquinavir/ritonavir versus lopinavir/ritonavir as initial HIV-1 therapy in adults. J Acquir Immune Defic Syndr. 2009;50(4):367–374. | |
McDonald CK, Martorell C, Ramgopal M, et al. Cobicistat-boosted protease inhibitors in HIV-infected patients with mild to moderate renal impairment. HIV Clin Trials. 2014;15(6):269–273. | |
Post FA, Winston J, Andrade-Villanueva JF, et al. Elvitegravir/cobicistat/emtricitabine/tenofovir DF in HIV-infected patients with mild-to-moderate renal impairment. J Acquir Immune Defic Syndr. 2015;68(3):310–313. | |
Prezcobix [package insert]. Titusville, NJ: Janssen Therapeutics; 2015. | |
Evotaz [package insert]. Princeton, NJ: Bristol-Myers Squibb Company; 2015. | |
Bam RA, Yant SR, Cihlar T. Tenofovir alafenamide is not a substrate for renal organic anion transporters (OATs) and does not exhibit OAT-dependent cytotoxicity. Antivir Ther. 2014;19(7):687–692. | |
Sax PE, Zolopa A, Brar I, et al. Tenofovir alafenamide vs tenofovir disoproxil fumarate in single tablet regimens for initial HIV-1 therapy: a randomized phase 2 study. J Acquir Immune Defic Syndr. 2014;67(1):52–58. | |
Mills A, Crofoot G Jr, McDonald C, et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate in the first protease inhibitor-based single-tablet regimen for initial HIV-1 therapy: a randomized phase 2 study. J Acquir Immune Defic Syndr. 2015;69(4):439–445. | |
Sax PE, Wohl D, Yin MT, et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate, coformulated with elvitegravir, cobicistat, and emtricitabine, for initial treatment of HIV-1 infection: two randomised, double-blind, phase 3, non-inferiority trials. Lancet. 2015;385(9987):2606–2615. | |
Molina JM. Efficacy and safety of once-daily regimens in the treatment of HIV infection. Drugs. 2008;68(5):567–578. | |
Sweet D, Song J, Zhong Y, Signorovitch J. Real-world medication persistence with single versus multiple tablet regimens for HIV-1 treatment. J Int AIDS Soc. 2014;17(4 Suppl 3):19537. | |
Custodio JM, Liu Y, Graham H, et al. Bioequivalence of two pediatric formulations vs adult tablet formulation of cobicistat. In: 21st conference on retroviruses and opportunistic infections (CROI); March 3–6, 2014; Boston, MA. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.